
Abstract
Intrinsic organic small molecule and polymer materials are insulators. The discovery that polymers can be made highly conductive by doping has therefore sparked strong interest in this novel class of conductors. More recently, efficient doping of small molecule materials has also been achieved and is now a key technology in the multi-billion dollar organic light emitting diode industry. Nevertheless, a comprehensive description of charge transport in the presence of doping is still missing for organic semiconductors with localized electronic states. Here, we present a theoretical and computational approach based on percolation theory and quantitatively predict experimental results from the literature for the archetype small molecule materials ZnPc, F8ZnPc and C60. We show that transport in the complex potential landscape that emerges from the presence of localized charges can be aptly analyzed by focusing on the network properties of transport paths instead of just the critical resistance. Specifically, we compute the activation energy of conductivity and the Seebeck energy and yield excellent agreement with experimental data. The previously unexplained increase of the activation energy at high doping concentrations can be clarified by our approach.
Similar content being viewed by others

Introduction
The astonishing finding that the conductivity of conjugated polymers can be tailored over many orders of magnitude by doping with additional charges1,2 has led to a vast amount of work on these materials since the 1970s3. While the possibility of electrically doping small-molecule organic materials was also known for a long time4, its technological breakthrough was delayed until the late 1990s5,6. Nowadays, doping of organic semiconductors with controlled impurities is essential for almost all of their device applications, due to the notoriously low conductivity of undoped organic materials.
Experimentally, doping of small molecule semiconductors is a comparatively simple procedure, since the dopant molecule is usually just co-evaporated with the matrix molecule in the desired mixing ratio. On the theoretical side, despite its paramount importance in the organic semiconductor field, the description of charge transport in the presence of doping is still far from complete. In the past decades, a number of significant advances7,8,9,10,11,12,13,14,15 have been made, but still open questions remain.
An example is the influence of the Coulomb potentials of dopants and charge carriers on the nature of charge transport. While these potentials also exist in doped inorganic semiconductors, there is an important difference: Charge carrier states in organic semiconductors are strongly localized in many cases16,17,18. This leads to weaker screening and complex potential landscapes with strong spatial correlations10,19,20. In such a landscape, what is the mode of charge transport, which paths will carriers take, what are the important length scales? What effect will a certain doping concentration have on a given material?
Spatially averaged techniques based on the transport energy (TE) model8,21 are too limited for these questions, as they assume translational symmetry of the available hopping target energies22. Correlated energy landscapes have been studied extensively, mostly using a dipolar glass model22,23,24,25, atomistically calculated molecular interactions or site energies smoothed with a moving average26,27. The focus has been primarily on calculating the mobility, analytically or by kinetic Monte Carlo (kMC) simulations. The combined energetic and spatial nature of the carrier trajectories has, to our knowledge, not been explicitly investigated previously, especially not for the correlations caused by different concentrations of dopant and carrier charges.
In this article, we show that valuable information is to be gained from the analysis of the transport networks that carry most of the electrical current. We investigate how the mode of transport changes with the doping concentration and with it its limiting factors. We show why the Seebeck energy ES can only be correctly obtained from averaging over the whole transport network and by doing so confirm the previously unexplained observation that ES is not generally equal to the activation energy of the conductivity EA.
While no analytical tools exist for treating this problem exactly, we computationally obtain quantitative results that can be compared to experiment. The tools we use are a percolation analysis combined with a simple model of the electrostatic potential landscape. The computations are parallelizable and our implementations in the Python programming language for the samples of this report have typical runtimes of a few minutes on 24-core machines, with room for improvement.
Percolation theory is a powerful technique for analyzing complex systems that has found applications in many scientific fields since its general formulation by Broadbent and Hammersley in 195728, ranging from solid state physics29 over climate physics30 to neuroscience31, language32 and epidemics33. As Broadbent and Hammersley28 put it, diffusion and percolation are two complementary perspectives on random motion of fluids through media: While for diffusion, the randomness is assigned to the fluid, percolation assigns it to the medium. This is the particular strength of a percolation analysis for hopping transport, where the questions asked are usually ultimately about the medium (like What is the conductivity of a given sample?) and not about the fluid (like Where does a particular charge carrier move to?).
Application of percolation theory to transport in organic semiconductors is not new, and has produced many interesting results15,34,35,36. However, most published work focuses on estimating the critical hop. As discussed above, we want to extend these analyses by utilizing information from the whole network of transport paths.
As a general workflow, we first model the energetic landscape in the sample by constructing a molecular geometry (lattice/amorphous) and placing charged dopants on randomly selected sites, as well as charge carriers in energetically optimized positions. We then identify the sample with a weighted complete graph, where the vertices correspond to the molecular sites and the edges to carrier hops, whose weights are given by the hopping rates. By using a variant of the resistor network model37,38 (see “Methods” section and Supplementary Note 3 for details), we reduce this huge graph to the critical conducting subnetworks, i.e. the networks of sites that carry almost all of the current.
Our basic assumptions are that (i) each site cannot contain more than one charge, (ii) charges are localized to one site, (iii) not all site energies are the same, and (iv) the sample structure is not affected by adding the dopants.
Results
Potential landscapes and the density of states (DOS)
We start by modeling the energetic landscape in the sample as a function of doping concentration. To achieve this, we place sites in a predefined three-dimensional structure (regular lattice or randomly with a minimal intersite distance to account for finite molecular volume), optionally with energies from a distribution. We then randomly replace sites with dopants, assigning them a positive charge. We finally place an equal number of electrons on the remaining sites, in a configuration that minimizes the total Coulomb energy of all charges (see the “Methods” section for more details).
Note that we do not consider the charge transfer (CT) process from dopant to matrix. For efficient host/dopant systems, it has been found that this CT adds negligibly to the temperature activation of the conductivity39,40. Here, we concentrate on the charge transport, given the doping CT was successful. Thus, by dopants we refer just to the ionized dopants.
