
Abstract
Transhydrogenation of pentane (P) and 1,5-hexadiene (1,5HD) and pentane and 2,4-hexadiene (2,4HD) was studied over a CrOx/alumina catalyst at 523–773 K. Thermodynamic stability differences between the conjugated (2,4-hexadiene) and non-conjugated (1,5-hexadiene) isomers indicated that transhydrogenation was favoured between pentane and 1,5-hexadiene but not pentane and 2,4-hexadiene (+ ve ∆G). At 773 K a significantly enhanced alkene yield was observed for the P/1,5HD system, clearly showing the effect of transhydrogenation. The yield of alkenes was ~ 50% and included alkylated and isomerized alkenes. Alkylation and isomerization were significant reactions under reaction conditions. Pentane was shown to affect the chemistry of 1,5HD and vice versa with the conversion of pentane significantly enhanced at all reaction temperatures, indicating a molecular interaction between the reactants even when transhydrogenation was not obvious. In contrast, no effect on the conversion of pentane was observed when the co-feed was 2,4HD. An unexpected effect of pentane on 2,4HD conversion was observed, with all reactions of cis-2,4-hexadiene (including alkylation and isomerization) being completely inhibited at low reaction temperatures (573 K and 523 K) by the presence of pentane, suggesting that pentane competes for the same sites as cis-2,4-hexadiene. Transhydrogenation activity between pentane and 1,5-hexadiene was less obvious at the lower reaction temperature, which appeared to be a kinetic effect. Direct hydrogenation of 1,5-hexadiene revealed that 1,5HD sampled the same hydrogen population for hydrogenation and transhydrogenation. Comparisons of transhydrogenation of 1-hexyne, 1,5-hexadiene, and 2,4-hexadiene with pentane have revealed significant differences in the adsorption and reaction chemistry of the three isomers.
Similar content being viewed by others

