
Deciphering the Role of Filamin B Calponin-Homology Domain in Causing the Larsen Syndrome, Boomerang Dysplasia, and Atelosteogenesis Type I Spectrum Disorders via a Computational Approach

 ,
,  , ,
, ,
Abstract
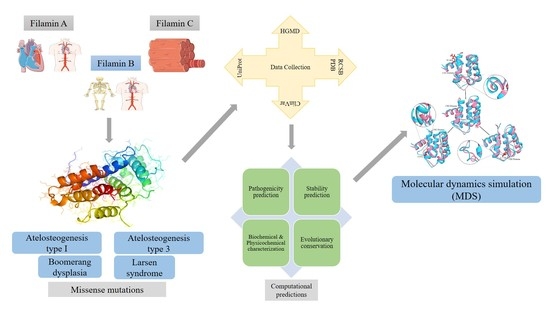
:
1. Introduction
2. Materials and Methods
2.1. Collection of Variants
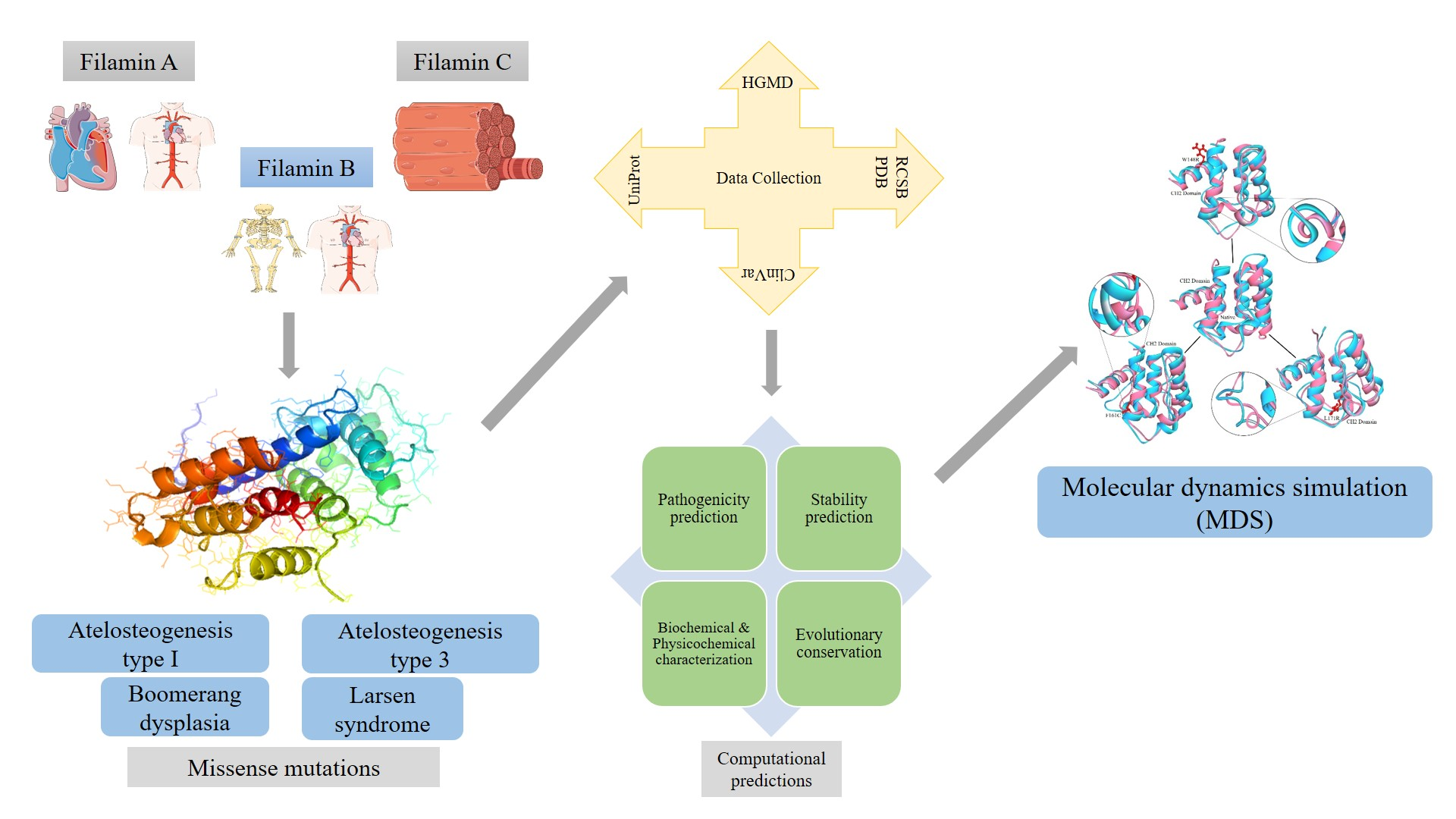
2.2. Evolutionary Conservation
2.3. Prediction of Pathogenicity
2.4. Stability Prediction
2.5. SNPeffect
2.6. Preparation of Variant Models
2.7. Molecular Dynamics Simulation (MDS)
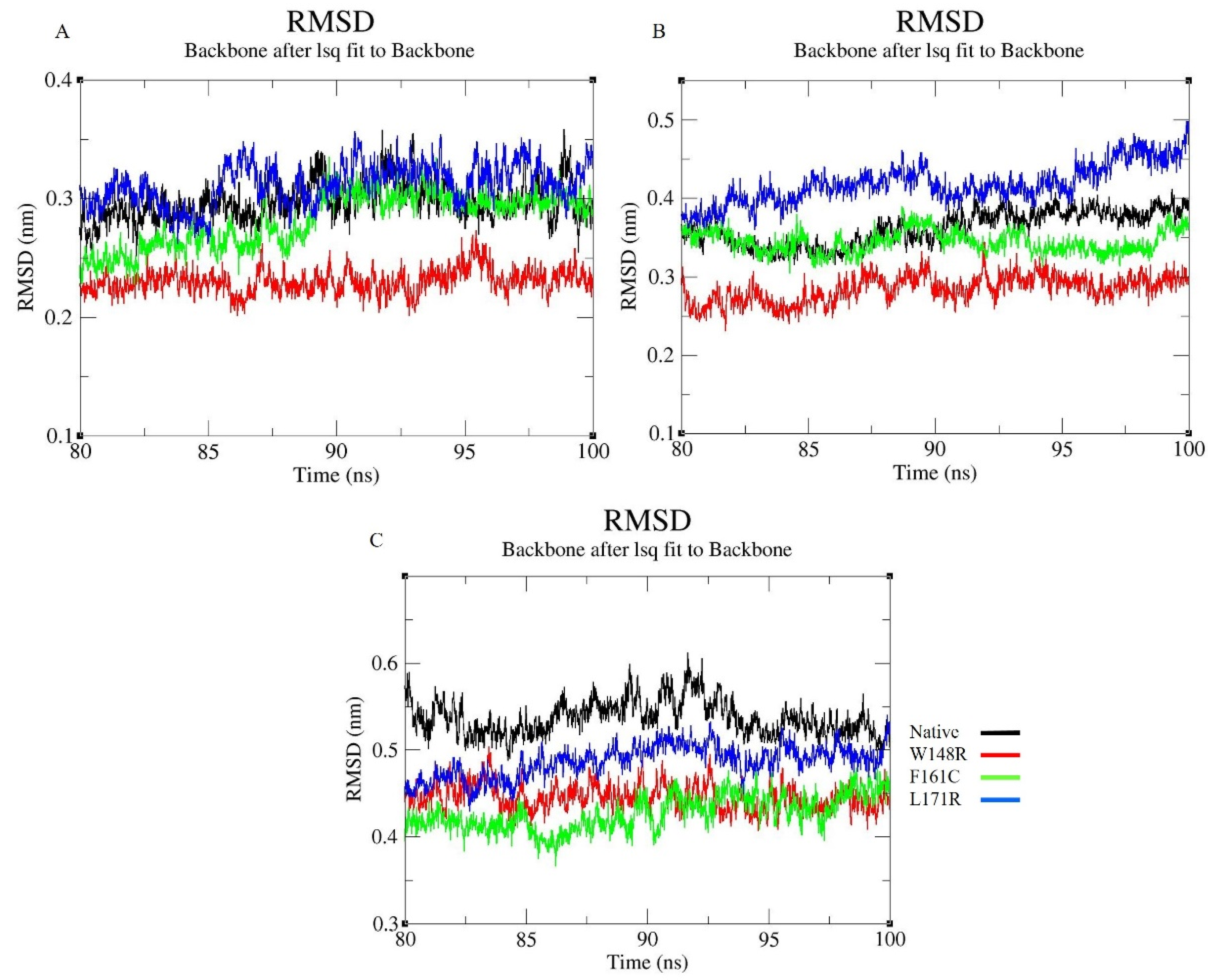
2.8. Trajectory Analysis
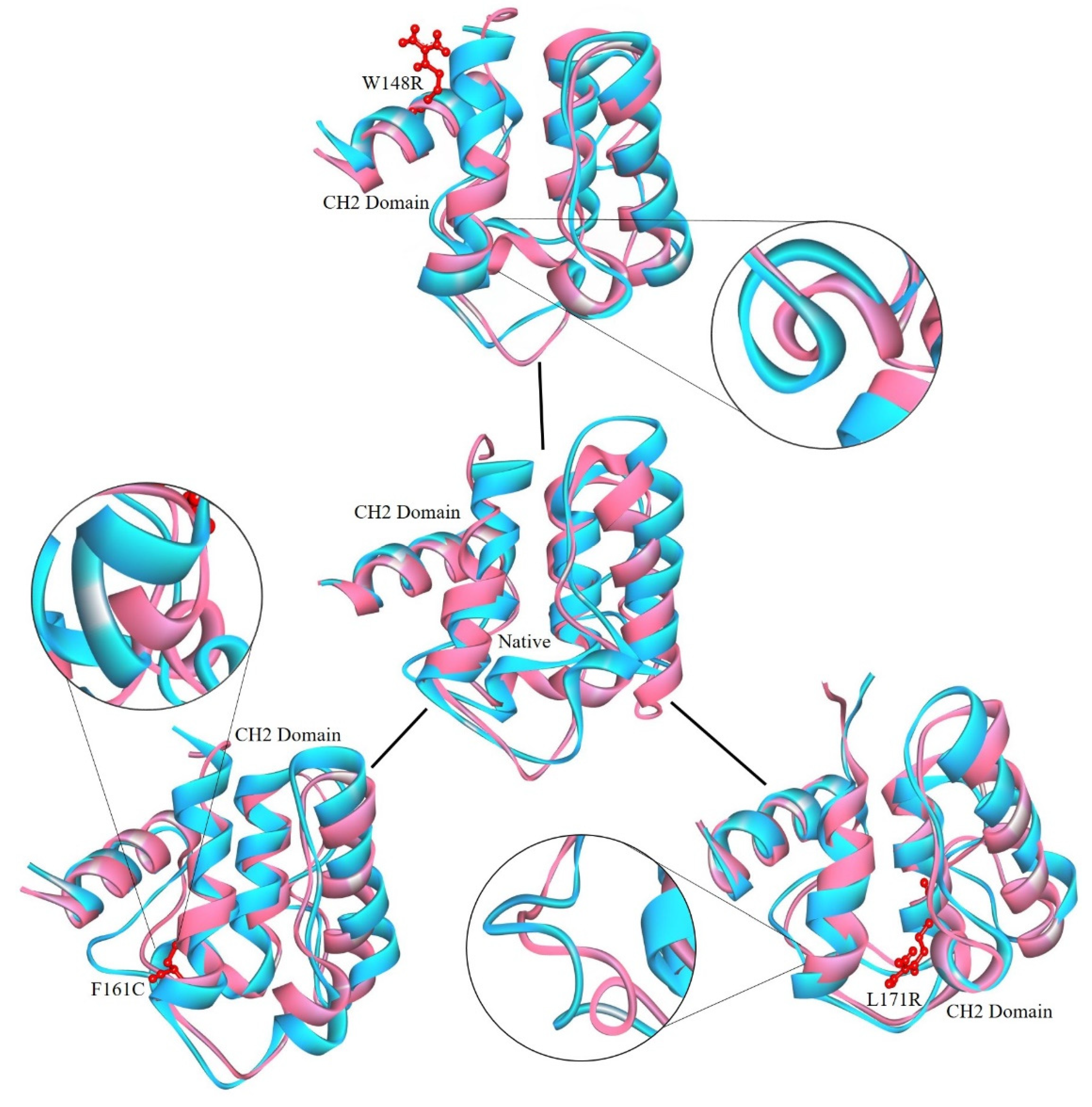
3. Results
3.1. Evolutionary Conservation Analysis
3.2. Pathogenicity Prediction Analysis
3.3. Stability Prediction Analysis
3.4. Biochemical and Physicochemical Characterization
3.5. SNPeffect and HOPE
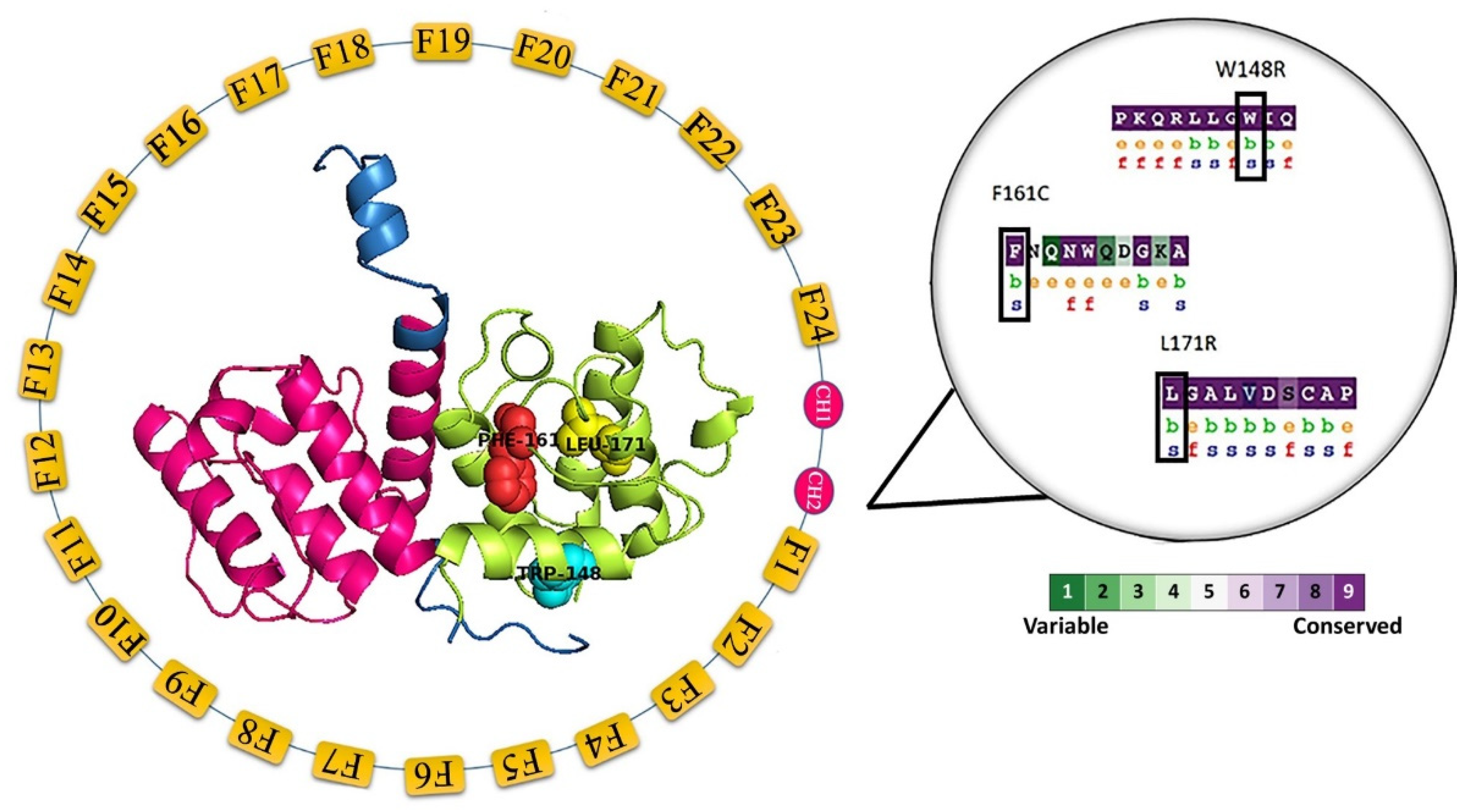
3.6. Preparation of Protein Model
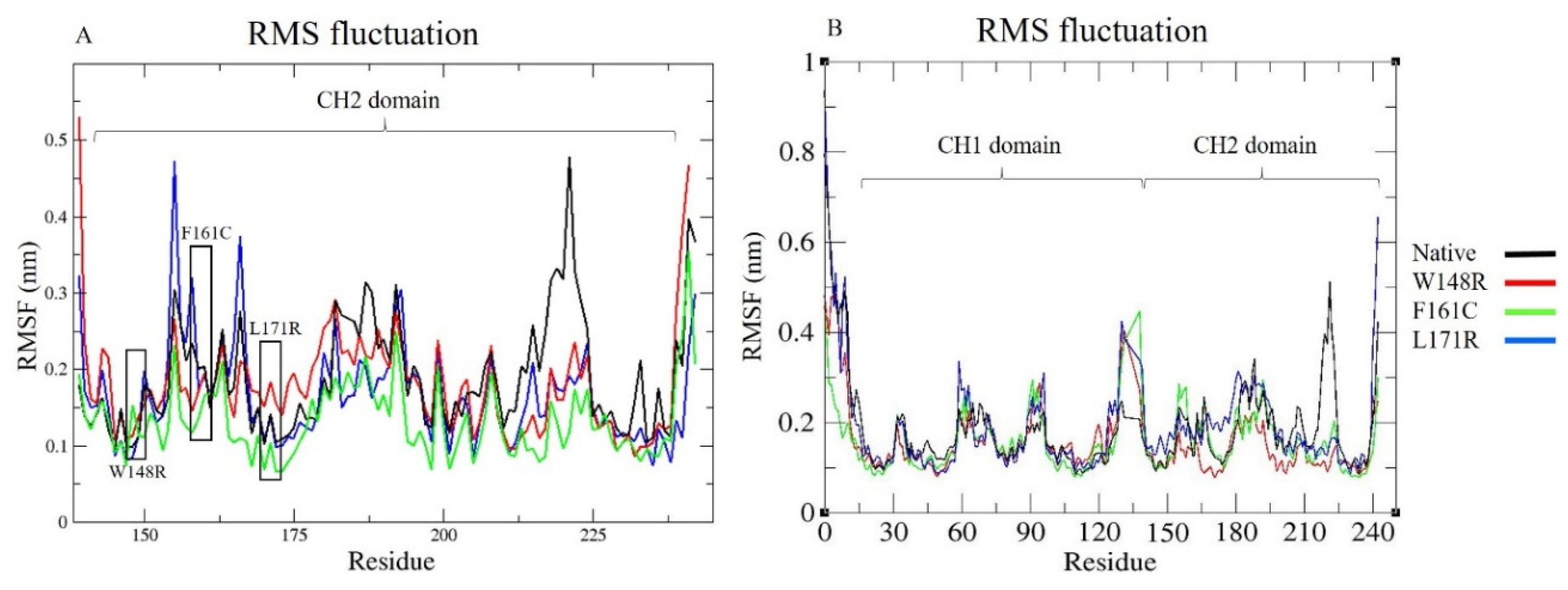
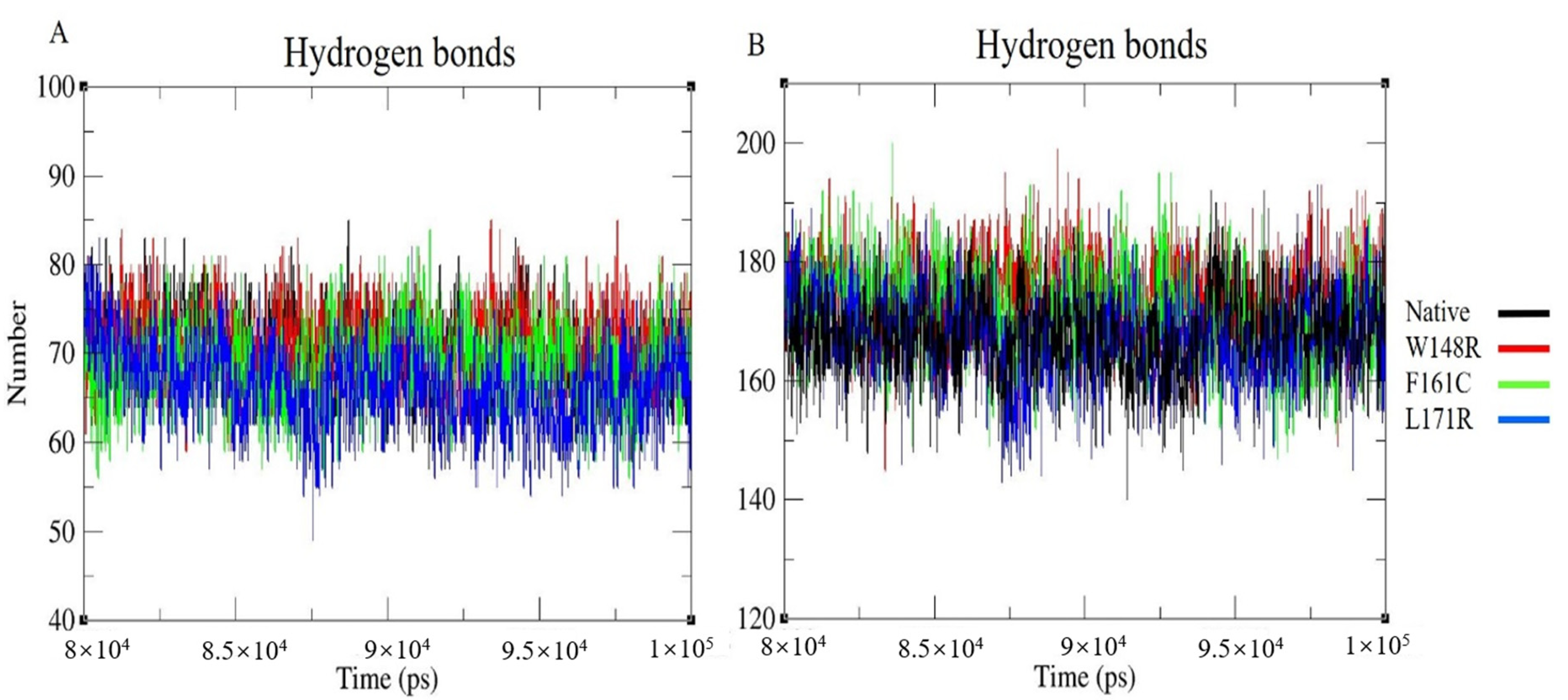
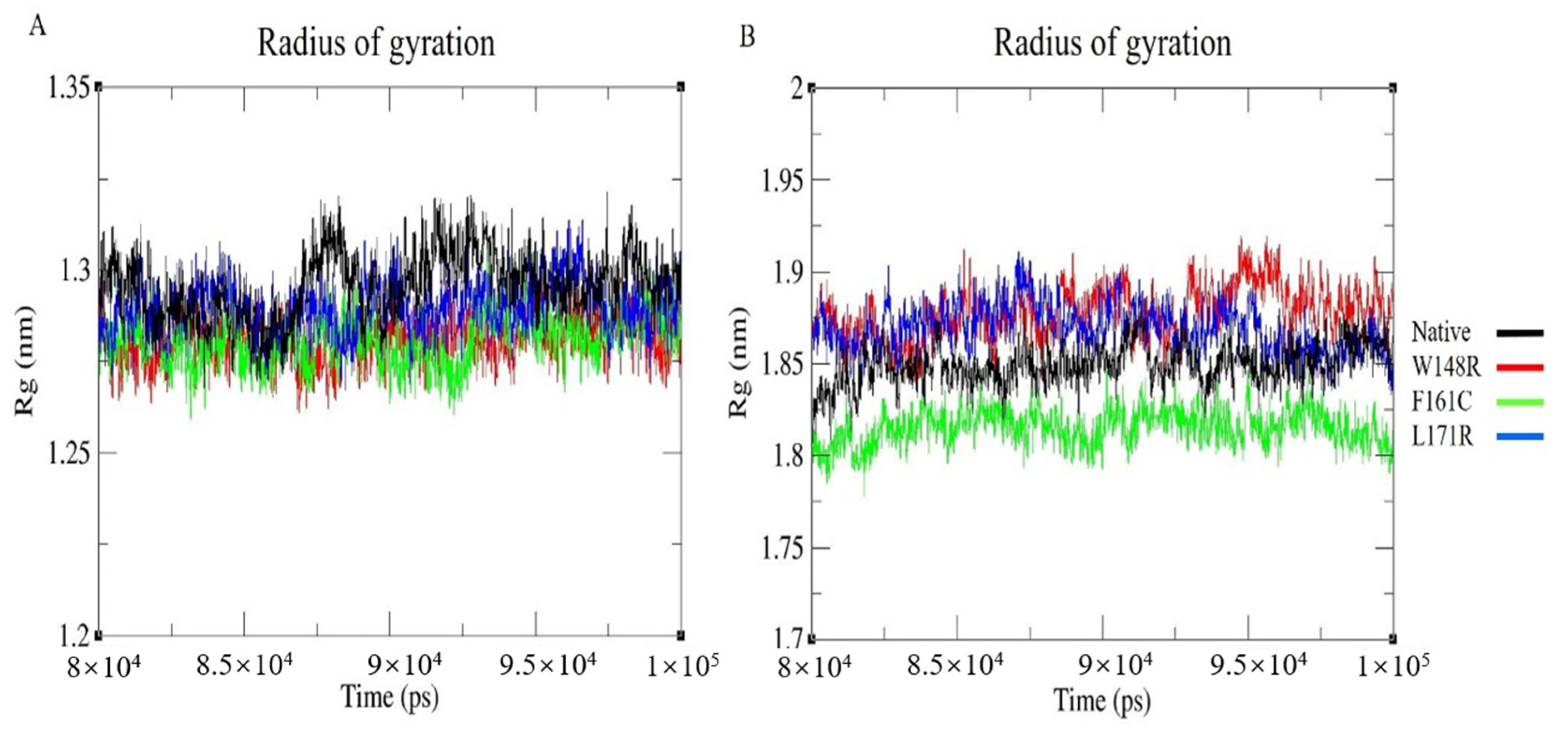
3.7. Molecular Dynamics Simulation (MDS) and Trajectory Analysis
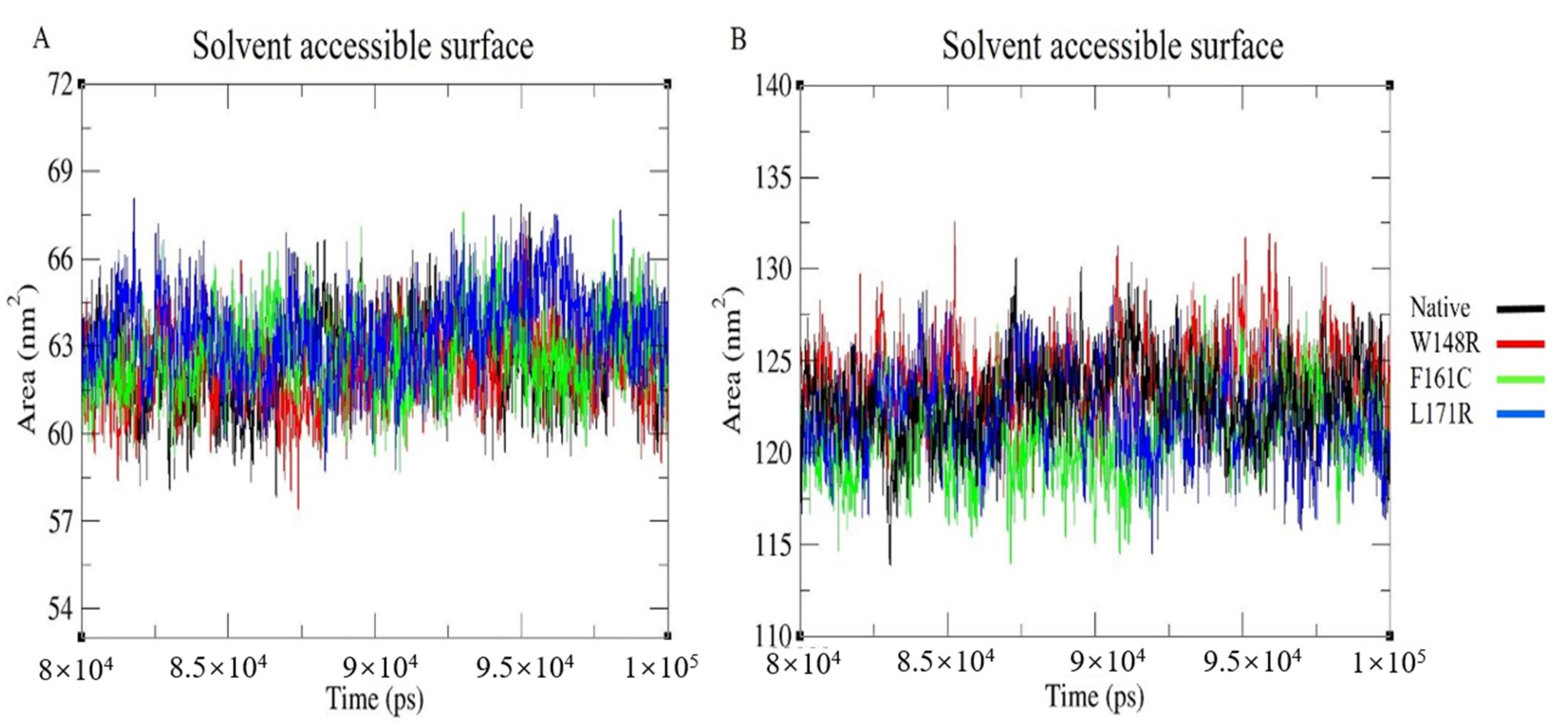
3.8. Solvent Accessible Surface
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Stossel, T.P.; Condeelis, J.; Cooley, L.; Hartwig, J.H.; Noegel, A.; Schleicher, M.; Shapiro, S.S. Filamins as integrators of cell mechanics and signaling. Nat. Rev. Mol. Cell Biol. 2001, 2, 138–145. [Google Scholar] [CrossRef]
- Lu, J.; Lian, G.; Lenkinski, R.; De Grand, A.; Vaid, R.R.; Bryce, T.; Stasenko, M.; Boskey, A.; Walsh, C.; Sheen, V. Filamin B mutations cause chondrocyte defects in skeletal development. Hum. Mol. Genet. 2007, 16, 1661–1675. [Google Scholar] [CrossRef] [Green Version]
- Hartwig, J.H.; Stossel, T.P. Isolation and properties of actin, myosin, and a new actinbinding protein in rabbit alveolar macrophages. J. Biol. Chem. 1975, 250, 5696–5705. [Google Scholar]
- Wang, K.; Ash, J.F.; Singer, S.J. Filamin, a new high-molecular-weight protein found in smooth muscle and non-muscle cells. Proc. Natl. Acad. Sci. USA 1975, 72, 4483–4486. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, F.; Stossel, T.P.; Hartwig, J.H. The filamins. Cell Adhes. Migr. 2011, 5, 160–169. [Google Scholar] [CrossRef] [Green Version]
- Popowicz, G.M.; Schleicher, M.; Noegel, A.A.; Holak, T.A. Filamins: Promiscuous organizers of the cytoskeleton. Trends Biochem. Sci. 2006, 31, 411–419. [Google Scholar] [CrossRef]
- Zhou, A.-X.; Hartwig, J.H.; Akyürek, L.M. Filamins in cell signaling, transcription and organ development. Trends Cell Biol. 2010, 20, 113–123. [Google Scholar] [CrossRef]
- Robertson, S. FLNB Disorders; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Stephens, K., Amemiya, A., Eds.; GeneReviews®; University of Washington: Seattle, WA, USA, 1993. Available online: http://www.ncbi.nlm.nih.gov/books/NBK2534/ (accessed on 19 July 2020).