When a sample is doped, an equal amount of localized positive and negative charges is introduced into the system, effecting their electrostatic potential on all other sites in the sample. This leads to a broadening of the DOS, as some states get raised and some get lowered by the one or the other type of charge (Fig. 1). In the light doping regime, the peak position of the unoccupied DOS stays constant, due to the equal amount of positive and negative charges. Some previous analytical treatments of this effect only included the dopant charges10,41, leading to qualitatively different results for the DOS and the energetic landscape.

a Amorphous material, i.e. with point-molecules placed randomly, but with a minimal distance of 0.5 nm. b Tetragonal lattice using the lattice spacings of ZnPc (see Supplementary Table 1 for details). The occupied states are shown in blue. With increasing doping concentration, the DOS broadens for both structures, as some states get raised and some get lowered by the one or the other type of charge. In the light doping regime, the peak position of the unoccupied DOS stays constant, due to the equal amount of positive and negative charges. Note that for the tetragonal lattice, an additional peak in the unoccupied DOS arises due to the the neutral ZnPc in the Coulomb potential of the, for n-doping, negatively charged ZnPc next to it and the positively charged dopant two places away (we do not account for a different shape of the dopant molecule). The doping concentrations are given in molar percentages and the plots are offset vertically for clarity.
As the doping concentration increases, the asymmetry between ionized dopants and charge carriers becomes evident: While the dopants are fixed and distributed randomly in space, the carriers can move and attain the configuration that minimizes their total potential energy. As the density ND of randomly positioned dopants increases, the amplitude of the fluctuations of their density also increases, namely like42\(\sqrt{{N}_{{\rm{D}}}}\). The carriers can partially screen these fluctuations, and sites in the regions with small dopant density see more of the carrier potential than of the dopant potential. Consequently, the peak position of the DOS shifts to positive energies at high doping concentrations, as seen in Fig. 1.
We want to point out that the DOS approaches zero around the Fermi level, i.e. at the high-energy edge of the density of occupied states (DOOS) (blue in Fig. 1). This so-called Coulomb gap has been studied previously and is a consequence of the electrostatic interaction of localized fermionic charges43. The appearance of this feature underlines the validity of our method. We will not go into detail about its origin, but instead refer the reader to the existing literature43,44,45,46,47.
The increase of density fluctuations upon increasing the doping concentration is also reflected in the spatial structure of the potential. Figure 2 shows 3-dimensional potential contour plots of a tetragonal matrix lattice at different doping concentrations. The blue/orange shapes are iso-surfaces of negative/positive potentials, respectively.

Blue/orange structures are regions with negative/positive potential for charge carriers, respectively (exemplary colorbar for 32%). See Supplementary Note 1 for parameter information. Upon increasing the concentration, there is a transition from rather isolated potential hills and valleys to large, fluctuating structures. Thus, at low concentrations, the spatial correlations of the site energies are stronger than at high concentrations. This transition is accompanied by a transition in the critical charge transport paths, i.e. from long-range hopping between the separated wells to hopping on all length scales through the fluctuating landscape (see Fig. 3c).
At low concentrations, the dopants as well as the charge carriers form separated potential wells, corresponding to the separated closed iso-surfaces for 0.32% in Fig. 2. There is a small dispersion of the individual energies, reflected in a distribution of the contour sizes. Carriers are mostly situated close to the dopants, forming short dipoles. Increasing the doping concentration, the potentials start to fluctuate more and more on all length scales. In the potential landscape, this is reflected in large-scale connected iso-surfaces which exhibit fluctuating structure down to small length scales. So-called Gaussian potential fluctuations caused by dopant charges have been studied for inorganic-doped compensated semiconductors previously48. Our systems are not equivalent to the systems studied in ref. 48 because the sites that accommodate the charge carriers (the acceptors in ref. 48, the matrix sites in this article) and the dopants are not spatially uncorrelated in our case: Our charge carriers position themselves in energy-minimizing configurations, while the acceptors in ref. 48 are fixed. However, the notion of 0-, 1-, and 2-complexes, where a dopant can host either zero, one, or two charge carriers still applies and leads to the same type of potential fluctuations.
The activation energy of conductivity
After the energetic landscape has been calculated, we transform the sample into a resistor network by assigning each pair of hopping sites (i, j) a resistivity given by the inverse rate of a hop between them, using Marcus hopping rates and Fermi–Dirac occupation statistics. The hopping rates pij have the form
with \({\xi }_{ij}=2\ {r}_{ij}/\alpha +{(\Delta {\varepsilon }_{ij}^{{\rm{eff}}}+\lambda )}^{2}/(4kT\lambda ),\) and \({p}_{0}=(2\pi /\hslash )({J}_{0}^{2}/\sqrt{4\pi \lambda kT})\). Here, rij is the distance between the sites, α the localization length, \(\Delta {\varepsilon }_{ij}^{{\rm{eff}}}\) their effective energy difference (see “Methods” section for definition), λ the reorganization energy, and J0 the transfer integral prefactor. We do not restrict the hopping distance, allowing for variable-range hopping (VRH).
We then numerically find the critical resistor and the corresponding critical subnetwork (see Fig. 3) for different temperatures: Starting from an empty network, the resistors are added, ordered by increasing resistivity until a connected path from one face of the sample cube to its opposing face exists for the first time (see “Methods” section for details). The critical resistor is the first one that completes such a path when it is inserted. The critical subnetwork is the collection of all resistors connected to that path at that instance.

