
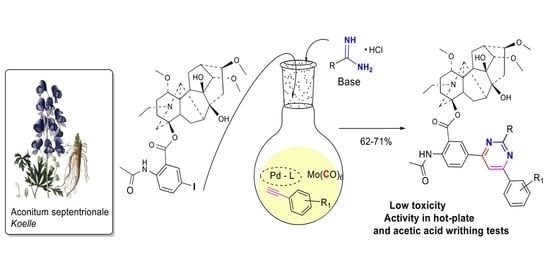
Hybrides of Alkaloid Lappaconitine with Pyrimidine Motif on the Anthranilic Acid Moiety: Design, Synthesis, and Investigation of Antinociceptive Potency

 , ,
, ,
Abstract
:
1. Introduction
2. Results and Discussion
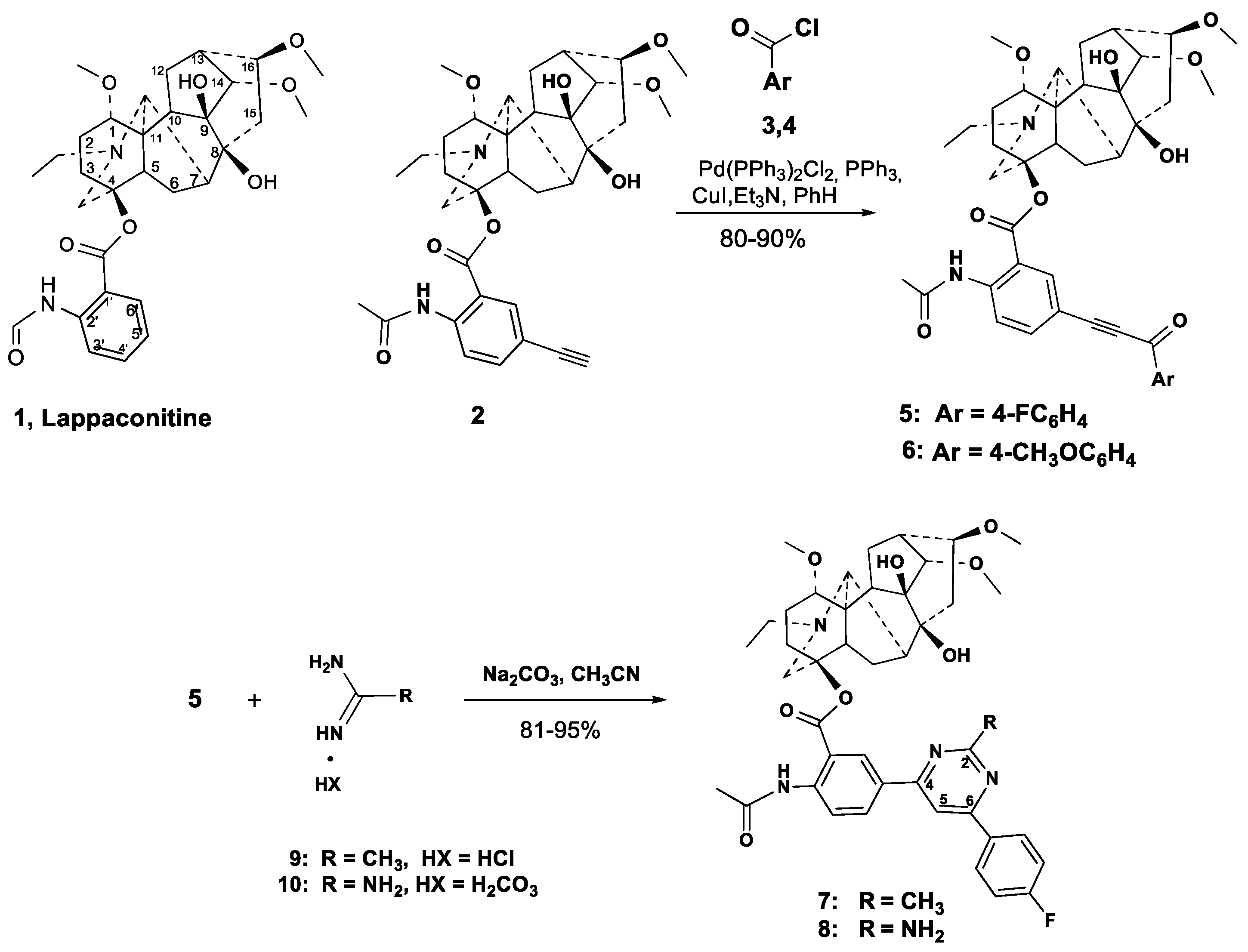
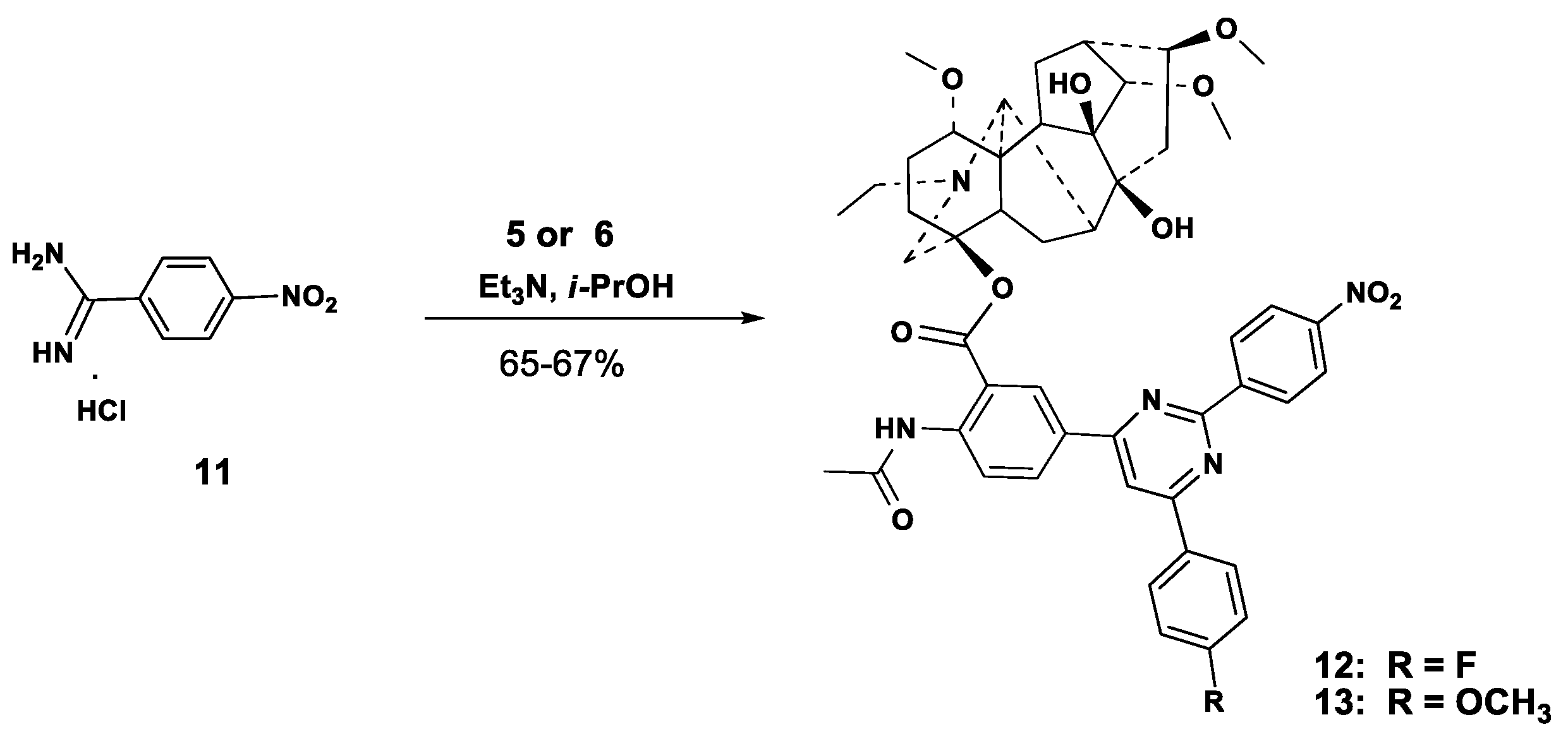
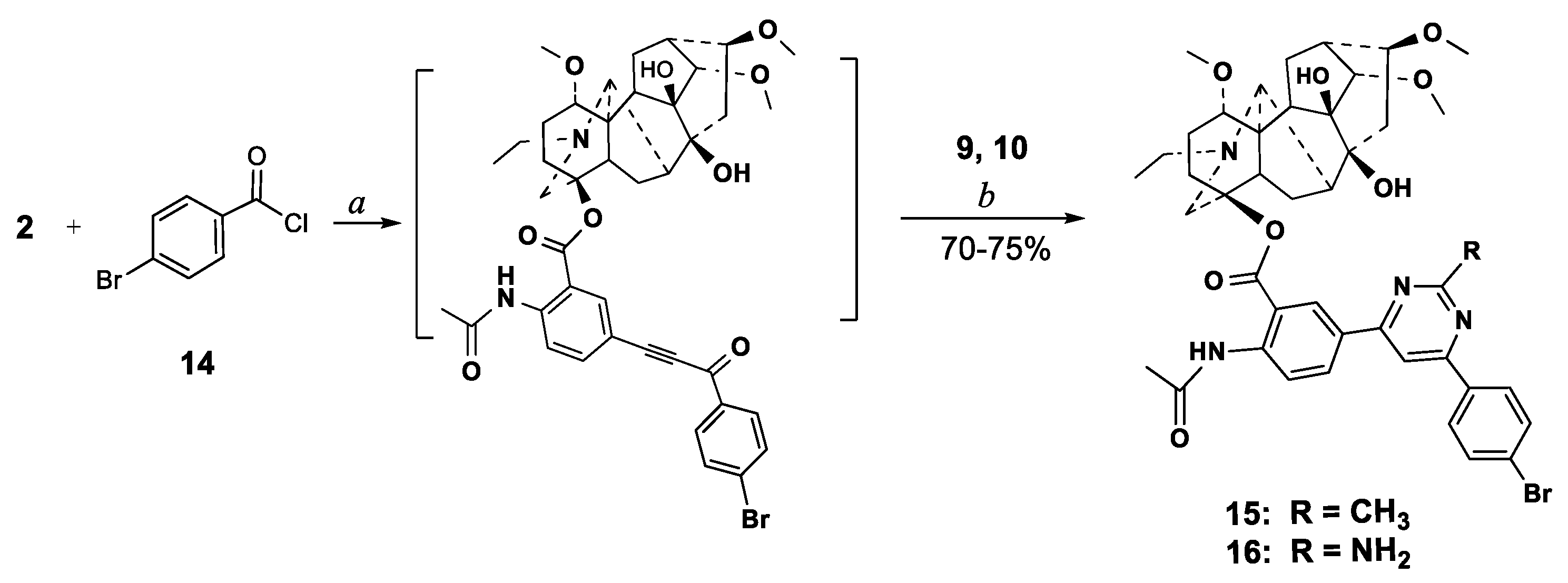
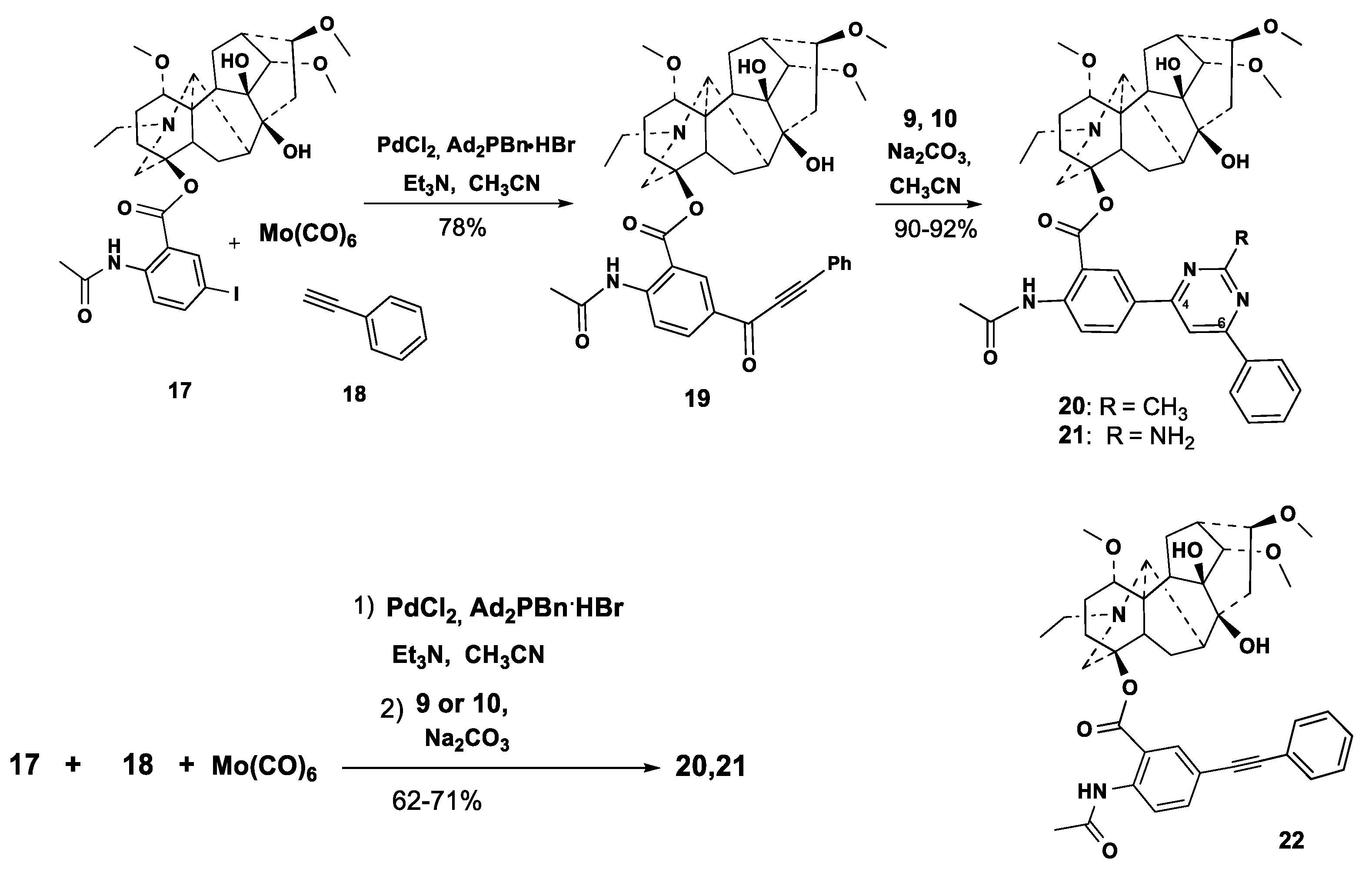
2.1. Chemical Synthesis
2.2. Biological Study
2.2.1. Analgesic Activity
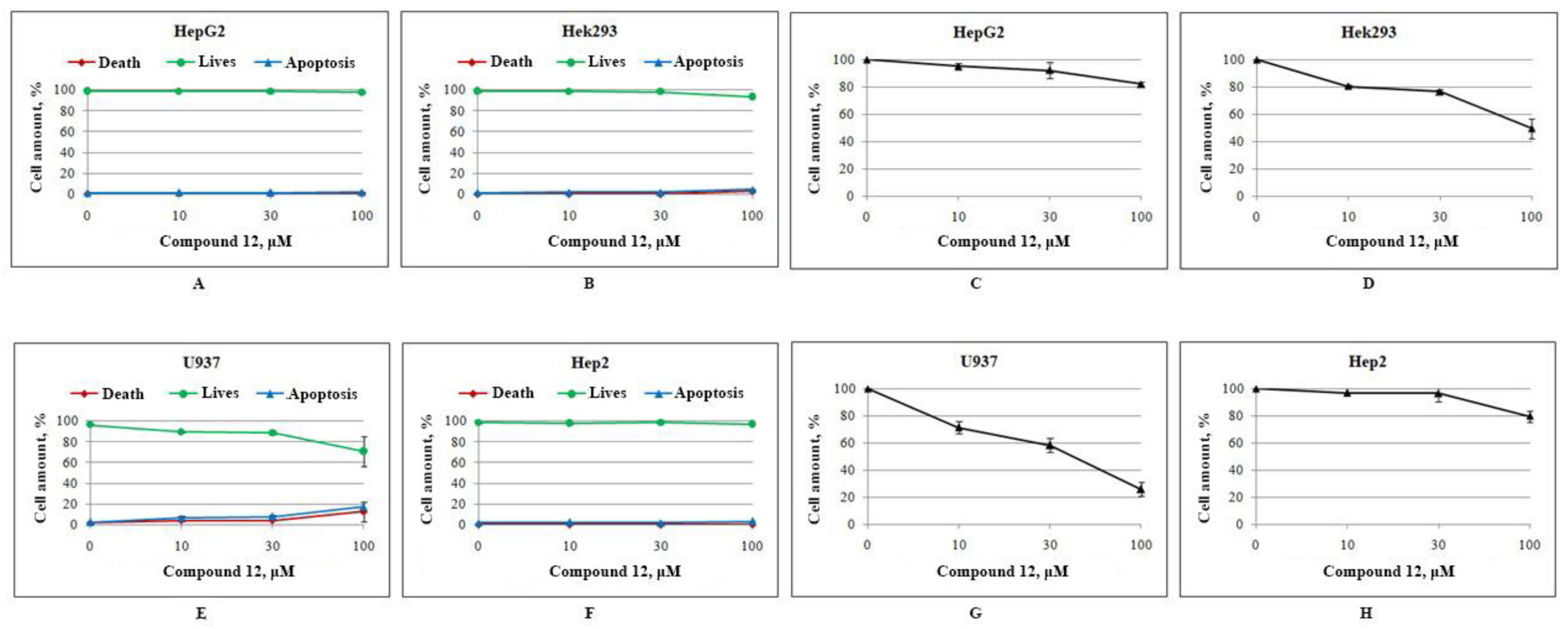
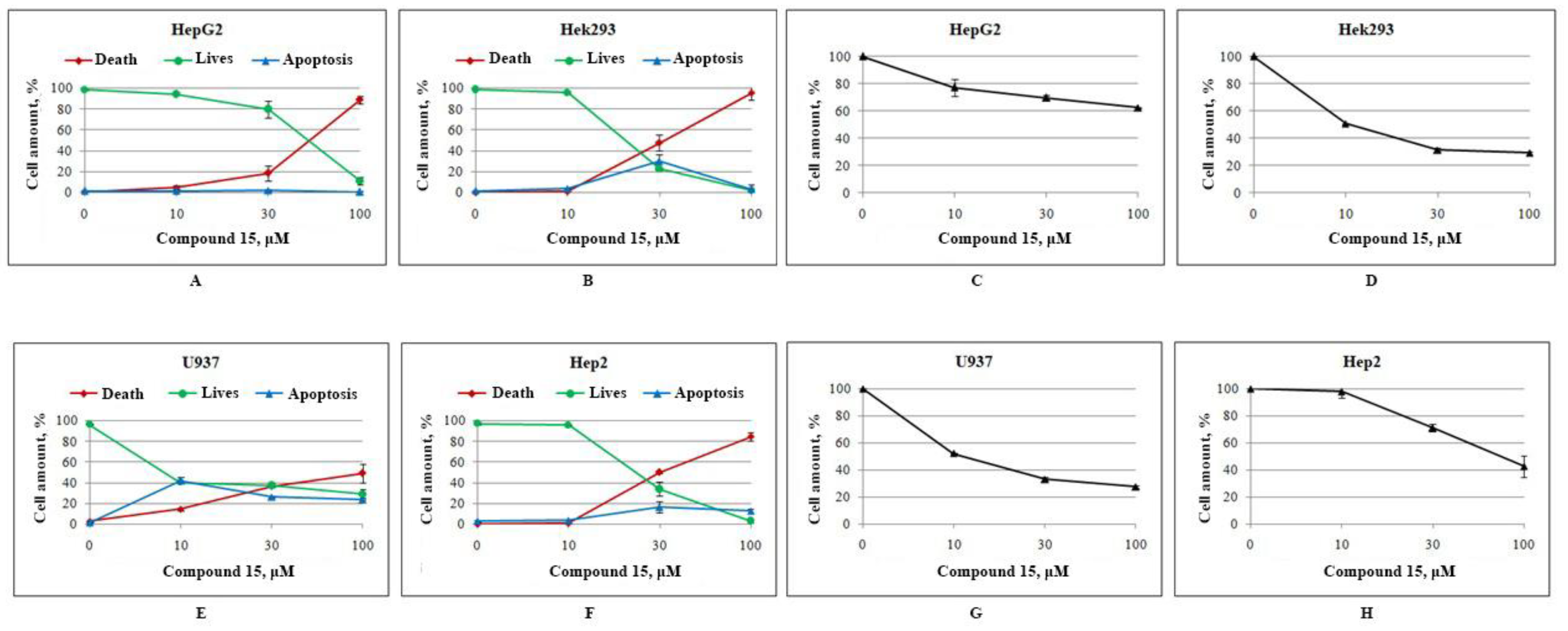
2.2.2. Toxicity and Cytotoxicity Studies
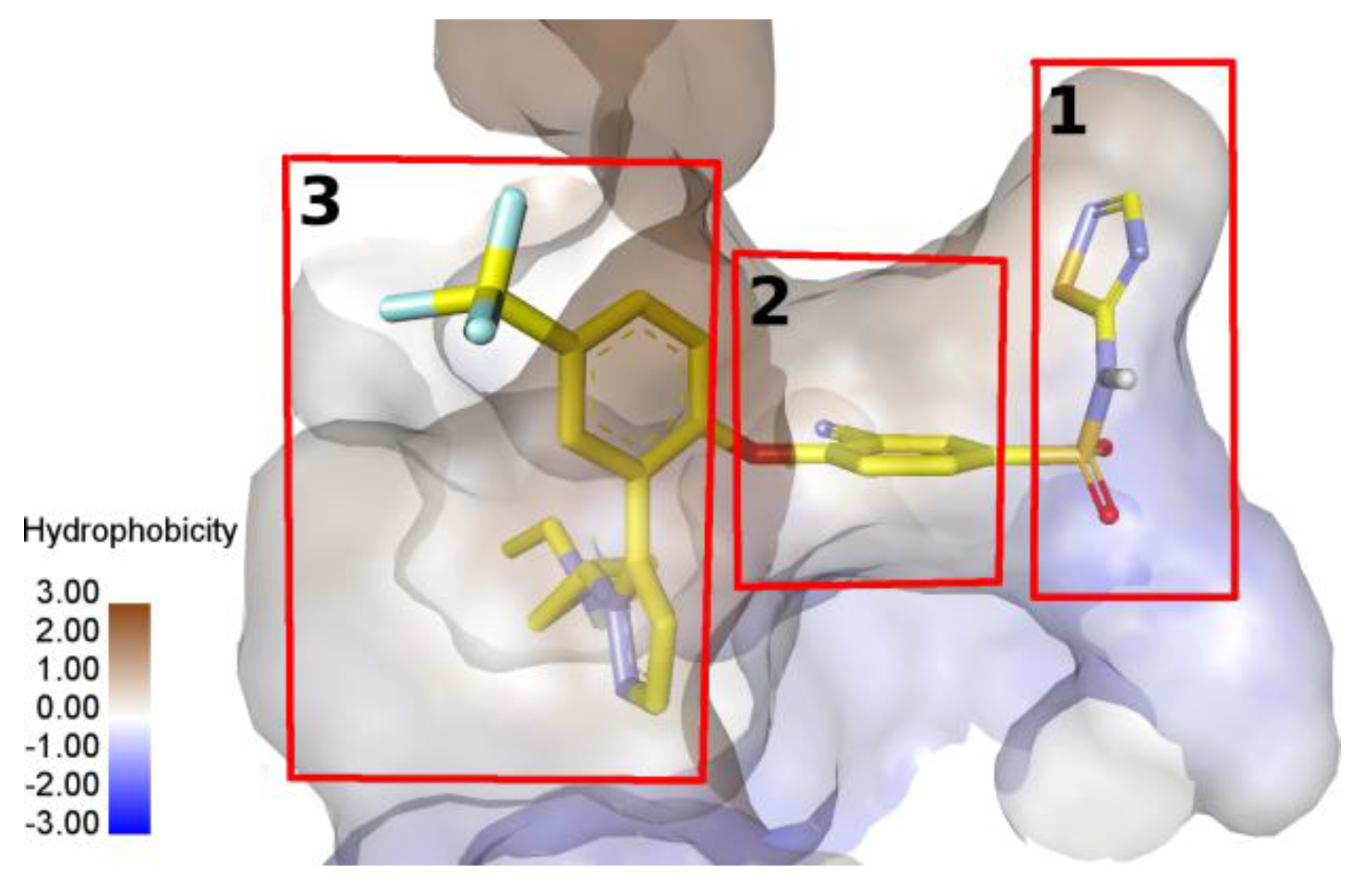
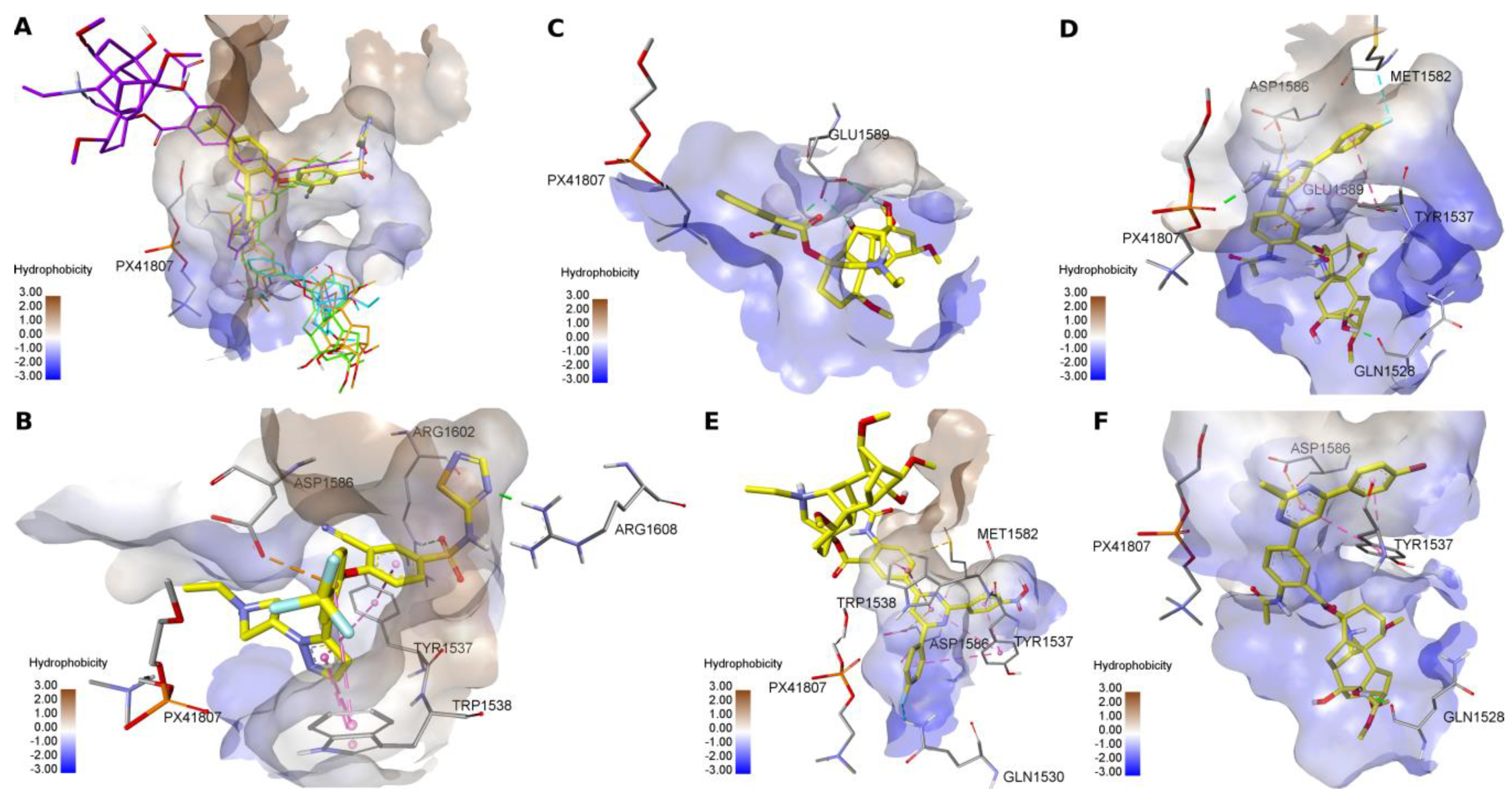
2.3. Molecular Modeling of a Possible Mechanism of Antagonistic Effect of Lappaconitine 1 and New Lappaconitine Derivatives 10, 12, 15, on the Voltage-Gated Sodium Channel 1.7
