
Combining High-Pressure Perturbation with NMR Spectroscopy for a Structural and Dynamical Characterization of Protein Folding Pathways



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. High Pressure NMR Instrumentation
3. “Global” Thermodynamic and Kinetic Parameters for Folding/Unfolding Reactions Obtained from 1D High-Pressure NMR Spectroscopy
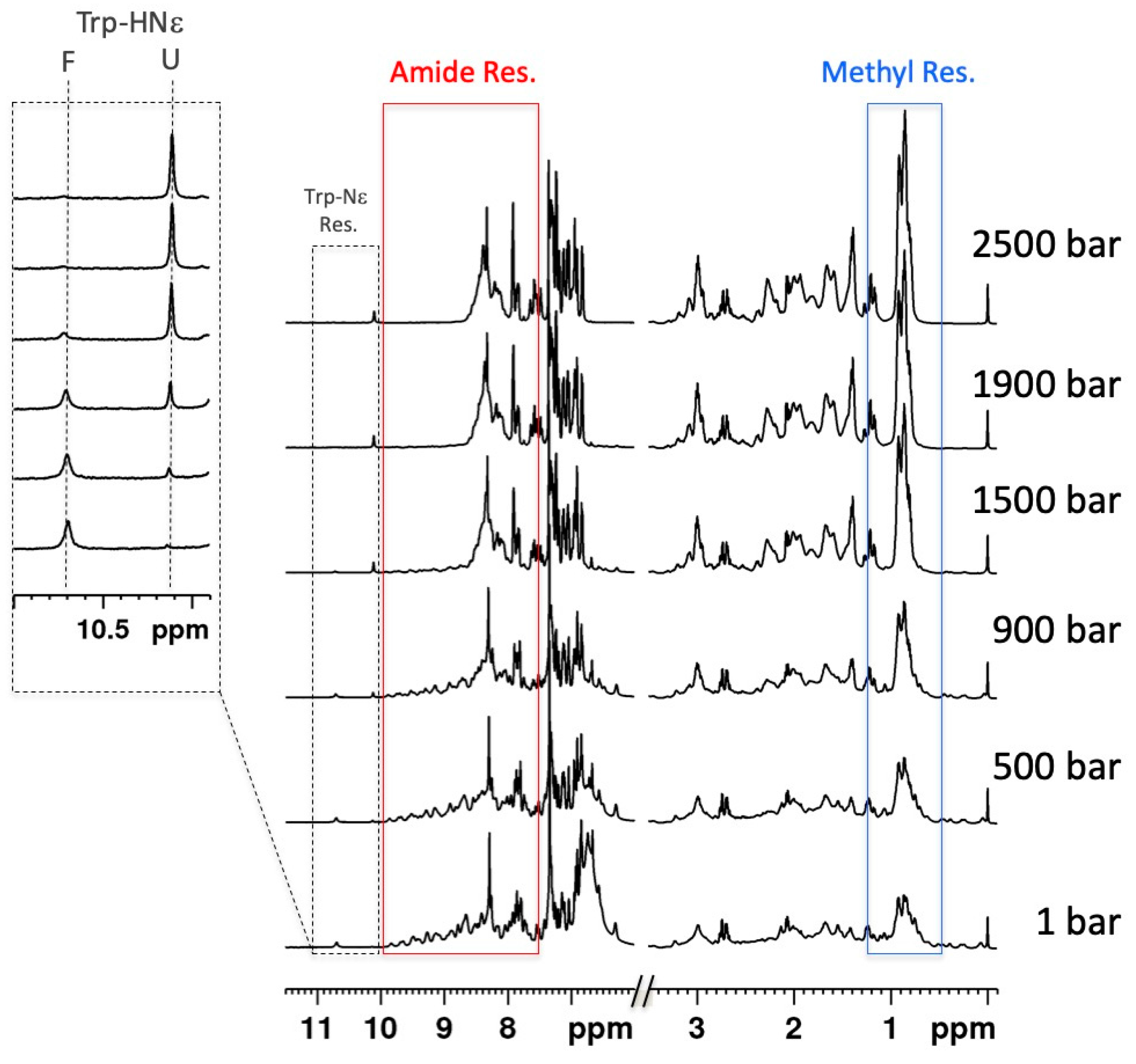
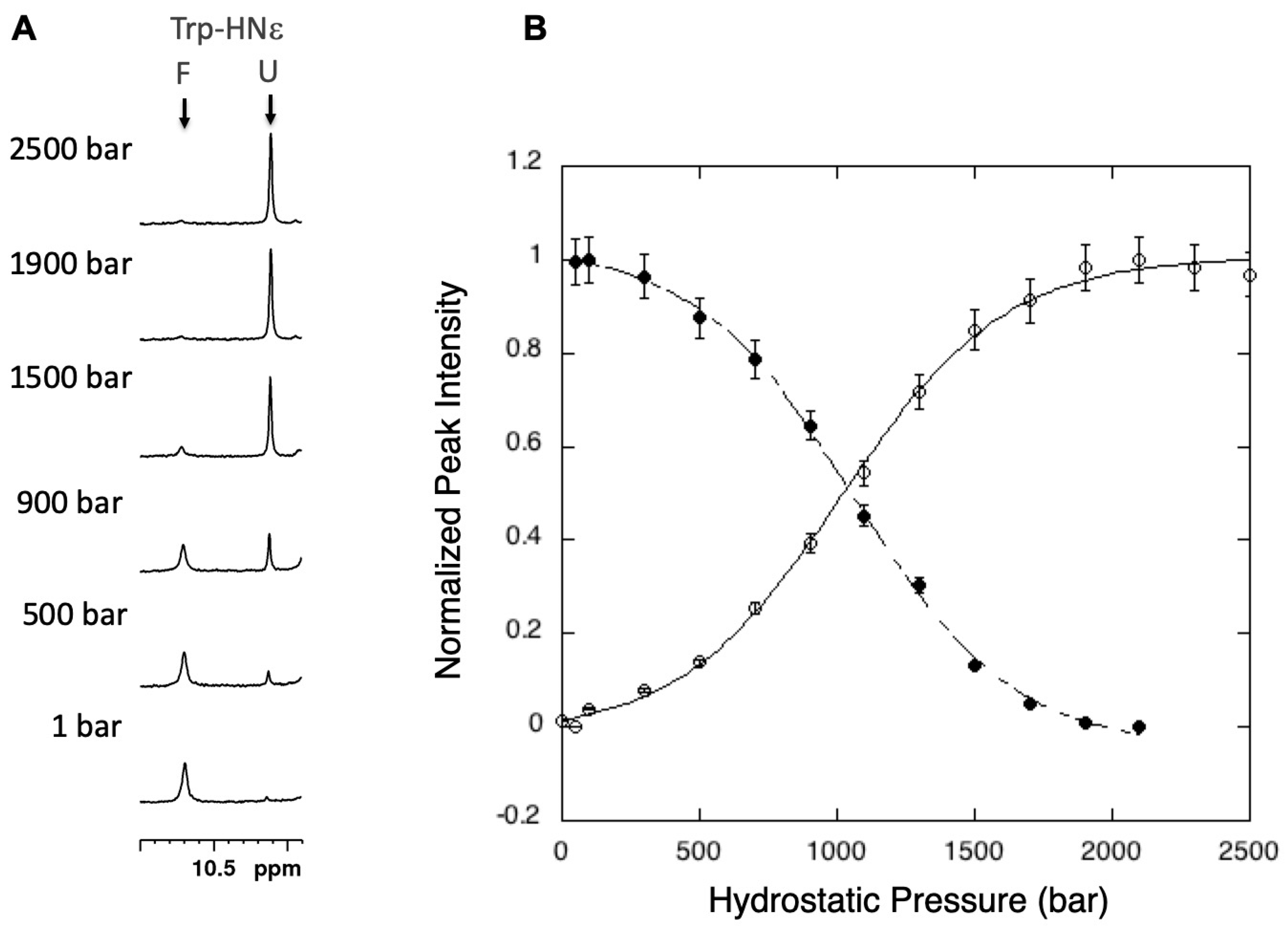
3.1. Steady-State Measurements of Global Thermodynamic Parameters with 1D High-Pressure NMR
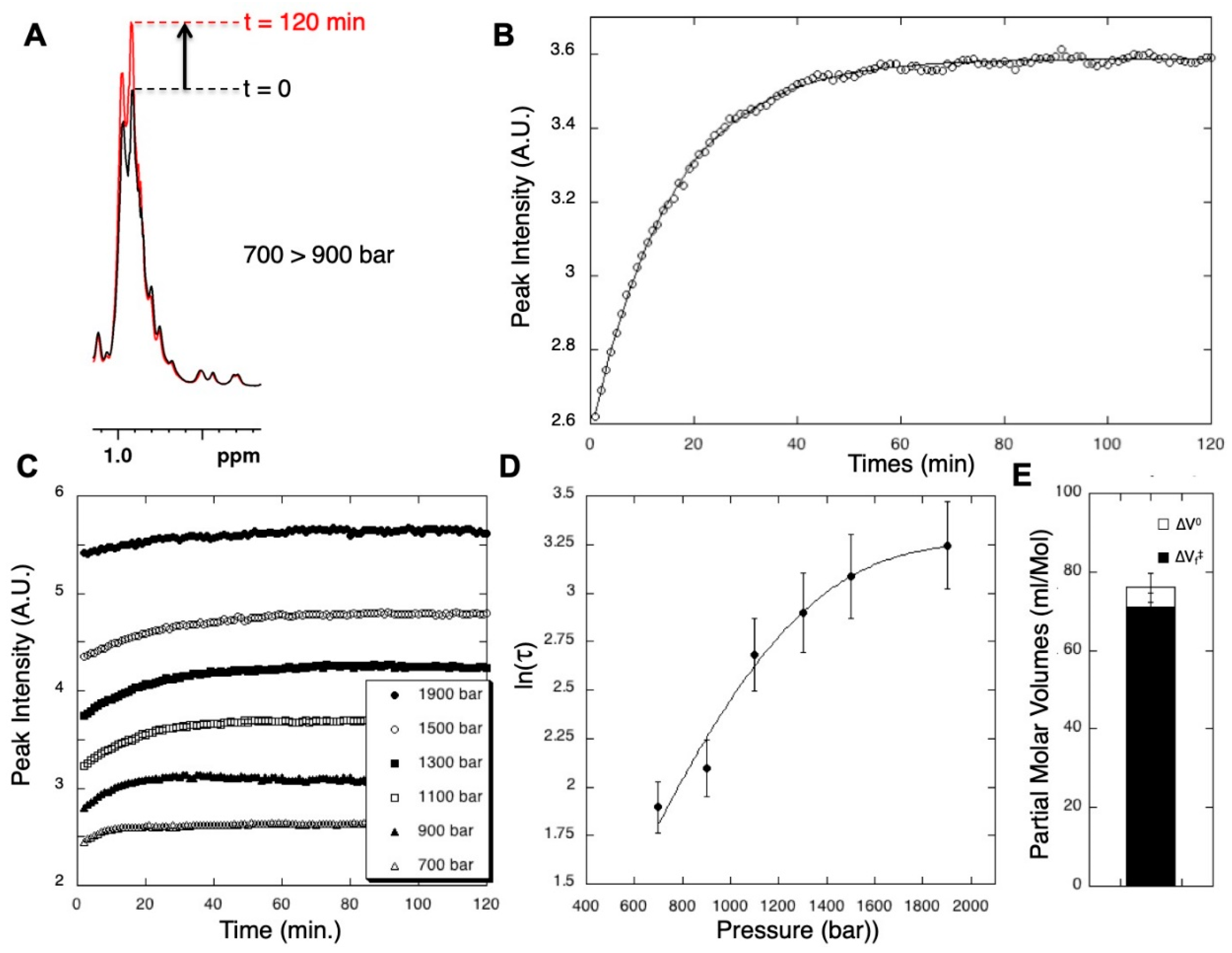
3.2. Measurements of Global Kinetic Parameters of the Folding/Unfolding Reaction with 1D High-Pressure NMR
4. “Local” Thermodynamic and Kinetic Parameters for Folding/Unfolding Reactions Obtained from 2D High-Pressure NMR Spectroscopy
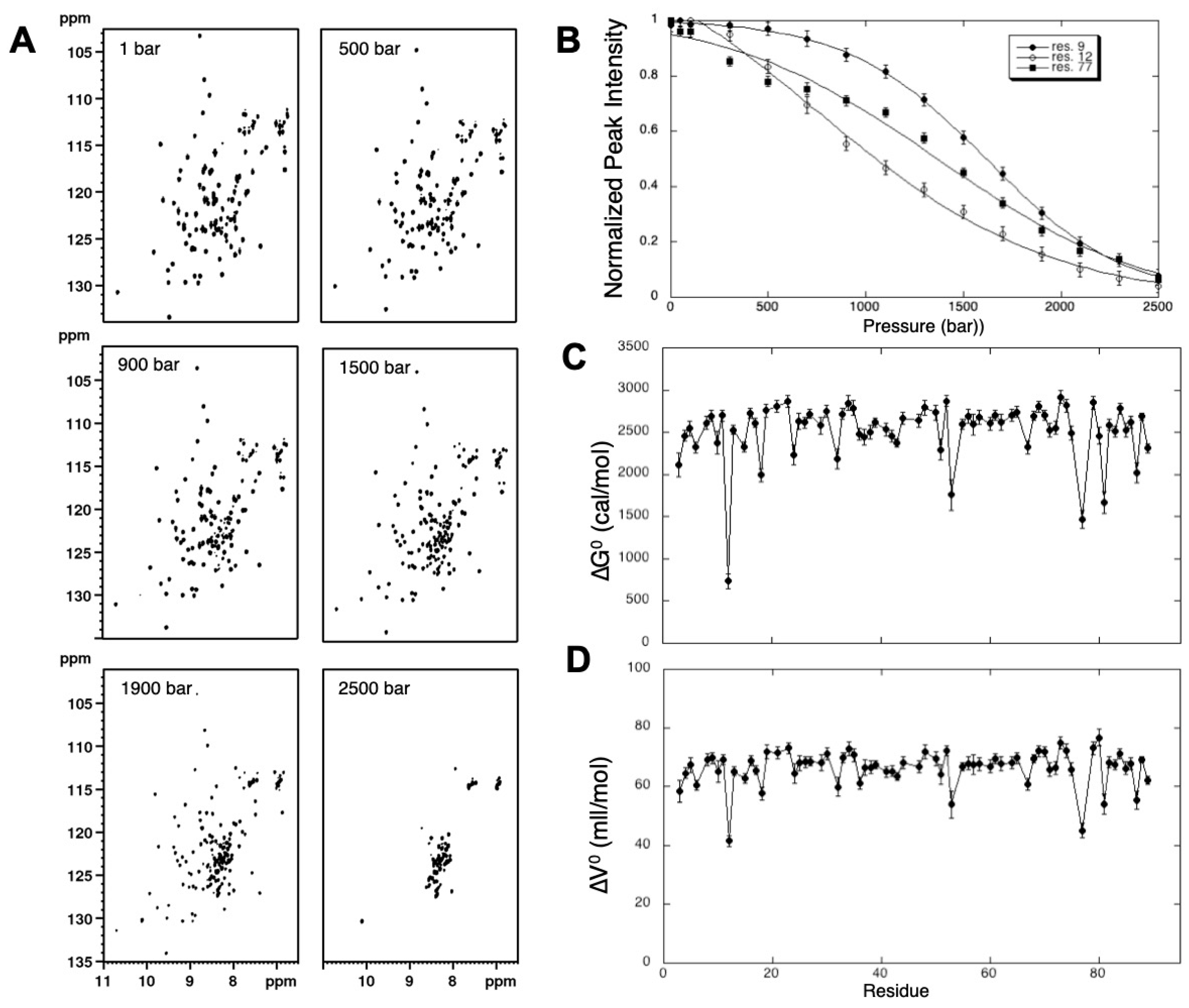
4.1. Measurement of “Local” Thermodynamic Parameters for Folding/Unfolding Reaction: Tracking Folding Intermediates in the Protein Energy Landscape with 2D High-Pressure NMR
4.2. Measurements of “Local” Kinetic Parameters of the Folding/Unfolding Reaction with 2D High-Pressure NMR
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations and Nomenclature
References
- Anfinsen, C.B. Principles that govern the folding of protein chains. Science 1973, 181, 223–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onuchic, J.N.; Nymeyer, H.; Garcia, A.E.; Chahine, J.; Socci, N.D. The energy landscape theory of protein folding: Insights into folding mechanisms and scenarios. Adv. Protein Chem. 2000, 53, 87–152. [Google Scholar] [PubMed]
- Bryngelson, J.D.; Wolynes, P.G. Spin glasses and the statistical mechanics of protein folding. Proc. Natl. Acad. Sci. USA 1987, 84, 7524–7528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shea, J.E.; Brooks, C.L., III. From folding theories to folding proteins: A review and assessment of simulation studies of protein folding and unfolding. Annu. Rev. Phys. Chem. 2001, 52, 499–535. [Google Scholar] [CrossRef] [PubMed]
- Leopold, P.E.; Montal, M.; Onuchic, J.N. Protein folding funnels: A kinetic approach to the sequence-structure relationship. Proc. Natl. Acad. Sci. USA 1992, 89, 8721–8725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daggett, V.; Fersht, A.R. Is there a unifying mechanism for protein folding? TIBS 2003, 28, 18–25. [Google Scholar] [CrossRef]