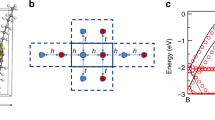
for F8ZnPc (see Supplementary Note 2 for parameter information). Blue dots represent the ionized dopant positions. In panels a and b, a low doping ratio of 0.32% is present. In the case of no energetic disorder a, the network consist of bunches of short transitions (which contribute almost nothing to global transport) in the vicinity of dopants and long transitions between the dopants. These long transitions are critical for the global conductivity. When intrinsic energetic disorder of σ0 = 120 meV is introduced b, the long transitions can be broken into several shorter ones, as some sites are randomly energetically lowered by the disorder. They can thus serve as bridging sites between the dopants and EA is reduced compared to the disorder-free case. c Shows a high doping ratio of 32%. The white lines represent the critical subnetwork in a 3D contour plot of the calculated bulk potential. Note that the critical subnetwork paths avoid regions of both high (red, where they are repelled) and low (cyan, where they would be bound) potential and mainly occupy potential-free regions. Charge carriers have to circumnavigate the regions of strong potential to traverse the sample, which increases EA at very high concentrations.
The derivative of the exponent ξc of the critical hopping rate with respect to inverse temperature equals the activation energy EA of the conductivity37,38. It has been repeatedly found that doped organic semiconductors follow such an Arrhenius behavior, where \(\mathrm{ln}\,\sigma \propto -1/T\), and not ∝−1/T2, as suggested by the Gaussian disorder model7,9,40,49.
We always randomly sample 48 configurations to find ξc. We thus account for the fact that the ground state carrier configuration is not unique and can even change with time. We, however, implicitly assume that the transitions occur adiabatically and only one carrier moves at a time.
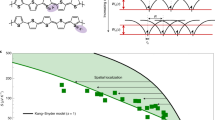
Figure 4 compares the experimentally obtained EA of ZnPc and octa-flourinated ZnPc (F8ZnPc), n-doped with (2-Cyc-DMBI)2, as a function of doping concentration40, with our calculated values, observing very good agreement (see Supplementary Table 2 for parameter values). Note that the only unknown parameters of our model are the intrinsic disorder width σ0 and the localization length α, which we fix to the same values of σ0 = 100 meV and α = 0.44 nm for both materials. All other parameters are taken from experiment, when available, or from independent calculations in the literature.

The main difference in molecular parameters between ZnPc and F8ZnPc is their CT-binding energy with the dopant EC (see Supplementary Table 2 for parameter values). Strikingly, the difference in EC is enough to accurately explain the difference in EA, which is especially pronounced at low-doping ratios. The reason is the deeper lying Fermi level for ZnPc due to the low EC. Error bars show one standard deviation. Experimental data reproduced from ref. 40.
The comparison between the two materials in Fig. 4 is particularly interesting, since they are very similar in all molecular parameters except for their quadrupole moment40. This difference leads to strongly different binding energies (with reference to the isolated matrix level) of the charge-transfer complex formed by the ionized dopant and the charge on the neighboring matrix molecule: 930 meV for ZnPc and 680 meV for F8ZnPc40. As can be seen from Fig. 4, the different binding energies lead to an EA which is about 150 meV lower for F8ZnPc at low-doping ratios around 3 × 10−2, in the experiment as well as in our calculations. The reason is the difference in Fermi level, which lies deeper for ZnPc due to its higher binding energy.
When we artificially increase the dielectric permittivity, all Coulomb interactions are weakened and the potential landscape flattens out. Consequentially, EA decreases significantly, about 100 meV for ZnPc with the relative permittivity set to 4 instead of 2.8, as seen in Supplementary Fig. 2. This is in line with findings that the binding energy of CT states decreases with increasing permittivity50,51. Thus, we confirm the design guideline that a high dielectric permittivity of the matrix leads to low activation energies. This is also a reason for the very low EA of C60 that is discussed below.
Characteristic length scales
As the doping concentration increases, EA drops. At higher concentrations, the distances between sites with similar energies decrease. Carriers thus do not have to hop as far to reach favorable target energies and paths with smaller critical barriers open up: EA decreases.
Interestingly, the decisive length scale for this effect is the localization length α and not the matrix site density N: Halving α shifts all activation energies up by 50–100 meV, and steepens the decay of EA as seen in Fig. 5a. Conversely, doubling the lattice distances (spacing between site centers of mass) does not change EA at all at low and moderate doping concentrations. The reason is the following: In the presence of a strong long-range potential, the optimal VRH hopping range is solely determined by the shape of that potential, regardless of whether a target site is available at that range. If the hopping distance is only large compared to the nearest-neighbor (NN) spacing, a site close to that optimal range likely exists, and the exact value of the NN spacing is irrelevant. The decisive length scales for the temperature-activated part of the charge transport at low and moderate doping concentrations are the typical dopant distance \({N}_{{\rm{d}}}^{-1/3}\) and the localization length α, while the site density N does not play a role.

a This plot underlines that the spatial and energetic aspects of transport are closely connected: Halving the localization radius α (blue circles) compared to a reference sample (green squares), i.e. doubling the penalty of long-distance hops, also increases the activation energy by up to 100 meV. This is not obvious, since the exponent of the hopping rate (Eq. (6)) that contains α does not even depend on temperature. The connection of α and EA only emerges due to the tradeoff of variable-range hopping: For stronger localization, low-energy hops are more likely to be traded for short-distance hops. Since the typical critical hopping distance is the distance between dopants at low doping ratios, lowering the dopant distance is the main reason for the decrease in activation energy upon increasing doping ratio. Interestingly, decreasing α is not equivalent to increasing the lattice distances d (black triangles) at low and moderate doping ratios. At high concentrations, however, where the charges get very close and the typical hopping distance saturates at the lattice distance, the effect of halving α and of doubling d becomes equivalent. b EA versus molar doping ratio for F8ZnPc with different widths σ0 of the intrinsic energetic disorder. Remarkably, the sample with the highest intrinsic disorder exhibits the lowest EA at low doping concentrations. The reason are matrix sites that are randomly lowered in energy by the intrinsic disorder and can serve as bridging sites between the dopant wells (see Fig. 3). This benefit disappears at high doping concentrations, where the notion of individual dopant wells vanishes. Error bars show one standard deviation.
