
Abstract
Aim and objective
Emerging translational evidence suggests that epigenetic alterations (DNA methylation, miRNA expression, and histone modifications) occur after external stimuli and may contribute to exacerbated inflammation and the risk of suffering several diseases including diabetes, cardiovascular diseases, cancer, and neurological disorders. This review summarizes the current knowledge about the harmful effects of high-fat/high-sugar diets, micronutrient deficiencies (folate, manganese, and carotenoids), obesity and associated complications, bacterial/viral infections, smoking, excessive alcohol consumption, sleep deprivation, chronic stress, air pollution, and chemical exposure on inflammation through epigenetic mechanisms. Additionally, the epigenetic phenomena underlying the anti-inflammatory potential of caloric restriction, n-3 PUFA, Mediterranean diet, vitamin D, zinc, polyphenols (i.e., resveratrol, gallic acid, epicatechin, luteolin, curcumin), and the role of systematic exercise are discussed.
Methods
Original and review articles encompassing epigenetics and inflammation were screened from major databases (including PubMed, Medline, Science Direct, Scopus, etc.) and analyzed for the writing of the review paper.
Conclusion
Although caution should be exercised, research on epigenetic mechanisms is contributing to understand pathological processes involving inflammatory responses, the prediction of disease risk based on the epigenotype, as well as the putative design of therapeutic interventions targeting the epigenome.
Similar content being viewed by others


Introduction
Inflammation encompasses a myriad of pathophysiological and immune responses to diverse environmental “insults”, such as toxins or pathogens, in order to facilitate tissue recovery and to maintain homeostasis [1]. These mitigation and reparation processes are mediated by the production and recruitment of cytokines, chemokines, adhesion molecules, and other autocrine/paracrine molecules focused on the local site of damage, thus inducing an acute inflammatory reaction [2]. However, when inflammation persists for a long time, it becomes a chronic condition, triggering a cascade of inflammatory events that eventually lead to durable cellular harms, permanent tissue injury, and organ dysfunction [3]. This state involves the induction of several pro-inflammatory mediators produced predominantly by activated macrophages, including Interleukin 1 beta (IL-1β), tumor necrosis factor-alpha (TNF-α), and interleukin 6 (IL-6) to perpetuate the inflammatory phenotype [4]. The inflammatory signaling is mediated by enzymes and adhesion molecules as well as the activation of nuclear factor kappa β (NF-κB) and other transcription factors as central regulators where the immune system and associated cells also play an orchestrated role [5]. The identification of factors involved in the onset and progression of inflammation is essential for the better understanding of inflammation-related disorders and the search for therapeutic targets.
Emerging evidence suggests that epigenetic processes affecting gene expression without changes in the nucleotide sequence may contribute to the pathophysiology of inflammatory processes [6]. In this context, it has been documented that epigenetic modifications (such as DNA methylation in CpG islands, chromatin remodeling by histone tail modifications, and non-coding RNA expression) occur after environmental stimuli and play a fundamental role in inflammatory gene transcription [7]. Indeed, integrative epigenome-wide association studies (EWAS) using large-scale bioinformatics analysis have reported different epigenetic marks related to several circulatory inflammation markers [8]. Therefore, epigenetic signature alterations may exacerbate inflammatory responses and influence the risk of chronic inflammatory disease, including diabetes, cardiovascular diseases, cancer, and neurological disorders [9]. However, elucidation of the specific epigenetic pathways involved in the modulation of the inflammasome and disease susceptibility remain largely unknown.
This review summarizes the current knowledge about the effects of obesity, infections (comprising bacterial and viral agents), smoking/excessive alcohol drinking, chronic stress, climate, pollution and other environmental factors including physical activity and the role of nutrients and dietary bioactive compounds on inflammation status through epigenetic mechanisms, and how these events may influence chronic disease development. This knowledge may allow the implementation of personalized nutrition based on inflammatory epigenetic signatures for the prevention and management of chronic inflammatory diseases.
Nutrition/dietary bioactive compounds
Nutritional factors have been related with a pro-inflammatory potential [10]. Particularly, the consumption of Western-type diets evokes a state of chronic metabolic inflammation “metainflammation” that contribute to the development of many prevalent non-communicable diseases [11]. In this context, complex interactions among food components and the epigenome modifications shape the cellular phenotype by a dynamic regulation of gene expression from some time ago [12]. Thus, epigenetic phenomena may account for the observed relationships between diet, inflammation, and diet‐related diseases [13].
Transgenerational animal studies have shown an increased inflammatory response after the consumption of high-fat/high calorie diets by altering miRNA expression and DNA methylation processes [14, 15]. In individuals at high-cardiovascular risk, the epigenetic signature associated with the consumption of fruit juice (rich in fructose) was enriched for pro-inflammatory pathways [16]. Consistently, compelling evidence supports the association of excessive fructose consumption with the presence of non-alcoholic fatty liver disease (NAFLD) involving alteration of transcriptomic and epigenetic mechanisms underlying lipid metabolism deregulation, increased liver fat accumulation, and inflammation [17]. Additionally, maternal low-dietary protein regulated miRNA expression targeting genes mapped to inflammatory-related pathways and metabolic health in offspring mice [18].
The role of dietary fatty acids in epigenetic and inflammatory processes has also been explored [19] and is summarized (Table 1). In this context, the trans fatty acid “elaidate” induced pro-inflammatory and adipogenic transcriptional profiles through methylation changes in cultured human monocytes [20]. Likewise, the saturated fatty acids “stearate” and “palmitate” were associated with DNA hypermethylation of the PPARγ1 gene promoter, which was identified as a critical determinant of pro-inflammatory activation and insulin resistance in macrophages [21]. Of note, DNA methylation levels of the TNF promoter were associated with adiposity measures and n-6 polyunsaturated fatty acid (PUFA) intake, suggesting a nutriepigenomic regulation of this recognized pro-inflammatory marker [22]. Remarkably, it has been reported that short-chain fatty acids (produced by the gut microbiota) can also target the epigenome to regulate host pathophysiological processes, including inflammation [23].
On the other hand, the anti-inflammatory potential of essential fatty acids mediated by epigenetic phenomena has also been analyzed (Table 1). For example, n-3 PUFA supplementation was associated with changes in DNA methylation profiles in blood leukocytes related to pathways involved in inflammatory and immune responses, among others [24]. In addition, in vitro analyses revealed that the anti-inflammatory effect of oleic acid, a monounsaturated fatty acid (MUFA), was related to DNA methylation signatures [25].
In relation to dietary fiber, an epigenome-wide association study in African American adolescents showed that DNA methylation levels at LPCAT1 and RASA3 genes (playing a role in colon cancer) were associated with fiber consumption, adiposity, and inflammation [26]. Concerning micronutrients, unbalanced intakes of folate [27], manganese [28], and carotenoids [29] have been associated with changes in the methylation status of candidate inflammatory genes in some populations.
On the other hand, dietary bioactive compounds are known to negatively regulate several inflammatory pathways [30]. Thus, it has been reported that plant-derived polyphenols exert anti-inflammatory properties by interfering with immune cell regulation, synthesis of pro-inflammatory cytokines, and gene expression, which are associated with health benefits for different chronic diseases related to inflammation such as obesity, type 2 diabetes (T2D), neurodegeneration, cardiovascular disease (CVD), and some types of cancer [31]. Polyphenols are compounds with phenolic structural features naturally distributed in fruits, vegetables, whole grains as well as in tea, chocolate, and red wine [32]. These include phenolic acids, flavonoids (i.e. epicatechin, luteolin, and fisetin) and phenolic amides, and other non-flavonoid polyphenols found in foods such as resveratrol, gallic acid and its derivatives, and curcumin [32]. Interestingly, studies have revealed that the protective effects of polyphenols on inflammation are partially modulated via epigenetic modifications, thus contributing to the current understanding of the molecular mechanisms of action of these biologically active compounds (Table 2).
For instance, the daily intake of grape extract resveratrol for 1 year modified the expression of a group of miRNAs involved in the regulation of the inflammatory response in PBMCs from hypertensive male patients with T2DM, including miR-21, miR-181b, miR-663, miR-30c2, miR-155, and miR-34a [33]. Moreover, mango (Mangifera indica L.) polyphenols, containing gallic acid and gallotanins, reduced inflammation in two in vitro and in vivo models of intestinal colitis by regulating the PI3K/AKT/mTOR pathway partially through up-regulation of miR-126 expression [34]. Additionally, the administration of (−)-epicatechin attenuated the high-glucose-induced inflammatory response in human monocytes by epigenetic modulation of H3K9 acetylation and H3K4 dimethylation [35], notably the combination of luteolin- and fisetin-induced anti-inflammatory effects in human monocytic cells under high-glucose concentrations involving histone acetyltransferase/histone deacetylase modifications [36]. In a similar high-glucose condition, curcumin decreased the production of pro-inflammatory cytokines by inhibiting histone acetylation in monocytes [37].
In addition to polyphenols, the anti-inflammatory role of other dietary factors and specific functional foods has also been assessed in different experimental models. In this regard, extra virgin olive oil (EVOO) and Nigella sativa oil displayed anti-inflammatory activities in lipopolysaccharide (LPS)-exposed human macrophages through epigenetic mechanisms [38]. In this study, the administration of both oils reverted the altered expressions of DNMT3A and HDAC1 to normal levels under inflammatory conditions, with an additional role of EVOO in the reduction of global methylation. Also, a nutritional intervention with Mediterranean diet plus EVOO influenced the methylation status of genes involved in inflammatory pathways in PBMCs [39]. Similarly, higher adherence to Mediterranean diet was positively associated with the methylation of a set of genes related to inflammation and immunocompetence in high cardiovascular risk volunteers [40].
Meanwhile, increased DNA methylation of the tumor necrosis factor (TNF) gene was found after supplementation of grapefruits extracts in rats, which may contribute to reduce chronic low-grade systemic inflammation in obesity [41]. Also, ginger extracts ameliorated obesity and inflammation in white adipose tissue of rats fed a high-fat diet via regulation of miR-21/132 expression and AMPK activation [42].
A maternal diet rich in methionine pathway metabolites induced global hypermethylation on T cells from F1 C57Bl/6 mice, which was associated with lower expression of inflammatory T cell chemokine receptors (CCR2, CCR5, CXCR3) and cytokines (TNF, IL2, and IL4) [43]. Furthermore, selenium and coenzyme Q10 supplementation reduced pro-inflammatory markers in healthy elderly participants through changes in plasma miRNA expression [44]. Vitamin D has been reported to down regulate inflammation-linked miRNA expression in adipocytes both in vitro and in vivo [45]. Besides, maternal high-zinc diet attenuated intestinal inflammation in offspring chicks by epigenetic changes [46].
Further, increasing research has provided evidence about the long-lasting epigenetic effects of calorie restriction which mediates expression of genes related to immuno-metabolic processes that may enhance quality of life and extend lifespan, with important applications for the prevention of chronic inflammatory diseases [47]. Interestingly, methylation profiles in inflammatory genes have been proposed to be used as epigenetic biomarkers concerning adiposity and metabolic outcomes in response to low-calorie diets (30% of energy restriction, 55% of energy as carbohydrates, 15% as proteins, and 30% as lipids) [48, 49]. Moreover, findings from the RESMENA and DIOGENES trials have evidenced a decline in inflammation [50, 51] and improvements of metabolic syndrome features [52, 53] after following an energy-restricted diet (− 30% energy of the calculated requirements, 40% total energy value from carbohydrates, 30% from proteins and 30% from lipids), which was partially explained by epigenetic signatures.
Physical activity
Studies have shown that some physical exercise may exert anti-inflammatory effects through epigenetic regulation depending on the type of activity, exercise duration, body composition, gender, and age [54]. However, excessive physical activity (i.e. in 10-km marathons, or treadmill runs for 120 min followed by a 5-km time trial in a fasted condition) can also induce inflammation [54]. Thus, in a population-based cohort study, substituting light-intensity physical activity (< 3 METs; acceleration intensities 1–3) for sedentary time was associated with higher methylation of the ASC gene, a potential biomarker of systemic inflammation [55]. Also, aerobic capacity, as measured by peak oxygen uptake (17.31 ml/kg/min), was positively associated with increased ASC methylation as well as with decreased plasma IL-1β levels in stable outpatients with heart failure, suggesting that inflammatory processes may influence aerobic capacity [56]. Furthermore, interval walking training (IWT) increased NFKB2 gene promoter methylation, indicating that this physical regime may epigenetically impact the susceptibility to inflammation [57]. IWT consisted of performing several continuous sets of 3-min low-intensity walking periods at 40% of the peak aerobic capacity, followed by 3-min high-intensity walking periods at > 70% peak aerobic capacity for as many days as possible over a period of 6 months [57]. Of note, diet could interact with physical activity since the supplementation of dried tofu during IWT enhanced NFKB2 gene methylation more than IWT alone, suggesting an immunomodulatory synergistic effect of diet and physical activity via epigenetic modulation [58]. Consistently, higher dose of dairy product intake (1 unit of cheese + 2 units of yogurt) plus IWT produced increases in NFKB1 and NFKB2 gene methylations in older women, suggesting a larger pro-inflammatory cytokine gene suppression effect [59].