- Cheung, M.S.; Chavez, L.L.; Onuchic, J.N. The energy landscape for protein folding and possible connections to function. Polymer 2004, 45, 547–555. [Google Scholar] [CrossRef]
- Onuchic, J.N.; Wolynes, P.G. Theory of protein Folding. Curr. Opin. Struct. Biol. 2004, 14, 70–75. [Google Scholar] [CrossRef]
- Kamatari, Y.O.; Kitahara, R.; Yamada, H.; Yokoyama, S.; Akasaka, K. High-pressure NMR spectroscopy for characterizing folding intermediates and denatured states of proteins. Methods 2004, 34, 133–143. [Google Scholar] [CrossRef]
- Akasaka, K. Probing conformational fluctuations of proteins by pressure perturbation. Chem. Rev. 2006, 106, 1814–1835. [Google Scholar] [CrossRef]
- Akasaka, K.; Kitahara, R.; Kamatari, Y.O. Exploring the folding energy landscape with pressure. Arch. Biochem. Biophys. 2013, 531, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Roche, J.; Caro, J.A.; Norberto, D.R.; Barthe, P.; Roumestand, C.; Schlessman, J.L.; Garcia, A.E.; Garcia-Moreno, B.; Royer, C.A. Cavities determine the pressure unfolding of proteins. Proc. Natl. Acad. Sci. USA 2012, 109, 6945–6950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rouget, J.B.; Aksel, T.; Roche, J.; Saldana, J.L.; Garcia, A.E.; Barrick, D.; Royer, C.A. Size and sequence and the volume change of protein folding. J. Am. Chem. Soc. 2011, 133, 6020–6027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lerch, M.T.; Horwitz, J.; McCoy, J.; Hubbell, W.L. Circular dichroism and site-directed spin labeling reveal structural and dynamical features of high-pressure states of myoglobin. Proc. Natl. Acad. Sci. USA 2013, 110, 4714–4722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dellarole, M.; Royer, C.A. High-pressure fluorescence applications. Methods Mol. Biol. 2014, 1076, 53–74. [Google Scholar]
- Dwyer, C.L. High Pressure NMR and IR Spectroscopy in Organometallic Chemistry. In Compréhensive Organometallic Chemistry III—From Fundamentals to Applications, 3rd ed.; Michael, D., Robert, P., Crabtree, H., Eds.; Elsevier Ltd.: Amsterdam, The Netherlands, 2007; Volume 1, pp. 483–507. [Google Scholar]
- Torrent, J.; Rubens, P.; Ribó, M.; Heremans, K.; Vilanova, M. Pressure versus temperature unfolding of ribonuclease A: An FTIR spectroscopic characterization of 10 variants at the carboxy-terminal site. Protein Sci. 2001, 10, 725–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolini, C.; Ravindra, R.; Ludolph, B.; Winter, R. Characterization of the temperature- and pressure-induced inverse and reentrant transition of the minimum elastin-like polypeptide GVG(VPGVG) by DSC, PPC, CD, and FT-IR spectroscopy. Biophys. J. 2004, 86, 1385–1392. [Google Scholar] [CrossRef]