- Takafuta, T.; Wu, G.; Murphy, G.F.; Shapiro, S.S. Human β-Filamin Is a New Protein That Interacts with the Cytoplasmic Tail of Glycoprotein Ibα. J. Biol. Chem. 1998, 273, 17531–17538. [Google Scholar] [CrossRef] [Green Version]
- Bröcker, F.; Bardenheuer, W.; Vieten, L.; Jülicher, K.; Werner, N.; Marquitan, G.; Michael, D.; Opalka, B.; Schütte, J. Assignment of human filamin gene FLNB to human chromosome band 3p14.3 and identification of YACs containing the complete FLNB transcribed region. Cytogenet. Cell Genet. 1999, 85, 267–268. [Google Scholar] [CrossRef]
- Gorlin, J.B.; Yamin, R.; Egan, S.; Stewart, M.; Stossel, T.P.; Kwiatkowski, D.J.; Hartwig, J.H. Human endothelial actin-binding protein (ABP-280, nonmuscle filamin): A molecular leaf spring. J. Cell Biol. 1990, 111, 1089–1105. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Zhang, S.-X.; Sokol, N.; Cooley, L.; Boulianne, G.L. Physical and genetic interaction of filamin with presenilin in Drosophila. J. Cell Sci. 2000, 113, 3499–3508. [Google Scholar] [PubMed]
- Takafuta, T.; Saeki, M.; Fujimoto, T.-T.; Fujimura, K.; Shapiro, S.S. A New Member of the LIM Protein Family Binds to Filamin B and Localizes at Stress Fibers. J. Biol. Chem. 2002, 278, 12175–12181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krakow, D.; Robertson, S.P.; King, L.M.; Morgan, T.; Sebald, E.T.; Bertolotto, C.; Wachsmann-Hogiu, S.; Acuna, D.; Shapiro, S.S.; Takafuta, T.; et al. Mutations in the gene encoding filamin B disrupt vertebral segmentation, joint formation and skeletogenesis. Nat. Genet. 2004, 36, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Farrington-Rock, C.; Kirilova, V.; Dillard-Telm, L.; Borowsky, A.D.; Chalk, S.; Rock, M.J.; Cohn, D.H.; Krakow, D. Disruption of the Flnb gene in mice phenocopies the human disease spondylocarpotarsal synostosis syndrome. Hum. Mol. Genet. 2007, 17, 631–641. [Google Scholar] [CrossRef] [Green Version]
- Zheng, L.; Baek, H.-J.; Karsenty, G.; Justice, M. Filamin B represses chondrocyte hypertrophy in a Runx2/Smad3-dependent manner. J. Cell Biol. 2007, 178, 121–128. [Google Scholar] [CrossRef] [Green Version]
- Bicknell, L.S.; Morgan, T.; Bonafé, L.; Wessels, M.W.; Bialer, M.G.; Willems, P.J.; Cohn, D.H.; Krakow, D.; Robertson, S.P. Mutations in FLNB cause boomerang dysplasia. J. Med. Genet. 2005, 42, e43. [Google Scholar] [CrossRef] [Green Version]
- Bonaventure, J.; Lasselin, C.; Mellier, J.; Cohen-Solal, L.; Maroteaux, P. Linkage studies of four fibrillar collagen genes in three pedigrees with Larsen-like syndrome. J. Med. Genet. 1992, 29, 465–470. [Google Scholar]
- Daniel, P.B.; Morgan, T.; Alanay, Y.; Bijlsma, E.; Cho, T.-J.; Cole, T.; Collins, F.; David, A.; Devriendt, K.; Faivre, L.; et al. Disease-associated mutations in the actin-binding domain of filamin B cause cytoplasmic focal accumulations correlating with disease severity. Hum. Mutat. 2012, 33, 665–673. [Google Scholar] [CrossRef]