In patients with obesity, physical activity (26 mixed aerobic and endurance training sessions of 90 min administered twice a week during 3 months) modulated the overexpression of the inflamma-miR-146a-5p, which was postulated as a biomarker and personalized predictor of the clinical response to physical activity weight-reduction programs in obesity [60]. Surprisingly, acute aerobic exercise (30 min, 75% VO2max) elicited higher elevation of inflammatory miRNAs in obese patients compared to lean individuals [61]. In basketball players, changes in circulating miR-146a after acute exhaustive exercise (the average playing time of every athlete was 260 min for 3 months, peak VO2 ≈35 ml/min/kg) showed linear correlations with levels of the inflammatory marker high-sensitivity C-reactive protein [62]. Moreover, distinct and specific circulating inflammatory miRNA (c-inflammamiRs) signatures were found in plasma samples from active middle-aged males following different doses of acute aerobic exercise (0 h, 24 h, 72 h) 10-km, half-marathon, and marathon races), suggesting an epigenetic mechanism controlling the exercise-induced inflammatory cascade [63]. The results of this study revealed a dose-dependent effect of aerobic exercise on systemic inflammation, with higher levels detected after 10-km race [63]. Compared to age-matched sedentary controls, master athletes (European Veterans Athletics Championships in 2010, Nyiregyhaza, Hungary) had decreasing ageing-related inflammation in skeletal muscle related to lower levels of miR-7, which has been suggested to be involved in chronic inflammation in the elderly [64]. Additionally, acute strenuous exercise (consisting of stepping up and down from a step until complete exhaustion) led to enhanced chronic low-grade inflammation in PBMCs from obese individuals via an imbalance on Histone H4 Acetylation/Histone Deacetylase 2 as compared to lean subjects [65]. Together, these findings suggest that the impact of physical activity in inflammation is dependent of type, intensity, and clinical settings of exercise interventions [54].
Obesity and associated diseases
Obesity is a metabolic condition associated with adipose tissue dysfunction and low-grade systemic inflammation that causally contributes to the development of chronic disorders such as T2D and CVD, where epigenetic mechanisms may be involved [66]. Indeed, it has been reported that the alteration of the adipocyte physiology in obesity might be related to specific alterations in the expression pattern of miRNAs related to inflammatory processes [67]. Also, the adverse effects of the inflammatory state include insulin resistance in the adipose tissue and pancreatic β-cell dysfunction, which may induce epigenetic changes that perpetuate inflammation [68]. In consequence, the resulting hyperglycemia and hyperlipidemia conditions as well as persistent inflammation involving epigenomic deregulation could cause damage to the vasculature with putative risk to develop CVD [69]. In the same time, excessive adiposity negatively impacts immune function and host defense in obese individuals, increasing the susceptibility to infection and related morbidity and mortality [70].
Epigenetic mechanisms involved in obesity-related inflammation are summarized (Table 3). For example, global DNA hypermethylation has been positively associated with increased expression of specific pro-inflammatory genes (including the CCL2 gene) in adipocytes from obese individuals [71]. BMI-discordant twin pair analyses detected methylome deregulations of subcutaneous adipose tissue in obesity that trigger inflammation and may contribute to the development of unhealthy obesity outcomes [72]. Thus, methylation analyses in obese individuals showed significantly lower methylation of four CpGs in the first exon of the TLR4 gene, suggesting epigenetic regulation of inflammatory processes in obesity [73]. Moreover, it was reported that aberrant methylation of the IL6 gene promoter may play a role in the etiology and pathogenesis of excessive body weight in humans [74]. Findings from the Methyl Epigenome Network Association (MENA) project revealed associations between methylation sites in peripheral blood mononuclear cells (PBMCs) and waist circumference, which were located in genes related to inflammation and obesity [75]. Likewise, it was reported that DNA methylation in adipose-derived stem cells was significantly modified by an obese environment, affecting pathways involved in adipogenesis, inflammation, and immunosuppression [76]. Genome-wide DNA methylation analysis in visceral adipose tissue of severely obese men with and without metabolic syndrome detected differentially methylated regions mapped to genes related to inflammation and immunity [77].
Of note, the expression of the NNMT gene, a major methyltransferase enzyme, was positively correlated with markers of inflammation in adipose tissue samples from morbidly obese patients [78]. In addition, a higher expression of DNMT3b methyltransferase was found in adipose tissue macrophages isolated from obese mice, supporting a role for DNMT3b in regulation of macrophage polarization and inflammation in obesity [79]. In adipose tissue of obese mice, gene expression levels of the Dnmt3a methyltransferase were markedly increased, as were many inflammatory cytokines, suggesting that increased expression of Dnmt3a may contribute to obesity-related inflammation [80]. Remarkably, DNA methylation changes of the Klf14 gene (a master regulator of gene expression) provided prediction for chronic inflammation in the adipose tissue of mice suffering obesity and diabetes conditions [81]. Furthermore, altered gene methylation profiles on immune cells were related to impaired metabolism and inflammatory response in a porcine model of obesity [82].
In mice, diet-induced obesity led to hypermethylation of the Ankrd26 gene (previously associated with the development of obesity and T2DM), which in turn, contributed to enhanced secretion of pro-inflammatory mediators in white adipose tissue [83]. Consistently, epigenetic silencing of the ANKRD26 gene by increased promoter methylation correlated with a pro-inflammatory profile and the presence of cardio-metabolic risk factors in peripheral leukocytes from obese individuals [84]. Transgenerational studies detected DNA methylation changes of key inflammatory genes in monocytes from neonates born of obese mothers, underlying an intrauterine epigenetic programming of immune function by maternal obesity [85]. Accordingly, maternal pregravid obesity has been associated with epigenetic modifications altering the inflammatory program of the offspring’s monocytes at birth [86].
A bioinformatic approach identified a total of 23 active microRNAs (miRNAs) and transcription factor regulatory pathways significantly associated with obesity-related inflammation [87]. Also, a set of exosomal miRNAs differentially expressed in abdominal obesity was associated with inflammation [88]. Overweight and obesity led to deregulation of circulating inflammatory miRNAs, which may contribute to the heightened inflammatory state associated with these conditions [89]. In adipocytes and macrophages, inflammation boosted a specific miRNA pattern, with a negative impact on the physiopathology of obesity-induced inflammation [90].
Particularly, circulating miR-374a-5p was characterized as a potential modulator of the inflammatory response in obesity [91]. In vitro analyses unveiled a key role of miR-326 expression in mediating obesity-induced adipose tissue inflammation through regulating the differentiation toward Th17 cells [92]. Also, miR-30 was identified as an important regulator of macrophage polarization in mice, indicating that miR-30 could be a therapeutic target for obesity-induced metabolic inflammation [93]. Besides, adipocyte-secreted exosomal miR-34 was progressively increased with the development of dietary obesity, transmitting signals of nutrient overload to adipose-resident macrophages for exacerbation of obesity-induced systemic inflammation and associated metabolic complications [94]. In the same way, obesity induced an imbalance in macrophage polarization in adipose tissue through miR-155 up-regulation, thus causing chronic inflammation and insulin resistance [95]. Accordingly, obesity-associated inflammation induced miR-155 expression in adipocytes resulting in an increased inflammatory state in these cells [96]. Using an obese mice model, it was observed that the expression of miR-27a increased concomitantly with the activation of pro-inflammatory pathways [97]. Meanwhile, miR-130b contributed to obesity-associated adipose tissue inflammation and insulin resistance in diabetic rodents [98]. Endoplasmic reticulum stress and inflammatory markers were up-regulated in obese patients, showing positive correlations with miR-320 expression in adipose tissue [99]. miR-221 triggered white adipose tissue inflammation and insulin resistance in obesity partially through suppressing SIRT1 [100]. Visceral adipose miR-223 up-regulation modulated macrophage-mediated inflammation in human and murine obesity models [101]. miR-126 and miR-193b were further identified as important regulators of adipose inflammation in human obesity through effects on CCL2 production [102].
In contrast, the anti-inflammatory miR-1934, miR532-5p, and miR-146a were down-regulated in obesity, which promoted inflammation in adipose tissues [103,104,105]. Moreover, down-regulation of miR-706 played a role in increasing adipose tissue inflammation in the offspring during maternal obesity in mice [106]. Likewise, decreased expression of miR-21 was associated with enhanced inflammatory cytokine production in PBMCs from obese individuals [107]. Decreased levels of miR-301a and miR-26a correlated with increased chronic inflammation in circulation in obese mouse models [108, 109].
Furthermore, transcriptional analyses showed associations between expressions of histone deacetylases (HDACs), adiposity indices, and obesity-induced inflammation in adipose tissues from obese women [110]. Interestingly, diet-induced and genetic mouse models of obesity displayed decreased expression of the histone demethylase (HDMs) KDM1A in adipose tissue, which promoted the expression of inflammatory genes, thus contributing to the development of obesity-associated inflammation [111]. Furthermore, it was evidenced that SirT1 expression, an essential nutrient-sensing HDAC, regulated adipose tissue inflammation during overnutrition in rodents and humans [112]. Also, it was demonstrated that lipin 1 (a bifunctional protein that regulates gene transcription and triacylglycerol synthesis) inhibited the secretion of inflammatory factors in adipocytes via repression of NFATc4 transcriptional activity and HDACs recruitment; however, this anti-inflammatory effect was attenuated during obesity [113].
Together, these insights support the role of epigenetic mechanisms underlying obesity-induced inflammation and accompanying chronic diseases, including T2D and CVD [66]. This knowledge opens new possibilities for a potential use of epigenetic signatures as biomarkers for diagnosis, prognosis, and personalization of obesity treatment as well as targets for disease management [67].
Endocrine disrupting chemicals
Endocrine disrupting chemicals (EDCs) are common environmental compounds (i.e. phthalates and bisphenol A) that may induce chronic disease through hormone dysfunction and inflammatory processes [114]. In this regard, exposure to many EDCs can influence the onset or progression of CVD by epigenetic modifications affecting lipid homeostasis and atherosclerosis [115]. Indeed, the association of bisphenol A exposure with CVD and hypertension encompasses endocrine disturbance, induction of oxidative stress and inflammation, and epigenetic phenomena [116].
Concerning the role of epigenetic phenomena in mediating EDCs-induced inflammation, it was reported that mono-(2-ethylhexyl)phthalate led to an increased inflammatory response by impairing important epigenetic regulators and inflammasome activation in macrophages [117]. Also, maternal exposure to butyl benzyl phthalate increased the risk for allergic airway inflammation in the offspring through methylation modifications [118]. Meanwhile, perinatal bisphenol A exposure enhanced the mast cell-mediated production of pro-inflammatory mediators of adult mice, which was associated with pulmonary inflammation and global DNA methylation levels [119]. Similarly, gestational exposure to bisphenol A increased inflammation/oxidative stress markers in sheep through epigenetic alterations [120].
Infections
Infectious diseases are caused by a broad range of pathogenic microorganisms including viruses, bacteria, parasites or fungi. In the past years, new infectious diseases have emerged, including the severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2/COVID-19) in December 2019, with serious public health implications worldwide due to rapid spread and the absence of specific treatment schemes or vaccine [121].
After invading the host, complex signaling pathways between immune cells are activated in order to coordinately initiate an inflammatory response against infection, where epigenetic mechanisms shaping the course of inflammation are involved [122]. In some cases, the immunological response to infection may be excessive, producing an inflammatory cytokine storm that eventually lead to extensive tissue damage and organ dysfunction [123].
For instance, it has been reported that inflammation triggered by Helicobacter pylori infection was related to differential DNA methylation patterns in human gastric mucosa [124]. Also, H. pylori-induced chronic inflammation played a direct role in the induction of aberrant DNA methylation, which correlated with gastric cancer risk [125]. In humans, the levels of methylation in gastric mucosae were associated with H. pylori virulence and measures of chronic inflammation [126]. During chronic H. pylori infection, inflammation-induced epigenetic silencing of miR-210 was identified as a mechanism of proliferation of gastric epithelium, with implications in gastric cancer development [127].
