


Abstract
In operando, ambient-pressure x-ray photoelectron spectroscopy (AP-XPS) has been used to evaluate surface states of gadolinia-doped ceria (GDC) thin-film electrodes during H2 oxidation and H2O electrolysis, on yttria-stabilized zirconia (YSZ)-supported solid oxide electrochemical cells (SOCs). Porous nickel (Ni) and gold (Au) overlayers deposited on separate GDC thin films served as current collectors and potential electrocatalysts to facilitate heterogeneous chemistry for H2 oxidation or H2O electrolysis. Electrochemical characterization of the GDC thin-film electrodes complemented in operando XPS measurements of the O 1s spectra to correlate electrochemical overpotentials with surface chemistry near the Ni/GDC and Au/GDC interfaces. Shifts in O 1s binding energies across the metal/GDC/YSZ interfaces signified changes of local surface potential and provided a means of estimating kinetic parameters associated with charge transfer reactions. Effective oxygen partial pressure and surface potential impacted oxide vacancy and ceria polaron concentrations in the GDC, resulting in different reactivities of the GDC under the tested conditions. Both the Ni/GDC and Au/GDC demonstrated much higher currents for H2O electrolysis vs. H2 oxidation for comparable metal/GDC overpotentials due to increased electronic conductivity of the GDC under positive potentials and associated spreading of the electrochemically active region away from the triple-phase boundary. Higher electrochemical activity of the Ni/GDC electrode is attributed to the increased H2 activation on Ni in promoting charge transfer reactions (particularly for H2 oxidation). These results provide a basis for developing more informed reaction mechanisms for both H2 oxidation and H2O electrolysis of GDC-based composite electrodes in SOCs.
Export citation and abstract BibTeX RIS
1. Introduction
Solid oxide electrochemical cells (SOCs) offer a means for clean and efficient conversion of chemical energy into electricity as fuel cells or a means of storing excess electrical energy into chemical fuels as electrolysis cells. Gadolinia-doped ceria (GDC) has been investigated widely for its potential as an electrolyte and electrode material in SOCs operating at intermediate temperatures between 500 °C and 700 °C [1–9]. Over this temperature range, GDC has an oxide-ion conductivity that is approximately 1–2 orders of magnitude higher than yttria-stabilized zirconia (YSZ) [2], which has been the most common SOC electrolyte material. GDC's higher ionic conductivity lowers ohmic resistances associated with bulk-ion transport in SOCs. Under reducing environments associated with the fuel electrode, reduction of some cerium cations (from Ce4+ to Ce3+) results in mixed ionic-electronic conductivity (MIEC) [10–12]. The MIEC behaviour lowers open circuit voltages (OCVs) due to electronic leakage, particularly at temperatures above 600 °C, which reduces efficiency in GDC-electrolyte fuel cells. On the other hand, the mixed conductivity enhances catalytic activity in composite metal/GDC electrodes for fuel oxidation (including carbonaceous fuels) and H2O electrolysis, in part by extending the surface area available for the electrochemical reactions [13–16].
Extensive studies have characterized bulk transport and thermodynamic properties of GDC under conditions relevant for its application as either an electrolyte or a component in a composite SOC fuel electrode [17–20]. Some studies have used thin films to indirectly resolve near-surface oxide vacancies for GDC as a function of effective O2 partial pressure (PO2) and temperature characteristic of SOC electrodes [21, 22]. Although these studies correlated partial GDC reduction with MIEC behaviour and electrochemical activity due to high concentration of near-surface oxide vacancies, development of reaction mechanisms for GDC-based composite fuel electrodes requires more quantitative surface measurements of GDC in operando with relevant composite materials.
Ambient-pressure x-ray photoelectron spectroscopy (AP-XPS) has received notable attention in recent years for studying high-temperature, electrochemical reactions in operando. Researchers have used this technique to study ceria and doped ceria under low effective PO2 characteristic of fuel electrodes in SOCs [14, 15, 23–25]. In operando AP-XPS can measure surface oxidation states associated with electrochemical reactions, as shown in recent noteworthy studies on ceria-based electrodes to explore H2/H2O or CO/CO2 electrochemistry [26–28]. These studies provided insight into the reducibility of ceria surface cations as a function of overpotential, temperature, and gaseous composition. On the other hand, they have not provided significant insight into the interactions between ceria and metal current collectors or within the metal/ceria-based composite electrodes (which can promote catalytic and charge transfer reactions). Understanding metal/GDC interactions is critical for developing quantitative electrochemical mechanisms for GDC-based electrodes such as Ni/GDC composites, which are now being used in intermediate temperature fuel cells. With appropriately designed metal/GDC electrodes, in operando AP-XPS can measure surface potentials and changes in some surface species fractions near the metal/GDC interfaces.