The decisive role of α for EA is counterintuitive as the term in the hopping rate containing α does not even depend on temperature. To rationalize the emergent dependence EA(α), we revisit a similar result that has been found for the dependence of carrier mobility on an electric field, using the concept of the effective temperature52,53,54,55. In the effective-temperature approach, which has been studied in several publications52,53,54, the field dependence can be described by replacing the laboratory temperature T in the expression for the mobility with the effective temperature Teff given by
with the constant prefactor γ, the elementary charge e and the electric field F. In Eq. (2), the electric field also only appears in combination with α and the site density does not enter. This at first sight counter-intuitive result is related to the fact that with increasing hopping distance in field direction also a proportionally increasing amount of electrical energy is gained. Thus, the greater difficulty of long-range hops is compensated by their greater energy gain and a scaling of the site distances has no net effect. As discussed above, this holds as long as the optimal hopping distance is large compared to the lattice distance.
This is very similar to our case, where the electric field is not external but originates from the ionized dopants and charge carriers in the sample. The optimal hopping ranges are given by the potential and as long as the sites are sufficiently dense, no significant changes in the transport networks happen. Changing only α on the other hand affects the spatial penalty of the hops without any compensation. This changes the optimal hopping range and the tolerated energy differences along the transport paths—and hence, EA—increase: Energy has become "cheaper” compared to distance.
Going to very high doping concentrations, the dopants are getting so close to each other that the typical hopping distance saturates at the lattice distance and the above arguments do not apply anymore. Then, the decisive parameters are N−1/3 and α. Consequently, EA for the sample with doubled lattice distances follows the reference sample at low and the half-α sample at high doping concentrations in Fig. 5a.
Since the potential fluctuations get stronger for higher charge densities, they even lead to an increase of EA with very high doping concentrations (see Figs. 4–6). Such an increase has been observed in various experimental reports in the literature, e.g. for n-doped C6049,56 or n-doped polymer samples57, but was not explained therein. We can now link it to the change in critical subnetwork topology in the following way: The steady-state transfer rate of a given connection decreases with increasing absolute energy difference of the connected sites, since in steady state, forward and backward transfer rates must balance. It is thus unlikely for the critical subnetwork to expand into regions with either largely positive or negative potential and the most important paths circumvent all potential structures, staying mostly in the regions with the potential around zero (Fig. 3c).
In this situation, one can think of the network of conducting paths as solving a discrete percolation problem on short scales and a continuum percolation problem on large scales. Similarly as described in ref. 48, the carriers have to be activated to an energy εc such that the regions with potential smaller than εc are connected.
Intrinsic disorder
As a further illustrative example for the power of the percolation method, let us investigate the influence of intrinsic disorder. By intrinsic disorder, we mean a distribution of site energies that is present without the introduction of charges, i.e. in an undoped sample. It is well-known that some degree of intrinsic energetic disorder is present in all molecular solids and it is in many cases modeled by a Gaussian distribution of energies16,58,59. Here, we also draw our initial site energies from a Gaussian distribution g0(ε) with standard deviation σ0:
In Fig. 5b, we compare the activation energy of the conductivity as a function of doping concentration for three different σ0, using the F8ZnPc structure parameters. Remarkably, the sample with the highest intrinsic disorder width (σ0 = 120 meV) exhibits the lowest activation energy of 250 meV at the lowest doping concentrations, about 150 meV less than for the disorder-free sample. This is in line with previous findings that energetic disorder can facilitate electron–hole pair dissociation60,61.
The reason for this counter-intuitive behavior is the fact that a broader intrinsic disorder increases the density of sites that have low energies by chance. These sites can then serve as bridges between the Coulomb wells created by the ionized dopants, facilitating the transitions from dopant to dopant. This mechanism can be transparently observed by inspection of the critical subnetworks. Figure 3a shows the disorder-free case: bundles of connections close to dopants are connected by long-distance hops between the dopant wells. When intrinsic disorder is introduced (Fig. 3b), there are still some bundles close to dopants, but the long-distance connections mostly contain multiple hopping nodes, which are the aforementioned bridging sites, with energies lowered by the intrinsic disorder. In this way, energetically more favorable paths open up and the activation energy of the conductivity is reduced with respect to the disorder-free case.
At high doping concentrations, the notion of an individual Coulomb well for each dopant vanishes in favor of a potential landscape fluctuating on all length scales. Bridging between the dopants becomes not advantageous anymore and the difference between the curves in Fig. 5b vanishes. Instead, there are extended (i.e. including more than one dopant on average) regions of the sample with negative and positive potentials, which now limit charge transport.
Activation energy vs. Seebeck energy
Interestingly, in ref. 56 it has been found that the activation energy of the conductivity increases at high doping concentrations (Fig. 6a), while the Seebeck energy ES continues to decrease (see Fig. 6). This previously unexplained discrepancy can be readily clarified with our approach: The Seebeck energy does not depend on the details of the transport mechanism, particularly not on the rate of the critical hop, but only on the average energy that is transported with each charge carrier. ES can thus be calculated from the DOS belonging to the critical subnetwork \(\widetilde{N}(\varepsilon )\) as62
where the energy ε is given with respect to the Fermi level EF.

a Experimental and calculated activation energy of conductivity of C60:W2(hpp)4 as a function of molar doping ratio. We used an amorphous structure of C60 for the whole range of doping ratios (see Supplementary Table 3 for other parameter values). The excellent agreement of our results with the experimental data shows that the assumption of structural changes upon doping of C60 is not necessary to explain the increase of activation energy at high concentrations, but it can be rationalized solely as the result of electrostatic fluctuations. b Seebeck energy ES of C60:W2(hpp)4 as a function of molar doping ratio. That ES, surprisingly, does not show an increase at high doping concentrations can now be understood: Although the critical hopping energy shows a stronger temperature activation, ES is determined by the average energy of the critical subnetwork with respect to the Fermi level. This average keeps decreasing as the Fermi level moves up in the DOS (see Supplementary Fig. 1 for the DOS plots). Error bars show one standard deviation. Experimental data reproduced from ref. 56, Figs. 3 and 4.
