
Abstract
Autism spectrum disorders (ASD) are subdivided into idiopathic (unknown) etiology and secondary, based on known etiology. There are hundreds of causes of ASD and most of them are genetic in origin or related to the interplay of genetic etiology and environmental toxicology. Approximately 30 to 50% of the etiologies can be identified when using a combination of available genetic testing. Many of these gene mutations are either core components of the Wnt signaling pathway or their modulators. The full mutation of the fragile X mental retardation 1 (FMR1) gene leads to fragile X syndrome (FXS), the most common cause of monogenic origin of ASD, accounting for ~ 2% of the cases. There is an overlap of molecular mechanisms in those with idiopathic ASD and those with FXS, an interaction between various signaling pathways is suggested during the development of the autistic brain. This review summarizes the cross talk between neurobiological pathways found in ASD and FXS. These signaling pathways are currently under evaluation to target specific treatments in search of the reversal of the molecular abnormalities found in both idiopathic ASD and FXS.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Autism spectrum disorders (ASD) is a term used to diagnose a behavioral disorder seen in individuals presenting with a combination of severe difficulties in the areas of social communication, social interaction, and repetitive behaviors and movements. This condition currently affects 1.7% of children between 3 and 17 years of age in the USA [1]. Surveillance data from the Centers for Disease Control and Prevention (CDC) reported a prevalence of 1 in 54 children before 8 years of age in 2016. Currently, for every 4 boys, only 1 girl is diagnosed with ASD [2]. To meet the diagnostic criteria established in 2013 by the Diagnostic and Statistical Manual of Mental Disorders (DSM), fifth edition, an individual must presently manifest or have a history of deficits in social interaction and communication across multiple contexts, as well as restricted or repetitive behavioral patterns (e.g., repetitive speech or motor movements such as hand flapping or hand biting) and perseveration in areas of interest [3]. Most individuals diagnosed with ASD fail to have a normal back-and-forth conversation or to develop and/or maintain social interactions and relationships [4]. Affected individuals also struggle with the integration of verbal and nonverbal communication and exhibit abnormal eye contact and body language [3]. About 25 to 30% of children with ASD remain minimally verbal after the preschool years [5, 6]. Furthermore, they might present with intellectual challenges as well as psychiatric symptoms such as aggression, sleep disturbances, and mood disorders [7, 8].
The umbrella term “spectrum” refers to the heterogeneous display of symptoms and variable severity found in affected individuals. There are hundreds of causes of ASD and most of them are genetic in origin with over 1000 gene mutations associated with ASD [9]. There is also significant evidence that an interplay between genetic predisposition to ASD and environmental exposures, such as heavy metals, endocrine disruptors, phthalates, bisphenol A, polychlorinated biphenyls, and even air pollution can lead to or exacerbate ASD through further disruption of CNS pathways important for normal development [9]. ASD is subdivided into idiopathic, in which the etiology is unknown, and secondary based on known etiology. With the advent of more detailed genetic testing including microarray testing assessing for deletions and duplications, in addition to fragile X DNA testing and when these are negative moving forward with whole exome sequencing (WES) or whole genome sequencing (WGS), then ~ 30 to 50 % of the genetic etiologies of ASD can be identified [10, 11]. Many of these gene mutations are either core components of the Wnt signaling pathway or their modulators [12, 13]. All individuals who are diagnosed with ASD require a full genetic evaluation [14].
Fragile X syndrome (FXS) is one of the well-documented causes of ASD; in fact, it is the most common cause of ASD as a result of a monogenic mutation. FXS is caused by the full mutation of the FMR1 gene, located in the X chromosome, leading to an abnormal expansion of the CGG trinucleotide repeats (greater than 200) in the 5′ noncoding region causing the gene’s methylation and reduction in transcription and a deficiency or absence of the fragile X mental retardation protein (FMRP), an important regulator involved in synaptic development and plasticity [15, 16]. Due to the absence of FMRP, individuals with FXS experience cognitive, behavioral, and physical involvement [17, 18]. Approximately 60% of individuals with FXS also receive an ASD diagnosis and these patients usually present with increased behavioral challenges and lower IQ compared to those with FXS without ASD [19,20,21]. In all cases of neurodevelopmental disorders, patients with a comorbid diagnosis of ASD present with more severe cognitive challenges compared to those affected only by the genetic syndrome alone.