In this study, simultaneous electrochemical measurements and AP-XPS were performed at the Advanced Light Source (ALS) at Lawrence Berkeley National Laboratory on thin-film Ni/GDC and Au/GDC electrodes operating for H2 oxidation and for H2O electrolysis [29, 30]. Spatially resolved measurements of the O 1s spectra and binding energy shifts for local surface potentials across the metal/GDC interfaces in combination with simultaneous electrochemical measurements provided critical information on the nature of the electrochemical reactions as a function of temperature and PO2 characteristic of fuel electrodes in SOCs. With the increased catalytic activity for Ni compared to Au, differences in the electrochemical performance of the two different electrodes provided an indication on how catalytic activity on the Ni surface can significantly impact the GDC surface chemistry under conditions where reduced electron conductivity confines reactions closer to the metal/GDC interface. The AP-XPS measurements with extensive electrochemical characterization for H2 oxidation and H2O electrolysis on the thin-film electrodes at similar operating conditions provided mechanistic insight into the nature or electrochemical reactions across metal/GDC under reducing environments.
2. Experimental
2.1. Electrochemical cell fabrication and mounting
Single-chamber SOCs in this work were fabricated by radio frequency (RF) sputtering (Kurt Lesker PVD 75) of thin-film GDC on polycrystalline YSZ-support substrates. The substrates were made from YSZ powder (Tosoh, 8% by mole of Y2O3) pressed in a cylindrical die at 1400 bar. The resulting disk-shape pellet was machined into a square shape with interconnected grooves across the length of one side to provide a location for subsequent deposition of a platinum (Pt) counter electrode (CE) on the opposing side of the GDC thin-film working electrodes (WEs). Figure 1(a) shows how the grooves provided the CE with adequate exposure to gases in the single-chamber cell tests at the AP-XPS facility, where a ceramic contact heater on the CE side controlled the cell operating temperature. The machined-YSZ substrate was sintered at 1450 °C for 3 h to densify with dimensions of 1 mm thick and 10 mm × 10 mm in top-side surface area. The low current density demanded by the thin-film electrodes permitted the use of the thick YSZ electrolyte support. Two 300 nm thick films (2.5 mm × 4.0 mm in area) of GDC (20% by mole of Gd2O3, Gd0.2Ce0.8O1.9) were deposited by RF-sputtering on the flat topside of the YSZ substrate. The two GDC thin films were separated by a 1.8 mm gap on the YSZ surface to avoid electrochemical interactions during independent electrochemical testing of each WE.

Figure 1. (a) Schematic of YSZ-supported solid oxide electrochemical cell with two thin-film Ni/GDC and Au/GDC working electrodes and a backside Pt counter electrode deposited in grooves of the YSZ. (b) Schematic showing operation of the electrochemical cells at external voltage bias (Vcell). For Vcell > 0, H2O electrolysis takes place on the Ni/GDC or Au/GDC working electrode (WE) and oxide ions are transported from the WE to the porous Pt counter electrode (CE) where H2 oxidation occurs. For Vcell < 0, H2 oxidation takes place on the Ni/GDC or Au/GDC WE and oxide ions are transported to the WE from the Pt CE where H2O electrolysis occurs. XPS spectral maps are taken across GDC from the metal/GDC border to the GDC/YSZ border. (c) Photographic image of the WE electrode side of a cell mounted on the ceramic surface heater before AP-XPS testing at the ALS.
Download figure:
Standard image High-resolution imageNi and Au (~300 nm thick) metal overlayers were sputter-deposited (AJA International, ATC 1800-V) separately on the two GDC films with a 300 μm border of exposed GDC around the edges of the overlayers. The deposited Ni and Au overlayers were annealed at 750 °C in 5% H2 in volume (balanced with Ar) such that the overlayers formed interconnected open structure exposing pockets of the GDC surface over the entire area of each WE as illustrated in scanning electronic microscopy (SEM) images for a GDC thin-film WE with a Ni overlayer in figure 2(a) and with an Au overlayer in figure 2(b). The annealed metal overlayers consisted of 1–2 μm wide structures with adequate interconnections to provide both good in-plane conductivity across the thin-film WE and sufficient exposure of the GDC surface for significant length of the triple-phase boundary (TPB) between the metal, GDC, and gas phases on the WE surface. As such, the interconnected Ni and Au overlayers served both as current collectors and as a possible facilitator of charge transfer reactions between the metal and GDC as illustrated in figure 1(b).

Figure 2. SEM images of surface morphologies of: (a) porous Ni overlayer and (b) porous Au overlayer (sputter-deposited) on thin-film GDC electrode surface after annealing in 5% H2 in volume (balanced with Ar) at 750 °C.
Download figure:
Standard image High-resolution imageThe substantial TPB can be characterized by the length per unit geometric area of the GDC surface (Ltpb). To characterize the Ltpb of both metal/GDC interfaces, scanning electron micrographs, such as those shown in figure 2(a) for the Ni overlayer and figure 2(b) for the Au overlayer, were processed to identify edges of the porous metal overlayer on the GDC surface. Image processing was done in MATLAB and the 'Edge' function was used with the Canny algorithm to detect edges with the porous metal on the GDC surface. In general, the annealing of the two different metal overlayers led to slightly smaller length scales and thus higher Ltpb for the Ni overlayer than for the Au. For this study, average Ltpb was 1.87 × 106 m for Ni/GDC and 1.23 × 106 m for Au/GDC, per unit surface area (in m2) of the GDC thin films covered by the Ni or Au overlayer. The 50% higher Ltpb for Ni/GDC would improve rates for any rate-limiting reactions involving charge and/or species transfer between the metal and the GDC surface. On the other hand, for reactions occurring completely on the GDC surface, Ltpb should play a very limited role in rates.