3. Experimental Section
3.1. General Information
3.2. Syntheses and Spectral Data
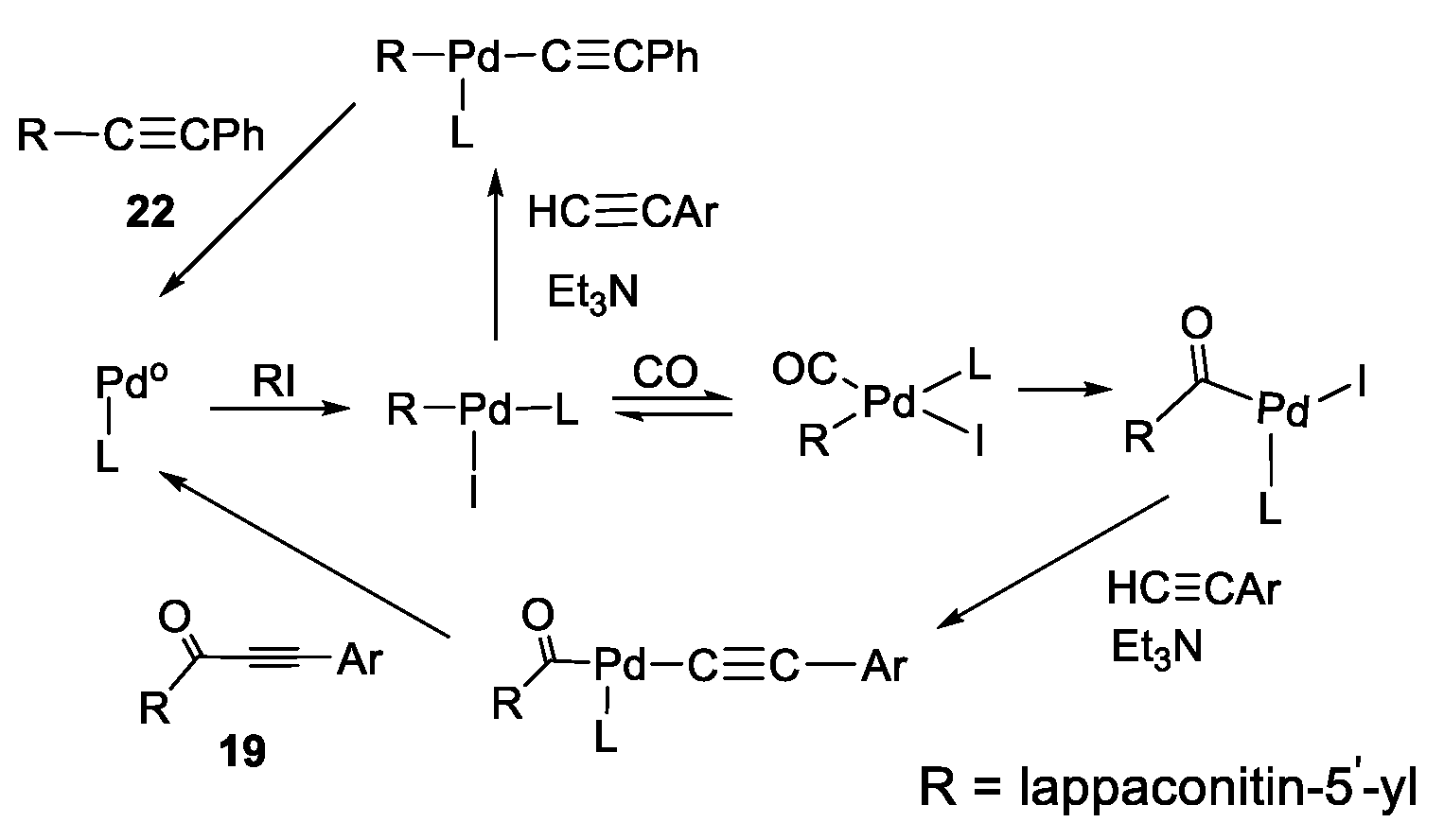
3.2.1. Carbonylative Sonogashira Reaction
3.2.2. Procedures for Cross-Coupling Reactions
- (a)
- A mixture of alkynyl ketone 5 or 6 (0.3 mmol), 4-nitrobenzamidine hydrochloride 11 (72.5 mg, 0.36 mmol) and Et3N (90.9 mg, 0.9 mmol) was reflux under stirring in i-PrOH (8 mL) 8 h (TLC). After cooling (about 10 h), the precipitate was filtered, washed with CHCl3 (10 mL), the combined organic layer was concentrated under reduced pressure, and the residue was subjected to column chromatography (chloroform-EtOH). Fraction with Rf 0.4 (chloroform-EtOH, 20:1), was evaporated and dried in vacuo to give compounds 12, 13 as a yellow powders.