- Sawyer, G.M.; Clark, A.R.; Robertson, S.P.; Sutherland-Smith, A.J. Disease-associated Substitutions in the Filamin B Actin Binding Domain Confer Enhanced Actin Binding Affinity in the Absence of Major Structural Disturbance: Insights from the Crystal Structures of Filamin B Actin Binding Domains. J. Mol. Biol. 2009, 390, 1030–1047. [Google Scholar] [CrossRef]
- Xu, Q.; Wu, N.; Cui, L.; Wu, Z.; Qiu, G. Filamin B: The next hotspot in skeletal research? J. Genet. Genom. 2017, 44, 335–342. [Google Scholar] [CrossRef]
- Xu, Q.; Wu, N.; Cui, L.; Lin, M.; Kumar, D.T.; Doss, C.G.P.; Wu, Z.; Shen, J.; Song, X.; Qiu, G. Comparative analysis of the two extremes of FLNB-mutated autosomal dominant disease spectrum: From clinical phenotypes to cellular and molecular findings. Am. J. Transl. Res. 2018, 10, 1400–1412. [Google Scholar]
- Zhang, D. Mutations responsible for Larsen syndrome cluster in the FLNB protein. J. Med. Genet. 2005, 43, e24. [Google Scholar] [CrossRef]
- Bicknell, L.S.; Farrington-Rock, C.; Shafeghati, Y.; Rump, P.; Alanay, Y.; Alembik, Y.; Al-Madani, N.; Firth, H.; Karimi-Nejad, M.H.; Kim, C.A.; et al. A molecular and clinical study of Larsen syndrome caused by mutations in FLNB. J. Med. Genet. 2006, 44, 89–98. [Google Scholar] [CrossRef] [Green Version]
- Farrington-Rock, C.; Firestein, M.H.; Bicknell, L.S.; Superti-Furga, A.; Bacino, C.A.; Cormier-Daire, V.; Le Merrer, M.; Baumann, C.; Roume, J.; Rump, P.; et al. Mutations in two regions ofFLNBresult in atelosteogenesis I and III. Hum. Mutat. 2006, 27, 705–710. [Google Scholar] [CrossRef]
- Schultz, C.; Langer, L.O.; Laxova, R.; Pauli, R.M. Atelosteogenesis type III: Long term survival, prenatal diagnosis, and evidence for dominant transmission. Am. J. Med. Genet. 1999, 83, 28–42. [Google Scholar] [CrossRef]
- Kumar, D.T.; Jain, N.; Kumar, S.U.; Doss, C.G.P.; Zayed, H. Identification of potential inhibitors against pathogenic missense mutations of PMM2 using a structure-based virtual screening approach. J. Biomol. Struct. Dyn. 2020, 1–17. [Google Scholar] [CrossRef]
- Kumar, D.T.; Jain, N.; Kumar, S.U.; Paramita Jena, P.; Ramamoorthy, S.; Doss, C.G.P.; Zayed, H. Molecular dynamics simulations to decipher the structural and functional consequences of pathogenic missense mutations in the galactosylceramidase (GALC) protein causing Krabbe’s disease. J. Biomol. Struct. Dyn. 2020, 1–16. [Google Scholar] [CrossRef]
- Siva, R.; Zayed, H. An extensive computational approach to analyze and characterize the functional mutations in the galactose-1-phosphate uridyl transferase (GALT) protein responsible for classical galactosemia. Comput. Biol. Med. 2020, 117, 103583. [Google Scholar] [CrossRef]
- Sneha, P.; Thirumal, D.K.; Tanwar, H.; Siva, R.; Doss, C.G.P.; Zayed, H. Structural Analysis of G1691S Variant in the Human Filamin B Gene Responsible for Larsen Syndrome: A Comparative Computational Approach. J. Cell. Biochem. 2017, 118, 1900–1910. [Google Scholar] [CrossRef]
- Kumar, D.T.; Kumar, U.; Laeeque, A.S.N.; Abhay, S.A.; Bithia, R.; Magesh, R.; Kumar, M.; Zayed, H.; Doss, C.G.P. Computational model to analyze and characterize the functional mutations of NOD2 protein causing inflammatory disorder—Blau syndrome. In Advances in Protein Chemistry and Structural Biology; Donev, R., Ed.; Academic Press: Cambridge, MA, USA, 2020; pp. 379–408. [Google Scholar]