There is an overlap of molecular mechanisms in those with idiopathic ASD and those with many genetic causes of ASD including those with a full mutation of FMR1. In this review, we present the shared neurobiological pathways currently being studied to target specific treatments that have the potential to reverse the abnormalities of these pathways.
Molecular Pathways and Novel/Potential Drugs
Wnt Signaling Pathway
Wingless (Wnt) signaling pathway is a transduction pathway involved in critical functions such as cell proliferation and differentiation and neuronal development. Wnt proteins are cysteine-rich glycosylated proteins that serve as intercellular signaling molecules [22]. Wnt proteins interact with transmembrane receptors, Frizzled proteins, leading to stimulation of various intracellular signal transduction cascades, including the β-catenin-dependent pathway, also known as the canonical pathway, that results in the transcription of Wnt-targeted genes [23,24,25].
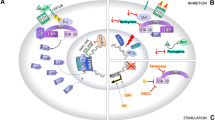
The Wnt signaling pathway governs the balance between proliferation and differentiation in cortical neural progenitors playing a fundamental role in embryonic development [26, 27]. The Wnt canonical signaling leads to the stabilization of β-catenin, allowing it to translocate to the nucleus and act as a transcription factor as seen in Fig. 1. The Wnt/β-catenin pathway plays a significant role in the etiology of ASD and is therefore of interest in the search for targeted treatments. Genes involved in the Wnt signaling pathway have an essential function in neurodevelopment and processes found to be altered in ASD, such as neurogenesis, interneuron development, and synaptic plasticity [13, 28, 29].

Canonical Wnt signaling pathway and activators. (a) Wnt proteins are cysteine-rich glycolipoproteins that act as ligands to receptors of the Wnt pathway. When Wnt is bound to the Frizzled receptor and its coreceptors, such as lipoprotein receptor-related protein (LRP), cytoplasmic disheveled (Dsh) protein is recruited and activated. Dsh regulates the inactivation of the destruction complex. This allows for β-catenin to be released from the destruction complex. Stabilized β-catenin can then enter the nucleus, displace Groucho/transducin-like enhancer of split (TLE) (not shown) and activate the transcription of Wnt target genes. (b) In the absence of Wnt, β-catenin is degraded by the destruction complex that includes molecules such as glycogen synthase kinase 3 beta (GSK-3β), casein kinase 1 alpha (CK1α), adenomatous polyposis coli (APC), and axin. The destruction complex phosphorylates β-catenin and facilitates ubiquitination by β-TrCP, leading to β-catenin’s proteosomal degradation. Dsh = disheveled; GSK-3β = glycogen synthase kinase 3 beta; APC = adenomatosis polyposis coli; LRP = lipoprotein receptor-related proteins; β-TrCP = beta-transducin repeats-containing proteins; SSRI’s = selective serotonin reuptake inhibitors; VA = valproic acid. Activators of the Wnt pathway: Lithium inhibits GSK-3β and therefore activates the Wnt pathway. Selective serotonin reuptake inhibitors (SSRIs) and valproic acid (VA) act by stimulating the expression of Wnt-targeted genes
The CTNNB1 gene is one of the critical regulators of the Wnt canonical pathway. CTNNB1 normally codes for β-catenin, which mediates cell adhesion and cell signaling. Mutations in CTNNB1, such as loss of function, result in disruption of normal Wnt pathway signaling and are associated with intellectual disability (ID) and ASD [30]. Dong et al. studied the impact of CTNNB1 loss of function in murine models. They found that CTNNB1 knockout (KO) mice displayed ASD-like behavior, including increased anxiety, impairments in cognition and social interactions, and repetitive behaviors [31].
CHD8 is a gene that codes for chromo-helicase-domain protein 8 (CHD8), a chromatin remodeling factor that is also a negative regulator of the Wnt pathway by directly inhibiting β-catenin [32]. Mutations leading to CHD8 insufficiency result in elevated canonical Wnt pathway signaling. The elevated Wnt signaling in embryonic neural progenitor cells leads to excessive proliferation and contributes to macrocephaly [33]. Furthermore, CHD8 regulates the expression of genes associated with a high risk of ASD [34]. Mutations in CHD8 result in the dysregulation of genes involved in neurodevelopment and synaptic function [35,36,37]. CHD8 haploinsufficiency in the mouse model results in autistic phenotype, including macrocephaly, motor delay, anxiety, and altered social behavior [35, 36, 38]. Patients with CHD8 mutations present a high incidence of ASD, macrocephaly, facial dysmorphisms, intellectual disability, and hypotonia [33, 39, 40]. Other genes involved in the Wnt signaling that are high-risk candidate genes for ASD include DEAD-box helices 3, X-linked (DDX3X), and transcription factor 7-like 2 (TCF72L) [41].