- (b)
- One-pot cross-coupling/cyclocondensation. To a stirred mixture of Pd(PPh3)2Cl2 (4.3 mg, 0.005 mmol), PPh3 (2.1 mg, 0.007 mmol), CuI (2.5 mg, 0.01 mmol) in benzene (5 mL) under argon flow was added Et3N (206 mg, 0.27 mL, 2 mmol) followed by freshly prepared 4-bromobenzoyl chloride 14 (132 mg, 0.6 mmol) in benzene (2 mL). The 5′-Ethynyllappaconitine 2 (304 mg, 0.5 mmol) in benzene (5 mL) was added dropwise during one hour. The reaction mixture was heated to 65 °C under stirring for 7 h (TLC-control). After removing the solvent in vacuo under argon, the crude material was dissolved in acetonitrile (7 mL), and acetamidine hydrochloride 9 (67 mg, 0.7 mmol) and anhydrous Na2CO3 (170 mg, 1.6 mmol), or guanidine hydrocarbonate 10 (41 mg, 0.7 mmol) and Et3N (162 mg, 1.6 mmol) were added. The reaction mixture was stirred under reflux for 8 h. After cooling (about 8 h) the precipitate was filtered, washed with CHCl3 (10 mL), the combined organic layer was washed with water (2 × 5 mL), dried over magnesium sulfate and filtered. The solvent was removed under reduced pressure, and the residue was subjected to column chromatography (chloroform-EtOH). The fraction with Rf 0.4 (chloroform-EtOH, 20:1), was collected evaporated and dried in vacuo to give compounds 15 or 16 as a yellow powders.
- (c)
- One-pot carbonylative cross-coupling/cyclocondensation. To solution of Et3N (101 mg, 1 mmol) in CH3CN (7 mL) PdCl2 (2 mg, 0.011 mmol), and Ad2PBn·HBr (7 mg, 0.017 mmol) were successively added under a stream of argon with stirring. The mixture was heated to 65 °C (bath) and Mo(CO)6 (158 mg, 0.59 mmol), phenylacetylene 18 (60 mg, 0.59 mmol) and 5′-iodolappaconitine 17 (236 mg, 0.33 mmol) were added. The reaction mixture was heated until 2 h at 65 °C, treated with amidinium salt 9 (0.46 mmol) and anhydrous Na2CO3 (106 mg, 1 mmol), or guanidine carbonate 10 (0.46 mmol) and Et3N (101 mg, 1 mmol) and stirred under reflux for 8 h. After completion based on TLC, the cooled mixture was filtered over Celite, the precipitate washed with CHCl3 (15 mL), the combined solvent was removed under reduced pressure, and the residue was subjected to column chromatography (chloroform-EtOH, 80:1). Fraction with Rf 0.4 (chloroform-EtOH, 20:1) was evaporated and dried in vacuo to give compounds 20, 21 as yellow powders.