- Kumar, S.U.; Priyanka, N.; Sneha, P.; Doss, C.G.P. Functional and structural characterization of missense mutations in PAX6 gene. Front. Biol. 2015, 10, 377–385. [Google Scholar] [CrossRef]
- Kumar, S.U.; Kumar, D.T.; Mandal, P.D.; Sankar, S.; Haldar, R.; Kamaraj, B.; Walter, C.E.J.; Siva, R.; Doss, C.G.P.; Zayed, H. Comprehensive in silico screening and molecular dynamics studies of missense mutations in Sjogren-Larsson syndrome associated with the ALDH3A2 gene. In Advances in Protein Chemistry and Structural Biology; Donev, R., Ed.; Academic Press: Cambridge, MA, USA, 2020; pp. 349–377. [Google Scholar]
- Kumar, U.; Rajan, B.; Kumar, T.; Doss, C.G.P.; Zayed, H. Mutational landscape of K-Ras substitutions at 12th position-a systematic molecular dynamics approach. J. Biomol. Struct. Dyn. 2020, 1–15. [Google Scholar] [CrossRef]
- Sneha, P.; Ahmad, E.E.; Ahmed, G.S.; Kumar, D.T.; Siva, R.; George, P.D.C.; Zayed, H. Structural analysis of missense mutations in galactokinase 1 (GALK1) leading to galactosemia type-2. J. Cell. Biochem. 2018, 119, 7585–7598. [Google Scholar] [CrossRef]
- The UniProt Consortium. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar] [CrossRef]
- The UniProt Consortium. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2018, 46, 2699. [Google Scholar] [CrossRef] [Green Version]
- The UniProt Consortium. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef] [Green Version]
- Cooper, D.N.; Ball, E.V.; Krawczak, M. The human gene mutation database. Nucleic Acids Res. 1998, 26, 285–287. [Google Scholar] [CrossRef] [Green Version]
- Burley, S.K.; Berman, H.M.; Bhikadiya, C.; Bi, C.; Chen, L.; Di Costanzo, L.; Christie, C.; Dalenberg, K.; Duarte, J.M.; Dutta, S.; et al. RCSB Protein Data Bank: Biological macromolecular structures enabling research and education in fundamental biology, biomedicine, biotechnology and energy. Nucleic Acids Res. 2019, 47, D464–D474. [Google Scholar] [CrossRef] [Green Version]
- Hicks, S.; Wheeler, D.A.; Plon, S.E.; Kimmel, M. Prediction of missense mutation functionality depends on both the algorithm and sequence alignment employed. Hum. Mutat. 2011, 32, 661–668. [Google Scholar] [CrossRef] [Green Version]
- Ashkenazy, H.; Erez, E.; Martz, E.; Pupko, T.; Ben-Tal, N. ConSurf 2010: Calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 2010, 38, W529–W533. [Google Scholar] [CrossRef] [Green Version]
- Glaser, F.; Pupko, T.; Paz, I.; E Bell, R.; Bechor-Shental, D.; Martz, E.; Ben-Tal, N. ConSurf: Identification of Functional Regions in Proteins by Surface-Mapping of Phylogenetic Information. Bioinformatics 2003, 19, 163–164. [Google Scholar] [CrossRef] [Green Version]
- Bendl, J.; Stourac, J.; Salanda, O.; Pavelka, A.; Wieben, E.D.; Zendulka, J.; Brezovsky, J.; Damborsky, J. PredictSNP: Robust and Accurate Consensus Classifier for Prediction of Disease-Related Mutations. PLoS Comput. Biol. 2014, 10, e1003440. [Google Scholar] [CrossRef] [PubMed]