Experimental periodontitis using systemic microbial challenge (Porphyromonas gingivalis gavage) led to distinct patterns of inflammatory and epigenetic features [128]. miR-181a-5p and miR-21a-5p influenced the expression of inflammatory mediators in macrophages infected with Brucella abortus, thus contributing to bacterial control in host cells [129]. In addition, bacterial vaginosis predicted the length of gestation through miRNA-related epigenetic programming of the inflammatory response [130]. Furthermore, histone H3K14 hyperacetylation and differentially expressed miRNAs regulated the host inflammatory response during Staphylococcus aureus infection in mice mammary tissue [131]. Similarly, Escherichia coli infection in murine mammary tissue promoted histone hyperacetylation at genes related to the expression of inflammatory genes and drastic immune response [132].
Sepsis is defined as life-threatening organ dysfunction caused by a deregulated systemic immune response to infection. Interestingly, DNA methylation changes have been associated with sepsis in monocytes, which correlated with inflammatory signals [133]. Besides, chromatin modifications have been implicated in the regulation of the cellular immune/inflammatory responses in patients with sepsis [134].
Regarding viral infections, it was demonstrated that TLR3 activation increased HIV-1 transactivation in primary human macrophages via the inflammatory JNK and NFκB pathways and histone acetylation [135]. In vivo analyses revealed that respiratory syncytial virus infection induced the H3K4 demethylase KDM5B to promote an altered immune/inflammatory response that contributed to the development of chronic disease [136]. Human cytomegalovirus infection resulted in profound effects on the host cell DNA methylation machinery and was associated with inflammation in vivo [137]. Hypermethylation of PPAR gamma (PPARG) promoter was associated with liver inflammation and fibrosis in chronic hepatitis B virus infection [138]. An in vitro assay showed that loss of TIMP-3 by hypermethylation promoted chronic inflammation and tumor invasion during human papillomavirus infection [139]. miR-155, an indicator of inflammation-induced hepatocyte damage, was up-regulated both in monocytes and in the serum of patients with chronic hepatitis C infection [140]. Remarkably, DNA methyltransferase inhibition of regulatory T cells (Tregs) accelerated resolution of influenza-induced lung inflammation and related injury repair in mice [141].
Indeed, the development of immunomodulatory therapies targeting the epigenome during infectious diseases have emerged in the past years [142]. In this context, histone H3 modulation in macrophages was proposed as a strategy to attenuate the NF-κB/NLRP3-mediated inflammatory response during infection by the parasite Leishmania donovani [143]. Also, in vitro inhibition of the epigenetically active bromodomain and extraterminal domain (BET) proteins suppressed inflammation induced by the fungal pathogens Candida albicans and Aspergillus fumigatus [144]. Moreover, the pharmacological inhibition of the histone Lys demethylase JMJD3 protected mice against early septic death and reduced pro-inflammatory cytokine production via up-regulation of miR-146a [145]. Similarly, histone demethylase KDM3C demonstrated an anti-inflammatory effect by suppressing NF-κB signaling against oral bacterial infection in mice [146].
Smoking and excessive alcohol drinking
Smoking and urban particulate air pollution may alter systemic immunologic and inflammatory reactions. Cigarette smoke-induced inflammation was related to significant changes in active and repressive gene markers on histone 3 and histone 4 involving the regulation of the NLRP10 molecule, both in vivo and in vitro [147]. In bronchoalveolar lavage cells, tobacco smoke exposure increased the activity of inflammatory pathways by inducing continuous active demethylation processes [148]. Also, exposure of human macrophages to cigarette smoke extract promoted pro-inflammatory cytokine release by activation of the NF-κB pathway and concomitant posttranslational modifications of HDACs [149]. Remarkably, the prenatal environmental tobacco smoke exposure in mice increased the risk of pulmonary inflammation in the offspring through altered DNA methylation patterns [150]. In humans, the smoking-induced hypomethylation of the GPR15 gene (a chemoattractant receptor involved in systemic inflammation) has been proposed as an epigenetic biomarker underlying the potential role of smoking in chronic inflammatory pathologies [151]. Bioinformatic analyses revealed that long-term chronic smoking in African American women was associated with altered promoter DNA methylation of genes mapped to critical sub-networks modulating inflammation and immune function [152].
Excessive alcohol drinking causes inflammation and impairs the body’s ability to regulate the inflammatory response. Integrative epigenetic profiling analysis in blood samples from individuals with chronic alcohol consumption identified DNA methylation changes in genes related to inflammation (including HERC5) [153]. Nevertheless, the treatment with S-adenosyl-l-methionine (SAM), the methyl donor for all methylation reactions, did not improve the histopathology scores for hepatocyte inflammation and damage in patients with alcohol liver disease [154].
Notably, alcohol exposure decreased miR-148a expression in human hepatocytes, leading to NLRP3 inflammasome activation and liver injury [155]. Molecular analyses identified a regulatory role for miR-155 in chronic alcohol-induced intestinal inflammation and barrier dysfunction in a knockout animal model [156]. In this regard, up-regulation of miR-155 by chronic alcohol-intake trigged the production of the inflammatory cytokine TNF-α in macrophages [157]. Similarly, alcohol-induced miR-155 and HDAC11 increased the responsiveness of Kupffer cells to LPS by disinhibiting the TLR4 inflammatory pathway [158]. Furthermore, chronic ethanol intake up-regulated miR-155 and contributed to neuro-inflammation in mice [159], whereas miR-339-5p had an inhibitory effect in this patho-phenotype [160].
Sleep patterns
Experimental and clinical observational studies suggest that disturbed sleep patterns may contribute to inflammatory processes linked to CVD and other metabolic diseases through a complex network of autonomic, endocrine, and cytokine signals [161]. Epigenetic signatures may be an important biological mechanism linking poor sleep to inflammation, although limited evidence exists. In this context, sleep deprivation (deprived of restful REM sleep using the flowerpot technique) was associated with a significant unbalance in histone activity as well as oxidative stress and ongoing inflammation in rat hippocampus [162]. Moreover, differentially expressed genes involved in inflammatory pathways and declined fertility were detected in male rats exposed to chronic sleep restriction, which was not related to DNA methylation mechanisms [163]. Also, sleep-disordered breathing (a common disorder inducing oxidative stress and inflammation) was associated with epigenetic age acceleration among individuals at high cardiovascular risk [164]. Furthermore, epigenetic modifications have been proposed to constitute an important determinant of inflammatory phenotype in obstructive sleep apnea [165].
Chronic stress and social features
Chronic stress has been linked to negative health outcomes, including increased inflammation. Findings from the Multi-Ethnic Study of Atherosclerosis (MESA) showed that chronic stress by living in unfavorable neighborhood conditions was associated with methylation changes in genes related to stress reactivity (AVP, BDNF, FKBP5, SLC6A4) and inflammation (CCL1, CD1D, F8, KLRG1, NLRP12, SLAMF7, TLR1) [166]. In this same sample, life course measures of socioeconomic status also correlated with the methylation status of stress- and inflammation-related genes using a multi-level modeling approach [167]. Indicators of socioeconomic status were also associated with DNA methylation of genes involved in inflammation in healthy Italian individuals [168]. Likewise, social environments (including household socioeconomic condition in childhood) predicted DNA methylation patterns of inflammatory genes in young adulthood in Asians [169]. Moreover, it was reported that exposure to trauma and adversity during early life (i.e. neighborhood violence during childhood) amplified the adult pro-inflammatory response to stress in African American men, which was related to epigenetic phenomena involving IL6 promoter hypomethylation [170]. Additionally, the miR-106b∼25 cluster seems to play a role in mediating inflammatory and behavioral responses to repeated social defeat stress in a mouse model of stress vulnerability [171].
Interestingly, a brief yoga intervention (8 weeks of twice-weekly, hour-long yoga classes) modified the methylation levels of inflammatory markers in a community population of women with psychological distress [172]. Indeed, a day of intensive practice of mindfulness meditation reduced the expression of chromatin modulatory and inflammatory genes in PBMCs from experienced meditators [173]. Further research confirmed a relationship between a short meditation intervention (a day of intensive meditation practice for 8 h) in trained subjects and methylated sites in genes involved in immune cell metabolism and inflammation [174]. Remarkably, life satisfaction was prospectively associated with promoter methylation of the inflammatory TLR2 gene [175].
Climate
Climatological conditions such as air pollution and oxygen level may be associated with susceptibility to chronic inflammatory diseases by affecting the epigenome. Regarding the effects of air pollution on inflammation by epigenetic modulation, it was reported that the long-term exposure (annual average in the preceding year) to fine particulate matter (PM2.5) was associated with increased TNF-α expression through a reduction in TNF-α gene methylation in women [176]. In addition, changes in TNF methylation were proposed to mediate acute inflammation following personal exposure to fine particulate air pollution [177]. Consistently, adverse cardiac autonomic dysfunctions by PM2.5 exposure was partially related to methylation changes in the inflammatory TLR2 gene [178]. Increases in primary and secondary air pollutant concentrations also involved TLR2 methylation modifications, which could be an epigenetic biomarker underlying the adverse effects of air contamination on health [179, 180]. Noteworthy, exposure to urban particular matter interacted with obesogenic nutrition to regulate inflammation and oxidative stress pathways involving tissue-differential DNA methylation effects [181]. Also, whole-genome analyses detected altered DNA methylation patterns in oxidative and inflammatory pathway genes associated with both air pollution and vascular disease risk in Italians [182]. An inverse association was correspondingly reported between the daily exposure to particulate matter (specifically PM10) and the DNA methylation status of inflammatory genes in peripheral blood of obese subjects [183].
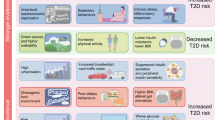
Besides, it was evidenced that the adverse cardiovascular and metabolic effects as consequence of air pollution inhalation may be mediated by miRNAs targeting key factors orchestrating coagulation and inflammatory pathways [184]. Later work disclosed positive correlations between PM2.5 exposure and the expression of several miRNAs (miR-21-5p, miR-187-3p, miR-146a-5p, miR-1-3p, and miR-199a-5p) predicted to target inflammatory markers [185]. Microarray analyses uncovered an underlying mechanism of PM2.5-induced airway inflammation involving regulation of non-coding RNAs co-targeting miR-3607-5p in bronchial cells [186]. Furthermore, inhalation of ozone (O3), another criteria air pollutant, disrupted the expression of miRNA profiles associated with inflammatory and immune response signaling [187]. Intriguingly, it was reported that particulate matter air pollution might attenuate the inflammatory response in Chinese children, in some extent mediated by miRNAs regulating pro-inflammatory genes [188]. These findings support the involvement of epigenetic phenomena in the induction of inflammatory processes after the exposure to environmental air pollutants (Fig. 1).

The interplay of obesity, infections, diet, stress, and environmental factors in inflammation through epigenetic mechanisms
The role of hypoxia as a main inducing factor of inflammation has been elucidated in recent years [189]. For example, low oxygen consumption was associated with IL6 gene hypomethylation and increased serum IL-6 concentrations in obese subjects with sleep apnea–hypopnea syndrome [190]. A mechanistic role of inflammasome activation in determining aerobic capacity (measured by peak oxygen uptake) was suggested since the percentage of methylation of the ASC gene and plasma IL-1β levels correlated with aerobic capacity in stable outpatients with heart failure [191]. Also, response to hypoxia in adipocytes was related to gene promoter hypomethylation and up-regulation of pro-inflammatory cytokines [192]. Moreover, gestational intermittent hypoxia induced endothelial dysfunction, triggered pro-inflammatory gene expression, and caused epigenetic changes in adult male offspring to increase the risk of developing cardiometabolic disease [193]. Similarly, epigenetic programming of pro-inflammatory phenotype in the heart development and vulnerability to disease later in life were associated with fetal hypoxia in rats [194]. Furthermore, hypoxia drove cardiac miRNAs profiles and inflammation processes in the right and left ventricle in a murine model [195].
Concluding remarks
Obesity and unhealthy diet as well as adverse environmental stimuli including sleep deprivation, chemical exposure, alcohol abuse, smoking, and climate pollution promote inflammatory processes in the host through epigenetic alterations, involving predominantly DNA methylation modifications in animal studies (Table 4). However, further studies in humans focused on other epigenetic mechanisms, such as histone acetylation/deacetylation processes and miRNA regulation affecting pro-inflammatory gene expression are required.