Introduction
It is well known that hydrocarbon feedstock streams, such as naphtha, LPG, or gas oil are cracked in a furnace to produce mixtures of hydrocarbons of varying molecular weight and functionality [1,2,3,4,5,6]. To optimise the value-added converting low-value, cracked hydrocarbons (e.g., alkanes) into valued distillate products is important. Although transhydrogenation is not a new technology for the production of olefins and other valuable petroleum distillate products, there has been limited scientific attention toward the invention. The transhydrogenation of alkanes and alkynes (or alkadienes) into alkenes (olefins) is a method for converting two low-value chemicals into two high-value chemicals.
Transhydrogenation is thermodynamically favoured at most temperatures for the reaction of alkanes with alkynes but that it is not always the case when alkadienes are the hydrogen acceptors [7]. Thus, if we calculate the free energy for the transhydrogenation of pentane with 1-hexyne, 1,5- hexadiene and 2,4-hexadiene, we find that the ∆G of the process moves from negative to positive (see Table 1). Therefore, there can be thermodynamic constraints on the process.
Alkane dehydrogenation is endothermic (~ 124 kJ mol−1) and a significant amount of energy is required to break C–H bonds in a molecule. The equilibrium conversion of the process is limited by the reaction temperature and as that increases so does conversion [8]. Hence, to accommodate the thermodynamic limitations, typical reaction temperatures are ≥ 823 K for reasonable conversions [9,10,11], at these temperatures all C–H bonds in an alkane have an equal chance to react, which tends to make the process less selective. The equilibrium conversion can be increased by continuous removal of hydrogen in accordance with Le Chatelier’s principle and indeed some on-demand dehydrogenation processes operate at partial pressure less than 1 atm, often with the use of a diluent in the alkane feed [12]. On the other hand, alkyne/alkadiene hydrogenation is exothermic (~ − 167 kJ mol−1) and produces a significant amount of heat, which can potentially be utilized in the dehydrogenation process. It also can act as an acceptor for the continuous supply of hydrogen produced by alkane dehydrogenation. Transhydrogenation combines both processes as a single process [13]
Recent literature has revealed transhydrogenation reactions on various catalysts such as supported vanadia, chromia and doped chromia, most of which involved the use of LPG hydrocarbons [7, 14,15,16,17], these research studies showed clearly that transhydrogenation occurs. Significant conversion and yield of the olefin are reported, for instance, Jackson et al. reported ~ 72% increase in propene from propyne and butane transhydrogenation using 1% vanadia/θ-alumina catalyst at 823 K. Most of this research [7, 14,15,16] has been limited to C2-C4 substrates. In our recent work [17], we extended this approach using a wider range of hydrocarbons (i.e., C-5 and C-6). Pentane and 1-hexyne transhydrogenation was investigated over chromia catalysts and revealed that transhydrogenation clearly occurred, especially at low temperatures [17]. In this paper, the behaviour of conjugated and non-conjugated hexadiene isomers (2,4-hexadiene and 1,5-hexadiene respectively) for transhydrogenation with pentane was investigated using chromia catalyst. The data obtained were compared with that from pentane/1-hexyne transhydrogenation to better understand the benefits and limitations of the transhydrogenation process.
Experimental
A ~ 4% chromium oxide/alumina catalyst was prepared by incipient wetness impregnation using a γ-alumina support. The alumina support (50 g) was pre-dried at 353 K for 16 h prior to impregnation. An aqueous solution of [NH4]2Cr2O7 (4.85 g, 99 + % Aldrich) was used to prepare the catalyst with a 4% w/w loading. Johnson Matthey, UK, supplied the γ-alumina support (surface area 208 m2 g−1, pore volume 0.52 ml g−1). After impregnation, the catalyst was dried for 2 h at 353 K and then at 393 K overnight. Finally, the sample catalyst was calcined at 873 K for 6 h. After calcination, the sample was ground using a mortar and pestle and sieved to a uniform particle size of 250–425 μm.
Thermo-gravimetric analysis was performed on the catalysts using a combined TGA/DSC SDT Q600 thermal analyser coupled to an ESS evolution mass spectrometer for evolved gas analysis (TGA/MS). The samples (~ 10–30 mg) were heated from ambient temperature to 1273 K using a heating ramp of 15 K min−1 in 2% H2/N2 or 2% O2/Ar. Relevant mass fragments were followed by online mass spectrometry. CHN analysis was performed by combustion using a CE-440 elemental analyser.
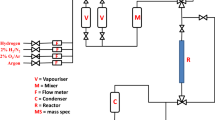
The activity and selectivity of the catalyst were determined using an all-glass, atmospheric pressure, flow, microreactor. The block schematic of the flow reactor has been reported earlier [17]. The catalyst (0.5 g) was reduced in situ with pure hydrogen (40 ml min−1) by heating to 823 K and holding at this temperature for 2 h. After reduction was completed, the flow was switched to argon (40 ml min−1) and the system purged for 0.5 h. Pentane (99 + % Aldrich) and 1,5-hexadiene (97 + % Aldrich) or 2,4-hexadiene (97 + % Aldrich) were introduced to the argon carrier gas (30 ml min−1) using temperature-controlled vapourisers. A molar feed ratio of ~ 5.5:1 for the pentane:hexadiene system was obtained. The ratio was set by controlling the vapour pressure for each reactant using bubbler vapourisers and maintaining the vapouriser temperature for 1,5HD, 2,4HD and pentane at 258, 273, and 273 K, respectively. The gas was then put through a static mixer before continuing to the reactor. The weight hourly space velocity (WHSV) was 2.8 h−1 for pentane, 0.59 h−1 for 1,5-hexadiene and 0.6 h−1 for 2,4-hexadiene. The reaction products were condensed and collected over a 2-h period in heptane at 253 K and then the cumulative volume was sampled and analysed by gas chromatography (Agilent series, FID detector) fitted with a 150-m Petrocol column. An online ESS mass spectrometer was also coupled to the reactor to continuously follow any non-condensed eluted products over the 2-h time period of the reactions [17]. The reaction was studied with each component individually and as a mixed feed, in the temperature range 523–773 K at atmospheric pressure. No hydrogen was co-fed in the process. The conversion for each reactant feed was calculated as:
The yield as:
Results
The catalyst was characterized using various techniques and has been reported elsewhere [17]. A standard TGA/TPR from ambient temperature to 1273 K on the catalyst gave single weight loss/hydrogen uptake at ~ 650 K, which is in keeping with the literature [18]. The XRD analysis of the alumina support and the catalyst showed only patterns for alumina: no chromia species were detected. This was expected as crystalline chromium oxide is typically not observed at this loading [19,20,21]. AAS and colorimetric analysis of the catalyst showed appreciable amounts of both Cr3+ and Cr6+ oxidation states at various treatment stages of the catalyst preparation. The as-prepared catalyst was found to contain 3.32% Cr by AAS of which ~ 97% was Cr(VI) with the rest Cr(III). However, after reduction at 873 K, 93% was Cr(III) and only 7% Cr(VI) [17]. Hence, the results confirmed that chromium in the catalyst was in mixed oxidation states even after reduction. The catalyst is designated as CrOx/Al2O3 to signify the chromium species as a non-stoichiometric oxide [18, 19, 21].
The reactions were initially performed with the alumina support and showed that the products were mainly formed via cracking and alkylation with low conversion. The details are reported elsewhere [17]. This behaviour from alumina is consistent with the literature [15, 16].
Pentane/1,5-Hexadiene (P/1,5HD) system
The reactant conversions were followed during the individually fed and co-fed reactions. A dramatic increase in the conversion of pentane was observed at all temperatures when pentane and 1,5-hexadiene were co-fed compared to pentane dehydrogenation, while for 1,5-hexadiene the conversion at the higher temperatures (> 573 K) showed a slight decrease when pentane was co-fed relative to 1,5-hexadiene with no added pentane. The results are presented in Fig. 1.