A 100 nm thick Pt CE was sputter-deposited (AJA International, ATC 1800-V) in the grooves on the backside of the YSZ as shown in figure 1(a). The Pt CE was fired at 800 °C to form a porous overlayer on the YSZ surface and thereby facilitate effective charge transfer for both H2O electrolysis and H2 oxidation on the CE. The grooves, in which the Pt was deposited, facilitated gas transport to and from Pt/YSZ/gas TPB on the CE, while allowing the cell to maintain good thermal contact with the surface heater during in operando XPS experiments. A Pt wire was attached to the Pt CE using a Pt paste and brought around to topside of the YSZ where a Pt mesh for electrical contact was fixed on an alumina blocking layer deposited on the topside of the YSZ substrate to facilitate an external electrical-lead connection. In the AP-XPS test station at ALS, the cell was mounted with three probes pressing separately onto the Ni overlayer, the Au overlayer and the Pt mesh to ensure good electrical connections as well as thermal contact between the backside of the cell and the ceramic heater as shown in figure 1(c).
2.2. Electrochemical characterizations
The SOC was connected to a potentiostat and frequency response analyser, BioLogic at ALS or Metrohm Autolab at the University of Maryland (UMD) for electrochemical characterization. Each WE with the Ni or Au overlayer underwent electrochemical characterization separately. A bias voltage (Vcell) was applied between the Pt CE and the connected metal overlayer (Ni or Au), which was grounded. Depending on the sign of the bias, the GDC WE facilitated either H2 oxidation or H2O electrolysis while the reverse reaction occurred on the Pt CE. For example, with a positive voltage bias (Vcell > 0) applied to the cell as illustrated in figure 1(b), oxide ions (O2−) were driven from the GDC WE, where H2O electrolysis occurred, to the Pt CE, where H2 was oxidized. On the other hand, a negative bias (Vcell < 0) drove H2 oxidation on the GDC WE and H2O electrolysis on Pt CE.
Electrochemical testing at cell operating temperatures (T) between 570 and 650 °C provided a range of conditions over which the electronic conductivity of GDC rose significantly at the effective PO2 in this study. For the ALS measurements, T was determined by the electrolyte bulk resistance (Rbulk), which was mapped in calibrated temperature experiments at UMD, where the cell-tube assembly was heated in a temperature-controlled vertical tubular furnace. This was important to the tests at ALS where the beamline test station did not provide direct thermocouple measurements on the cell. Due to limited beamline access at ALS, electrochemical characterization of the SOC at UMD was performed at Ptot = 1 bar over a broad range of H2 and H2O partial pressures (PH2 and PH2O) ranging from 2.6 mbar to 26 mbar diluted by an Ar balance. The low PH2 and PH2O conditions simulated the pressures available in the AP-XPS facility at ALS.
For the tests at ALS, H2 (PH2 = 0.26 mbar) and H2O (PH2O = 0.26 or 0.052 mbar) gases were backfilled into the XPS test chamber through leak valves to the desired composition. PH2 and PH2O were monitored via dedicated quadrupole mass spectrometers for each gas, and the measurements were observed to be stable throughout the experiments even during electrochemical activation of the cells. The H2/H2O mixtures enabled the GDC WE to perform as a fuel electrode for H2 oxidation and H2O electrolysis at different effective PO2.
2.3. X-ray photoelectron spectroscopy measurements
In operando AP-XPS characterization at ALS Beamline 9.3.2 provided quantitative spectroscopic surface chemical analysis [23, 27, 30–34]. The synchrotron-based XPS with differential pumping stages enabled simultaneous electrochemical analysis and surface chemical measurements in the presence of gases up to a few mbar [32]. Effective gas-phase PO2 was varied by the PH2/PH2O ratios and the cell operating T. XPS incident radiation fixed at 650 eV permitted measurements of the O 1s spectra with peaks around 532, 534, and 536 eV associated with oxide ion (O2−), surface hydroxyl (OH−), and near-surface/adsorbed water (H2O), respectively [15, 35]. At these energy levels, the inelastic mean free path is expected to be approximately 0.65 nm [28], which is only slightly above the GDC lattice constant (0.543 nm).
The beamline is maintained under UHV between periods of ambient pressure operation to reduce risks of contamination. XPS survey scans showed negligibly small C 1s (see supplementary material figure S1 (available online at stacks.iop.org/JPENERGY/3/014004/mmedia)) spectra which verified that there were not significant amounts of carbonaceous surface species. The survey scans up to the 650 eV limit confirmed no significant binding energy shifts resulting from charging effects for relevant elements and valence bands during conditions without any applied voltage bias to the cell. The grounded metal overlayer, in particular the Au current collector, did not show any binding energy shift (on the Au 4f peaks in supplementary material figure S2) and thus served as a reference for assessing the observed binding energy shifts for the O 1s peaks during conditions of applied bias.