- Smeller, L. Pressure-temperature phase diagrams of biomolecules. Biochim. Biophys. Acta 2002, 1595, 11–29. [Google Scholar] [CrossRef]
- Benedek, G.B.; Purcell, E.M. Nuclear magnetic resonance in liquids under high pressure. J. Chem. Phys. 1954, 22, 2003–2012. [Google Scholar] [CrossRef]
- Ballard, L.; Reiner, C.; Jonas, J. High-resolution NMR probe for experiments at high-pressures. J. Magn. Res. 1996, 123, 81–86. [Google Scholar] [CrossRef]
- Ballard, L.; Yu, A.; Reiner, C.; Jonas, J. A high-pressure, high-resolution NMR probe for experiments at 500 MHz. J. Magn. Res. 1998, 133, 190–193. [Google Scholar] [CrossRef] [PubMed]
- Jonas, J. High-resolution nuclear magnetic resonance studies of proteins. Biochem. Biophys. Acta 2002, 1595, 145–159. [Google Scholar] [CrossRef]
- Jiri, J. High-Pressure Studies Using NMR Spectroscopy. In Encyclopedia of Spectroscopy and Spectrometry, 2nd ed.; Lindon, J.C., Tranter, G.E., Koppenaal, D., Eds.; Academic Press (Elsevier Ltd.): Cambridge, MA, USA, 2010; pp. 854–864. [Google Scholar]
- Castro, P.; Delsuc, M.A. An NMR probe with a high-pressure chamber made from composite materials. Magn. Reson. Chem. 1998, 36, 833–838. [Google Scholar] [CrossRef]
- Meier, T.; Khandarkhaeva, S.; Petitgirard, S.; Körber, T.; Lauerer, A.; Rössler, E.; Dubrovinsky, L. NMR at pressures up to 90 GPa. J. Magn Reson. 2018, 292, 44–47. [Google Scholar] [CrossRef]
- Fourme, R.; Girard, E.; Akasaka, K. High-pressure macromolecular crystallography and NMR: Status, achievements and prospects. Curr. Opin. Struct. Biol. 2012, 22, 636–642. [Google Scholar] [CrossRef] [PubMed]
- Vanni, H.; Earl, W.L.; Merbach, A.E. Two approaches to high-resolution high-pressure nuclear magnetic resonance. J. Magn. Reson. 1978, 29, 11–19. [Google Scholar] [CrossRef]
- Roe, D.J. Sapphire NMR tube for high-resolution studies at elevated pressure. J. Magn. Reson. 1985, 63, 388–391. [Google Scholar]
- Yamada, H. Pressure-resisting glass cell for high-pressure, high resolution NMR measurements. Rev. Sci. Instrum. 1974, 45, 640–642. [Google Scholar] [CrossRef]