Scientific advances concerning the epigenetic mechanisms underlying inflammation-related chronic diseases such as diabetes, cardiovascular diseases, cancer, and neurodegenerative disorders are providing a better understanding of the molecular bases for the implicated pathological processes, and the prediction of individual disease risk based on the epigenotype. Nevertheless, it is necessary to integrate this knowledge with other emerging factors influencing the susceptibility/resistance to inflammation including the genetic background, microbiota composition, and metabolomic profiles using systems biology and large-scale bioinformatics tools.
Also, the fact that diverse pathogens, including respiratory viruses, induce epigenome modifications to promote systemic infection, opens opportunities for the development of efficient medications for specific targets. This finding is of current relevance for emerging widespread viral infections such as SARS-CoV-2/COVID-19, with a common fatal inflammatory lung condition without current effective therapies or available vaccine. Moreover, the suppressive effect of obesity and other environmental mediators of the immune function also needs to be addressed.
Progress in the identification of epigenetically active dietary components and lifestyle factors will contribute to the design of therapeutic interventions alleviating persistent inflammation by targeting the epigenome. In this regard, dietary bioactive compounds (i.e. polyphenols), n-3 PUFA, and regular physical activity have demonstrated anti-inflammatory properties through epigenetic phenomena. Nevertheless, the heterogeneity of the existing literature and the scarcity of studies in humans makes it difficult to propose specific recommendations about the amounts of polyphenols and n-3PUFA consumptions as well as the type, intensity, or duration of exercise that could counteract inflammatory processes. However, current available knowledge highlights the importance of anti-inflammatory dietary and exercise patterns for health and evidence the need of performing more nutriepigenetic investigations through randomized controlled clinical trials in order to prescribe precision nutritional and lifestyle recommendations for specific population and diseased groups.
Although further scientific advances in these research areas are needed, these insights are paving the way for the design of innovative strategies aimed to the prevention, management, prognosis, and treatment of chronic inflammatory diseases through personalized approaches (including precision nutrition) based on inflammatory epigenetic signatures.
Conclusions
Obesogenic and health-damaging environments can drive persistent inflammation by modifying some specific epigenetic mechanisms and negatively impact the development of chronic inflammatory diseases. The prescription of nutritional therapies using epigenetically active nutrients and physical activities with anti-inflammatory properties may help to revert the adverse effects of chronic inflammation.
References
Medzhitov R. Inflammation 2010: new adventures of an old flame. Cell. 2010;140:771–6.
Bennett JM, Reeves G, Billman GE, Sturmberg JP. Inflammation-nature’s way to efficiently respond to all types of challenges: implications for understanding and managing “the epidemic” of chronic diseases. Front Med (Lausanne). 2018;5:316.
Chen L, Deng H, Cui H, Fang J, Zuo Z, Deng J, Li Y, Wang X, Zhao L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget. 2017;9:7204–18.
Kany S, Vollrath JT, Relja B. Cytokines in inflammatory disease. Int J Mol Sci. 2019;20:6008.
Bondia-Pons I, Ryan L, Martinez JA. Oxidative stress and inflammation interactions in human obesity. J Physiol Biochem. 2012;68:701–11.
Shanmugam MK, Sethi G. Role of epigenetics in inflammation-associated diseases. Subcell Biochem. 2013;61:627–57.
Bayarsaihan D. Epigenetic mechanisms in inflammation. J Dent Res. 2011;90:9–17.
Gonzalez-Jaramillo V, Portilla-Fernandez E, Glisic M, Voortman T, Ghanbari M, Bramer W, Chowdhury R, Nijsten T, Dehghan A, Franco OH, Nano J. Epigenetics and inflammatory markers: a systematic review of the current evidence. Int J Inflam. 2019;2019:6273680.
Stylianou E. Epigenetics of chronic inflammatory diseases. J Inflamm Res. 2018;12:1–14.
Ramallal R, Toledo E, Martínez JA, Shivappa N, Hébert JR, Martínez-González MA, Ruiz-Canela M. Inflammatory potential of diet, weight gain, and incidence of overweight/obesity: the SUN cohort. Obesity (Silver Spring). 2017;25:997–1005.
Christ A, Lauterbach M, Latz E. Western diet and the immune system: an inflammatory connection. Immunity. 2019;51:794–811.
Milagro FI, Mansego ML, De Miguel C, Martínez JA. Dietary factors, epigenetic modifications and obesity outcomes: progresses and perspectives. Mol Aspects Med. 2013;34:782–812.
Martínez JA, Cordero P, Campión J, Milagro FI. Interplay of early-life nutritional programming on obesity, inflammation and epigenetic outcomes. Proc Nutr Soc. 2012;71:276–83.
Zheng J, Zhang Q, Mul JD, Yu M, Xu J, Qi C, Wang T, Xiao X. Maternal high-calorie diet is associated with altered hepatic microRNA expression and impaired metabolic health in offspring at weaning age. Endocrine. 2016;54:70–80.
Ding Y, Li J, Liu S, Zhang L, Xiao H, Li J, Chen H, Petersen RB, Huang K, Zheng L. DNA hypomethylation of inflammation-associated genes in adipose tissue of female mice after multigenerational high fat diet feeding. Int J Obes (Lond). 2014;38:198–204.
Nicodemus-Johnson J, Sinnott RA. Fruit and juice epigenetic signatures are associated with independent immunoregulatory pathways. Nutrients. 2017;9:752.
DiStefano JK. Fructose-mediated effects on gene expression and epigenetic mechanisms associated with NAFLD pathogenesis. Cell Mol Life Sci. 2020;77:2079–90.
Zheng J, Xiao X, Zhang Q, Wang T, Yu M, Xu J. Maternal low-protein diet modulates glucose metabolism and hepatic microRNAs expression in the early life of offspring†. Nutrients. 2017;9:205.
González-Becerra K, Ramos-Lopez O, Barrón-Cabrera E, Riezu-Boj JI, Milagro FI, Martínez-López E, Martínez JA. Fatty acids, epigenetic mechanisms and chronic diseases: a systematic review. Lipids Health Dis. 2019;18(1):178.
Flores-Sierra J, Arredondo-Guerrero M, Cervantes-Paz B, Rodríguez-Ríos D, Alvarado-Caudillo Y, Nielsen FC, Wrobel K, Wrobel K, Zaina S, Lund G. The trans fatty acid elaidate affects the global DNA methylation profile of cultured cells and in vivo. Lipids Health Dis. 2016;15:75.
Wang X, Cao Q, Yu L, Shi H, Xue B, Shi H. Epigenetic regulation of macrophage polarization and inflammation by DNA methylation in obesity. JCI Insight. 2016;1:e87748.
Hermsdorff HH, Mansego ML, Campión J, Milagro FI, Zulet MA, Martínez JA. TNF-α promoter methylation in peripheral white blood cells: relationship with circulating TNFα, truncal fat and n-6 PUFA intake in young women. Cytokine. 2013;64:265–71.
Cuevas-Sierra A, Ramos-Lopez O, Riezu-Boj JI, Milagro FI, Martinez JA. Diet, gut microbiota, and obesity: links with host genetics and epigenetics and potential applications. Adv Nutr. 2019;10:S17–30.
Tremblay BL, Guénard F, Rudkowska I, Lemieux S, Couture P, Vohl MC. Epigenetic changes in blood leukocytes following an omega-3 fatty acid supplementation. Clin Epigenetics. 2017;9:43.
Silva-Martínez GA, Rodríguez-Ríos D, Alvarado-Caudillo Y, Vaquero A, Esteller M, Carmona FJ, Moran S, Nielsen FC, Wickström-Lindholm M, Wrobel K, Wrobel K, Barbosa-Sabanero G, Zaina S, Lund G. Arachidonic and oleic acid exert distinct effects on the DNA methylome. Epigenetics. 2016;11:321–34.
Chen L, Dong Y, Wang X, Hao G, Huang Y, Gutin B, Zhu H. Epigenome-Wide Association Study of dietary fiber intake in African American adolescents. Mol Nutr Food Res. 2018;62:e1800155.
Ramos-Lopez O, Samblas M, Milagro FI, Zulet MA, Mansego ML, Riezu-Boj JI, Martinez JA. Association of low dietary folate intake with lower CAMKK2 gene methylation, adiposity, and insulin resistance in obese subjects. Nutr Res. 2018;50:53–62.
Kresovich JK, Bulka CM, Joyce BT, Vokonas PS, Schwartz J, Baccarelli AA, Hibler EA, Hou L. The inflammatory potential of dietary manganese in a cohort of elderly men. Biol Trace Elem Res. 2018;183:49–57.
Bollati V, Favero C, Albetti B, Tarantini L, Moroni A, Byun HM, Motta V, Conti DM, Tirelli AS, Vigna L, Bertazzi PA, Pesatori AC. Nutrients intake is associated with DNA methylation of candidate inflammatory genes in a population of obese subjects. Nutrients. 2014;6:4625–39.
Ricker MA, Haas WC. Anti-inflammatory diet in clinical practice: a review. Nutr Clin Pract. 2017;32:318–25.
Yahfoufi N, Alsadi N, Jambi M, Matar C. The immunomodulatory and anti-inflammatory role of polyphenols. Nutrients. 2018;10:1618.
Tsao R. Chemistry and biochemistry of dietary polyphenols. Nutrients. 2010;2:1231–46.
Tomé-Carneiro J, Larrosa M, Yáñez-Gascón MJ, Dávalos A, Gil-Zamorano J, Gonzálvez M, García-Almagro FJ, Ruiz Ros JA, Tomás-Barberán FA, Espín JC, García-Conesa MT. One-year supplementation with a grape extract containing resveratrol modulates inflammatory-related microRNAs and cytokines expression in peripheral blood mononuclear cells of type 2 diabetes and hypertensive patients with coronary artery disease. Pharmacol Res. 2013;72:69–82.
Kim H, Banerjee N, Barnes RC, Pfent CM, Talcott ST, Dashwood RH, Mertens-Talcott SU. Mango polyphenolics reduce inflammation in intestinal colitis-involvement of the miR-126/PI3K/AKT/mTOR axis in vitro and in vivo. Mol Carcinog. 2017;56:197–207.
Cordero-Herrera I, Chen X, Ramos S, Devaraj S. (–)-Epicatechin attenuates high-glucose-induced inflammation by epigenetic modulation in human monocytes. Eur J Nutr. 2017;56:1369–73.
Kim A, Yun JM. Combination treatments with luteolin and fisetin enhance anti-inflammatory effects in high glucose-treated THP-1 cells through histone acetyltransferase/histone deacetylase regulation. J Med Food. 2017;20(8):782–9.
Yun JM, Jialal I, Devaraj S. Epigenetic regulation of high glucose-induced proinflammatory cytokine production in monocytes by curcumin. J Nutr Biochem. 2011;22:450–8.
Bordoni L, Fedeli D, Fiorini D, Gabbianelli R. Extra virgin olive oil and nigella sativa oil produced in central Italy: a comparison of the nutrigenomic effects of two mediterranean oils in a low-grade inflammation model. Antioxidants (Basel). 2019;9:20.
Arpón A, Milagro FI, Razquin C, Corella D, Estruch R, Fitó M, Marti A, Martínez-González MA, Ros E, Salas-Salvadó J, Riezu-Boj JI, Martínez JA. Impact of consuming extra-virgin olive oil or nuts within a mediterranean diet on DNA methylation in peripheral white blood cells within the PREDIMED-navarra randomized controlled trial: a role for dietary lipids. Nutrients. 2017;10:15.
Arpón A, Riezu-Boj JI, Milagro FI, Marti A, Razquin C, Martínez-González MA, Corella D, Estruch R, Casas R, Fitó M, Ros E, Salas-Salvadó J, Martínez JA. Adherence to Mediterranean diet is associated with methylation changes in inflammation-related genes in peripheral blood cells. J Physiol Biochem. 2016;73:445–55.
de la Garza AL, Etxeberria U, Palacios-Ortega S, Haslberger AG, Aumueller E, Milagro FI, Martínez JA. Modulation of hyperglycemia and TNFα-mediated inflammation by helichrysum and grapefruit extracts in diabetic db/db mice. Food Funct. 2014;5:2120–8.
Kim S, Lee MS, Jung S, Son HY, Park S, Kang B, Kim SY, Kim IH, Kim CT, Kim Y. Ginger extract ameliorates obesity and inflammation via regulating microRNA-21/132 expression and AMPK activation in white adipose tissue. Nutrients. 2018;10:1567.
Delaney C, Hoeltzel M, Garg SK, Warner R, Johnson K, Yung R. Maternal micronutrient supplementation suppresses T cell chemokine receptor expression and function in F1 mice. J Nutr. 2012;142:1329–35.