Two-dimensional XPS spectral maps (binding energy versus spatial location along a line across the surface of the WE) were recorded with a dispersive hemispherical analyser with two multi-channel plates coupled to a phosphor screen and a charge-coupled device camera installed on the beamline [29]. XPS analyser settings are provided in the supplementary material. The measurements provided spatial resolution of a 16 μm diameter spot size. Linear scans covered a spectral range for the O 1s spectra (525–540 eV) along a line across the exposed GDC from the metal/GDC border to the GDC/YSZ border. Linear scans at different Vcell (1.0, 0.6, 0, −0.6, and −1.0 V) provided measurements of surface species and overpotentials for conditions of both H2O electrolysis and H2 oxidation on the WE. Analysis of relative shifts of the primary (O2−) and satellite (OH− and H2O) peaks in the O 1s spectra associated with Vcell provided a basis for determining the local potential at specific location based on binding energy shifts as described in previous AP-XPS studies on solid oxide cells [14, 15].
As for reduction of GDC, past AP-XPS studies [14, 15, 23, 24] have shown changes in the Ce 3d spectra (880–920 eV) to assess the reduction of ceria under operation conditions similar to this study. The Ce 3d peak was beyond the excitation range of the beamline used in this study. As such, we could not readily access the relevant Ce 3d bands and deemed it unnecessary to show Ce 3d spectra in current study. From our previous studies and that of others [36], we provided thermodynamics calculation and analysis of the reduction state of GDC to help explain how the O 1s observations correlated with changes in Ce reduction.
3. Results and discussion
Figures 3(a) and (b) compare the electrochemical performances for the GDC WEs with the Ni overlayer (i.e. Ni/GDC electrode) and with the Au overlayer (i.e. Au/GDC electrode) during in operando XPS measurements at ALS for different T.

Figure 3. Electrochemical characterization of the thin-film GDC electrochemical cells operating with in operando XPS mapping. IRbulk-corrected V–I curves are plotted for T = 570 and 620 °C: (a) Ni/GDC WE operating at different PH2/PH2O ratios and (b) Au/GDC WE operating at PH2/PH2O = 1.0.
Download figure:
Standard image High-resolution imageThe IRbulk-corrected potentials (Vcell − IRbulk) subtract the ohmic overpotential from Vcell, where Rbulk is extracted from high-frequency impedance measurements. Because the tests were in a single chamber filled with mixtures of H2 and H2O, OCV was very close to 0 V for the H2/H2O electrochemistry. Negative Vcell − IRbulk (positive/anodic current) indicated H2 oxidation on the WE whereas positive Vcell − IRbulk (negative/cathodic current) indicated H2O electrolysis on the WE. Since both WEs shared the same YSZ electrolyte support and Pt CE, differences between V–I curves in figures 3(a) and (b) at the same operating conditions were attributed to differences in electrochemical behaviour of the Ni/GDC and Au/GDC electrodes.
Comparing figures 3(a) and (b) reveals that the Ni/GDC electrode achieved significantly higher current with increasing magnitude of Vcell, both for H2O electrolysis (Vcell > 0) and particularly for H2 oxidation (Vcell < 0). This ratio of currents for large positive and negative Vcell is much greater than the 50% increase in Ltpb of the Ni/GDC electrode in comparison to the Au/GDC electrode. For H2O electrolysis, the Ni/GDC electrode shows smaller activation overpotentials at small Vcell than the Au/GDC electrode. The Ni/GDC interface enhances charge transfer reaction rates due to the improved ability of Ni to adsorb H and OH on its surface much more effectively than Au [37]. At PH2 = PH2O = 0.26 mbar and T = 620 °C, the Ni/GDC WE supported current densities of −8.7 mA cm−2 at Vcell − IRbulk = 0.5 V and 4.0 mA cm−2 at −0.5 V (based on the area of the porous Ni overlayer). At the same conditions, the Au/GDC WE supported current densities of −1.9 mA cm−2 at +0.5 V and 0.7 mA cm−2 at −0.5 V. Under all conditions tested at the ALS, the Ni/GDC maintained significantly higher currents than the Au/GDC electrode with the exception of highly negative cell voltages at the lower T = 570 °C where the current densities of Ni/GDC and Au/GDC electrodes are within a factor of 2 which is close to the differences in Ltpb per area of the two WEs. The similar performance at highly oxidizing conditions (large negative Vcell) may be due to significant build-up of oxygenated species on the Ni surface reducing Ni/GDC charge transfer [38]. Non-oxidized H+ transfer across the Ni/GDC interface has been assessed to be the principal charge transfer reaction for H2 oxidation and H2O electrolysis [39, 40], and the increased affinity for H on Ni surfaces (vs. Au) provides higher exchange current densities and thus, lower activation overpotentials (ηact,WE) for both forward and reverse reactions.