- Yamada, H.; Nishikawa, K.; Honda, M.; Shimura, T.; Akasaka, K.; Tabayashi, K. Pressure-resisting cell for high-pressure, high-resolution nuclear magnetic resonance measurements at very high magnetic fields. Rev. Sci. Instrum. 2001, 72, 1463–1471. [Google Scholar] [CrossRef]
- Kitahara, R.; Royer, C.; Yamada, H.; Boyer, M.; Saldana, J.L.; Akasaka, K.; Roumestand, C. Equilibrium and pressure-jump relaxation studies of the conformational transitions of P13MTCP1. J. Mol. Biol. 2002, 320, 609–628. [Google Scholar] [CrossRef]
- Vajpai, N.; Nisius, L.; Wiktor, M.; Grzesiek, S. High-pressure NMR reveals close similarity between cold and alcohol protein denaturation in ubiquitin. Proc. Natl. Acad. Sci. USA 2013, 110, 368–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lassalle, M.W.; Akasaka, K. The use of high-pressure nuclear magnetic resonance to study protein folding. Methods Mol. Biol. 2007, 350, 21–38. [Google Scholar] [PubMed]
- Akasaka, K. High pressure NMR spectroscopy. Subcell. Biochem. 2015, 72, 707–721. [Google Scholar] [PubMed]
- Urbauer, J.L.; Ehrhardt, M.R.; Bieber, R.J.; Flynn, P.F.; Wand, A.J. High-resolution triple-resonance NMR spectroscopy of a novel calmodulin-peptide complex at kilobar pressures. J. Am. Chem. Soc. 1996, 118, 11329–11330. [Google Scholar] [CrossRef]
- Wand, A.J.; Earhardt, M.R.; Urbauer, J.L. Apparatus and Method for High Pressure NMR Spectroscopy. U.S. Patent 6,362,624, 26 March 2002. [Google Scholar]
- Arnold, M.R.; Kalbitzer, H.R.; Kremer, W. High-sensitivity sapphire cells for high pressure NMR spectroscopy on proteins. J. Magn. Reson. 2003, 161, 127–131. [Google Scholar]
- Erlach, M.B.; Munte, C.E.; Kremer, W.; Hartl, R.; Rochelt, D.; Niesner, D.; Kalbitzer, H.R. Ceramic cells for high pressure NMR spectroscopy of proteins. J. Magn. Reson. 2010, 204, 196–199. [Google Scholar] [CrossRef]
- Peterson, R.W.; Wand, J.A. Self-contained high-pressure cell, apparatus, and procedure for the preparation of encapsulated proteins dissolved in low viscosity fluids for nuclear magnetic resonance spectroscopy. Rev. Sci. Instrum. 2005, 76, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Brunner, E.; Arnold, M.R.; Kremer, W.; Kalbitzer, H.R. Pressure stability of phospholipid bicelles: Measurement of residual dipolar couplings under extreme conditions. J. Biomol. NMR 2001, 21, 173–176. [Google Scholar] [CrossRef]