Conversion comparison of pentane (P), 1,5-hexadiene (1,5HD) and a 5.5:1 P:1,5HD mixture over CrOx/Al2O3
The product yields were calculated and are presented in Figs. 2, 3, 4, 5 and 6 (these results represent the cumulative results of products collected over 2 h, detailed analysis of the products is reported in Supplementary information, Tables S1–S5). As the temperature was decreased, the yield of olefin also decreased. At 773 K traces of benzene were observed, which subsequently decrease at lower reaction temperatures.

Yield of products at 773 K with feeds of pentane, 1,5-hexadiene and 5.5:1 P:1,5HD feed over CrOx/alumina

Yield of products at 673 K with feeds of pentane, 1,5-hexadiene and 5.5:1 P:1,5HD feed over CrOx/alumina

Yield of products at 623 K with feeds of pentane, 1,5-hexadiene and 5.5:1 P:1,5HD feed over CrOx/alumina

Yield of products at 573 K with feeds of pentane, 1,5-hexadiene and 5.5:1 P:1,5HD feed over CrOx/alumina

Yield of products at 523 K with feeds of pentane, 1,5-hexadiene and 5.5:1 P:1,5HD feed over CrOx/alumina
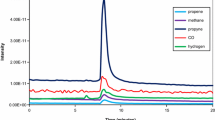
Gaseous product analysis using on-line mass spectrometry confirmed the evolution of C3H8 and C4H8 initially before hydrogen is detected. The C3H8 and C4H8 response decays rapidly suggesting that the sites responsibly are rapidly deactivated. There was a significant reduction of the hydrogen between 10 and 15 min before a more gradual decline. This same gas evolution trend was observed at 773–623 K reaction temperatures. The result obtained at 773 K is presented in Fig. 7.

Profile of the evolved gases over CrOx/Al2O3 with a P/1,5HD feed at 773 K
Hydrogenation of 1,5-hexadiene was also performed to determine and compare the catalyst efficacy between hydrogenation and transhydrogenation. The reaction was performed using a limited hydrogen feed (2% H2/N2) in the ratio 2:1 hexadiene:2% H2/N2. The results are presented in Fig. 8. The hydrogenation process showed similar reaction products and distributions to that obtained during transhydrogenation but the hydrogenation process exhibited a much higher percentage yield of alkenes. This was a marked difference from that found with hydrogenation of 1-hexyne, where alkanes were the principal product [17].

Comparison of hydrogenation and transhydrogenation over CrOx/alumina at 623 K
The catalysts were analysed post-reaction by CHN analysis to determine whether there had been any carbon laydown during the reaction. The results are shown in Fig. 9. Catalysts, which have been subjected to 1,5HD as the sole reactant, show a higher amount of carbon deposition at temperatures > 573 K but at 523 K and 573 K there is little difference between P/1,5HD co-feed and 1,5HD alone.

Carbon laydown, as measured by CHN analysis, on the spent CrOx/Al2O3 catalysts as a function of temperature
The catalyst used for the P/1,5HD reaction was subjected to a temperature-programmed oxidation (TPO) using the TGA/MS system. The results are shown in Figs. 10, 11.