Alehagen U, Johansson P, Aaseth J, Alexander J, Wågsäter D. Significant changes in circulating microRNA by dietary supplementation of selenium and coenzyme Q10 in healthy elderly males. A subgroup analysis of a prospective randomized double-blind placebo-controlled trial among elderly Swedish citizens. PLoS ONE. 2017;12:e0174880.
Karkeni E, Bonnet L, Marcotorchino J, Tourniaire F, Astier J, Ye J, Landrier JF. Vitamin D limits inflammation-linked microRNA expression in adipocytes in vitro and in vivo: a new mechanism for the regulation of inflammation by vitamin D. Epigenetics. 2018;13:156–62.
Li C, Guo S, Gao J, Guo Y, Du E, Lv Z, Zhang B. Maternal high-zinc diet attenuates intestinal inflammation by reducing DNA methylation and elevating H3K9 acetylation in the A20 promoter of offspring chicks. J Nutr Biochem. 2015;26:173–83.
Hernández-Saavedra D, Moody L, Xu GB, Chen H, Pan YX. Epigenetic regulation of metabolism and inflammation by calorie restriction. Adv Nutr. 2019;10:520–36.
Cordero P, Campion J, Milagro FI, Goyenechea E, Steemburgo T, Javierre BM, Martinez JA. Leptin and TNF-alpha promoter methylation levels measured by MSP could predict the response to a low-calorie diet. J Physiol Biochem. 2011;67:463–70.
Campión J, Milagro FI, Goyenechea E, Martínez JA. TNF-α promoter methylation as a predictive biomarker for weight-loss response. Obesity (Silver Spring). 2009;17:1293–7.
Perez-Cornago A, de la Iglesia R, Lopez-Legarrea P, Abete I, Navas-Carretero S, Lacunza CI, Lahortiga F, Martinez-Gonzalez MA, Martinez JA, Zulet MA. A decline in inflammation is associated with less depressive symptoms after a dietary intervention in metabolic syndrome patients: a longitudinal study. Nutr J. 2014;13:36.
Gögebakan O, Kohl A, Osterhoff MA, van Baak MA, Jebb SA, Papadaki A, Martinez JA, Handjieva-Darlenska T, Hlavaty P, Weickert MO, Holst C, Saris WH, Astrup A, Pfeiffer AF. DiOGenes. Effects of weight loss and long-term weight maintenance with diets varying in protein and glycemic index on cardiovascular risk factors: the diet, obesity, and genes (DiOGenes) study: a randomized, controlled trial. Circulation. 2011;124:2829–38.
de la Iglesia R, Mansego ML, Sánchez-Muniz FJ, Zulet MA, Martinez JA. Arylesterase activity is associated with antioxidant intake and paraoxonase-1 (PON1) gene methylation in metabolic syndrome patients following an energy restricted diet. EXCLI J. 2014;13:416–26.
Lopez-Legarrea P, Mansego ML, Zulet MA, Martinez JA. SERPINE1, PAI-1 protein coding gene, methylation levels and epigenetic relationships with adiposity changes in obese subjects with metabolic syndrome features under dietary restriction. J Clin Biochem Nutr. 2013;53:139–44.
Barrón-Cabrera E, Ramos-Lopez O, González-Becerra K, Riezu-Boj JI, Milagro FI, Martínez-López E, Martínez JA. Epigenetic modifications as outcomes of exercise interventions related to specific metabolic alterations: a systematic review. Lifestyle Genom. 2019;12:25–44.
Nishida Y, Hara M, Higaki Y, Taguchi N, Nakamura K, Nanri H, Horita M, Shimanoe C, Yasukata J, Miyoshi N, Yamada Y, Higashimoto K, Soejima H, Tanaka K. Habitual light-intensity physical activity and ASC methylation in a middle-aged population. Int J Sports Med. 2019;40:670–7.
Butts B, Butler J, Dunbar SB, Corwin E, Gary RA. Effects of exercise on ASC methylation and IL-1 cytokines in heart failure. Med Sci Sports Exerc. 2018;50:1757–66.
Zhang Y, Hashimoto S, Fujii C, Hida S, Ito K, Matsumura T, Sakaizawa T, Morikawa M, Masuki S, Nose H, Higuchi K, Nakajima K, Taniguchi S. NFκB2 gene as a novel candidate that epigenetically responds to interval walking training. Int J Sports Med. 2015;36:769–75.
Morikawa M, Nakano S, Mitsui N, Murasawa H, Masuki S, Nose H. Effects of dried tofu supplementation during interval walking training on the methylation of the NFKB2 gene in the whole blood of older women. J Physiol Sci. 2018;68:749–57.
Masuki S, Nishida K, Hashimoto S, Morikawa M, Takasugi S, Nagata M, Taniguchi S, Rokutan K, Nose H. Effects of milk product intake on thigh muscle strength and NFKB gene methylation during home-based interval walking training in older women: a randomized, controlled pilot study. PLoS ONE. 2017;12:e0176757.
Russo A, Bartolini D, Mensà E, Torquato P, Albertini MC, Olivieri F, Testa R, Rossi S, Piroddi M, Cruciani G, De Feo P, Galli F. Physical activity modulates the overexpression of the inflammatory miR-146a-5p in obese patients. IUBMB Life. 2018;70:1012–22.
Bao F, Slusher AL, Whitehurst M, Huang CJ. Circulating microRNAs are upregulated following acute aerobic exercise in obese individuals. Physiol Behav. 2018;197:15–21.
Li Y, Yao M, Zhou Q, Cheng Y, Che L, Xu J, Xiao J, Shen Z, Bei Y. Dynamic regulation of circulating micrornas during acute exercise and long-term exercise training in basketball athletes. Front Physiol. 2018;9:282.
de Gonzalo-Calvo D, Dávalos A, Montero A, García-González Á, Tyshkovska I, González-Medina A, Soares SM, Martínez-Camblor P, Casas-Agustench P, Rabadán M, Díaz-Martínez ÁE, Úbeda N, Iglesias-Gutiérrez E. Circulating inflammatory miRNA signature in response to different doses of aerobic exercise. J Appl Physiol. 1985;2015(119):124–34.
Koltai E, Bori Z, Osvath P, Ihasz F, Peter S, Toth G, Degens H, Rittweger J, Boldogh I, Radak Z. Master athletes have higher miR-7, SIRT3 and SOD2 expression in skeletal muscle than age-matched sedentary controls. Redox Biol. 2018;19:46–51.
Dorneles GP, Boeira MCR, Schipper LL, Silva IRV, Elsner VR, Dal Lago P, Peres A, Romão PRT. Acute strenuous exercise induces an imbalance on histone H4 acetylation/histone deacetylase 2 and increases the proinflammatory profile of PBMC of obese individuals. Oxid Med Cell Longev. 2017;2017:1530230.
Crujeiras AB, Cordero P, Garcia-Diaz DF, Stachowska E, González-Muniesa P. Molecular basis of the inflammation related to obesity. Oxid Med Cell Longev. 2019;2019:5250816.
Lorente-Cebrián S, González-Muniesa P, Milagro FI, Martínez JA. MicroRNAs and other non-coding RNAs in adipose tissue and obesity: emerging roles as biomarkers and therapeutic targets. Clin Sci (Lond). 2019;133:23–40.
Ahmed M, de Winther MPJ, Van den Bossche J. Epigenetic mechanisms of macrophage activation in type 2 diabetes. Immunobiology. 2017;222:937–43.
Keating ST, Plutzky J, El-Osta A. Epigenetic changes in diabetes and cardiovascular risk. Circ Res. 2016;118:1706–22.
Milner JJ, Beck MA. The impact of obesity on the immune response to infection. Proc Nutr Soc. 2012;71:298–306.
Petrus P, Bialesova L, Checa A, Kerr A, Naz S, Bäckdahl J, Gracia A, Toft S, Dahlman-Wright K, Hedén P, Dahlman I, Wheelock CE, Arner P, Mejhert N, Gao H, Rydén M. Adipocyte expression of SLC19A1 links DNA hypermethylation to adipose tissue inflammation and insulin resistance. J Clin Endocrinol Metab. 2018;103:710–21.
Pietiläinen KH, Ismail K, Järvinen E, Heinonen S, Tummers M, Bollepalli S, Lyle R, Muniandy M, Moilanen E, Hakkarainen A, Lundbom J, Lundbom N, Rissanen A, Kaprio J, Ollikainen M. DNA methylation and gene expression patterns in adipose tissue differ significantly within young adult monozygotic BMI-discordant twin pairs. Int J Obes (Lond). 2016;40:654–61.
Remely M, Aumueller E, Jahn D, Hippe B, Brath H, Haslberger AG. Microbiota and epigenetic regulation of inflammatory mediators in type 2 diabetes and obesity. Benef Microbes. 2014;5:33–43.
Na YK, Hong HS, Lee WK, Kim YH, Kim DS. Increased methylation of interleukin 6 gene is associated with obesity in Korean women. Mol Cells. 2015;38:452–6.
Arpón A, Milagro FI, Ramos-Lopez O, Mansego ML, Riezu-Boj JI, Martínez JA, MENA Project. Methylome-Wide Association Study in peripheral white blood cells focusing on central obesity and inflammation. Genes (Basel). 2019;10:444.
Ejarque M, Ceperuelo-Mallafré V, Serena C, Maymo-Masip E, Duran X, Díaz-Ramos A, Millan-Scheiding M, Núñez-Álvarez Y, Núñez-Roa C, Gama P, Garcia-Roves PM, Peinado MA, Gimble JM, Zorzano A, Vendrell J, Fernández-Veledo S. Adipose tissue mitochondrial dysfunction in human obesity is linked to a specific DNA methylation signature in adipose-derived stem cells. Int J Obes (Lond). 2019;43:1256–68.
Guénard F, Tchernof A, Deshaies Y, Pérusse L, Biron S, Lescelleur O, Biertho L, Marceau S, Vohl MC. Differential methylation in visceral adipose tissue of obese men discordant for metabolic disturbances. Physiol Genomics. 2014;46:216–22.
Crujeiras AB, Pissios P, Moreno-Navarrete JM, Diaz-Lagares A, Sandoval J, Gomez A, Ricart W, Esteller M, Casanueva FF, Fernandez-Real JM. An epigenetic signature in adipose tissue is linked to nicotinamide N-Methyltransferase gene expression. Mol Nutr Food Res. 2018;62:e1700933.
Yang X, Wang X, Liu D, Yu L, Xue B, Shi H. Epigenetic regulation of macrophage polarization by DNA methyltransferase 3b. Mol Endocrinol. 2014;28:565–74.
Kamei Y, Suganami T, Ehara T, Kanai S, Hayashi K, Yamamoto Y, Miura S, Ezaki O, Okano M, Ogawa Y. Increased expression of DNA methyltransferase 3a in obese adipose tissue: studies with transgenic mice. Obesity (Silver Spring). 2010;18:314–21.
Iwaya C, Kitajima H, Yamamoto K, Maeda Y, Sonoda N, Shibata H, Inoguchi T. DNA methylation of the Klf14 gene region in whole blood cells provides prediction for the chronic inflammation in the adipose tissue. Biochem Biophys Res Commun. 2018;497:908–15.
Jacobsen MJ, Mentzel CM, Olesen AS, Huby T, Jørgensen CB, Barrès R, Fredholm M, Simar D. Altered methylation profile of lymphocytes is concordant with perturbation of lipids metabolism and inflammatory response in obesity. J Diabetes Res. 2016;2016:8539057.
Raciti GA, Spinelli R, Desiderio A, Longo M, Parrillo L, Nigro C, D’Esposito V, Mirra P, Fiory F, Pilone V, Forestieri P, Formisano P, Pastan I, Miele C, Beguinot F. Specific CpG hyper-methylation leads to Ankrd26 gene down-regulation in white adipose tissue of a mouse model of diet-induced obesity. Sci Rep. 2017;7:43526.
Desiderio A, Longo M, Parrillo L, Campitelli M, Cacace G, de Simone S, Spinelli R, Zatterale F, Cabaro S, Dolce P, Formisano P, Milone M, Miele C, Beguinot F, Raciti GA. Epigenetic silencing of the ANKRD26 gene correlates to the pro-inflammatory profile and increased cardio-metabolic risk factors in human obesity. Clin Epigenetics. 2019;11:181.
Cifuentes-Zúñiga F, Arroyo-Jousse V, Soto-Carrasco G, Casanello P, Uauy R, Krause BJ, Castro-Rodríguez JA. IL-10 expression in macrophages from neonates born from obese mothers is suppressed by IL-4 and LPS/INFγ. J Cell Physiol. 2017;232:3693–701.