3.2.3. 4β-{2′-Acetylamino-5′-[11′-(4-fluorophenyl)-9′-(4-nitrophenyl)-pyrimidine-7′-yl]benzoate}-1α,14α,16β-trimethoxy-20-ethylaconitane-8,9-diol (12)
3.2.4. 4β-{2′-Acetylamino-5′-[11′-(4-methoxyphenyl)-9′-(4-nitrophenyl)-pyrimidine-7′-yl]benzoate}-1α,14α,16β-trimethoxy-20-ethylaconitane-8,9-diol (13)
3.2.5. 4β-{2′-Acetylamino-5′-[11′-(4-bromophenyl)-9′-(methyl)pyrimidine-7′-yl]benzoate}-1α,14α,16β-trimethoxy-20-ethylaconitane-8,9-diol (15)
3.2.6. 4β-{2′-Acetylamino-5′-[9′-(amino)-11′-(4-bromophenyl)pyrimidine-7′-yl]benzoate}-1α,14α,16β-trimethoxy-20-ethylaconitane-8,9-diol (16)
3.2.7. 4β-{2′-Acetylamino-5′-[9′-(methyl)-11′-(phenyl)pyrimidine-7′-yl]benzoate}-1α,14α,16β-trimethoxy-20-ethylaconitane-8,9-diol (20)
3.2.8. 4β-{2′-Acetylamino-5′-[9′-(amino)-11′-(phenyl)pyrimidine-7′-yl]benzoate}-1α,14α,16β-trimethoxy-20-ethylaconitane-8,9-diol (21)
3.3. Biological Evaluation
3.3.1. Pharmacology
Animals
Analgesic Tests
3.4. Cell Culture and Determination of Cytotoxicity
3.5. Molecular Docking Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ameri, A. The effects of Aconitum alkaloids on the central nervous system. Progr. Neurobiol. 1998, 56, 211–223. [Google Scholar] [CrossRef]
- Ono, M.; Satoh, T. Pharmacological studies of lappaconitine. Analgesic activities. Arzneimittelforschung 1988, 38, 892–895. [Google Scholar] [PubMed]
- Tang, X.C.; Zhu, M.Y.; Feng, J.; Wang, Y.E. The pharmacological action of Lappaconitine hydrobromide. Acta Pharm. Sin. 1983, 18, 579–584. [Google Scholar]
- Guo, X.; Tang, X.C. Lappaconitine and N-deacetyllappaconitine potentiate footshock-induced analgesia in rats. Life Sci. 1991, 48, 1365–1370. [Google Scholar] [CrossRef]
- Wang, Y.Z.; Xiao, Y.Q.; Zhang, C.; Sun, X.M. Study of analgesic and anti-inflammatory effects of lappaconitine gelata. J. Tradit. Chin. Med. 2009, 29, 141–145. [Google Scholar] [CrossRef]
- Pereira, F. Polypharmacology of Aconitum and Delphinium sp. Diterpene alkaloids: Antiarrhythmic, analgesic and anti-inflammatory effects. Mini Rev. Org. Chem. 2017, 14, 304–310. [Google Scholar] [CrossRef]
- Zheng, W.S.; Sheng, Y.X.; Zhang, Y.J.; Fang, X.Q.; Wang, L.L. Pharmacokinetic study of lappaconitine hydrobromide transfersomes in rats by LC-MS. Pharmaceut. Anal. Acta 2011, 4, 2153–2435. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.C.; Ge, C.T.; Wang, P.; Zhang, J.L.; Yu, Y.Y.; Fu, C.Y. Analgesic effects of lappaconitine in leukemia bone pain in a mouse model. Peer J. 2015, 3, e936. [Google Scholar] [CrossRef] [Green Version]
- Ono, M.; Satoh, T. Pharmacological studies of lappaconitine. Occurrence of analgesic effect without opioid receptor. Res. Commun. Chem. Pathol. Pharmacol. 1989, 63, 13–25. [Google Scholar]
- Zhang, L.; Liu, X.; Wang, Y.; Wu, J. Clinical observation of Oxy Contin combined with Lappaconitine for pain on moderate and severe chronic cancer pain. J. Mod. Oncol. 2016, 24, 2961–2964. [Google Scholar]
- Guo, X.; Tang, X.C. Effects of reserpine and 5-HT on analgesia induced by lappaconitine and N-deacetyllappaconitine. Acta Pharmacol. Sin. 1990, 11, 14–17. [Google Scholar]
- Ono, M.; Satoh, T. Pharmacological studies on lappaconitine: Possible interaction with endogenous noradrenergic and serotonergic pathways to induce antinociception. Jpn. J. Pharmacol. 1992, 58, 251–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ou, S.; Zhao, Y.D.; Xiao, Z.; Wen, H.Z.; Cui, J.; Ruan, H.Z. Effect of lappaconitine on neuropathic pain mediated by P2X3 receptor in rat dorsal root ganglion. Neurochem. Int. 2011, 58, 564–573. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.-L.; Ao, J.-P.; Wang, Y.-R.; Huang, Q.; Li, T.-F.; Li, X.-Y.; Wang, Y.-X. Lappaconitine, a C18-diterpenoid alkaloid, exhibits antihypersensitivity in chronic pain through stimulation of spinal dynorphin A expression. Psychopharmacology 2018, 235, 2559–2571. [Google Scholar] [CrossRef] [PubMed]
- Friese, J.; Gleitz, J.; Gutser, U.T.; Heubach, J.F.; Mathiesen, T.; Wilffert, B.; Selve, N. Aconitum sp. alkaloids: The modulation of voltage-dependent Na+ channels, toxicity and antinociceptive properties. Eur. J. Pharmacol. 1997, 337, 165–174. [Google Scholar] [CrossRef]
- Valeev, A.E.; Verkhratskii, A.N.; Dzhakhangirov, F.N. Effects of allapinin on sodium currents in isolated neurons of the trigeminal ganglia and cardiomyocytes of rats. Bull. Eksp. Biol. Med. 1991, 111, 388–390. [Google Scholar]
- Seitz, U.; Ameri, A. Different effects on [3H]-noradrenaline uptake of the Aconitum alkaloids aconitine, 3-acetylaconitine, lappaconitine, and N-desacetyllappaconitine in rat hippocampus. Biochem. Pharmacol. 1998, 55, 883–888. [Google Scholar] [CrossRef]
- Catterall, W.A. Voltage-gated sodium channels at 60: Structure, function and pathophysiology. J. Physiol. 2012, 590, 2577–2589. [Google Scholar] [CrossRef]
- Li, Y.F.; Zheng, Y.M.; Yu, Y.; Gan, Y.; Gao, Z.B. Inhibitory effects of lappaconitine on the neuronal isoforms of voltagegated sodium channels. Acta Pharmacol. Sin. 2019, 40, 451–459. [Google Scholar] [CrossRef]
- Mueller, A.; Dekan, Z.; Kaas, Q.; Agwa, A.J.; Starobova, H.; Alewood, P.F.; Schroeder, C.I.; Mobli, M.; Deuis, J.R.; Vetter, I. Mapping the Molecular Surface of the Analgesic NaV1.7-Selective Peptide Pn3a Reveals Residues Essential for Membrane and Channel Interactions. ACS Pharmacol. Transl. Sci. 2020, 3, 535–546. [Google Scholar] [CrossRef] [Green Version]
- Guo, T.; Zhang, Y.; Zhao, J.; Zhu, C.; Feng, N. Nanostructured lipid carriers for percutaneous administration of alkaloids isolated from Aconitum sinomontanum. J. Nanobiotechnol. 2015, 13, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, F.; Wang, H.; Shu, H.; Li, J.; Jiang, J.; Chang, J.; Hsieh, Y.J. Separation and characterization of the metabolic products of lappaconitine in rat urine by high-performance liquid chromatography. Chromatogr. B Biomed. Sci. Appl. 1990, 526, 109–118. [Google Scholar] [CrossRef]
- Xie, F.; Wang, H.C.; Li, J.H.; Shu, H.L.; Jiang, J.R.; Chang, J.P.; Hsieh, Y.Y. Studies on the metabolism of lappaconitine in humans. Identification of the metabolites. Biomed. Chromatogr. 1990, 4, 43–48. [Google Scholar] [CrossRef]