- Fu, Y.; Wand, A.J. Partial alignment and measurement of residual dipolar couplings of proteins under high hydrostatic pressure. J. Biomol. NMR 2013, 56, 353–357. [Google Scholar] [CrossRef]
- Sibille, N.; Dellarole, M.; Royer, C.; Roumestand, C. Measuring residual dipolar couplings at high hydrostatic pressure: Robustness of alignment media to high pressure. J. Biomol. NMR 2014, 58, 9–16. [Google Scholar] [CrossRef]
- Herrada, I.; Barthe, P.; Vanheusden, M.; DeGuillen, K.; Mammri, L.; Delbecq, S.; Rico, F.; Roumestand, C. Monitoring Unfolding of Titin I27 Single and Bi Domain with High-Pressure NMR Spectroscopy. Biophys. J. 2018, 11, 341–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lassalle, M.W.; Yamada, H.; Akasaka, K. The pressure-temperature free energy-landscape of staphylococcal nuclease monitored by (1)H-NMR. J. Mol. Biol. 2000, 298, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Hata, H.; Kono, R.; Fujidawa, M.; Kitahara, R.; Kamatari, Y.O.; Akasaka, K.; Xu, Y. High pressure NMR study of dihydrofolate reductase from deep-sea bacterium Moritella profunda. Cell Mol. Biol. 2004, 50, 311–316. [Google Scholar] [PubMed]
- Ravindra, R.; Winter, R. On the temperature-pressure free-energy landscape of proteins. ChemPhysChem 2003, 4, 359–365. [Google Scholar] [CrossRef]
- Fersht, A.R. Characterizing transition states in protein folding: An essential step in the puzzle. Curr. Opin. Struct. Biol. 1995, 5, 79–84. [Google Scholar] [CrossRef]
- Saotome, T.; Doret, M.; Kulkarni, M.; Yang, Y.S.; Barthe, P.; Kuroda, Y.; Roumestand, C. Folding of the Ig-Like Domain of the Dengue Virus Envelope Protein Analyzed by High-Hydrostatic-Pressure NMR at a Residue-Level Resolution. Biomolecules 2019, 9, 309. [Google Scholar] [CrossRef] [Green Version]
- Vidugiris, G.J.A.; Markley, J.L.; Royer, C.A. Evidence for a molten globule-like transition state in protein folding from determination of activation volumes. Biochemistry 1995, 34, 4909–4912. [Google Scholar] [CrossRef]
- Kremer, W.; Arnold, M.; Munte, C.E.; Hartl, R.; Erlach, M.B.; Koehler, J.; Meier, A.; Kalbitzer, H.R. Pulsed pressure perturbations, an extra dimension in NMR spectroscopy of proteins. J. Am. Chem. Soc. 2011, 133, 13646–13651. [Google Scholar] [CrossRef]
- Englander, S.W.; Mayne, L.; Krishna, M.M.G. Protein folding and misfolding: Mechanism and principles. Q. Rev. Biophys. 2007, 40, 287–326. [Google Scholar] [CrossRef] [Green Version]