Sureshchandra S, Wilson RM, Rais M, Marshall NE, Purnell JQ, Thornburg KL, Messaoudi I. Maternal pregravid obesity remodels the DNA methylation landscape of cord blood monocytes disrupting their inflammatory program. J Immunol. 2017;199:2729–44.
Zhang XM, Guo L, Chi MH, Sun HM, Chen XW. Identification of active miRNA and transcription factor regulatory pathways in human obesity-related inflammation. BMC Bioinf. 2015;16:76.
Yang Z, Wei Z, Wu X, Yang H. Screening of exosomal miRNAs derived from subcutaneous and visceral adipose tissues: determination of targets for the treatment of obesity and associated metabolic disorders. Mol Med Rep. 2018;18:3314–24.
Hijmans JG, Diehl KJ, Bammert TD, Kavlich PJ, Lincenberg GM, Greiner JJ, Stauffer BL, DeSouza CA. Influence of overweight and obesity on circulating inflammation-related microRNA. Microrna. 2018;7:148–54.
Ortega FJ, Moreno M, Mercader JM, Moreno-Navarrete JM, Fuentes-Batllevell N, Sabater M, Ricart W, Fernández-Real JM. Inflammation triggers specific microRNA profiles in human adipocytes and macrophages and in their supernatants. Clin Epigenetics. 2015;7:49.
Doumatey AP, He WJ, Gaye A, Lei L, Zhou J, Gibbons GH, Adeyemo A, Rotimi CN. Circulating MiR-374a-5p is a potential modulator of the inflammatory process in obesity. Sci Rep. 2018;8:7680.
Vega-Cárdenas M, Uresti-Rivera EE, Cortés-García JD, Briones-Espinoza M, Ruíz-Rodríguez VM, Reynaga-Hernández E, Mendez-Mancilla A, Portales-Pérez DP. Increased levels of adipose tissue-resident Th17 cells in obesity associated with miR-326. Immunol Lett. 2019;211:60–7.
Miranda K, Yang X, Bam M, Murphy EA, Nagarkatti PS, Nagarkatti M. MicroRNA-30 modulates metabolic inflammation by regulating Notch signaling in adipose tissue macrophages. Int J Obes (Lond). 2018;42:1140–50.
Pan Y, Hui X, Hoo RLC, Ye D, Chan CYC, Feng T, Wang Y, Lam KSL, Xu A. Adipocyte-secreted exosomal microRNA-34a inhibits M2 macrophage polarization to promote obesity-induced adipose inflammation. J Clin Invest. 2019;129:834–49.
Zhang Y, Mei H, Chang X, Chen F, Zhu Y, Han X. Adipocyte-derived microvesicles from obese mice induce M1 macrophage phenotype through secreted miR-155. J Mol Cell Biol. 2016;8:505–17.
Karkeni E, Astier J, Tourniaire F, El Abed M, Romier B, Gouranton E, Wan L, Borel P, Salles J, Walrand S, Ye J, Landrier JF. Obesity-associated inflammation induces microRNA-155 expression in adipocytes and adipose tissue: outcome on adipocyte function. J Clin Endocrinol Metab. 2016;101:1615–26.
Yao F, Yu Y, Feng L, Li J, Zhang M, Lan X, Yan X, Liu Y, Guan F, Zhang M, Chen L. Adipogenic miR-27a in adipose tissue upregulates macrophage activation via inhibiting PPARγ of insulin resistance induced by high-fat diet-associated obesity. Exp Cell Res. 2017;355:105–12.
Zhang M, Zhou Z, Wang J, Li S. MiR-130b promotes obesity associated adipose tissue inflammation and insulin resistance in diabetes mice through alleviating M2 macrophage polarization via repression of PPAR-γ. Immunol Lett. 2016;180:1–8.
Liu L, Li X. Downregulation of miR-320 alleviates endoplasmic reticulum stress and inflammatory response in 3T3-L1 adipocytes. Exp Clin Endocrinol Diabetes. 2019. https://doi.org/10.1055/a-1012-8420.
Peng J, Zhou Y, Deng Z, Zhang H, Wu Y, Song T, Yang Y, Wei H, Peng J. miR-221 negatively regulates inflammation and insulin sensitivity in white adipose tissue by repression of sirtuin-1 (SIRT1). J Cell Biochem. 2018;119:6418–28.
Deiuliis JA, Syed R, Duggineni D, Rutsky J, Rengasamy P, Zhang J, Huang K, Needleman B, Mikami D, Perry K, Hazey J, Rajagopalan S. Visceral adipose microRNA 223 is upregulated in human and murine obesity and modulates the inflammatory phenotype of macrophages. PLoS ONE. 2016;11:e0165962.
Arner E, Mejhert N, Kulyté A, Balwierz PJ, Pachkov M, Cormont M, Lorente-Cebrián S, Ehrlund A, Laurencikiene J, Hedén P, Dahlman-Wright K, Tanti JF, Hayashizaki Y, Rydén M, Dahlman I, van Nimwegen E, Daub CO, Arner P. Adipose tissue microRNAs as regulators of CCL2 production in human obesity. Diabetes. 2012;61:1986–93.
Liu L, Li Q, Xiao X, Wu C, Gao R, Peng C, Li D, Zhang W, Du T, Wang Y, Yang S, Zhen Q, Ge Q. miR-1934, downregulated in obesity, protects against low-grade inflammation in adipocytes. Mol Cell Endocrinol. 2016;428:109–17.
Ge Q, Gérard J, Noël L, Scroyen I, Brichard SM. MicroRNAs regulated by adiponectin as novel targets for controlling adipose tissue inflammation. Endocrinology. 2012;153:5285–96.
Runtsch MC, Nelson MC, Lee SH, Voth W, Alexander M, Hu R, Wallace J, Petersen C, Panic V, Villanueva CJ, Evason KJ, Bauer KM, Mosbruger T, Boudina S, Bronner M, Round JL, Drummond MJ, O’Connell RM. Anti-inflammatory microRNA-146a protects mice from diet-induced metabolic disease. PLoS Genet. 2019;15:e1007970.
Alfaradhi MZ, Kusinski LC, Fernandez-Twinn DS, Pantaleão LC, Carr SK, Ferland-McCollough D, Yeo GS, Bushell M, Ozanne SE. Maternal obesity in pregnancy developmentally programs adipose tissue inflammation in young. Lean Male Mice Offspring Endocrinol. 2016;157:4246–56.
Mazloom H, Alizadeh S, Esfahani EN, Razi F, Meshkani R. Decreased expression of microRNA-21 is associated with increased cytokine production in peripheral blood mononuclear cells (PBMCs) of obese type 2 diabetic and non-diabetic subjects. Mol Cell Biochem. 2016;419:11–7.
Li H, Xue M, Xu J, Qin X. MiR-301a is involved in adipocyte dysfunction during obesity-related inflammation via suppression of PPARγ. Pharmazie. 2016;71:84–8.
Xie Q, Wei M, Kang X, et al. Reciprocal inhibition between miR-26a and NF-κB regulates obesity-related chronic inflammation in chondrocytes. Biosci Rep. 2015;35:e00204.
Shanaki M, Omidifar A, Shabani P, Toolabi K. Association between HDACs and pro-inflammatory cytokine gene expressions in obesity. Arch Physiol Biochem. 2020. https://doi.org/10.1080/13813455.2020.1734843.
Hanzu FA, Musri MM, Sánchez-Herrero A, Claret M, Esteban Y, Kaliman P, Gomis R, Párrizas M. Histone demethylase KDM1A represses inflammatory gene expression in preadipocytes. Obesity (Silver Spring). 2013;21:E616–25.
Gillum MP, Kotas ME, Erion DM, Kursawe R, Chatterjee P, Nead KT, Muise ES, Hsiao JJ, Frederick DW, Yonemitsu S, Banks AS, Qiang L, Bhanot S, Olefsky JM, Sears DD, Caprio S, Shulman GI. SirT1 regulates adipose tissue inflammation. Diabetes. 2011;60:3235–45.
Kim HB, Kumar A, Wang L, Liu GH, Keller SR, Lawrence JC Jr, Finck BN, Harris TE. Lipin 1 represses NFATc4 transcriptional activity in adipocytes to inhibit secretion of inflammatory factors. Mol Cell Biol. 2010;30:3126–39.
Makene VW, Pool EJ. The effects of endocrine disrupting chemicals on biomarkers of inflammation produced by lipopolysaccharide stimulated RAW264.7 macrophages. Int J Environ Res Public Health. 2019;16:2914.
Helsley RN, Zhou C. Epigenetic impact of endocrine disrupting chemicals on lipid homeostasis and atherosclerosis: a pregnane X receptor-centric view. Environ Epigenet. 2017;3:dvx017.
Han C, Hong YC. Bisphenol A, hypertension, and cardiovascular diseases: epidemiological, laboratory, and clinical trial evidence. Curr Hypertens Rep. 2016;18:11.
Park MH, Gutiérrez-García AK, Choudhury M. Mono-(2-ethylhexyl) phthalate aggravates inflammatory response via Sirtuin regulation and inflammasome activation in RAW 264.7 cells. Chem Res Toxicol. 2019;32:935–42.
Jahreis S, Trump S, Bauer M, Bauer T, Thürmann L, Feltens R, Wang Q, Gu L, Grützmann K, Röder S, Averbeck M, Weichenhan D, Plass C, Sack U, Borte M, Dubourg V, Schüürmann G, Simon JC, von Bergen M, Hackermüller J, Eils R, Lehmann I, Polte T. Maternal phthalate exposure promotes allergic airway inflammation over 2 generations through epigenetic modifications. J Allergy Clin Immunol. 2018;141:741–53.
O’Brien E, Dolinoy DC, Mancuso P. Perinatal bisphenol A exposures increase production of pro-inflammatory mediators in bone marrow-derived mast cells of adult mice. J Immunotoxicol. 2014;11:205–12.
Song W, Puttabyatappa M, Zeng L, Vazquez D, Pennathur S, Padmanabhan V. Developmental programming: prenatal bisphenol A treatment disrupts mediators of placental function in sheep. Chemosphere. 2020;243:125301.
Hamid S, Mir MY, Rohela GK. Novel coronavirus disease (COVID-19): a pandemic (epidemiology, pathogenesis and potential therapeutics). New Microbes New Infect. 2020;35:100679.
Zhang Q, Cao X. Epigenetic regulation of the innate immune response to infection. Nat Rev Immunol. 2019;19:417–32.
Teijaro JR. Cytokine storms in infectious diseases. Semin Immunopathol. 2017;39:501–3.
Yamashita S, Nanjo S, Rehnberg E, Iida N, Takeshima H, Ando T, Maekita T, Sugiyama T, Ushijima T. Distinct DNA methylation targets by aging and chronic inflammation: a pilot study using gastric mucosa infected with Helicobacter pylori. Clin Epigenetics. 2019;11:191.
Maeda M, Moro H, Ushijima T. Mechanisms for the induction of gastric cancer by Helicobacter pylori infection: aberrant DNA methylation pathway. Gastric Cancer. 2017;20:8–15.
Schneider BG, Piazuelo MB, Sicinschi LA, Mera R, Peng DF, Roa JC, Romero-Gallo J, Delgado AG, de Sablet T, Bravo LE, Wilson KT, El-Rifai W, Peek RM Jr, Correa P. Virulence of infecting Helicobacter pylori strains and intensity of mononuclear cell infiltration are associated with levels of DNA hypermethylation in gastric mucosae. Epigenetics. 2013;8:1153–61.
Kiga K, Mimuro H, Suzuki M, Shinozaki-Ushiku A, Kobayashi T, Sanada T, Kim M, Ogawa M, Iwasaki YW, Kayo H, Fukuda-Yuzawa Y, Yashiro M, Fukayama M, Fukao T, Sasakawa C. Epigenetic silencing of miR-210 increases the proliferation of gastric epithelium during chronic Helicobacter pylori infection. Nat Commun. 2014;5:4497.
Palioto DB, Finoti LS, Kinane DF, Benakanakere M. Epigenetic and inflammatory events in experimental periodontitis following systemic microbial challenge. J Clin Periodontol. 2019;46:819–29.
Corsetti PP, de Almeida LA, Gonçalves ANA, Gomes MTR, Guimarães ES, Marques JT, Oliveira SC. miR-181a-5p regulates TNF-α and miR-21a-5p influences gualynate-binding protein 5 and IL-10 expression in macrophages affecting host control of Brucella abortus infection. Front Immunol. 2018;9:1331.