TGA analysis of post-reaction P/1,5HD samples

Gas evolved during TPO of P/1,5HD spent catalyst samples, m/e 44 (carbon dioxide)
There is a single weight loss that moved to slightly higher temperatures with increasing reaction temperature as is reflected in the carbon dioxide evolution, e.g., at a reaction temperature of 523 K, carbon dioxide maximum evolution is at 622 K, whereas at a reaction temperature of 773 K the carbon dioxide maximum is at 663 K.
Pentane/2,4-Hexadiene (P/2,4HD) system
In contrast to the P/1,5HD system, no increase in conversion was observed with pentane when reacting in the presence of 2,4HD. Indeed, at most temperatures lower conversions of pentane were obtained. The results are presented in Fig. 12. It is also clear that the conversion of 2,4HD in the P/2,4HD mix is, generally, less than that found for 2,4HD reacting alone.

Conversion comparison of pentane (P), 2,4-hexadiene (2,4HD) and a 5.5:1 P:2,4HD mixture over CrOx/Al2O3
The reaction using 2,4HD was performed with a 2:1 cis/trans mixture. It was observed that each isomer of 2,4HD reacted differently. Trans-2,4HD reacted faster than cis-2,4HD resulting in a higher conversion of trans-2,4HD at all reaction temperatures compared to cis-2,4HD. However, the most striking result was the absence of any conversion of cis-2,4HD at 523 K and 573 K when reacted with pentane, revealing that some interaction was taking place. The results are presented in Fig. 13.

Conversion comparison of cis- and trans-2,4HD using CrOx/Al2O3
The product yields were calculated and are presented in Figs. 14, 15, 16, 17 and 18 (detailed analysis of the products is reported in Supplementary information, Tables S6–S10). The yield of olefin was low but the yield of alkadienes also decreased relative to that found with 2,4HD alone. Traces of benzene were observed, which subsequently decreased at lower temperatures.

Yield of products at 773 K with feeds of pentane, 2,4-hexadiene and 5.5:1 P:2,4HD feed over CrOx/alumina

Yield of products at 673 K with feeds of pentane, 2,4-hexadiene and 5.5:1 P:2,4HD feed over CrOx/alumina

Yield of products at 623 K with feeds of pentane, 2,4-hexadiene and 5.5:1 P:2,4HD feed over CrOx/alumina

Yield of products at 573 K with feeds of pentane, 2,4-hexadiene and 5.5:1 P:2,4HD feed over CrOx/alumina

Yield of products at 523 K with feeds of pentane, 2,4-hexadiene and 5.5:1 P:2,4HD feed over CrOx/alumina
The evolved gases produced from the P/2,4 HD reaction were the same as from the P/1,5HD system (H2, C3H6 and C4H8) with hydrogen evolution from the start of the reaction. After the initial pulse, hydrogen evolution gradually declined, whereas butene and propene maintained a low-level evolution. The results are presented in Fig. 19.

Profile of the evolved gases over CrOx/Al2O3 using P/2,4HD mixed feed at 723 K
The catalysts were analyzed after use by CHN analysis to determine the extent of carbon laydown. The results presented in Fig. 20 show that the overall extent of the carbon deposition is small and less than that found with P/1,5HD system.

Carbon laydown, as measured by CHN analysis, on the spent CrOx/Al2O3 catalysts as a function of temperature with 2,4HD system
The nature of the carbon was probed by TPO and the results are shown in Figs. 21 and 22. The TPO analysis during the TGA revealed carbon dioxide as the main species evolved with the P/2,4HD system. This was determined by mass spectrometry (m/e 44). Like the P/1,5HD system, there was an observed shift in the CO2 desorption peak with reaction temperature, from 630 to 693 K with increasing reaction temperature. A significant difference, however, was observed in the TPO of the catalyst reacted at 623 K with a second weight loss/evolution of CO2 at 915 K.