- Lindberg, M.O.; Oliveberg, M. Malleability of protein folding pathways: A simple reason for complex behavior. Curr. Opin. Struct. Biol. 2007, 17, 21–29. [Google Scholar] [CrossRef]
- Englander, S.W.; Kallenbach, N.R. Hydrogen exchange and structural dynamics of proteins and nucleic acids. Q. Rev. Biophys. 1983, 4, 521–655. [Google Scholar] [CrossRef] [PubMed]
- Fowler, S.B.; Clarke, J. 2001. Mapping the folding pathway of an immunoglobulin domain: Structural detail from Phi value analysis and movement of the transition state. Structure 2001, 9, 355–366. [Google Scholar] [CrossRef]
- Marszalek, P.E.; Lu, H.; Li, H.; Carrion-Vazquez, M.; Oberhauser, A.F.; Schulten, K.; Fernandez, J.M. Mechanical unfolding intermediates in titin modules. Nature 1999, 402, 100–103. [Google Scholar] [CrossRef] [PubMed]
- Rico, F.; Gonzalez, L.; Casuso, I.; Puig-Vidal, M.; Scheuring, S. High-speed force spectroscopy unfolds titin at the velocity of molecular dynamics simulations. Science 2013, 342, 741–743. [Google Scholar] [CrossRef] [Green Version]
- Roche, J.; Dellarole, M.; Caro, J.A.; Guca, E.; Norberto, D.R.; Yang, Y.S.; Garcia, A.E.; Roumestand, C.; García-Moreno, B.; Royer, C.A. Remodeling of the folding free-energy landscape of staphylococcal nuclease by cavity-creating mutations. Biochemistry 2012, 51, 9535–9546. [Google Scholar] [CrossRef]
- de Oliveira, G.A.; Silva, J.L. A hypothesis to reconcile the physical and chemical unfolding of proteins. Proc. Natl. Acad. Sci. USA 2015, 112, E2775–E2784. [Google Scholar] [CrossRef] [Green Version]
- Fossat, M.J.; Dao, T.P.; Jenkins, K.; Dellarole, M.; Yang, Y.S.; McCallum, S.A.; Garcia, A.E.; Barrick, D.; Roumestand, C.; Royer, C.A. High-Resolution Mapping of a Repeat Protein Folding Free Energy Landscape. Biophys. J. 2016, 111, 2368–2376. [Google Scholar] [CrossRef]
- Roder, H.; Elove, G.A.; Englander, S.W. Structural characterization of folding intermediates in cytochrome-c by H-exchange labeling and proton NMR. Nature 1988, 335, 700–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, Y.; Sosnick, T.R.; Mayne, L.; Englander, S.W. Protein folding intermediates-native state hydrogen exchange. Science 1995, 269, 192–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, Y.; Milne, J.S.; Mayne, L.; Englander, S.W. Primary structure effects on peptide group hydrogen exchange. Proteins 1993, 17, 75–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuentes, E.J.; Wand, A.W. Local stability and dynamics of apocytochrome b562 examined by the dependence of hydrogen exchange on hydrostatic pressure. Biochemistry 1998, 37, 9877–9883. [Google Scholar] [CrossRef] [PubMed]