- Stone, E.A. Physicochemical constraint violation by missense substitutions mediates impairment of protein function and disease severity. Genome Res. 2005, 15, 978–986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, L.; Zhou, M.; Cui, Y. nsSNPAnalyzer: Identifying disease-associated nonsynonymous single nucleotide polymorphisms. Nucleic Acids Res. 2005, 33, W480–W482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, H.; Thomas, P.D. PANTHER-PSEP: Predicting disease-causing genetic variants using position-specific evolutionary preservation. Bioinformatics 2016, 32, 2230–2232. [Google Scholar] [CrossRef]
- Capriotti, E.; Calabrese, R.; Casadio, R. Predicting the insurgence of human genetic diseases associated to single point protein mutations with support vector machines and evolutionary information. Bioinformatics 2006, 22, 2729–2734. [Google Scholar] [CrossRef] [Green Version]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting Functional Effect of Human Missense Mutations Using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 76, 7.20.1–7.20.41. [Google Scholar] [CrossRef] [Green Version]
- Sim, N.-L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef]
- Hecht, M.; Bromberg, Y.; Rost, B. Better prediction of functional effects for sequence variants. BMC Genom. 2015, 16 (Suppl. 8), S1. [Google Scholar] [CrossRef] [Green Version]
- Venselaar, H.; Beek, T.A.H.T.; Kuipers, R.K.P.; Hekkelman, M.L.; Vriend, G. Protein structure analysis of mutations causing inheritable diseases. An e-Science approach with life scientist friendly interfaces. BMC Bioinform. 2010, 11, 548. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.-W.; Lin, J.; Chu, Y.-W. iStable: Off-the-shelf predictor integration for predicting protein stability changes. BMC Bioinform. 2013, 14, S5. [Google Scholar] [CrossRef] [Green Version]
- De Baets, G.; Van Durme, J.; Reumers, J.; Maurer-Stroh, S.; Vanhee, P.; Dopazo, J.; Schymkowitz, J.; Rousseau, F. SNPeffect 4.0: On-line prediction of molecular and structural effects of protein-coding variants. Nucleic Acids Res. 2012, 40, D935–D939. [Google Scholar] [CrossRef] [PubMed]
- Guex, N.; Peitsch, M.C. SWISS-MODEL and the Swiss-Pdb Viewer: An environment for comparative protein modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar] [CrossRef] [PubMed]
- Van Gunsteren, W.F. Biomolecular Simulation: The GROMOS96 Manual and User Guide; Biomos: Zürich, Switzerland, 1996. [Google Scholar]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Schmid, N.; Eichenberger, A.P.; Choutko, A.; Riniker, S.; Winger, M.; Mark, A.E.; Van Gunsteren, W.F. Definition and testing of the GROMOS force-field versions 54A7 and 54B7. Eur. Biophys. J. 2011, 40, 843–856. [Google Scholar] [CrossRef]
- Mosaeilhy, A.; Mohamed, M.M.; George, P.D.C.; El Abd, H.S.A.; Gamal, R.; Zaki, O.; Zayed, H. Genotype-phenotype correlation in 18 Egyptian patients with glutaric acidemia type I. Metab. Brain Dis. 2017, 32, 1417–1426. [Google Scholar] [CrossRef]