Weight loss profile from TPO of P/2,4HD mixed feed over CrOx/Al2O3 catalyst

Gas evolved during TPO of P/2,4HD spent catalyst samples, m/e 44 (carbon dioxide)
Discussion
In a previous paper [17] the transhydrogenation of pentane with 1-hexyne was examined. In that study, over the same CrOx/Al2O3 catalyst as used in this study, transhydrogenation clearly occurred at temperatures below 625 K, where the yield of alkenes was higher for the co-fed system than for a combination of the individual yields. This study expands the investigation to consider the transhydrogenation of pentane with 1,5-hexadiene and 2,4-hexadiene. Although 1-hexyne, 1.5-hexadiene and 2,4-hexadiene are isomers, as can be seen from Table 1, the change in bonding results in significant changes in stability and hence, overall reaction thermodynamics. The conjugated 2,4-hexadiene molecule is the most stable of the isomers and transhydrogenation with pentane is not thermodynamically favoured (positive ∆G), which results in conversion limits for transhydrogenation of ~ 8.5% at 673 K, ~ 12.5% at 773 K and ~ 17.5% at 873 K. In comparison, the transhydrogenation reaction between pentane and 1,5-hexadiene (1,5HD) has a small negative ∆G resulting in a maximum conversion of ~ 80% at most temperatures.
From Fig. 1 when pentane and 1,5HD were reacted together the conversion of pentane increased dramatically for all temperatures showing the effect on pentane of having 1,5HD present in the reaction mix. The effect of pentane on 1,5HD reactivity was similar, with an increase in 1,5HD conversion from ~ 48% to 100% at 523 K. Similar behaviour was observed for the pentane/1-hexyne system [17] and is in keeping with the thermodynamic analysis, implying that both species are adsorbing and interacting. In contrast, when pentane and 2,4HD were reacted the conversion of pentane showed no significant effect (Fig. 12). The absence of an effect on pentane is as would be expected from the thermodynamics—any impact would be small and limited to no more than 10% even at 773 K—if the only reaction was transhydrogenation. However, alkylation, isomerization and hydrogenation are all taking place (see Table S6 in the supplementary information). Rather surprisingly the effect on 2,4HD is to reduce the conversion and the effect is specific to the cis-2,4HD isomer, with no conversion of cis-2,4HD at lower temperatures, < 623 K (Fig. 11). This reflects the slight difference in stability between the cis- and trans-2,4HD isomers and hence, their strength of adsorption, suggesting that pentane inhibited the reaction of cis-2,4HD through competitive adsorption. Interestingly, one specific product, 2-methyl-1,3-pentadiene, was lost when cis-2,4HD conversion goes to zero. Therefore, we can assign this product to structural isomerization of cis-2,4HD and this confirms that the inhibition of cis-2,4HD applies to reactions other than just transhydrogenation. Zaera and co-workers [22] have shown that the adsorption of cis- and trans-alkenes is preferred on different crystal faces and so it is possible for pentane to disrupt the chemistry of one isomer without affecting the other isomer. This result also suggests that pentane and cis-2,4HD share a preference for the same sites.
In the reaction between pentane and 1,5HD, between 573 and 773 K the yield of alkenes was enhanced and there was also a low-level formation of benzene. The yield of alkenes at 773 K was 50%, which was a significant increase over that found with 1,5HD in the absence of pentane and even higher than the alkene yield found with the pentane/1-hexyne and P/2,4HD reactions, indicating that transhydrogenation is more effective with non-conjugated dienes than alkynes or conjugated dienes at this temperature [17]. Note that this is not a thermodynamic effect. However, this clear-cut benefit was only observed at 773 K and as the temperature was decreased the difference between feeding 1,5HD alone and in a mix with pentane was significantly reduced, such that by 623 K there was no effect on the selectivity profile. Given the enhancement in conversion, this is surprising and shows a significant difference between the terminal diene and the terminal alkyne in terms of transhydrogenation reactivity, as transhydrogenation was more pronounced at low temperature with the terminal alkyne [17]. In this case, the difference is kinetic, as distinct from thermodynamic in nature reflecting a difference in reactivity of alkyne and alkene. As expected from the preceding discussion, the P/2,4HD system shows little transhydrogenation benefit, however, this effect was thermodynamic rather than kinetic.