- Yang, S.; Zhang, H.; Beier, R.C.; Sun, F.; Cao, H.; Zhen, J.; Wang, Z.; Zhang, S. Comparative metabolism of Lappaconitine in rat and human livermicrosomes and in vivo of rat using ultra high-performance liquidchromatography–quadrupole/time-of-flight mass spectrometry. J. Pharm. Biomed. Anal. 2015, 110, 1–11. [Google Scholar] [CrossRef]
- Yunusov, M.S. Diterpenoid alkaloids. Nat. Prod. Rep. 1993, 10, 471–486. [Google Scholar] [CrossRef]
- Bryzgalov, A.O.; Tolstikova, T.G.; Shults, E.E.; Petrova, K.O. Natural products as a source of antiarrhythmic drugs. Mini Rev. Med. Chem. 2018, 18, 345–362. [Google Scholar] [CrossRef] [PubMed]
- Joshi, V.D.; Kshirsagar, M.D.; Singhal, S. Synthesis and pharmacological study of some novel pyrimidines. Der Pharm. Sin. 2012, 3, 343–348. [Google Scholar]
- Lan, Y.; Chen, Y.; Cao, X.; Zhang, J.; Wang, J.; Xu, X.; Qiu, Y.; Zhang, T.; Liu, X.; Liu, B.-F.; et al. Synthesis and biological evaluation of novel sigma-1 receptor antagonists based on pyrimidine scaffold as agents for treating neuropathic pain. J. Med. Chem. 2014, 57, 10404–10423. [Google Scholar] [CrossRef]
- Andhale, G.S.; Giles, D.; Gurubasavrajswamy, P.M.; Rishikesh, V.A. Design, synthesis and pharmacological evaluation of pyrimidine fused indane-1,3-dione derivatives. Pharma. Chem. 2017, 9, 145–151. [Google Scholar]
- Patel, S.; Modi, P.; Ranjan, V.; Chhabria, M. Structure-based design, synthesis and evaluation of 2,4-diaminopyrimidine derivatives as novel caspase-1 inhibitors. Bioorg. Chem. 2018, 78, 258–268. [Google Scholar] [CrossRef]
- Farag, A.K.; Elkamhawy, A.; Londhe, A.M.; Lee, K.-T.; Pae, A.N.; Roh, E.J. Novel LCK/FMS inhibitors based on phenoxypyrimidine scaffold as potential treatment for inflammatory disorders. Eur. J. Med. Chem. 2017, 141, 657–675. [Google Scholar] [CrossRef] [PubMed]
- Shipe, W.D.; Sharik, S.S.; Barrow, J.C.; McGaughey, G.D.; Theberge, C.R.; Uslaner, J.M.; Yan, Y.; Renger, J.J.; Smith, S.M.; Coleman, P.J.; et al. Discovery and Optimization of a series of pyrimidine-based phosphodiesterase 10A (PDE10A) inhibitors through fragment screening, structure-based design, and parallel synthesis. J. Med. Chem. 2015, 58, 7888–7894. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, M.; Lecca, D.; Abbracchio, M.P.; Ceruti, S. Pathophysiological role of purines and pyrimidines in neurodevelopment: Unveiling new pharmacological approaches to congenital brain diseases. Front. Pharmacol. 2017, 8, 941. [Google Scholar] [CrossRef] [PubMed]
- Alachouzos, G.; Lenselink, E.B.; Mulder-Krieger, T.; de Vries, H.; IJzerman, A.P.; Louvel, J. Synthesis and evaluation of N-substituted 2-amino-4,5-diarylpyrimidines as selective adenosine A1 receptor antagonists. Eur. J. Med. Chem. 2017, 125, 586–602. [Google Scholar] [CrossRef] [PubMed]
- Janse van Rensburg, H.D.; Terre’Blanche, G.; van der Walt, M.M.; Legoabe, L.J. 5-Substituted 2-benzylidene-1-tetralone analogues as A 1 and/or A 2A antagonists for the potential treatment of neurological conditions. Bioorg. Chem. 2017, 74, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Robinson, S.J.; Petzer, J.P.; Terre’Blanche, G.; Petzer, A.; van der Walt, M.M.; Bergh, J.J.; Lourens, A.C.U. 2-Aminopyrimidines as dual adenosine A1/A2A antagonists. Eur. J. Med. Chem. 2015, 104, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.-M.; Ma, X.; Paira, P.; Tan, A.; Herr, D.R.; Lim, K.L.; Ng, C.H.; Venkatesan, G.; Klotz, K.-N.; Federico, S.; et al. Discovery of indolylpiperazinylpyrimidines with dual-target profiles at adenosine A2A and dopamine D2 receptors for Parkinson’s disease treatment. PLoS ONE 2018, 13, e0188212. [Google Scholar] [CrossRef] [Green Version]
- Cheremnykh, K.P.; Savel’ev, V.; Shkurko, O.P.; Shults, E.E. Synthesis of hybrid molecules containing pyrimidine and diterpene alkaloid lappaconitine fragments. Chem. Heterocycl. Compd. 2018, 54, 1131–1138. [Google Scholar] [CrossRef]
- Osadchii, S.A.; Shul’ts, E.E.; Polukhina, E.V.; Vasil’ev, V.G.; Tolstikov, G.A. Study of alkaloids of the flora of Siberia and Altai: Synthesis of bivalent ligands of the aconitane type. Dokl. Chem. 2007, 416, 251–256. [Google Scholar] [CrossRef]
- Levi, L.; Müller, T.J.J. Multicomponent syntheses of functional chromophores. Chem. Soc. Rev. 2016, 45, 2825–2846. [Google Scholar] [CrossRef] [Green Version]
- Willy, B.; Müller, T.J.J. Consecutive multi-component syntheses of heterocycles via palladium-copper catalyzed generation of alkynones. Arkivoc 2008, 1, 195–208. [Google Scholar] [CrossRef] [Green Version]
- Albano, G.; Aronica, L.A. Acyl Sonogashira Cross-Coupling: State of the art and application to the synthesis of heterocyclic compounds. Catalysts 2020, 10, 25. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, T.; Tanaka, M. Carbonylation of organic halides in the presence of terminal acetylenes; novel acetylenic ketone synthesis. JCS Chem. Commun. 1981, 7, 333–334. [Google Scholar] [CrossRef]
- Wu, X.-F.; Neumann, H.; Beller, M. A General and convenient palladium-catalyzed carbonylative Sonogashira coupling of aryl bromides. Chem. Eur. J. 2010, 16, 12104–12107. [Google Scholar] [CrossRef]
- Vasilevskii, S.F.; Osadchii, S.A.; Shults, E.E.; Polukhina, E.V.; Stepanov, A.A.; Tolstikov, G.A. Synthesis of acetylene derivatives of lappaconitine. Dokl. Chem. 2007, 415, 181–185. [Google Scholar] [CrossRef]
- Åkerbladh, L.; Odell, L.R.; Larhed, M. Palladium-Catalyzed molybdenum hexacarbonyl-mediated gas-free carbonylative reactions. Synlett 2019, 30, 141–155. [Google Scholar]
- Köllhofer, A.; Pullmann, T.; Plenio, H. A versatile catalyst for the Sonogashira coupling of aryl chlorides. Angew. Chem. Int. Ed. 2003, 42, 1056–1058. [Google Scholar] [CrossRef] [PubMed]
- Nordmann, J.; Breuer, N.; Müller, T.J.J. Efficient consecutive four-component synthesis of 5-acylpyrid-2-ones initiated by copper-free alkynylation. Eur. J. Org. Chem. 2013, 20, 4303–4310. [Google Scholar] [CrossRef]
- Cheremnykh, K.P.; Savelyev, V.A.; Pokrovskii, M.A.; Baev, D.S.; Tolstikova, T.G.; Pokrovskii, A.G.; Shults, E.E. Design, synthesis, cytotoxicity, and molecular modeling study of 2,4,6-trisubstituted pyrimidines with anthranilate ester moiety. Med. Chem. Res. 2019, 28, 545–558. [Google Scholar] [CrossRef]
- Larhed, M.; Hallberg, A. Handbook of Organopalladium Chemistry for Organic Synthesis; John Wiley and Sons Inc.: New York, NY, USA, 2002; p. 1133. [Google Scholar]