To explore the role of the metal and GDC surfaces on H2 electrochemical oxidization and H2O electrolysis, AP-XPS measurements were taken during electrochemical excitations to probe surface states on the metal and GDC as a function of Vcell. AP-XPS of electrochemically active surfaces measures shifts in core-level binding energies of specific spectral peaks, which correlates with changes of surface electric potentials [14, 15]. In this study, shifts of the O 1s binding energy spectra across the metal/GDC/YSZ interfaces around the uncoated GDC film border measured changes in surface potentials as a function of Vcell. Since the metal electrodes were grounded, the measured binding energy shifts provided measures of ηact,WE associated with charge transfer reactions across the metal/GDC interface phases. Figures 4(a)–(c) show linear mapping of the relative counts per second (CPS) of the O 1s spectra as a function of position along the Ni/GDC/YSZ border for WEs operating at PH2 = PH2O = 0.26 mbar and T = 620 °C. For highly positive Vcell (= 1.0 V) in figure 4(a), positive shifts of the O 1s peak on the GDC were small but non-negligible. At these conditions (as discussed further below), the GDC is significantly reduced and thus provides higher polaron concentrations, which facilitates electron transport from as well as H+ transport to the Ni electrode. On the other hand, much larger negative binding energy shifts with negative Vcell (−1.0 V) in figure 4(c) revealed a larger ηact,WE for a similar current across the Ni/GDC interface, indicating increased transfer resistance for H2 oxidation (vs. H2O electrolysis) on the Ni/GDC electrode. At negative Vcell, the GDC has lower electronic conductivity and thus likely relies on H+ surface transport to enhance charge transfer.

Figure 4. (a)–(c) 2-D mapping of O 1s spectra (with brightness proportional to counts per second). The maps plot spectral intensity vs. binding energy across the exposed Ni/GDC/YSZ edge and show the binding energy shift as a function of Vcell associated with local potential change. Plots are shown for cells operating at PH2 = PH2O = 0.26 mbar and T = 620 °C under three potential biases (Vcell = 1.0, 0, and −1.0 V). (d) Analysis of the O 1s spectra by fitting to Gaussian/Lorentzian line-shapes peaks with Shirley-background corrected in CasaXPS (e.g. Vcell = 1.0 V). The fitted peaks compose of three oxygen-related species: oxide ion (O2−), surface hydroxyl (OH−), and near-surface/adsorbed water (H2O).
Download figure:
Standard image High-resolution imageThe O 1s spectra for each location were fitted to Gaussian/Lorentzian line-shape peaks (GL(30) with a product formula) with Shirley-background corrections using standard XPS data analysis techniques in CasaXPS. The complex peak shapes were fitted with the three peaks related to three oxygen-containing species (O2−, OH−, and H2O), exemplified at Vcell = 1.0 V in figure 4(d). The fits of the overlapping O2− and OH− peaks assumed identical full widths at half maximum with only a very small variation between the two different electrodes (FWHM(O2−) = FWHM(OH−) = 1.79 for Ni/GDC and FWHM(O2−) = FWHM(OH−) = 1.81 for Au/GDC). The integrated percentage area (percentage ratio relative to sum of O2−, OH− and H2O areas) for each component was then utilized for estimating surface species fractions. Figures 5(a) and (b) plot fitted O 1s spectra on the GDC surface at three representative Vcell and T = 620 °C near the Ni/GDC electrode and the Au/GDC electrode, respectively. In general, the local surface concentrations of OH− and O2− do not change significantly with Vcell for either WE, but Ni/GDC shows significantly higher concentration of OH−, which is likely tied to the higher activities (currents) for both H2O electrolysis and H2 oxidation. The higher OH− occupancy on the GDC can serve as a marker for higher activity because key charge transfer steps in both H2O electrolysis (reverse direction of reaction R1) and H2 oxidation (forward direction of reaction R2) as shown here depend on OH− activity on the GDC surface.
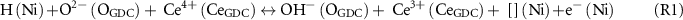
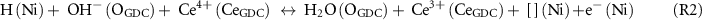

Figure 5. Analyses of the O 1s spectra on GDC surface (close to metal overlayer) with three surface species (O2−, OH−, and H2O) under Vcell = 1.0, 0, and −1.0 V, at PH2 = PH2O = 0.26 mbar and T = 620 °C: (a) Ni/GDC electrode and (b) Au/GDC electrode. Integrated area percentage relative to sum of O2−, OH− and H2O areas reflects concentration of each species.
Download figure:
Standard image High-resolution imagewhere (OGDC) and (CeGDC) represent oxide and ceria sites respectively on the GDC surface. The ability for the Ni surface to supply and to receive adsorbed H atoms plays a significant role in enhancing the electrochemical activity of the WE. The weak interactions of adsorbed H2O with lattice oxygen on the GDC surface [41] implies the importance of the metal surface in providing mobile H species to support more activity on the GDC surface. With the higher affinity of Ni for adsorbing H2O than Au [41], it is not surprising that the Ni/GDC electrode shows higher intensity for near-surface/adsorbed H2O in figure 5.