To examine the difference between hydrogenation and transhydrogenation 1,5HD was hydrogenated with a limited amount of gas-phase hydrogen. The results were surprising and revealed differences between the 1,5HD system and the 1-hexyne (1HY) system [17]. Hydrogenation of 1HY over this catalyst resulted in a significant alkane yield of ~ 20%, in contrast, hydrogenation of 1,5HD produces < 1% alkane, whereas with the formation of alkene the position is reversed with ~ 50% alkene from 1,5HD hydrogenation but only ~ 6% from 1HY. With 1-hexyne hydrogenation, it was proposed that the difference between transhydrogenation and hydrogenation may relate to 1-hexyne sampling different hydrogen populations that had been shown to be present on the catalyst [15, 17]. In contrast, 1,5HD appears to sample the same hydrogen population, whether undergoing hydrogenation or transhydrogenation. This can be understood in terms of the hydrogenation process. It has been shown that, over palladium, the hydrogen population responsible for direct hydrogenation of an alkyne to an alkane is surface hydrogen, whereas that for selective hydrogenation to the alkene is a sub-surface species. In the oxide system, a sub-surface species is unlikely and indeed hydrogenation of 1-hexyne gave principally alkane product [17] consistent with surface hydrogen. With 1,5-hexadiene however, it is unlikely that both C=C double bonds would be hydrogenated simultaneously, therefore a high yield of alkene would be expected as hydrogenation through to the alkane requires re-adsorption in competition with alkadienes.
Analysis of the gas phase revealed that both P/1,5HD and P/2,4HD systems produced hydrogen, butene, and propene. Formation of the same light alkenes by cracking of alkenes with the same backbone but with the double bonds in different positions is well known [23, 24]. The evolution of all three gasses goes through a peak and decay suggesting the deactivation of the sites responsible for their production. This would be in keeping with the carbon deposition data. Nevertheless, this is in contrast with the P/1HY system, which produced hydrogen, methane, and ethene with only hydrogen decaying. The difference in cracking products from alkenes and alkynes has been documented previously for the C3 system with propyne also producing methane by cracking [14, 15].
Temperature programmed oxidation of the spent catalysts used for P/1,5HD transhydrogenation, revealed a single weight loss over the temperature range 520–700 K. As the reaction temperature increased, a higher temperature was required to combust the carbon on the surface, suggesting that the carbonaceous residue was becoming more recalcitrant with a lower H:C ratio. Nevertheless, given the temperature of the weight loss, it is likely the principal surface species is aliphatic in nature. Similar behaviour was observed with the P/2,4HD system.
Conclusions
The transhydrogenation of pentane and 1,5-hexadiene and pentane and 2,4-hexdiene was studied over a CrOx/alumina catalyst. Thermodynamic stability differences between the conjugated (2,4-hexadiene) and non-conjugated (1,5-hexadiene) isomers indicated that transhydrogenation was favoured between pentane and 1,5-hexadiene but not pentane and 2,4-hexadiene. At 773 K a significantly enhanced alkene yield was observed for the P/1,5HD system clearly showing the beneficial effect of transhydrogenation. The yield of alkenes was ~ 50% and included alkylated and isomerized alkenes. Alkylation and isomerization were significant reactions under reaction conditions. Pentane was shown to affect the chemistry of 1,5HD and vice versa with the conversion of pentane significantly enhanced at all reaction temperatures, indicating a molecular interaction between the reactants even when transhydrogenation was not obvious. In contrast, no effect on the conversion of pentane was observed when the co-feed was 2,4HD confirming the absence of interaction. However, a very clear and unexpected effect of pentane on 2,4HD conversion was observed, with all reactions of cis-2,4-hexadiene (including alkylation and isomerization) being completely inhibited at low reaction temperatures (573 K and 523 K) by the presence of pentane. This suggests that pentane inhibits cis-2,4-hexadiene adsorption and indicates that cis-2,4-hexadiene and trans-2,4-hexadiene adsorbed on different sites. As reaction temperature was decreased, obvious transhydrogenation activity between pentane and 1,5-hexadiene was lost, which was in contrast to that found with transhydrogenation between pentane and 1-hexyne, where transhydrogenation was more obvious as reaction temperature decreased. This difference is kinetic in nature rather than thermodynamic. Direct hydrogenation of 1,5-hexadiene revealed that 1,5HD sampled the same hydrogen population for hydrogenation and transhydrogenation, whereas alkynes (propyne and 1-hexyne) sampled different populations [15, 17]. Comparisons of the transhydrogenation of 1-hexyne, 1,5-hexadiene and 2,4-hexadiene with pentane have revealed significant differences in the adsorption and reaction chemistry of the three isomers.