- Pires, A.S.; Porto, W.F.; Franco, O.L.; Alencar, S.A. In silico analyses of deleterious missense SNPs of human apolipoprotein E3. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.T.; Emerald, L.J.; George, P.D.C.; Sneha, P.; Siva, R.; Jebaraj, W.C.E.; Zayed, H. Computational approach to unravel the impact of missense mutations of proteins (D2HGDH and IDH2) causing D-2-hydroxyglutaric aciduria 2. Metab. Brain Dis. 2018, 33, 1699–1710. [Google Scholar] [CrossRef]
- Kumar, D.T.; Eldous, H.G.; Mahgoub, Z.A.; Doss, C.G.P.; Zayed, H. Computational modelling approaches as a potential platform to understand the molecular genetics association between Parkinson’s and Gaucher diseases. Metab. Brain Dis. 2018, 33, 1835–1847. [Google Scholar] [CrossRef]
- Agrahari, A.K.; Muskan, M.; George, P.D.C.; Siva, R.; Zayed, H. Computational insights of K1444N substitution in GAP-related domain of NF1 gene associated with neurofibromatosis type 1 disease: A molecular modeling and dynamics approach. Metab. Brain Dis. 2018, 33, 1443–1457. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; Van Gunsteren, W.F.; Hermans, J. Interaction Models for Water in Relation to Protein Hydration. In Intermolecular Forces, Proceedings of the Fourteenth Jerusalem Symposium on Quantum Chemistry and Biochemistry, Jerusalem, Israel, 13–16 April 1981; Pullman, B., Ed.; Springer: Dordrecht, The Netherlands, 1981; pp. 331–342. [Google Scholar] [CrossRef]
- Darden, T.; York, D.M.; Pedersen, L. Particle mesh Ewald: AnN⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Petrova, S.S.; Solov’Ev, A.D. The Origin of the Method of Steepest Descent. Hist. Math. 1997, 24, 361–375. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, S.; Kollman, P.A. Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; Van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Bresnick, A.R.; Warren, V.; Condeelis, J. Identification of a short sequence essential for actin binding by Dictyostelium ABP-120. J. Biol. Chem. 1990, 265, 9236–9240. [Google Scholar]
- Kainulainen, T.; Pender, A.; D’Addario, M.; Feng, Y.; Lekic, P.; McCulloch, C.A. Cell Death and Mechanoprotection by Filamin A in Connective Tissues after Challenge by Applied Tensile Forces. J. Biol. Chem. 2002, 277, 21998–22009. [Google Scholar] [CrossRef] [Green Version]
- Landrum, M.J.; Lee, J.M.; Riley, G.R.; Jang, W.; Rubinstein, W.S.; Church, D.M.; Maglott, D.R. ClinVar: Public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014, 42, D980–D985. [Google Scholar] [CrossRef] [Green Version]
- Ramensky, V. Human non-synonymous SNPs: Server and survey. Nucleic Acids Res. 2002, 30, 3894–3900. [Google Scholar] [CrossRef]