Sanders AP, Gennings C, Svensson K, Motta V, Mercado-Garcia A, Solano M, Baccarelli AA, Tellez-Rojo MM, Wright RO, Burris HH. Bacterial and cytokine mixtures predict the length of gestation and are associated with miRNA expression in the cervix. Epigenomics. 2017;9:33–45.
Modak R, Das Mitra S, Vasudevan M, Krishnamoorthy P, Kumar M, Bhat AV, Bhuvana M, Ghosh SK, Shome BR, Kundu TK. Epigenetic response in mice mastitis: role of histone H3 acetylation and microRNA(s) in the regulation of host inflammatory gene expression during Staphylococcus aureus infection. Clin Epigenetics. 2014;6:12.
Modak R, Das Mitra S, Krishnamoorthy P, Bhat A, Banerjee A, Gowsica BR, Bhuvana M, Dhanikachalam V, Natesan K, Shome R, Shome BR, Kundu TK. Histone H3K14 and H4K8 hyperacetylation is associated with Escherichia coli-induced mastitis in mice. Epigenetics. 2012;7:492–501.
Lorente-Sorolla C, Garcia-Gomez A, Català-Moll F, Toledano V, Ciudad L, Avendaño-Ortiz J, Maroun-Eid C, Martín-Quirós A, Martínez-Gallo M, Ruiz-Sanmartín A, Del Campo ÁG, Ferrer-Roca R, Ruiz-Rodriguez JC, Álvarez-Errico D, López-Collazo E, Ballestar E. Inflammatory cytokines and organ dysfunction associate with the aberrant DNA methylome of monocytes in sepsis. Genome Med. 2019;11:66.
Carson WF 4th, Kunkel SL. Regulation of cellular immune responses in sepsis by histone modifications. Adv Protein Chem Struct Biol. 2017;106:191–225.
Bhargavan B, Woollard SM, Kanmogne GD. Toll-like receptor-3 mediates HIV-1 transactivation via NFκB and JNK pathways and histone acetylation, but prolonged activation suppresses Tat and HIV-1 replication. Cell Signal. 2016;28:7–22.
Ptaschinski C, Mukherjee S, Moore ML, Albert M, Helin K, Kunkel SL, Lukacs NW. RSV-induced H3K4 demethylase KDM5B leads to regulation of dendritic cell-derived innate cytokines and exacerbates pathogenesis in vivo. PLoS Pathog. 2015;11:e1004978.
Esteki-Zadeh A, Karimi M, Strååt K, Ammerpohl O, Zeitelhofer M, Jagodic M, Mehrab-Mohseni M, Sjöholm L, Rahbar A, Söderberg-Nauclér C, Ekström TJ. Human cytomegalovirus infection is sensitive to the host cell DNA methylation state and alters global DNA methylation capacity. Epigenetics. 2012;7:585–93.
Zhao Q, Fan YC, Zhao J, Gao S, Zhao ZH, Wang K. DNA methylation patterns of peroxisome proliferator-activated receptor gamma gene associated with liver fibrosis and inflammation in chronic hepatitis B. J Viral Hepat. 2013;20:430–7.
Wu DW, Tsai LH, Chen PM, Lee MC, Wang L, Chen CY, Cheng YW, Lee H. Loss of TIMP-3 promotes tumor invasion via elevated IL-6 production and predicts poor survival and relapse in HPV-infected non-small cell lung cancer. Am J Pathol. 2012;181:1796–806.
Bala S, Tilahun Y, Taha O, Alao H, Kodys K, Catalano D, Szabo G. Increased microRNA-155 expression in the serum and peripheral monocytes in chronic HCV infection. J Transl Med. 2012;10:151.
Singer BD, Mock JR, Aggarwal NR, Garibaldi BT, Sidhaye VK, Florez MA, Chau E, Gibbs KW, Mandke P, Tripathi A, Yegnasubramanian S, King LS, D’Alessio FR. Regulatory T cell DNA methyltransferase inhibition accelerates resolution of lung inflammation. Am J Respir Cell Mol Biol. 2015;52:641–52.
Cole J, Morris P, Dickman MJ, Dockrell DH. The therapeutic potential of epigenetic manipulation during infectious diseases. Pharmacol Ther. 2016;167:85–99.
Lecoeur H, Prina E, Rosazza T, Kokou K, N’Diaye P, Aulner N, Varet H, Bussotti G, Xing Y, Milon G, Weil R, Meng G, Späth GF. Targeting macrophage histone H3 modification as a leishmania strategy to dampen the NF-κB/NLRP3-mediated inflammatory response. Cell Rep. 2020;30:1870–82 ((e4)).
Domínguez-Andrés J, Ferreira AV, Jansen T, Smithers N, Prinjha RK, Furze RC, Netea MG. Bromodomain inhibitor I-BET151 suppresses immune responses during fungal-immune interaction. Eur J Immunol. 2019;49:2044–50.
Pan Y, Wang J, Xue Y, Zhao J, Li D, Zhang S, Li K, Hou Y, Fan H. GSKJ4 protects mice against early sepsis via reducing proinflammatory factors and up-regulating MiR-146a. Front Immunol. 2018;9:2272.
Lee JY, Mehrazarin S, Alshaikh A, Kim S, Chen W, Lux R, Gwack Y, Kim RH, Kang MK. Histone Lys demethylase KDM3C demonstrates anti-inflammatory effects by suppressing NF-κB signaling and osteoclastogenesis. FASEB J. 2019;33:10515–27.
Kaur G, Bagam P, Pinkston R, Singh DP, Batra S. Cigarette smoke-induced inflammation: NLRP10-mediated mechanisms. Toxicology. 2018;398–399:52–67.
Ringh MV, Hagemann-Jensen M, Needhamsen M, Kular L, Breeze CE, Sjöholm LK, Slavec L, Kullberg S, Wahlström J, Grunewald J, Brynedal B, Liu Y, Almgren M, Jagodic M, Öckinger J, Ekström TJ. Tobacco smoking induces changes in true DNA methylation, hydroxymethylation and gene expression in bronchoalveolar lavage cells. EBioMedicine. 2019;46:290–304.
Yang SR, Chida AS, Bauter MR, Shafiq N, Seweryniak K, Maggirwar SB, Kilty I, Rahman I. Cigarette smoke induces proinflammatory cytokine release by activation of NF-κB and posttranslational modifications of histone deacetylase in macrophages. Am J Physiol Lung Cell Mol Physiol. 2006;291:L46-57.
Lee JW, Jaffar Z, Pinkerton KE, Porter V, Postma B, Ferrini M, Holian A, Roberts K, Cho YH. Alterations in DNA methylation and airway hyperreactivity in response to in utero exposure to environmental tobacco smoke. Inhal Toxicol. 2015;27:724–30.
Kõks G, Uudelepp ML, Limbach M, Peterson P, Reimann E, Kõks S. Smoking-induced expression of the GPR15 gene indicates its potential role in chronic inflammatory pathologies. Am J Pathol. 2015;185:2898–906.
Dogan MV, Shields B, Cutrona C, Gao L, Gibbons FX, Simons R, Monick M, Brody GH, Tan K, Beach SR, Philibert RA. The effect of smoking on DNA methylation of peripheral blood mononuclear cells from African American women. BMC Genomics. 2014;15:151.
Weng JT, Wu LS, Lee CS, Hsu PW, Cheng AT. Integrative epigenetic profiling analysis identifies DNA methylation changes associated with chronic alcohol consumption. Comput Biol Med. 2015;64:299–306.
Medici V, Virata MC, Peerson JM, Stabler SP, French SW, Gregory JF 3rd, Albanese A, Bowlus CL, Devaraj S, Panacek EA, Richards JR, Halsted CH. S-adenosyl-l-methionine treatment for alcoholic liver disease: a double-blinded, randomized, placebo-controlled trial. Alcohol Clin Exp Res. 2011;35:1960–5.
Heo MJ, Kim TH, You JS, Blaya D, Sancho-Bru P, Kim SG. Alcohol dysregulates miR-148a in hepatocytes through FoxO1, facilitating pyroptosis via TXNIP overexpression. Gut. 2019;68:708–20.
Lippai D, Bala S, Catalano D, Kodys K, Szabo G. Micro-RNA-155 deficiency prevents alcohol-induced serum endotoxin increase and small bowel inflammation in mice. Alcohol Clin Exp Res. 2014;38:2217–24.
Bala S, Marcos M, Kodys K, Csak T, Catalano D, Mandrekar P, Szabo G. Up-regulation of microRNA-155 in macrophages contributes to increased tumor necrosis factor alpha (TNFα) production via increased mRNA half-life in alcoholic liver disease. J Biol Chem. 2011;286:1436–44.
Bala S, Csak T, Kodys K, Catalano D, Ambade A, Furi I, Lowe P, Cho Y, Iracheta-Vellve A, Szabo G. Alcohol-induced miR-155 and HDAC11 inhibit negative regulators of the TLR4 pathway and lead to increased LPS responsiveness of Kupffer cells in alcoholic liver disease. J Leukoc Biol. 2017;102:487–98.
Lippai D, Bala S, Csak T, Kurt-Jones EA, Szabo G. Chronic alcohol-induced microRNA-155 contributes to neuroinflammation in a TLR4-dependent manner in mice. PLoS ONE. 2013;8:e70945.
Zhang Y, Wei G, Di Z, Zhao Q. miR-339-5p inhibits alcohol-induced brain inflammation through regulating NF-κB pathway. Biochem Biophys Res Commun. 2014;452:450–6.
Motivala SJ. Sleep and inflammation: psychoneuroimmunology in the context of cardiovascular disease. Ann Behav Med. 2011;42:141–52.
Andrabi M, Andrabi MM, Kunjunni R, Sriwastva MK, Bose S, Sagar R, Srivastava AK, Mathur R, Jain S, Subbiah V. Lithium acts to modulate abnormalities at behavioral, cellular, and molecular levels in sleep deprivation-induced mania-like behavior. Bipolar Disord. 2020;22:266–80.
Chen W, Guo X, Jin Z, Li R, Shen L, Li W, Cai W, Zhang G. Transcriptional alterations of genes related to fertility decline in male rats induced by chronic sleep restriction. Syst Biol Reprod Med. 2020;66:99–111.
Li X, Joehanes R, Hoeschele I, Rich SS, Rotter JI, Levy D, Liu Y, Redline S, Sofer T. Association between sleep disordered breathing and epigenetic age acceleration: evidence from the Multi-Ethnic Study of Atherosclerosis. EBioMedicine. 2019;50:387–94.
Kim J, Bhattacharjee R, Khalyfa A, Kheirandish-Gozal L, Capdevila OS, Wang Y, Gozal D. DNA methylation in inflammatory genes among children with obstructive sleep apnea. Am J Respir Crit Care Med. 2012;185:330–8.
Smith JA, Zhao W, Wang X, Ratliff SM, Mukherjee B, Kardia SLR, Liu Y, Roux AVD, Needham BL. Neighborhood characteristics influence DNA methylation of genes involved in stress response and inflammation: the Multi-Ethnic Study of Atherosclerosis. Epigenetics. 2017;12:662–73.
Needham BL, Smith JA, Zhao W, Wang X, Mukherjee B, Kardia SL, Shively CA, Seeman TE, Liu Y, Diez Roux AV. Life course socioeconomic status and DNA methylation in genes related to stress reactivity and inflammation: the Multi-Ethnic Study of Atherosclerosis. Epigenetics. 2015;10:958–69.
Stringhini S, Polidoro S, Sacerdote C, Kelly RS, van Veldhoven K, Agnoli C, Grioni S, Tumino R, Giurdanella MC, Panico S, Mattiello A, Palli D, Masala G, Gallo V, Castagné R, Paccaud F, Campanella G, Chadeau-Hyam M, Vineis P. Life-course socioeconomic status and DNA methylation of genes regulating inflammation. Int J Epidemiol. 2015;44:1320–30.
McDade TW, Ryan C, Jones MJ, MacIsaac JL, Morin AM, Meyer JM, Borja JB, Miller GE, Kobor MS, Kuzawa CW. Social and physical environments early in development predict DNA methylation of inflammatory genes in young adulthood. Proc Natl Acad Sci USA. 2017;114:7611–6.
Janusek LW, Tell D, Gaylord-Harden N, Mathews HL. Relationship of childhood adversity and neighborhood violence to a proinflammatory phenotype in emerging adult African American men: an epigenetic link. Brain Behav Immun. 2017;60:126–35.