- Roche, J.; Dellarole, M.; Caro, J.A.; Norberto, D.R.; Garcia, A.E.; Garcia-Moreno, B.; Roumestand, C.; Royer, C.A. Effect of internal cavities on folding rates and routes revealed by real-time pressure-jump NMR spectroscopy. J. Am. Chem. Soc. 2013, 135, 14610–14618. [Google Scholar] [CrossRef] [PubMed]
- Frydman, L.; Scherf, T.; Lupulescu, A. The acquisition of multidimensional NMR spectra within a single scan. Proc. Natl. Acad. Sci. USA 2002, 99, 15858–15862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schanda, P.; Brutscher, B. Very fast two-dimensional NMR spectroscopy for real-time investigation of dynamic events in proteins on the time scale of seconds. J. Am. Chem Soc. 2005, 127, 8014–8015. [Google Scholar] [CrossRef]
- Gal, M.; Schanda, P.; Brutscher, B.; Frydman, L. UtraSOFAST HMQC-NMR and repetitive acquisition of 2D protein spectra at Hz rates. J. Am. Chem. Soc. 2007, 129, 1372–1377. [Google Scholar] [CrossRef]
- Schanda, P.; Forge, V.; Brutscher, B. Protein folding and unfolding studied at atomic resolution by fast two-dimensional NMR spectroscopy. Proc. Natl. Acad. Sci. USA 2007, 104, 11257–11262. [Google Scholar] [CrossRef] [Green Version]
- Hyberts, S.G.; Arthanari, H.; Wagner, G. Applications of Non-Uniform Sampling and Processing. In Novel Sampling Approaches in Higher Dimensional NMR (Topics in Current Chemistry, vol. 316); Billeter, M., Orekhov, V., Eds.; Springer: Berlin, Germany, 2012; pp. 125–128. [Google Scholar]
- Hyberts, S.G.; Arthanari, H.; Robson, S.A.; Wagner, G. Perspectives in magnetic resonance: NMR in the post-FFT era. J. Magn. Reson. 2014, 241, 60–73. [Google Scholar] [CrossRef] [Green Version]
- Palmer, M.R.; Suiter, C.L.; Henry, G.E.; Rovnyak, J.; Hoch, J.C.; Polenova, T.; Rovnyak, D. Sensitivity of Nonuniform Sampling NMR. J. Phys. Chem. B 2015, 119, 6502–6515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Kitazawa, S.; Peran, I.; Stenzoski, N.; McCallum, S.A.; Raleigh, D.P.; Royer, C.A. High pressure ZZ-exchange NMR reveals key features of protein folding transition states. J. Am. Chem. Soc. 2016, 138, 15260–15266. [Google Scholar] [CrossRef] [Green Version]