- Johnson, A.D.; Handsaker, R.E.; Pulit, S.L.; Nizzari, M.M.; O’Donnell, C.J.; De Bakker, P.I.W. SNAP: A web-based tool for identification and annotation of proxy SNPs using HapMap. Bioinformatics 2008, 24, 2938–2939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capriotti, E.; Fariselli, P.; Casadio, R. I-Mutant2.0: Predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res. 2005, 33, W306–W310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agrahari, A.K.; Kumar, A.; Siva, R.; Zayed, H.; George, P.D.C. Substitution impact of highly conserved arginine residue at position 75 in GJB1 gene in association with X-linked Charcot–Marie-tooth disease: A computational study. J. Theor. Biol. 2018, 437, 305–317. [Google Scholar] [CrossRef] [PubMed]
- Capra, J.A.; Singh, M. Predicting functionally important residues from sequence conservation. Bioinformatics 2007, 23, 1875–1882. [Google Scholar] [CrossRef] [Green Version]
- Del Sol, A.; Fujihashi, H.; Amoros, D.; Nussinov, R. Residue centrality, functionally important residues, and active site shape: Analysis of enzyme and non-enzyme families. Protein Sci. 2006, 15, 2120–2128. [Google Scholar] [CrossRef] [Green Version]
- Larsen, L.J.; Schottstaedt, E.R.; Bost, F.C. Multiple congenital dislocations associated with characteristics facial abnormality. J. Pediatr. 1950, 37, 574–581. [Google Scholar] [CrossRef]
- Zhao, Y.; Shapiro, S.S.; Eto, M. F-actin clustering and cell dysmotility induced by the pathological W148R missense mutation of filamin B at the actin-binding domain. Am. J. Physiol. Physiol. 2016, 310, C89–C98. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Lu, J.; Lian, G.; Zhang, J.; Hecht, J.L.; Sheen, V. Filamin B Regulates Chondrocyte Proliferation and Differentiation through Cdk1 Signaling. PLoS ONE 2014, 9, e89352. [Google Scholar] [CrossRef] [Green Version]
- Wilson, S.G.; Jones, M.R.; Mullin, B.H.; Dick, I.M.; Richards, J.B.; Pastinen, T.; Grundberg, E.; Ljunggren, Ö.; Surdulescu, G.; Dudbridge, F.; et al. Common Sequence Variation inFLNBRegulates Bone Structure in Women in the General Population andFLNBmRNA Expression in Osteoblasts In Vitro. J. Bone Miner. Res. 2009, 24, 1989–1997. [Google Scholar] [CrossRef] [Green Version]
- Maroteaux, P.; Spranger, J.; Stanescu, V.; Le Marec, B.; Pfeiffer, R.A.; Beighton, P.; Mattei, J.F.; Opitz, J.M. Atelosteogenesis. Am. J. Med. Genet. 1982, 13, 15–25. [Google Scholar] [CrossRef]
- Sillence, D.; Worthington, S.; Dixon, J.; Osborn, R.; Kozlowski, K. Atelosteogenesis syndromes: A review, with comments on their pathogenesis. Pediatr. Radiol. 1997, 27, 388–396. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S.No | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| Variant | W148R | F161C | W165C | G168C | L171Q | L171R |
| PredictSNP | D1 | D1 | D1 | D1 | D1 | D1 |
| MAPP | D1 | D1 | D1 | D1 | D1 | D1 |
| PhD-SNP | D1 | D1 | D1 | D1 | D1 | D1 |
| PolyPhen-1 | D1 | D1 | D1 | D1 | D1 | D1 |
| PolyPhen-2 | D1 | D1 | D1 | D1 | D1 | D1 |
| SIFT | D1 | D1 | D1 | D1 | D1 | D1 |
| SNAP | D1 | D1 | D1 | D1 | D1 | D1 |
| i-Mutant2.0 | D2 | D2 | D2 | D2 | D2 | D2 |
| MUpro | D2 | D2 | D2 | D2 | D2 | D2 |
| iStable | D2 | D2 | D2 | D2 | D2 | D2 |
| S.No | Variant | Tango | Waltz | Limbo | FoldX |