Pfau ML, Menard C, Cathomas F, Desland F, Kana V, Chan KL, Shimo Y, LeClair K, Flanigan ME, Aleyasin H, Walker DM, Bouchard S, Mack M, Hodes GE, Merad MM, Russo SJ. Role of monocyte-derived microRNA106b∼25 in resilience to social stress. Biol Psychiatry. 2019;86:474–82.
Harkess KN, Ryan J, Delfabbro PH, Cohen-Woods S. Preliminary indications of the effect of a brief yoga intervention on markers of inflammation and DNA methylation in chronically stressed women. Transl Psychiatry. 2016;6:e965.
Kaliman P, Alvarez-López MJ, Cosín-Tomás M, Rosenkranz MA, Lutz A, Davidson RJ. Rapid changes in histone deacetylases and inflammatory gene expression in expert meditators. Psychoneuroendocrinology. 2014;40:96–107.
Chaix R, Fagny M, Cosin-Tomás M, Alvarez-López M, Lemee L, Regnault B, Davidson RJ, Lutz A, Kaliman P. Differential DNA methylation in experienced meditators after an intensive day of mindfulness-based practice: implications for immune-related pathways. Brain Behav Immun. 2020;84:36–44.
Kim D, Kubzansky LD, Baccarelli A, Sparrow D, Spiro A 3rd, Tarantini L, Cantone L, Vokonas P, Schwartz J. Psychological factors and DNA methylation of genes related to immune/inflammatory system markers: the VA Normative Aging Study. BMJ Open. 2016;6:e009790.
Wang C, O’Brien KM, Xu Z, Sandler DP, Taylor JA, Weinberg CR. Long-term ambient fine particulate matter and DNA methylation in inflammation pathways: results from the Sister Study. Epigenetics. 2020;15:524–35.
Wang C, Chen R, Shi M, Cai J, Shi J, Yang C, Li H, Lin Z, Meng X, Liu C, Niu Y, Xia Y, Zhao Z, Kan H, Weinberg CR. Possible mediation by methylation in acute inflammation following personal exposure to fine particulate air pollution. Am J Epidemiol. 2018;187:484–93.
Zhong J, Colicino E, Lin X, Mehta A, Kloog I, Zanobetti A, Byun HM, Bind MA, Cantone L, Prada D, Tarantini L, Trevisi L, Sparrow D, Vokonas P, Schwartz J, Baccarelli AA. Cardiac autonomic dysfunction: particulate air pollution effects are modulated by epigenetic immunoregulation of toll-like receptor 2 and dietary flavonoid intake. J Am Heart Assoc. 2015;4:e001423.
Lepeule J, Bind MA, Baccarelli AA, Koutrakis P, Tarantini L, Litonjua A, Sparrow D, Vokonas P, Schwartz JD. Epigenetic influences on associations between air pollutants and lung function in elderly men: the normative aging study. Environ Health Perspect. 2014;122:566–72.
Bind MA, Lepeule J, Zanobetti A, Gasparrini A, Baccarelli A, Coull BA, Tarantini L, Vokonas PS, Koutrakis P, Schwartz J. Air pollution and gene-specific methylation in the Normative Aging Study: association, effect modification, and mediation analysis. Epigenetics. 2014;9:448–58.
Pardo M, Kuperman Y, Levin L, Rudich A, Haim Y, Schauer JJ, Chen A, Rudich Y. Exposure to air pollution interacts with obesogenic nutrition to induce tissue-specific response patterns. Environ Pollut. 2018;239:532–43.
Fiorito G, Vlaanderen J, Polidoro S, Gulliver J, Galassi C, Ranzi A, Krogh V, Grioni S, Agnoli C, Sacerdote C, Panico S, Tsai MY, Probst-Hensch N, Hoek G, Herceg Z, Vermeulen R, Ghantous A, Vineis P, Naccarati A. EXPOsOMICS consortium‡. Oxidative stress and inflammation mediate the effect of air pollution on cardio- and cerebrovascular disease: a prospective study in nonsmokers. Environ Mol Mutagen. 2018;59:234–46.
Cantone L, Iodice S, Tarantini L, Albetti B, Restelli I, Vigna L, Bonzini M, Pesatori AC, Bollati V. Particulate matter exposure is associated with inflammatory gene methylation in obese subjects. Environ Res. 2017;152:478–84.
Pavanello S, Bonzini M, Angelici L, Motta V, Pergoli L, Hoxha M, Cantone L, Pesatori AC, Apostoli P, Tripodi A, Baccarelli A, Bollati V. Extracellular vesicle-driven information mediates the long-term effects of particulate matter exposure on coagulation and inflammation pathways. Toxicol Lett. 2016;259:143–50.
Chen R, Li H, Cai J, Wang C, Lin Z, Liu C, Niu Y, Zhao Z, Li W, Kan H. Fine particulate air pollution and the expression of microRNAs and circulating cytokines relevant to inflammation, coagulation, and vasoconstriction. Environ Health Perspect. 2018;126:017007.
Li X, Jia Y, Nan A, Zhang N, Zhou H, Chen L, Pan X, Qiu M, Zhu J, Zhang H, Ling Y, Jiang Y. CircRNA104250 and lncRNAuc001.dgp.1 promote the PM2.5-induced inflammatory response by co-targeting miR-3607–5p in BEAS-2B cells. Environ Pollut. 2020;258:113749.
Fry RC, Rager JE, Bauer R, Sebastian E, Peden DB, Jaspers I, Alexis NE. Air toxics and epigenetic effects: ozone altered microRNAs in the sputum of human subjects. Am J Physiol Lung Cell Mol Physiol. 2014;306:L1129–37.
Li J, Wang T, Wang Y, Xu M, Zhang L, Li X, Liu Z, Gao S, Jia Q, Fan Y, Wang Z, Wu N, Zhang X, Dai Y, Kong F, Wang W, Duan H. Particulate matter air pollution and the expression of microRNAs and pro-inflammatory genes: association and mediation among children in Jinan. China J Hazard Mater. 2020;389:121843.
González-Muniesa P, Garcia-Gerique L, Quintero P, Arriaza S, Lopez-Pascual A, Martinez JA. Effects of hyperoxia on oxygen-related inflammation with a focus on obesity. Oxid Med Cell Longev. 2015;2015:8957827.
Lopez-Pascual A, Lasa A, Portillo MP, Arós F, Mansego ML, González-Muniesa P, Martinez JA. Low oxygen consumption is related to a hypomethylation and an increased secretion of IL-6 in obese subjects with sleep apnea-hypopnea syndrome. Ann Nutr Metab. 2017;71:16–25.
Butts B, Butler J, Dunbar SB, Corwin EJ, Gary RA. ASC methylation and interleukin-1β are associated with aerobic capacity in heart failure. Med Sci Sports Exerc. 2017;49:1072–8.
Ali MM, Phillips SA, Mahmoud AM. HIF1α/TET1 pathway mediates hypoxia-induced adipocytokine promoter hypomethylation in human adipocytes. Cells. 2020;9:134.
Badran M, Yassin BA, Lin DTS, Kobor MS, Ayas N, Laher I. Gestational intermittent hypoxia induces endothelial dysfunction, reduces perivascular adiponectin and causes epigenetic changes in adult male offspring. J Physiol. 2019;597:5349–64.
Huang L, Chen X, Dasgupta C, Chen W, Song R, Wang C, Zhang L. Foetal hypoxia impacts methylome and transcriptome in developmental programming of heart disease. Cardiovasc Res. 2019;115:1306–19.
Chouvarine P, Legchenko E, Geldner J, Riehle C, Hansmann G. Hypoxia drives cardiac miRNAs and inflammation in the right and left ventricle. J Mol Med (Berl). 2019;97:1427–38.
Boddicker RL, Koltes JE, Fritz-Waters ER, Koesterke L, Weeks N, Yin T, Mani V, Nettleton D, Reecy JM, Baumgard LH, Spencer JD, Gabler NK, Ross JW. Genome-wide methylation profile following prenatal and postnatal dietary Ω-3 fatty acid supplementation in pigs. Anim Genet. 2016;47:658–71.
Bigagli E, Cinci L, Paccosi S, Parenti A, D’Ambrosio M, Luceri C. Nutritionally relevant concentrations of resveratrol and hydroxytyrosol mitigate oxidative burst of human granulocytes and monocytes and the production of pro-inflammatory mediators in LPS-stimulated RAW 264.7 macrophages. Int Immunopharmacol. 2017;43:147–55.
Izquierdo V, Palomera-Ávalos V, López-Ruiz S, Canudas AM, Pallàs M, Griñán-Ferré C. Maternal resveratrol supplementation prevents cognitive decline in senescent mice offspring. Int J Mol Sci. 2019;20:1134.
Maugeri A, Barchitta M, Mazzone MG, Giuliano F, Basile G, Agodi A. Resveratrol modulates SIRT1 and DNMT functions and restores LINE-1 methylation levels in ARPE-19 cells under oxidative stress and inflammation. Int J Mol Sci. 2018;19:2118.
Isac S, Panaitescu AM, Spataru A, Iesanu M, Totan A, Udriste A, Cucu N, Peltecu G, Zagrean L, Zagrean AM. Trans-resveratrol enriched maternal diet protects the immature hippocampus from perinatal asphyxia in rats. Neurosci Lett. 2017;653:308–13.
Carpi S, Scoditti E, Massaro M, Polini B, Manera C, Digiacomo M, Esposito Salsano J, Poli G, Tuccinardi T, Doccini S, Santorelli FM, Carluccio MA, Macchia M, Wabitsch M, De Caterina R, Nieri P. The extra-virgin olive oil polyphenols oleocanthal and oleacein counteract inflammation-related gene and mirna expression in adipocytes by attenuating NF-κB activation. Nutrients. 2019;11:2855.
Scoditti E, Carpi S, Massaro M, Pellegrino M, Polini B, Carluccio MA, Wabitsch M, Verri T, Nieri P, De Caterina R. Hydroxytyrosol modulates adipocyte gene and miRNA expression under inflammatory condition. Nutrients. 2019;11:2493.
Otton R, Bolin AP, Ferreira LT, Marinovic MP, Rocha ALS, Mori MA. Polyphenol-rich green tea extract improves adipose tissue metabolism by down-regulating miR-335 expression and mitigating insulin resistance and inflammation. J Nutr Biochem. 2018;57:170–9.
Gentile D, Fornai M, Colucci R, Pellegrini C, Tirotta E, Benvenuti L, Segnani C, Ippolito C, Duranti E, Virdis A, Carpi S, Nieri P, Németh ZH, Pistelli L, Bernardini N, Blandizzi C, Antonioli L. The flavonoid compound apigenin prevents colonic inflammation and motor dysfunctions associated with high fat diet-induced obesity. PLoS ONE. 2018;13:e0195502.
Heyman-Lindén L, Seki Y, Storm P, Jones HA, Charron MJ, Berger K, Holm C. Berry intake changes hepatic gene expression and DNA methylation patterns associated with high-fat diet. J Nutr Biochem. 2016;27:79–95.
Kim HJ, Lee W, Yun JM. Luteolin inhibits hyperglycemia-induced proinflammatory cytokine production and its epigenetic mechanism in human monocytes. Phytother Res. 2014;28:1383–91.
Lee W, Lee SY, Son YJ, Yun JM. Gallic acid decreases inflammatory cytokine secretion through histone acetyltransferase/histone deacetylase regulation in high glucose-induced human monocytes. J Med Food. 2015;18:793–801.
Kim HJ, Kim SH, Yun JM. Fisetin inhibits hyperglycemia-induced proinflammatory cytokine production by epigenetic mechanisms. Evid Based Complement Alternat Med. 2012;2012:639469.
Fan R, You M, Toney AM, Kim J, Giraud D, Xian Y, Ye F, Gu L, Ramer-Tait AE, Chung S. Red raspberry polyphenols attenuate high-fat diet-driven activation of NLRP3 inflammasome and its paracrine suppression of adipogenesis via histone modifications. Mol Nutr Food Res. 2020;64:e1900995.
Yun JM, Jialal I, Devaraj S. Effects of epigallocatechin gallate on regulatory T cell number and function in obese v. lean volunteers. Br J Nutr. 2010;103:1771–7.
Acknowledgements
The support from CIBERobn, IMDEA-Food Institute, and the Government of Navarra is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Additional information
Responsible Editor: John Di Battista.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Ramos-Lopez, O., Milagro, F.I., Riezu-Boj, J.I. et al. Epigenetic signatures underlying inflammation: an interplay of nutrition, physical activity, metabolic diseases, and environmental factors for personalized nutrition. Inflamm. Res. 70, 29–49 (2021). https://doi.org/10.1007/s00011-020-01425-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00011-020-01425-y