Wnt Signaling in FXS
Glycogen synthase kinase-3 β (GSK-3β) is part of the β-catenin destruction complex and, therefore, a negative regulator of the canonical Wnt signaling pathway [42]. It is also essential for neurogenesis, neuronal migration and differentiation, and synaptic development [43]. There is dysregulation of GSK-3β in Fmr1 KO mice with GSK-3β protein being abnormally elevated [44, 45]. GSK-3β is regulated by serine phosphorylation, which inhibits its activity [45, 46]. In Fmr1 KO mice, a deficit in serine phosphorylation of GSK-3β contributes to the increased GSK-3β activity [47]. The dysregulation of GSK-3 and Wnt pathway in FXS murine models explain some of the behavioral phenotypes associated with ASD.
Drugs Modulating the Wnt Pathway
Many drugs interact with genes involved in the Wnt signaling pathway and, therefore, represent a potential targeted treatment for many types of ASD.
Valproic acid is an anticonvulsant and mood-stabilizing drug used for conditions such as epilepsy and bipolar disorder. It has been shown to inhibit GSK-3 and enhance the canonical Wnt signaling pathway [48,49,50]. Prenatal exposure of valproic acid in mice results in autism-like phenotypes in the offspring, such as repetitive stereotypic-like movements and decreased number of social behaviors [51]. Sulindac, a nonsteroidal anti-inflammatory drug, has been shown to attenuate valproic acid-induced upregulation of the Wnt/β-catenin signaling pathway, improving autism-like phenotype in mice that have been prenatally exposed to valproic acid [52,53,54]. Lithium and selective serotonin reuptake inhibitors (SSRIs) also result in Wnt activation [55, 56]. Lithium acts as an agonist of the canonical Wnt signaling pathway by inhibiting GSK-3β [57] and has also been shown to be helpful for the behavioral problems including aggression in FXS [58].
Data in young children with ASD have demonstrated in positron emission tomography (PET) studies that serotonin synthesis is delayed in children under 5 years of age [59, 60]. This finding led to the use of a partial agonist to the 5HT1A receptor, specifically buspirone, in children ages 2 to 6 years old with ASD. A controlled trial of low dose (2.5 to 5.0 mg twice a day) buspirone compared to placebo was carried out in 166 young children with ASD lasting 24 weeks. The results demonstrated only limited improvement seen only in repetitive behaviors with the 2.5-mg dose and not for the 5.0-mg dose [61].
The original PET studies in children with ASD stimulated a trial of sertraline in young children with FXS ages 2 to 6 years old [62]. Such a treatment was thought to benefit the emerging anxiety that occurs in those with FXS around this early age and stimulate language, as was documented in an open-label trial of sertraline in children with FXS between 1 and 5 years of age in which both the expressive and receptive language trajectory was improved compared to those not treated with sertraline [63]. A controlled randomized double-blind trial of 2.5 to 5.0 mg of sertraline/day over 6 months was carried out in 52 children 2 to 6 years old with FXS, and 32 also had a concomitant diagnosis of ASD. The results demonstrated improvements in the Mullen Scales of Early Learning (MSEL) in the areas of fine motor coordination, visual reception, and the composite development T score in those treated with sertraline compared to placebo. Only those with FXS plus ASD demonstrated a significant benefit in the expressive language raw scores on sertraline versus placebo (23.5 vs 17.6; P < 0.005) [62]. This study has led to the common use of low-dose sertraline in young children with FXS. However, a similar study in young children with ASD but without FXS did not demonstrate benefit of sertraline versus placebo for the group [64]. Rajaratnam et al. have compared these studies and suggested that the greater degree of anxiety in those with FXS compared to those with idiopathic ASD may explain the better response to sertraline [65]. With an improvement in anxiety, there can be improvement in expressive language in those with FXS. This is yet another reason to identify those with ASD who have FXS.