In Fig. 6b, we observe two regimes: For low doping concentrations, EF is pinned far from the DOS peak due to the low energy of the CT states and overall, only a small fraction of sites is occupied. Increasing the doping concentration steeply decreases ES by creating more deep unoccupied states, which can be reached with less energy investment compared to the states at the DOS peak. This lowers EA in the same way as ES and a comparison of Fig. 6a and b, shows that both are very similar up to a molar ratio of about 3 × 10−2.
Increasing the concentration further starts to shift EF significantly through the DOS distribution and \(\widetilde{N}(\varepsilon )\) becomes more and more symmetrical around EF (see Supplementary Fig. 1 for plots of the distributions). In this regime, ES approaches zero with a smaller slope than before. The activation energy of the conductivity, on the other hand, is given only by the energy of the critical hop as outlined above and increases due to the increasing potential fluctuations at high doping concentrations.
One can even generally state that EA is always equal to or greater than ES, as for EA, the absolute distance ∣ε∣ from the Fermi level is decisive, while for ES, one has to average ε as in Eq. (4)62. In other words, EA is big, if the sites that are important for conduction are far from EF, regardless of them lying below or above EF. On the other hand, ES is increased only by those sites of the critical subnetwork that lie above EF.
With the knowledge of the critical subnetworks, we are able to calculate both EA and ES with the same set of parameters and closely reproduce the measured data (Fig. 6, see Supplementary Table 3 for parameter values). We use an amorphous structure of C60 throughout, showing that the assumption of sudden structural changes of C60 at high doping concentrations is not necessary to explain the observed behavior. Note that the widely used TE model8 predicts EA and ES to be equal. For a Coulombically interacting system like this, the TE model cannot be applied22. The assumption of spatially uncorrelated site energies, which is necessary for the TE model, is not fulfilled due to the presence of long-range Coulomb interactions.
Finally, as seen in Fig. 6, doped C60 shows a very low activation energy (around 60 meV at the minimum) compared to e.g. ZnPc, which has an EA above 200 meV for all doping concentrations (Fig. 4). The main reason for this is its comparably high relative dielectric constant63, which we set to 4 for our calculations, while for ZnPc a value of 2.8 was reported63.
Discussion
We present a comprehensive analysis of hopping transport in doped molecular semiconductor based on percolation theory and VRH. The analysis of the critical conducting subnetworks, which contain those transitions that carry almost all of the current, is the key strength of the percolation approach and sets it apart from effective-medium calculations or kMC simulations. We accurately reproduce experimental results for different materials from different experimental studies and explain the difference between the conductivity’s activation energy and the Seebeck energy. By analyzing the critical conducting subnetworks, we show that intrinsic energetic disorder can facilitate long-range charge transport at low, but not high doping concentrations. For engineering a minimal activation energy, an optimal doping concentration exists and is of the order of a molar ratio of 10% for the materials studied here. As expected, the activation energies are generally lowered by large dielectric constants, high localization radii and low CT-binding energies between host and dopant.
As a closing remark, it is also possible to analyze doped polymers within the presented framework. The delocalization of charges along their chains adds another dimension of connectivity to the spatial model. The analysis of the interplay of spatial and energetic structure of polymers with a percolation approach thus promises valuable insight into tailoring the electronic properties of those materials.
Methods
Potential landscape
Spatial distribution
The system is initialized as a three-dimensional collection of uncharged point sites. The spatial structure of this collection can be arbitrarily chosen. For this report, we employ random positions with a minimum distance and two tetragonal lattices for ZnPc and F8ZnPc. For the random with minimum distance configuration, to account for a finite molecule volume, sites are randomly placed in the sample volume and kept, if their distance to every existing site is larger than the prescribed minimum distance (molecule diameter), otherwise deleted again. This process is repeated until the desired number of sites has been placed, which is around 3 × 104 for the present report. In the literature45,64, it has been confirmed that this site number is sufficient for our widths of energetic dispersion (which do not significantly exceed 0.1 eV), in the sense that increasing the number of sites does not change the results significantly anymore. Initially, all sites are assigned zero energy.
Energetic distribution
An intrinsic energetic distribution can be imposed on the sites. We use a Gaussian intrinsic DOS according to Eq. (3), with different widths σ0, including σ0 = 0.
Dopant placement
Nd of the sites are randomly chosen to be active dopants (donors, for concreteness). They are assigned a positive charge and the electrical potential due to their charge is added to all other site energies. Dopants do not count as transport sites anymore.
Charge carrier placement
Nd electrons (corresponding to the Nd donors) are placed in a configuration as close to the ground state as possible. To achieve this, we place one electron after another at the site with the currently lowest energy and add the potential due to its charge to all other sites. After all electrons have been placed, we take out the first electron again (subtracting the potential due to its charge everywhere) and place it in the state that is now the lowest in energy. We iterate this loop five times and take the configuration that out of these five states has the lowest total energy. We assume that by this procedure we end up with a state that is optimized with respect to single-electron transitions and close to the ground state, since the Coulomb interactions are strong, making the ground state very attractive. Indeed, the resulting optimized total energy does not depend on the (random) donor starting configuration, which makes it unlikely that the algorithm gets stuck in local minima. This is reflected in the error bars in the main text figures, where each point for each temperature was averaged over 48 realizations of the sample.