Primary peak (O2−) binding energies after the spectra fitting were plotted as a function of the cell position across the metal/GDC/YSZ border at different Vcell as shown in figures 6(a) and (b) for Ni/GDC/YSZ and Au/GDC/YSZ borders, respectively. Binding energy shifts on the GDC surface indicate changes in surface potential associated with the applied bias and resulting reactions. Due to surface charging, shifts in the YSZ surface do not necessarily correspond to surface potentials beneath the GDC film where the O2− charge transfer occurred between those two phases. The relatively flat binding energy distribution across the GDC surface suggests sufficient electronic conductivity to sustain uniform potential distribution on the GDC surface at the measured current densities. Even though the Ni/GDC and Au/GDC WEs have similar magnitudes of ηact,WE at a given Vcell, much higher output currents associated with Ni/GDC electrode indicated superior electrochemical activity of the Ni/GDC electrode. Both WEs showed significantly higher currents for H2O electrolysis than for H2 oxidation at similar magnitude of ηact,WE. Plotting ηact,WE across the metal/GDC interface as a function of current per Ltpb (i') in figure 7 shows that for H2O electrolysis, ηact,WE for the Au/GDC WE was only slightly higher than for Ni/GDC at the same i'. On the other hand, for H2 oxidation, ηact,WE for the Au/GDC WE grew much faster with increasing magnitude of i' than for H2O electrolysis. This is unlike Ni which had more similar slopes ηact,WE vs. i' for H2O electrolysis and H2 oxidation. Assuming that the two charge transfer reactions R1 and R2 determine the activation overpotential, Zhu et al [42] derived the following expression in equation (1) for a relationship between ηact,WE and i' across the Ni/oxide interface for H2 oxidation based on the assumption that (R1) is in equilibrium and (R2) is rate limiting.
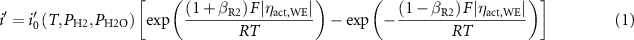

Figure 6. Binding energy of O 1s primary peak (associated with O2−) across the GDC/YSZ interface at PH2 = PH2O = 0.26 mbar, T = 620 °C: (a) for the Ni/GDC electrode and (b) for the Au/GDC electrode. The shifts of the local binding energy under potential bias relative to the OCV binding energy indicate near-surface electric potential changes.
Download figure:
Standard image High-resolution image
Figure 7. Comparison of measured overpotentials (circular markers) for metal/GDC interface as a function of current per TPB length for both Ni/GDC and Au/GDC electrodes. Overpotentials (with error bars) are determined by the binding energy shifts under different Vcell at PH2 = PH2O = 0.26 mbar and T = 620 °C. The curves represent independent fits of the H2O electrolysis and H2 oxidation activation overpotentials for each of the metal/GDC electrodes.
Download figure:
Standard image High-resolution imagewhere i'0 is the exchange current density which is a function of T and the partial pressures PH2 and PH2O [42]. Fitting equation (1) to measured ηact,WE plotted in figure 7 provided a basis for estimating fundamental charge transfer parameters such as i'0 and βR2 as shown in table 1. Least squared fits of i'0 and βR2 for H2O electrolysis currents and for H2 oxidation currents were performed independently for each WE, and the results in table 1 (which are plotted as curves in figure 7) show that with the exception of H2 oxidation all fits provide values of βR2 close to 0.5. Exchange current densities i'0 at 620 °C and PH2 = PH2O = 0.26 mbar are approximately 2.33 × 10−7 A m−1 of Ltpb with the exception of H2 oxidation on Au which has an i'0 that is approximately one order of magnitude lower. While it cannot be presumed that these values represent specific rates of charge transfer reactions, they do provide a basis for estimating orders of magnitudes of specific reaction rate constants that might be used in a more detailed charge transfer reaction rate expression [39, 40].
Table 1. Parameters from fits of current per unit length of Ltpb to measured metal/GDC activation overpotentials for both Ni/GDC and Au/GDC working electrodes.
| H2O electrolysis | H2 oxidation | |||
|---|---|---|---|---|
| Electrode | i'0 (A m−1 of Ltpb) | βR2 | i'0 (A m−1 of Ltpb) | βR2 |
| Ni/GDC @ 620 °C | 2.09 × 10−7 | 0.499 | 2.33 × 10−7 | 0.832 |
| Au/GDC @ 620 °C | 2.01 × 10−7 | 0.570 | 2.61 × 10−8 | 0.542 |
The charge transfer relationship in equation (1) assumes that the GDC oxide surface is largely equilibrated with the gas phase and that the low effective PO2 associated with the H2/H2O gas mixtures has not significantly impacted the GDC oxide vacancy site concentrations [VO]L and cation site concentrations of reduced ceria polarons [Ce3+]L. Surface oxide vacancies are critical in providing exchange of H2O with the gas phase through the following surface reactions.
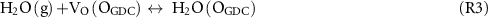
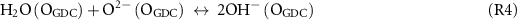
In addition, concentrations of reduced ceria polaron site [Ce3+]L play a role in increasing electronic conductivity of the GDC and thereby extending the region of effective reaction area for charge transfer reactions [35]. Thus, effective PO2 were calculated from chemical equilibrium with the H2/H2O mixtures and for the test conditions at the ALS that effective PO2 ranged from 1.2 × 10−24 bar at 570 °C to 6.3 × 10−23 bar at 620 °C. A calibrated Gd0.1Ce0.9O1.95 defect equilibrium model [36] was adopted for Gd0.2Ce0.8O1.9 and used to calculate [VO]L and cation site concentrations of reduced ceria polarons [Ce3+]L to assess whether changes in GDC defect concentrations significantly impact the electrochemical activity under the conditions tested. 20% Gd3+ doping on the Ce4+ sites results in lower boundary of [VO]L = 0.1 (out of a maximum of 2) at high PO2. Figure 8 plots the change in [VO]L and [Ce3+]L vs. effective PO2 at OCV and at ηact,WE = +0.2 V. Circular markers show predicted [VO]L and [Ce3+]L site fractions of tested conditions (both at ALS and UMD) at OCV. In general, for these relatively low T of solid oxide electrochemical cell operation, at open circuit, the calculated bulk [VO]L and [Ce3+]L remain very close to their fully oxidized limits. While it is understood that surface site concentrations of [VO]L and [Ce3+]L will be much higher [14], those concentrations will likely still close to equilibrated with the bulk and gas phase. On the other hand, when the GDC film experiences potentials ηact,WE, the voltage will shift surface and bulk concentrations as if it experienced a reduction or increase in effective PO2 (PO2,eff (ηact,WE)) according to the following equation.