Many other medications used to treat behavioral symptoms of individuals with ASD also interact with the Wnt pathway [12]. Antipsychotics such as haloperidol and clozapine have been shown to modulate this pathway. Haloperidol leads to the inhibition of GSK-3β [66] and promotes the expression of components that increase Wnt canonical signaling such as dishevelled-3 and β-catenin [67]. Studies in rat models have shown that antipsychotics may act on dishevelled-3 and other components of the Wnt pathway through D2 dopamine receptor antagonism [67, 68].
Methylphenidate is sometimes prescribed in individuals with ASD, FXS, and attention deficit hyperactivity disorder (ADHD). Methylphenidate and other stimulants have also been shown to modulate GSK-3β activity [69]. In contrast to D2-antagonism-mediated inactivation of GSK-3, acute amphetamine administration activates GSK-3 by decreasing inhibitory serine phosphorylation. Activation of D2 receptors by stimulants in mice led to decreased inhibitory phosphorylation of GSK-3 mediated by Akt. It was shown in the mouse model that the modulation of GSK-3 signaling after prolonged exposure to stimulants is different across brain regions [45, 69]. In FXS, a controlled trial was carried out years ago [70] comparing methylphenidate, dextroamphetamine, and placebo. Methylphenidate was found to be helpful over placebo and amphetamine. Because ADHD symptoms are a problem for ~ 90% of males and 50% of females with FXS, the use of stimulants can be beneficial for the majority of patients; however, when stimulants are used under the age of 5 years, irritability can increase [71]. Therefore, it is recommended that the clinician should wait until 5 years of age or older before the use of stimulants and if needed under age 5 years that guanfacine should be used to treat ADHD [71].
The challenges with modulating the Wnt signaling pathway involve the temporal and spatial effects of this pathway. For instance, increased activity of the Wnt pathway during embryonal development is associated with ASD, whereas decreased signaling and decreased β-catenin expression are associated with age-related disorders including Alzheimer’s disease [72]. Furthermore, in neurons, the different Frizzled receptors subtypes are expressed over different times and locations during development [73]. Another challenge involves the cross signaling between the Wnt pathway and other signaling pathways [25, 74] and the highly differential modulation of the pathway after exposure to drugs. These challenges explain the complexity of targeting the Wnt pathway in the search for targeted treatments in ASD.
The UBE3A Gene
Function and Expression
The overexpression of ubiquitin protein ligase E3A (UBE3A) gene influences the canonical Wnt and the retinoic acid (RA) signaling pathways, whereas its loss of function affects the bone morphogenetic protein (BMP) signaling [53]. UBE3A is the only gene in the 15q11–13 region that is expressed predominantly from the maternal allele in mature neurons [75, 76]. UBE3A encodes a homologous to E6AP C-terminus (HECT) domain E3 ubiquitin ligase and transfers ubiquitin to protein substrates, including itself, in the ubiquitin–proteasome proteolytic pathway [77], and it also serves in nonproteolytic critical processes, including the maintenance of stem cells and the activation of several signaling cascades [78]. The ubiquitin ligase activity of UBE3A must be properly maintained to promote normal brain development. In addition, phosphorylation of the gene by protein kinase A (PKA) at residue T485 regulates the gene’s activity. Missense mutations at the phosphorylation site can cause an increase in the activity leading to disruption of the gene control [79]. In humans, there are three different UBE3A protein isoforms localized in pre- and postsynaptic neuronal compartments and in both the cytoplasm and the nucleus [80]. Many proteins have been identified as substrates for UBE3A, but only a few have the potential to become targets for treatment opportunities in ASD [81]. LaSalle and colleagues have nicely summarized all known targeted proteins and interactors of UBE3A in their 2015 review [82].