Adjacent molecules in a molecular solid can form so-called CT states, if the energy levels of the two molecules are suitably situated, as is the case for a dopant–matrix dimer. We thus assign the next-neighbor sites of the donors the CT state energy EC. We assume that all interactions of sites that are further apart are just Coulomb interactions of point charges. EC is a parameter of our model. We find that this simple model for CT state formation explains the experimental features of material combinations with different EC already very well, as seen in Fig. 4, where the values for EC are taken from density functional theory calculations in ref. 40, i.e. were not fitting parameters.
The Fermi level EF is finally extracted as the midpoint between the highest occupied and the lowest unoccupied energy levels.
Percolation threshold
Following ref. 45, we calculate the percolation threshold of the system to obtain the conductivity of the sample in the following way.
For each pair of sites i and j, there is a probability pij of an electron transit from i to j, which is proportional to the probability f(i) that i is occupied, the probability 1−f(j) that j is unoccupied and the transition rate νij:
For the rate νij we choose the Marcus expression65,66
with the transfer integral prefactor J0, the reorganization energy λ, the distance rij between sites i and j, the localization length α and the energy barrier Δεij = εj−εi.
In thermal equilibrium, f(i) only depends on the energy εi of site i and is given by the Fermi–Dirac distribution
with the Fermi level εF. Detailed balance then requires that all transition rates are symmetric, i.e. pij = pji. Thus, pij is proportional to the probability that site i is occupied and site j is unoccupied as well as the probability for the opposite situation. If both site energies lie above or below the Fermi level, the rate is consequentually limited by the site energy that is the furthest from the Fermi level45.
We further account for the hopping carrier’s own Coulomb field by reducing the energy barrier by its self–Coulomb interaction, if the hopping site energies lie on opposite sides of the Fermi level45. This is necessary since the energy of the target site includes the Coulomb potential of the hopping carrier itself, evaluated at the target position. However, the carrier does not interact with itself, so this contribution to the target energy must not be included into the energy barrier of the hop.
Summarizing the last two paragraphs, we define the effective energy difference \(\Delta {\varepsilon }_{ij}^{{\rm{eff}}}\) as45
with the elementary charge e and the dielectric permittivity ϵ.
Collecting Eqs. (5), (6) and (8), we finally obtain the symmetric transition rates
with \({\xi }_{ij}=2\ {r}_{ij}/\alpha +{(\Delta {\varepsilon }_{ij}^{{\rm{eff}}}+\lambda )}^{2}/(4kT\lambda )\) and \({p}_{0}=(2\pi /\hslash )({J}_{0}^{2}/\sqrt{4\pi \lambda kT})\).
It has been shown37,38 that the conductivity of a resistor network with broadly (e.g. exponentially) distributed resistances like in Eq. (9) is given by the conductivity at the critical parameter ξc, where removing all connections with ξij > ξc just allows for percolation:
For a given spatial structure (e.g. crystal lattice, amorphous) and a random, uncorrelated placement of the resistances, ξc can be analytically obtained from the known percolation threshold of the bond percolation problem xc, assuming that the ξij are distributed uniformly in an interval [ξ0, ξ1]:
Upon introducing Coulomb interactions in our system we have to relax the assumption of the resistances being spatially uncorrelated and cannot expect Eq. (11) to hold. The basic idea of the percolation approach however, i.e. that the conductivity of the sample is determined by the critical resistance at ξc, still applies, since it only relies on the observation that if you turn on the resistances in increasing order, the resistor that just connects the critical subnetwork cannot be shunted by smaller resistances and shunts all higher resistances. We thus calculate ξc numerically for each realization of our sample and readily use it in Eq. (10) to obtain the conductivity and its temperature activation, similar to ref. 45.
However, in contrast to ref. 45, where just hopping transport between the states bound to dopants is investigated, we look for connections between all the sites, since we assume that in molecular solids all molecules (also in the undoped case) contribute localized states that can take part in hopping events. Thus, there is the limiting case where only states close to dopants take part in hopping transport, while in general, all molecules can function as hopping sites. This is an extension of the approach for doped crystalline semiconductors, where rather an interplay of localized dopant states and extended bulk states is present. This extension is especially essential at high doping concentrations, where the notion of an individual dopant potential vanishes and hopping takes place all over the system.
Since we do not introduce any artificial cut-off radius for neither the Coulomb interactions nor the possible transitions, we account for potential variations on all length scales up to the sample size and allow for VRH.
Data availability
The data that were computed in this work are available from the author upon request.
Code availability
The computer code used in this work is available at https://doi.org/10.5281/zenodo.407733667.
References
Shirakawa, H., Louis, E. J., MacDiarmid, A. G., Chiang, C. K. & Heeger, A. J. Synthesis of electrically conducting organic polymers: halogen derivatives of polyacetylene, (CH)x. Chem. Commun. https://doi.org/10.1039/C39770000578, 578 (1977).
Chiang, C. K. et al. Electrical conductivity in doped polyacetylene. Phys. Rev. Lett. 39, 1098 (1977).
Heeger, A. J. Semiconducting and metallic polymers: the fourth generation of polymeric materials (Nobel lecture). Angew. Chem. Int. Ed. 40, 2591–2611 (2001).
Kearns, D. R., Tollin, G. & Calvin, M. Electrical properties of organic solids. II. Effects of added electron acceptor on metal-free phthalocyanine. J. Chem. Phys. 32, 1020 (1960).
Blochwitz, J., Pfeiffer, M., Fritz, T. & Leo, K. Low voltage organic light emitting diodes featuring doped phthalocyanine as hole transport material. Appl. Phys. Lett. 73, 729 (1998).
Walzer, K., Maennig, B., Pfeiffer, M. & Leo, K. Highly efficient organic devices based on electrically doped transport layers. Chem. Rev. 107, 1233–1271 (2007).
Maennig, B. et al. Controlled p-type doping of polycrystalline and amorphous organic layers: self-consistent description of conductivity and field-effect mobility by a microscopic percolation model. Phys. Rev. B 64, 195208 (2001).