- Koster, R.; Anderson, M.; De Beer, E.J. Acetic acid for analgesic screening. Fed. Proc. 1959, 18, 412–415. [Google Scholar]
- Alexandrou, A.J.; Brown, A.R.; Chapman, M.L.; Estacion, M.; Turner, J.; Mis, M.A.; Wilbrey, A.; Payne, E.C.; Gutteridge, A.; Cox, P.J.; et al. Subtype-selective small molecule inhibitors reveal a fundamental role for Nav1.7 in nociceptor electrogenesis, axonal conduction and presynaptic release. PLoS ONE 2016, 11, e0152405. [Google Scholar] [CrossRef] [PubMed]
- Pankrushina, N.A.; Nikitina, I.A.; Anferova, N.V.; Osadchii, S.A.; Shakirov, M.M.; Shults, E.E.; Tolstikov, G.A. Study of alkaloids of the Siberian and Altai flora. 10. Synthesis of N(20)-deethyllappaconitine derivatives. Russ. Chem. Bull. Int. Ed. 2003, 52, 2490–2499. [Google Scholar] [CrossRef]
- Goerlich, J.; Schmutzler, R. Organophosphorus compounds with tertiary alkyl substituents. VI: A convenient method for the preparation of di-1-adamantylphosphine and di-1-adamantylchlorophosphine. Phosphorus Sulfur Silicon 1995, 102, 211–215. [Google Scholar] [CrossRef]
- Tewari, A.; Hein, M.; Zarf, A. General synthesis and catalytic applications of di(1-adamantyl)alkylphosphines and their phosphonium salts. Synthesis 2004, 6, 935–941. [Google Scholar] [CrossRef]
- Eddy, N.B.; Leimbach, D. Synthetic analgesics. II. Dithienylbutenyl- and dithienylbutylamines. J. Pharmacol. Exp. Ther. 1953, 107, 385–393. [Google Scholar]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge structural database. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mat. 2016, 72, 171–179. [Google Scholar] [CrossRef]
- Taylor, R.; Cole, J.; Korb, O.; McCabe, P. Knowledge-based libraries for predicting the geometric preferences of druglike molecules. J. Chem. Inform. Model. 2014, 54, 2500–2514. [Google Scholar] [CrossRef]
- Ahuja, S.; Mukund, S.; Deng, L.; Khakh, K.; Chang, E.; Ho, H.; Shriver, S.; Young, C.; Lin, S.; Johnson, J.P., Jr.; et al. Structural basis of Nav1.7 inhibition by an isoform-selective small-molecule antagonist. Science 2015, 350, aac5464. [Google Scholar] [CrossRef] [Green Version]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [Green Version]
- Biovia, D.S.; Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Richmond, T.J. Dassault Systèmes BIOVIA, Discovery Studio Visualizer, v.17.2, San Diego: Dassault Systèmes, 2016. J. Chem. Phys. 2000, 10, 21–9991. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Dose (mg/kg) | Acetic Acid-Induced Writhing Test | Dose (mg/kg) | Acetic Acid-Induced Writhing Test | ||
|---|---|---|---|---|---|---|
| Control | Mean ± SD (MPE, %) a | Control | Mean ± SD (MPE, %) a | |||
| 9 | 25 | 11.5 ± 0.63 | 11.2 ± 1.25 | 5 | 12.0 ± 0.4 | 10.5 ± 0.67 |
| 10 | 25 | 11.5 ± 0.63 | 8.2 ± 0.86 | 5 | 12.0 ± 0.4 | 4.3 ± 1.21 * (63) |
| 12 | 25 | 11.5 ± 0.63 | 4.8 ± 1.2 * (60) | 5 | 12.0 ± 0.4 | 4.4 ± 0.9 * (64) |
| 13 | 25 | 11.5 ± 0.63 | 5.5 ± 1.4 * (54) | 5 | 12.0 ± 0.4 | 4.6 ± 0.5 * (61) |
| 15 | 25 | 11.5 ± 0.63 | 7.2 ± 0.98 * (37) | 5 | 12.0 ± 0.4 | 3.4 ± 1.28 * (70) |
| 16 | 25 | 11.5 ± 0.63 | 8.0 ± 1.61 | 5 | 12.0 ± 0.4 | 8.9 ± 1.08 |
| 20 | 25 | 11.5 ± 0.63 | 10.7 ± 0.28 | 5 | 12.0 ± 0.4 | 9.7 ± 0.8 |
| 21 | 25 | 11.5 ± 0.63 | 7.0 ± 1.69 * (39) | 5 | 12.0 ± 0.4 | 10.4 ± 0.84 |
| 1 | 25 | - | ND | 5 | 12.0 ± 0.4 | 5.7 ± 1.25 * (50) |
| Diclofenac sodium | 10 | 11.5 ± 0.63 | 3.1 ± 0.7 * (74) | 10 | 11.5 ± 0.63 | 3.1 ± 0.7 * (74) |
| Compound | Dose (mg/kg) | Acetic Acid-Induced Writhing Test (Oral Administration) | Dose (mg/kg) | Acetic Acid-Induced Writhing Test (Intraperitoneal) | ||
|---|---|---|---|---|---|---|
| Control | Mean ± SD (MPE, %) a | Control | Mean ± SD (MPE, %) a | |||
| 10 | 1 | 11.0 ± 0.87 | 6.6 ± 1.75 * (40) | 1 | 11.5 ± 0.63 | 7.4 ± 0.80 * (63) |
| 12 | 1 | 11.0 ± 0.87 | 9.5 ± 1.0 * (21) | 1 | 11.5 ± 0.63 | 9.0 ± 1.1 * (28) |
| 13 | 1 | 11.0 ± 0.87 | 8.3 ± 0.8 * (31) | 1 | 11.5 ± 0.63 | 8.5 ± 1.3 * (29) |
| 15 | 1 | 11.0 ± 0.87 | 3.6 ± 1.82 * (67) | 1 | 11.5 ± 0.63 | 9.0 ± 1.5 |
| 1 | 1 | 11.0 ± 0.87 | 9.1 ± 1.05 | 5 (for 15) | 11.5 ± 0.63 | 4.0 ± 0.89 * (64) |
| Compound | Dose (mg/kg) | Hot Plate Test | Dose (mg/kg) | Hot Plate Test | ||
|---|---|---|---|---|---|---|
| Control | Mean ± SD (Protection, %) a | Control | Mean ± SD (Protection, %) a | |||
| 9 | 25 | 7.3 ± 0.71 | 8.9 ± 1.27 | 5 | 7.3 ± 0.71 | 8.6 ± 0.82 |
| 10 | 25 | 7.3 ± 0.71 | 11.2 ± 1.41 * (36) | 5 | 7.3 ± 0.71 | 11.7 ± 0.91 * (38) |
| 12 | 25 | 7.3 ± 0.71 | 10.3 ± 0.8 * (34) | 5 | 7.3 ± 0.71 | 10.3 ± 0.61 * (31) |
| 13 | 25 | 7.3 ± 0.71 | 10.1 ± 1.2 * (33) | 5 | 7.3 ± 0.71 | 10.0 ± 0.9 * (32) |
| 15 | 25 | 7.3 ± 0.71 | 9.3 ± 1.1 | 5 | 7.3 ± 0.71 | 11.0 ± 1.01 * (34) |
| 16 | 25 | 7.3 ± 0.71 | 8.4 ± 0.82 | 5 | 7.3 ± 0.71 | 8.1 ± 0.86 |
| 20 | 25 | 7.3 ± 0.71 | 8.0 ± 0.48 | 5 | 7.3 ± 0.71 | 8.9 ± 1.01 |
| 21 | 25 | 7.3 ± 0.71 | 10.6 ± 1.88 | 5 | 7.3 ± 0.71 | 9.7 ± 1.92 |
| 1 | - | - | - | 5 | 7.3 ± 0.71 | 12.3 ± 1.13 * (41) |
| Diclofenac sodium | 10 | 7.3 ± 0.71 | 10.8 ± 0.9 * (37) | 10 | 7.3 ± 0.71 | 10.8 ± 0.9* (37) |
Sample Availability: Samples of the compounds 7, 8, 12, 13, 15, 16, 20–22 are available from the authors. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheremnykh, K.P.; Savelyev, V.A.; Borisov, S.A.; Ivanov, I.D.; Baev, D.S.; Tolstikova, T.G.; Vavilin, V.A.; Shults, E.E. Hybrides of Alkaloid Lappaconitine with Pyrimidine Motif on the Anthranilic Acid Moiety: Design, Synthesis, and Investigation of Antinociceptive Potency. Molecules 2020, 25, 5578. https://doi.org/10.3390/molecules25235578
Cheremnykh KP, Savelyev VA, Borisov SA, Ivanov ID, Baev DS, Tolstikova TG, Vavilin VA, Shults EE. Hybrides of Alkaloid Lappaconitine with Pyrimidine Motif on the Anthranilic Acid Moiety: Design, Synthesis, and Investigation of Antinociceptive Potency. Molecules. 2020; 25(23):5578. https://doi.org/10.3390/molecules25235578
Chicago/Turabian StyleCheremnykh, Kirill P., Victor A. Savelyev, Sergey A. Borisov, Igor D. Ivanov, Dmitry S. Baev, Tatyana G. Tolstikova, Valentin A. Vavilin, and Elvira E. Shults. 2020. "Hybrides of Alkaloid Lappaconitine with Pyrimidine Motif on the Anthranilic Acid Moiety: Design, Synthesis, and Investigation of Antinociceptive Potency" Molecules 25, no. 23: 5578. https://doi.org/10.3390/molecules25235578