|---|---|---|---|---|---|
| 1 | W148R | T.1 | W.2 | L.1 | Reduces the protein stability. |
| 2 | F161C | T.1 | W.1. | L.1 | Reduces the protein stability. |
| 3 | W165C | T.1 | W.1. | L.1 | Severely reduces the protein stability. |
| 4 | G168C | T.1 | W.1. | L.1 | Severely reduces the protein stability. |
| 5 | L171Q | T.1 | W.1. | L.1 | Reduces the protein stability. |
| 6 | L171R | T.1 | W.1. | L.1 | Severely reduces the protein stability. |
| S.No | Variant | Change in AA Property | Mutation Impact |
|---|---|---|---|
| 1 | W148R | The mutation introduces an amino acid with different properties | The mutant residue is smaller than the wild-type residue. The mutation will cause a possible loss of external interactions |
| 2 | F161C | The mutation introduces an amino acid with different properties | The mutant residue is smaller than the wild-type residue. The mutation will cause a possible loss of external interactions |
| 3 | W165C | The mutation introduces an amino acid with different properties | The mutant residue is smaller than the wild-type residue. The mutation will cause a possible loss of external interactions |
| 4 | G168C | Mutation of this glycine can abolish the protein function. | The wild-type residue was buried in the core of the protein. The mutant residue is bigger and probably will not fit. |
| 5 | L171Q | The mutation introduces an amino acid with different properties | The wild-type residue was buried in the core of the protein. The mutant residue is bigger and probably will not fit. |
| 6 | L171R | The mutation introduces an amino acid with different properties | The wild-type residue was buried in the core of the protein. The mutant residue is bigger and probably will not fit. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
S., U.K.; Sankar, S.; Younes, S.; D., T.K.; Ahmad, M.N.; Okashah, S.S.; Kamaraj, B.; Al-Subaie, A.M.; C., G.P.D.; Zayed, H. Deciphering the Role of Filamin B Calponin-Homology Domain in Causing the Larsen Syndrome, Boomerang Dysplasia, and Atelosteogenesis Type I Spectrum Disorders via a Computational Approach. Molecules 2020, 25, 5543. https://doi.org/10.3390/molecules25235543
S. UK, Sankar S, Younes S, D. TK, Ahmad MN, Okashah SS, Kamaraj B, Al-Subaie AM, C. GPD, Zayed H. Deciphering the Role of Filamin B Calponin-Homology Domain in Causing the Larsen Syndrome, Boomerang Dysplasia, and Atelosteogenesis Type I Spectrum Disorders via a Computational Approach. Molecules. 2020; 25(23):5543. https://doi.org/10.3390/molecules25235543
Chicago/Turabian StyleS., Udhaya Kumar, Srivarshini Sankar, Salma Younes, Thirumal Kumar D., Muneera Naseer Ahmad, Sarah Samer Okashah, Balu Kamaraj, Abeer Mohammed Al-Subaie, George Priya Doss C., and Hatem Zayed. 2020. "Deciphering the Role of Filamin B Calponin-Homology Domain in Causing the Larsen Syndrome, Boomerang Dysplasia, and Atelosteogenesis Type I Spectrum Disorders via a Computational Approach" Molecules 25, no. 23: 5543. https://doi.org/10.3390/molecules25235543