Schmechel, R. Hopping transport in doped organic semiconductors: a theoretical approach and its application to p-doped zinc-phthalocyanine. Phys. Rev. B 93, 4653–4660 (2003).
Shen, Y. et al. Charge transport in doped organic semiconductors. Phys. Rev. B 68, 081204 (2003).
Arkhipov, V., Emelianova, E., Heremans, P. & Bässler, H. Analytic model of carrier mobility in doped disordered organic semiconductors. Phys. Rev. B 72, 235202 (2005).
Li, L., Meller, G. & Kosina, H. Analytical conductivity model for doped organic semiconductors. J. Appl. Phys. 101, 033716 (2007).
Tietze, M. L., Burtone, L., Riede, M., Lüssem, B. & Leo, K. Fermi level shift and doping efficiency in p-doped small molecule organic semiconductors: a photoelectron spectroscopy and theoretical study. Phys. Rev. B 86, 035320 (2012).
Abdalla, H., Zuo, G. & Kemerink, M. Range and energetics of charge hopping in organic semiconductors. Phys. Rev. B 96, 241202 (2017).
Gaul, C. et al. Insight into doping efficiency of organic semiconductors from the analysis of the density of states in n-doped C60 and ZnPc. Nat. Mater. 17, 439–444 (2018).
Zuo, G., Abdalla, H. & Kemerink, M. Conjugated polymer blends for organic thermoelectrics. Adv. Electron. Mater. 5, 1800821 (2019).
Bässler, H. Localized states and electronic transport in single component organic solids with diagonal disorder. Phys. Status Solidi B 107, 9–54 (1981).
Pollak, M. & Shklovskii, B. I. (eds) Hopping Transport in Solids, Hopping Conduction in Electrically Conducting Polymers, 377 (Elsevier, 1991).
Pope, M. & Swenberg, C. E. Electronic Processes in Organic Crystals and Polymers (Oxford University Press, Oxford, 1999).
Baranovski, S. Charge Transport in Disordered Solids with Applications in Electronics, Vol. 17 (John Wiley & Sons, 2006).
Zuo, G., Abdalla, H. & Kemerink, M. Impact of doping on the density of states and the mobility in organic semiconductors. Phys. Rev. B 93, 235203 (2016).
Nenashev, A. V., Oelerich, J. O. & Baranovskii, S. D. Theoretical tools for the description of charge transport in disordered organic semiconductors. J. Phys.: Condens. Matter 27, 093201 (2015).
Novikov, S. V. & Malliaras, G. G. Transport energy in disordered organic materials. Phys. Status Solidi B 243, 387–390 (2006).
Gartstein, Y. N. & Conwell, E. High-field hopping mobility in molecular systems with spatially correlated energetic disorder. Chem. Phys. Lett. 245, 351–358 (1995).
Dunlap, D. H., Parris, P. E. & Kenkre, V. M. Charge-dipole model for the universal field dependence of mobilities in molecularly doped polymers. Phys. Rev. Lett. 77, 542 (1996).
Novikov, S. V., Dunlap, D. H., Kenkre, V. M., Parris, P. E. & Vannikov, A. V. Essential role of correlations in governing charge transport in disordered organic materials. Phys. Rev. Lett. 81, 4472 (1998).
Sin, J. & Soos, Z. Hopping transport in molecularly doped polymers: joint modelling of positional and energetic disorder. Philos. Mag. 83, 901–928 (2003).
Mendels, D. & Tessler, N. Thermoelectricity in disordered organic semiconductors under the premise of the gaussian disorder model and its variants. J. Phys. Chem. Lett. 5, 3247–3253 (2014).
Broadbent, S. R. & Hammersley, J. M. Percolation processes: I. Crystals and mazes. In (ed. Green, B.), Mathematical Proceedings of the Cambridge Philosophical Society, Vol. 53, 629–641 (Cambridge University Press, 1957).
Ambegaokar, V., Halperin, B. I. & Langer, J. S. Hopping conductivity in disordered systems. Phys. Rev. B 4, 2612–2620 (1971).
Hua, L., Lu, Z., Chen, L., Yu, Y. & Wang, L. Percolation phase transition of surface air temperature networks: a new test bed for el niño/la niña simulations. Sci. Rep. 7, 8324 (2017).
Del Ferraro, G. et al. Finding influential nodes for integration in brain networks using optimal percolation theory. Nat. Commun. 9, 2274 (2018).
Stella, M. & Brede, M. Patterns in the English language: phonological networks, percolation and assembly models. J. Stat. Mech. 5, 05006 (2015).
Moore, C. & Newman, M. E. J. Epidemics and percolation in small-world networks. Phys. Rev. E 61, 5678–5682 (2000).
Vissenberg, M. & Matters, M. Theory of the field-effect mobility in amorphous organic transistors. Phys. Rev. B 57, 12964 (1998).
Baranovskii, S., Zvyagin, I., Cordes, H., Yamasaki, S. & Thomas, P. Percolation approach to hopping transport in organic disordered solids. Phys. Status Solidi (b) 230, 281–288 (2002).
Nenashev, A. et al. Advanced percolation solution for hopping conductivity. Phys. Rev. B 87, 235204 (2013).
Miller, A. & Abrahams, E. Impurity conduction at low concentrations. Phys. Rev. 120, 745–755 (1960).
Shklovskii, B. I. & Efros, A. L. Impurity band and conductivity of compensated semiconductors. Zh. Eksper. Teor. Fiz. 33, 468–474 (1971).
Tietze, M. L. et al. Elementary steps in electrical doping of organic semiconductors. Nat. Commun. 6, 1182 (2018).
Schwarze, M. et al. Molecular parameters responsible for thermally activated transport in doped organic semiconductors. Nat. Mater. 18, 242 (2019).