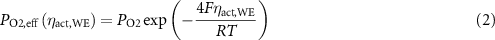

Figure 8. Plot of thermodynamically predicted Ce3+ (polaron) site fractions and VO (oxide vacancy) site fractions as a function of PO2 (in bar) for the different test conditions. Solid lines show curves of equilibrium site fractions at T = 570 and 620 °C of a given PO2 at OCV. Dashed lines shows curves equilibrium site fractions at T = 570 and 620 °C of a given PO2 and ηact,WE = +0.2 V on the GDC during H2O electrolysis. Circular markers show equilibrium site fractions of tested conditions (both at ALS and UMD) at OCV.
Download figure:
Standard image High-resolution imageFor ηact,WE < 0 during H2 oxidation at the conditions in this study, the increase in PO2,eff did not significantly impact [VO]L and [Ce3+]L, but for ηact,WE > 0 during H2O electrolysis, the drop in PO2,eff was significant. For an ηact,WE = 0.2 V during H2O electrolysis will shift the PO2,eff down by a factor of ~10−5 and thereby increase [Ce3+]L by a factor of 10 as indicated by the plots for ηact,WE = 0.2 V at the relevant T in figure 8. However, figure 8 also shows that [VO]L at these lower T remains close to the high PO2 limit. As such, the effects of ηact,WE on surface chemistry are not likely due to an increase in H2O exchange but may be due to an extension of the reaction region away from the Ni/GDC electrode TPB through improved electronic conductivity of the more reduced GDC surface. This explains the higher currents for positive ηact,WE in both the Ni/GDC and Au/GDC electrodes.
To more fully explore the surface chemistry as a function of ηact,WE, the fraction of O 1s peak area attributed to OH− was evaluated on the GDC surface very near the Ni overlayer. Figure 9 plots the measured fraction of the O 1s peak attributed to OH− which correlates with oxide surface site fractions. Because the AP-XPS detected oxygen a few nm beneath the surface, the surface site fractions of OH− were likely higher than the area fractions plotted in figure 9. Figure 9 indicates a few interesting trends. Firstly, voltage does not seem to greatly impact the OH− surface site fractions. Higher water partial pressure (PH2O) and lower T both increased OH− presence on the surface, but because these values do not represent actual site fractions, it remains unclear if surface OH− creation or removal remains a rate limiting step. The relative site fractions from figure 9 provide valuable data for surface mechanism calibration relevant for Ni/GDC electrodes in solid oxide electrochemical cell modelling.

Figure 9. Measured area percentage (with error bars) of OH− (ratio of integrated OH− area relative to sum of O2−, OH− and H2O areas) on the surface of the GDC near the Ni/GDC interface plotted as function of Vcell at different operation conditions at the ALS beamline endstation.
Download figure:
Standard image High-resolution imageDifferences between binding energy shifts of OH− peaks relative to shifts in the O2− peak provided an indicator of electrochemically active region of the oxide surface [28]. Since the OH− represents exclusively surface adsorbates and a large fraction of the O2− peak comes from subsurface contributions, differences of binding energy shifts between OH− and O2− correlated with a local potential difference between the surface adsorbates and lattice oxygen in the GDC film during electrochemical reaction. Figures 10(a) and (b) plot the binding energy difference across the metal/GDC/YSZ interfaces at different Vcell for both Ni/GDC and Au/GDC, respectively. Under non-zero Vcell, no significant differences were seen for the region of exposed GDC surface and YSZ edge compared to OCV. Binding energies of O2− and OH− shifting by the same amount implies minimal surface charge build-up in that region associated with electrochemistry. At the metal/GDC interface, the results showed no divergence of binding energy difference except for H2 oxidation reaction (Vcell = −1.0 V). This is consistent with the conclusion that surface polarization of the adsorbed OH− on GDC was minimal when the GDC has higher conductivity (i.e. during electrolysis) or when charge transfer across the metal/GDC interface was low (such as the Au/GDC WE for H2 oxidation). However, the increased charge transfer reactions at the Ni/GDC interface during H2 oxidation indicates a significant change in the binding energy difference of OH− and O2−. This indicates a significant surface charge build-up for ~40–50 μm from the Ni/GDC interface. This suggests the importance of charge transfer reactions R1 and R2 in facilitating electrochemistry and further supports the importance of activity of the metallic component in solid oxide fuel cell anodes [43].

Figure 10. Difference of binding energy shifts between O 1s primary peak (O2−) and secondary peak (OH−) across: (a) Ni/GDC/YSZ interfaces and (b) Au/GDC/YSZ interfaces, under Vcell = 1.0, 0, and −1.0 V, at PH2 = PH2O = 0.26 mbar, and T = 620 °C.