- Charlier, C.; Alderson, T.R.; Courtney, J.M.; Ying, J.; Anfinrud, P.; Bax, A. Study of protein folding under native conditions by rapidly switching the hydrostatic pressure inside an NMR cell. Proc. Natl. Acad. Sci. USA 2018, 115, E4169–E4178. [Google Scholar] [CrossRef] [Green Version]
- Kuwata, K.; Li, H.; Yamada, H.; Legname, G.; Prusiner, S.B.; Akasaka, K.; James, T.L. Locally disordered conformer of the hamster prion protein: A crucial intermediate to PrPSc? Biochemistry 2002, 41, 12277–12283. [Google Scholar] [CrossRef] [PubMed]
- Niraula, T.N.; Konno, T.; Li, H.; Yamada, H.; Akasaka, K.; Tachibana, H. Pressure-dissociable reversible assembly of intrinsically denatured lysozyme is a precursor for amyloid fibrils. Proc. Natl. Acad. Sci. USA 2004, 101, 4089–4093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamatari, Y.O.; Yokoyama, S.; Tachibana, H.; Akasaka, K. Pressure-jump NMR study of dissociation and association of amyloid protofibrils. J. Mol. Biol. 2005, 349, 916–921. [Google Scholar] [CrossRef] [PubMed]
- Munte, C.E.; Beck-Erlach, M.; Kremer, W.; Koehler, J.; Kalbitzer, H.R. Distinct conformational states of the alzheimer b-amyloid peptide can be detected by high-pressure NMR spectroscopy. Angew. Chem. Int. Ed. Engl. 2013, 52, 8943–8947. [Google Scholar] [CrossRef]
- Louis, J.M.; Roche. Evolution under drug pressure remodels the folding free-energy landscape of mature HIV-1 protease. J. Mol. Biol 2016, 428, 2780–2792. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dubois, C.; Herrada, I.; Barthe, P.; Roumestand, C. Combining High-Pressure Perturbation with NMR Spectroscopy for a Structural and Dynamical Characterization of Protein Folding Pathways. Molecules 2020, 25, 5551. https://doi.org/10.3390/molecules25235551
Dubois C, Herrada I, Barthe P, Roumestand C. Combining High-Pressure Perturbation with NMR Spectroscopy for a Structural and Dynamical Characterization of Protein Folding Pathways. Molecules. 2020; 25(23):5551. https://doi.org/10.3390/molecules25235551
Chicago/Turabian StyleDubois, Cécile, Isaline Herrada, Philippe Barthe, and Christian Roumestand. 2020. "Combining High-Pressure Perturbation with NMR Spectroscopy for a Structural and Dynamical Characterization of Protein Folding Pathways" Molecules 25, no. 23: 5551. https://doi.org/10.3390/molecules25235551