Zuo, G., Abdalla, H. & Kemerink, M. Erratum: Impact of doping on the density of states and the mobility in organic semiconductors [phys. rev. b 93, 235203 (2016)]. Phys. Rev. B 97, 079902 (2018).
Landau, L. D. & Lifshitz, E. M. Course of Theoretical Physics, Vol. 5: Statistical Physics 3rd edn (Butterworth-Heinemann, 1980).
Efros, A. L. & Shklovskii, B. I. Coulomb gap and low temperature conductivity of disordered systems. J. Phys. C 8, L49 (1975).
Baranovskii, S. D., Efros, A. L., Gelmont, B. L. & Shklovskii, B. I. Coulomb gap in disordered systems: computer simulation. J. Phys. C 12, 1023–1034 (1978).
Shklovskii, B. I. & Efros, A. L. Electronic Properties of Doped Semiconductors (Springer-Verlag, Berlin, Heidelberg, 1984).
Lee, M., Massey, J. G., Nguyen, V. L. & Shklovskii, B. I. Coulomb gap in a doped semiconductor near the metal-insulator transition: tunneling experiment and scaling ansatz. Phys. Rev. B 60, 1582–1591 (1999).
Müller, M. & Ioffe, L. B. Glass transition and the coulomb gap in electron glasses. Phys. Rev. Lett. 93, 256403 (2004).
Efros, A. L., Shklovskii, B. I. & Yanchev, I. Y. Impurity conductivity in low compensated semiconductors. Phys. Status Solidi B 50, 45 (1972).
Olthof, S. et al. Ultralow doping in organic semiconductors: evidence of trap filling. Phys. Rev. Lett. 109, 176601 (2012).
Koster, L. J. A., Shaheen, S. E. & Hummelen, J. C. Pathways to a new efficiency regime for organic solar cells. Adv. Energy Mater. 2, 1246–1253 (2012).
Torabi, S. et al. Strategy for enhancing the dielectric constant of organic semiconductors without sacrificing charge carrier mobility and solubility. Adv. Funct. Mater. 25, 150–157 (2015).
Marianer, S. & Shklovskii, B. I. Effective temperature of hopping electrons in a strong electric field. Phys. Rev. B 46, 13100 (1992).
Baranovskii, S., Cleve, B., Hess, R. & Thomas, P. Effective temperature for electrons in band tails. J. Non-cryst. Solids 164, 437–440 (1993).
Arkhipov, V. & Bässler, H. Field-dependent effective temperature of localized charge carriers in hopping systems with a random energy distribution. Philos. Mag. Lett. 69, 241–246 (1994).
Nenashev, A. V., Oelerich, J. O., Dvurechenskii, A. V., Gebhard, F. & Baranovskii, S. D. Fundamental characteristic length scale for the field dependence of hopping charge transport in disordered organic semiconductors. Phys. Rev. B 96, 035204 (2017).
Menke, T., Ray, D., Meiss, J., Leo, K. & Riede, M. In-situ conductivity and seebeck measurements of highly efficient n-dopants in fullerene C60. Appl. Phys. Lett. 100, 093304 (2012).
Higgins, A., Mohapatra, S. K., Barlow, S., Marder, S. R. & Kahn, A. Dopant controlled trap-filling and conductivity enhancement in an electron-transport polymer. Appl. Phys. Lett. 106, 163301 (2015).
Tessler, N., Preezant, Y., Rappaport, N. & Roichmann, Y. Charge transport in disordered organic materials and its relevance to thin- film devices: a tutorial review. Adv. Mater. 21, 2741–2761 (2009).
Baranovskii, S. D. Mott lecture: description of charge transport in disordered organic semiconductors: analytical theories and computer simulations. Phys. Status Solidi A 215, 1700676 (2018).
Albrecht, U. & Bässler, H. Yield of geminate pair dissociation in an energetically random hopping system. Chem. Phys. Lett. 235, 389–393 (1995).
Mityashin, A. et al. Unraveling the mechanism of molecular doping in organic semiconductors. Adv. Mater. 24, 1535–1539 (2012).
Zvyagin, I. P. On the theory of hopping transport in disordered semiconductors. Phys. Status Solidi B 58, 443 (1973).
Scholz, R. et al. Quantifying charge transfer energies at donor-acceptor interfaces in small-molecule solar cells with constrained DFTB and spectroscopic methods. J. Phys.: Condens. Matter 25, 473201 (2013).
Kordt, P., Speck, T. & Andrienko, D. Finite-size scaling of charge carrier mobility in disordered organic semiconductors. Phys. Rev. B 94, 014208 (2016).
Marcus, R. A. Electron transfer reactions in chemistry. theory and experiment. Rev. Mod. Phys. 65, 599–610 (1993).
Fishchuk, I. I., Kadashchuk, A., Bässler, H. & Nešpůrek, S. Nondispersive polaron transport in disordered organic solids. Phys. Rev. B 67, 224303 (2003).
Hofacker, A. Computer Code to “Critical charge transport networks in doped organic semiconductors”. https://doi.org/10.5281/zenodo.4077336 (2020).
Acknowledgements
We thank Prof. Karl Leo for helpful suggestions and helping to improve the manuscript. Financial support by the German Federal Ministry of Education and Research (Project UNVEiL) and the German Research Foundation (Project LE 747/48-1) and Open Access Funding by the Publication Fund of the TU Dresden is gratefully acknowledged. We thank the Center for Information Services and High Performance Computing (ZIH) at TU Dresden for generous allocations of computer time.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
A.H. motivated the study, developed the theoretical framework, developed and executed the computer code, analyzed and visualized the results and wrote the article text.
Corresponding author
Ethics declarations
Competing interests
The author declares no competing interests.
Additional information
Peer review information Primary handling editor: John Plummer.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hofacker, A. Critical charge transport networks in doped organic semiconductors. Commun Mater 1, 88 (2020). https://doi.org/10.1038/s43246-020-00091-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s43246-020-00091-1