Download figure:
Standard image High-resolution imageDue to the limited access at the ALS, further electrochemical characterization of the two WEs were performed over a range of T (up to 650 °C) and effective PO2 using a range of H2/H2O mixtures. A higher range of PH2/PH2O ratios up to 10 were tested, and Ar dilution allowed low PH2 and PH2O values to be achieved comparable to those tested at ALS. Similar trends were observed between the Ni/GDC and Au/GDC electrode as shown in figures 11(a) and (b) compared to figures 3(a) and (b), respectively. The IRbulk-corrected V–I curves in figure 11 showed higher currents for H2O electrolysis for both WEs, but at 600 °C, the Ni/GDC electrode exhibited much lower polarization resistance than the Au/GDC electrode. At the higher T = 650 °C where electronic conductivity of the GDC significantly increased, the Au/GDC electrode showed significant increase in electrochemical activity. However, the much lower currents during H2 oxidation suggests that the negative ηact,WE suppresses the increased conductivity and associated regions of electrochemical activity. In general, increased reduction of ceria on the GDC surface at higher T and under cathodic current extends the regions of electrochemical activity on the GDC surface, but such effects are suppressed by the negative ηact,WE during high anodic current.

Figure 11. Comparison of electrochemical performance (IRbulk-corrected V–I curves) of the SOCs: (a) Ni/GDC electrode and (b) Au/GDC electrode, at fixed PH2/PH2O = 1.0 ratio for T from 570 to 650 °C.
Download figure:
Standard image High-resolution imageDependency on the effective PO2 of the electrochemical activity of the Ni/GDC and Au/GDC electrodes was further explored by increasing PH2/PH2O ratios at a constant T = 600 °C. Figures 12(a) and (b) plot IRbulk-corrected V–I curves for both WEs ratios for different PH2/PH2O up to a ratio of 10 (corresponding to an effective PO2 = 10−25 bar at 600 °C). This lower PO2 enables a substantial increase in [Ce3+]L for positive ηact,WE as suggested in figure 8, but the improvement in H2O electrolysis activity is not significant. Nonetheless, the increased reduction more than offsets the increased H2 product concentration and the higher PH2/PH2O ratios improved H2O electrolysis activity. Raising the PH2/PH2O ratio more significantly increased activity for H2 oxidation, particularly for the Ni/GDC electrode. The higher PH2/PH2O ratios reduce the risk for buildup of oxide species on the Ni surface at high anodic current. The improvement of H2 oxidation on Ni/GDC electrode at higher anodic current provides further proof of the role of the Ni activity on providing a key role in H2 oxidation for fuel cell conditions.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 12. Comparison of electrochemical performance (IRbulk-corrected V–I curves) of the SOCs: (a) Ni/GDC electrode and (b) Au/GDC electrode, for a range of PH2/PH2O ratios at T = 600 °C.
Download figure:
Standard image High-resolution image{kind=link}
4. Conclusion
The presented study explored surface potentials, active surface species, and associated electrochemical performance of the Ni/GDC and Au/GDC electrodes in H2 oxidation and H2O electrolysis. Ambient pressure XPS measurements of electrochemically active GDC thin-film SOCs provided a means of measuring local surface potential through shift of characteristic binding energy and thereby overpotentials across the metal/GDC interface associated with charge transfer reactions. Specifically, investigation of O 1s core-level XPS of the GDC electrodes under H2/H2O atmosphere has revealed how near-surface oxidation states as well as active surface-absorbed species relate to electrochemical activity in this study. Fundamental charge transfer parameters such as i'0 and βR2 were derived to provide a basis for estimating specific reaction rate constants that can be used in a more detailed kinetics. Our study revealed that [VO]L and [Ce3+]L in the GDC were affected by effective PO2 and surface potential which significantly impacted the reactivity of the GDC. The relative site fractions of OH− provided valuable data for surface mechanism calibration relevant for Ni/GDC electrodes. Differences between binding energy shifts of OH− peaks relative to shifts in the O2− peak revealed the region of electrochemically activity on the GDC away from the Ni/GDC interface.
Further electrochemical characterization of the Ni/GDC and Au/GDC showed the importance of the metal surface activity for increasing H2 oxidation. The Ni electrode provides much higher H2 oxidation currents than the less active Au electrode. On the other hand, the increased electronic conductivity of the GDC in ceria reduction-promoted voltages decreased the importance of the Ni activity, and both the Ni/GDC and Au/GDC electrodes showed similar and higher electrochemical activity for H2O electrolysis. The simultaneous electrochemical measurements and surface characterization with XPS has provided key data to assist in developing a more mechanistic understanding of GDC electrodes for both solid oxide fuel cells and solid oxide H2O-electrolysis cells.
Acknowledgments
This work was funded through the support of the Office of Naval Research through Contract # N000140510711 (Dr. Michele Anderson program manager). The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract # DE-AC02-05CH11231. We thank the University of Maryland Nanocenter and the University of Maryland Energy Research Center (UMERC) for support. We would also like to thank Dr. Chunjuan Zhang (now at BASF) who helped with the sputter deposition of the GDC films and the thin-film current collectors, and Drs. Sandrine Ricote and Huayang Zhu (Colorado School of Mines) who provided valuable feedback on the GDC kinetics.