References
Givens EN, Charles PJ, Rosinski EJ (1976) Converting low molecular weight olefins over zeolites. US 3960978
Coleman ST (2014) Process for cracking heavy hydrocarbon feed. US 8658023
Ward AM, Oprins AM, Housmans THM (2015) Process for the production of light olefins and aromatics from a hydrocarbon feedstock. WO 2015000842
OSHA (2014) Petroleum refining process. In: Labor U (ed). OSHA, USA. http://www.osha.gov/dts/osta/otm/otm_iv/otm_iv_2.html
Garba MD, Galadima A (2015) Efficiencies of green chemistry metrics in the activities of petroleum refinery process. Int Sci Invest J 4:65–87
Dessau R, Partridge M, Randall D (1995) Production of Olefins by Transhydrogenation. WO 1995030635
Garba MD, Jackson SD (2016) Catalytic upgrading of refinery cracked products by transhydrogenation: a review. Appl Petrochem Res 7:1–8
Bhasin MM, McCain JH, Vora BV, Imai T, Pujado PR (2001) Dehydrogenation and oxydehydrogenation of paraffins to olefins. Appl Catal A 221:397–419
Alvin HW (1970) The manufacture of propylene. In: Robert FG (ed) Refining petroleum for chemicals. American Chemical Society, New York, pp 153–178
Craig RG, Spence DC (2007) Catalytic dehydrogenation of liquefied petroleum gas by the Houdry Catofin and Catadiene processes. McGraw-Hill Education, New York
Bipin VV, Peter RP (2007) Catalytic dehydrogenation. In: Lee S (ed) Encyclopedia of chemical processing. Taylor and Francis, New York, pp 379–395
Erofeev VI, Khomyakov IS, Egorova LA (2014) Production of high-octane gasoline from straight-run gasoline on ZSM-5 modified zeolites. Theor Found Chem Eng 48:71–76
Song H, Wang N, Song H-L, Li F (2015) La–Ni modified S2O82-/ZrO2-Al2O3 catalyst in n-pentane hydroisomerization. Catal Commun 59:61–64
Jackson SD, Matheson IM, Webb G (2003) Carbon deposition during transhydrogenation over chromia catalysts. Am Chem Soc Div Pet Chem 49:50–53
Jackson SD, Matheson IM, Webb G (2005) An isotopic study of the transhydrogenation of propyne with propane over a potassium-doped chromia/alumina catalyst. Appl Catal A 289:16–21
Wigzell F, Rugmini S, Jackson SD (2015) Transhydrogenation of propyne with butane over a vanadia/θ-alumina catalyst. Appl Petrochem Res 7:9–21
Garba MD, Jackson SD (2019) Transhydrogenation of pentane and 1-hexyne over CrOx/Al2O3 and potassium-doped CrOx/Al2O3 catalysts. Appl Petroleum Res 9:113–125
Edussuriya M, Jackson SD, Rugmini S (2010) Deactivation and regeneration of chromia and vanadia catalysts in alkane dehydrogenation. Curr Top Catal 9:15–24
Fridman VZ, Xing R, Severance M (2016) Investigating the CrOx/Al2O3 dehydrogenation catalyst model: I. Identification and stability evaluation of the Cr species on the fresh and equilibrated catalysts. Appl Catal A 523:39–53
De Rossi S, Caselleto MP, Ferraris G, Cimino A, Minelli G (1998) Chromia/zirconia catalysts with Cr content exceeding a monolayer. Appl Catal A 167:257–270
Jackson SD, Stitt EH (2003) Propane dehydrogenation over chromia catalysts: micro-catalysis, deactivation, macro-kinetics, and reactor modelling. Curr Top Catal 3:245–265
Lee I, Delbecq F, Morales R, Albiter MA, Zaera F (2009) Tuning selectivity in catalysis by controlling particle shape. Nat Mat 8:132–138
Kissin YV (1996) Chemical mechanism of hydrocarbon cracking over solid acidic catalysts. J Catal 163(1):50–62
Kissin YV (2001) Chemical mechanisms of catalytic cracking over solid acidic catalysts: alkanes and alkenes. Catal Rev 43(1):85–146
Acknowledgements
The authors would like to fully acknowledge the funding provided, for one of us (MDG), by the Petroleum Technology Development Funds (PTDF) through the Government of Nigeria and the University of Glasgow for the placement.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Garba, M.D., Jackson, S.D. Transhydrogenation of pentane with 1,5- and 2,4-hexadiene over CrOx/Al2O3. Appl Petrochem Res 11, 79–88 (2021). https://doi.org/10.1007/s13203-020-00259-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13203-020-00259-3