UBE3A and ASD
About 1 to 3% of ASD cases worldwide have the maternal 15q11.2–q13.3 duplication (Dup 15q syndrome) [83]. These duplications are the second most prevalent genetic cause of ASD next to FXS [84]. The major contributor for this de novo copy number variation is the UBE3A gene. In Dup15q syndrome, UBE3A is found to be 1.5- to 2-fold overexpressed at both the transcriptional and protein levels [84]. A mutation in UBE3A or deletion of the maternal UBE3A copy leads to Angelman syndrome, whereas deletion of the paternal copy of this 15q11.2 to q13.3 region leads to Prader–Willi syndrome [82, 85]. Both excess and deficient syndromes share overlapping clinical features with FXS. Including, but not limited to, the development of seizures during childhood, excessive chewing behavior, hand stereotypies, reduced expressive language, and high rates of comorbid ASD (50–80%) found most commonly in those patients with maternally derived defects [86]. A subgroup of patients with FXS (< 10%) have a Prader–Willi phenotype (PWP) with hyperphagia, lack of satiation after meals, severe obesity, delayed puberty, and a small phallus [87], but they do not have a 15q deletion. Instead, Nowicki and colleagues documented in 2007 a 2- to 4-fold reduction of cytoplasmic FMRP-interacting protein 1 (CYFIP1) in patients with FXS with PWP compared to patients with FXS without PWP and controls [88]; this protein partners with FMRP and is encoded by a gene located on the 15q11.2 region [89]. It is not known why the levels of CYFIP1 are lower in the PWP of FXS, but this phenotype has a higher rate of ASD than in FXS alone. CYFIP2 has also shown its involvement in fragile X-like behaviors and binge eating [90, 91]. Both CYFIP1 and CYFIP2 have been linked to other neurological disorders besides ASD including schizophrenia, Alzheimer disease, and epilepsy [89, 92].
Modulating the RA Signaling Pathway
UBE3A negatively regulates the expression of the rate-limiting enzyme aldehyde dehydrogenase 1A2 (ALDH1A2), affecting the retinoic acid (RA) signaling, causing the impairment of RA-mediated synaptic plasticity in the autistic brain [81]. The retinoic acid-related orphan receptors alpha, beta, and gamma (ROR α γ, RORA-C, or NR1F1–3) have also been studied for their involvement in many neuropsychiatric disorders including ASD [93, 94]. RORα is associated with the promoter region of a few ASD-associated genes. These genes are downregulated in RORα-deficient neurons and may increase the risk of ASD [95,96,97,98,99]. RORA is positively regulated by female and negatively regulated by male sex hormones, through one of its transcriptional targets, aromatase (CYP19A1), involving the mechanism of sex bias in autism [96, 98].
Retinol (vitamin A) is an important nutrient for embryonic development [100, 101] and also for maintaining overall health. Vitamin A deficiency (VAD) is the leading cause of night blindness and it also impairs immunity causing an increased risk of infections. In a recent nutritional study conducted in China, ~ 77% of children with autism had VAD [102]. Followed by a controlled study including 332 children with ASD diagnosis and 197 age- and gender-matched controls; they described that patients with severe ASD had lower serum retinol than their controls (P < 0.05). Further analysis showed a significant interaction between VA levels and the Autism Behavior Checklist (ABC) total score (p = 0.001) [103]. Retinol is converted to RA through a sequence of oxidative steps catalyzed by the ALDH family enzymes [104]. RA is the functional metabolite of VA and it is essential for cortical generation of neurons, neuronal differentiation, synaptic plasticity, brain development, brain function, and the establishment of the neurotransmitter systems [105,106,107]. The ganglionic eminence is a unique transitory proliferative structure of very high neuronal RA synthesis; located in the ventral telencephalon in the human fetal brain, it contributes to at least 35% to the population of cortical GABAergic interneurons for the cerebral cortex [108, 109]. Studies in animal models have detected significant reductions of some GABAergic populations in embryonic mouse forebrains due to VA perturbations. Both excess and deficiency in RA signaling causes interneuron reduction in the brain cortex [109]. The subpopulation of GABAergic interneurons, particularly parvalbumin +, is significantly reduced in the human autistic brain [110, 111]. Exposure to known teratogenic medications like valproic acid and isotretinoin during embryogenesis may increase RA levels and contribute to autism [109]. RA treatment is estimated to regulate expression of several thousand genes by 2-fold or more based on in vitro studies on human cell lines [112], but where and when endogenous RA influences gene expression in vivo depends on the cellular and developmental context and is poorly understood. Oral replenishment of RA in the overexpressing UBE3A mouse model alleviated autistic-type behavior and recovered cellular RA homeostasis [81]. Liu et al. conducted a pilot study in 2017 in which 64 children with ASD, aged 1 to 8 years old completed a 6-month follow-up study of VA supplementation; however, neither the Aberrant Behavior Checklist (ABC), the Childhood Autism Rating Scale (CARS), nor the Social Responsiveness Scale (SRS) showed significant changes after 6 months of treatment [113].
In FXS, studies in Fmr1 KO mice indicated defects in synaptic RA signaling contributing to cognitive impairment [114, 115]. These findings were recently corroborated by Zhang and colleagues in 2018. They analyzed neurons generated from two FXS patient-derived induced pluripotent stem (iPS) cell lines and confirmed that the inactivation of FMR1 blocks the synaptic RA signaling in FXS resulting in a loss of RA-dependent homeostatic plasticity and circuit dysfunction. Homeostatic plasticity is a form of synaptic plasticity that maintains neural network stability [116]. RA signaling is suggested by the results of this study to be impaired by the absence of FMRP expression in patients with FXS. To date, no open-label or controlled trials of oral intake of RA have been conducted in patients with FXS. The results from animal models and iPS cell research open the possibility for genomic medicine to find ways to reverse the abnormalities found in the RA signaling pathway during embryonic brain development aiming to improve brain plasticity and functionality in ASD and FXS.
PI3K/AKT/mTOR Pathway
The PI3K/AKT/mTOR (phosphoinositide 3-kinase/protein kinase B/mechanistic target of rapamycin) pathway is often abnormally regulated in those with ASD, so it is a pathway of great interest for targeted treatments of ASD [117]. The mTOR protein is an evolutionarily conserved checkpoint that affects cell growth and proliferation by controlling many cellular processes [118]. It is involved in regulating protein synthesis, transcription, autophagy, and cellular metabolism. In the nervous system, mTOR is thought to be involved in the development of neuronal circuitry, synaptic plasticity, and regulation of complex behaviors [119]. This pathway has been found to be important in many human conditions, from cancer to brain development [118].
There are several genetic abnormalities that directly affect the mTOR pathway and are associated with ASD [119,120,121,122]. Tuberous sclerosis complex (TSC) results from a mutation in either the TSC1 or TSC2 genes [123]. PTEN hamartoma tumor syndrome (PHTS) results from a mutation in the PTEN gene [124]. Both of these conditions are examples of genetic disorders which impact the mTOR pathway signaling and are associated with ASD [124].
Drugs Targeting PI3K/AKT/mTOR Pathway
Therapies targeting the PI3K/AKT/mTOR pathway were initially developed to prevent solid organ transplant rejection, for cancer treatment, and to prevent neovascularization [119]. Even though the relationship between mTOR dysregulation and ASD is not fully understood, there is interest to see if these therapies are useful for individuals with ASD.
Sirolimus (rapamycin) and everolimus are mTOR inhibitors which have several approved indications, including immunosuppression and treating malignancies [122]. The use of mTOR inhibitors for improving neurocognitive outcomes has been studied in patients with TSC. TSC is a multisystem, autosomal dominant, and genetic disorder which results in overactivation of mTOR [123]. The estimated prevalence of ASD is 40–50% in patients with TSC [125]. In the TSC mouse model, treatment with rapamycin improved behavioral deficits [126, 127] and social interactions [128]. An additional trial using a rat model of TSC showed improvement in social deficit behaviors after treatment with everolimus [129].
Everolimus has been approved to treat the common neoplasms associated with TSC, specifically subependymal giant cell astrocytoma and renal angiomyolipoma [130]. A double-blind randomized placebo-controlled trial of everolimus was conducted to evaluate neurocognition and behavioral improvements with the treatment. There were no significant improvements in these outcomes with everolimus [131]. An additional double-blind randomized placebo-controlled trial also did not find improvement in cognitive functioning, autism, or neuropsychological deficits in children with TSC. The authors of this study hypothesized that the medication may need to be initiated in very young patients prior to the permanent alteration in neurodevelopment caused by TSC [132]. Another study from Japan did show a trend toward improvement of ASD symptoms with everolimus [133]. More studies will need to be conducted to determine if treatment with everolimus impacts ASD.
Idelalisib is a PI3K inhibitor targeting the PI3Kẟ isoform found in hematopoietic cells, which has been approved for certain types of B cell malignancy [134]. Additional treatments which target this pathway include copanlisib [135] and alpelisib [136], which have been approved for use in cancer patients. There is interest in studying the use of PI3K inhibitors in treating PTEN hamartoma tumor syndrome (PHTS) [137]. To date, no clinical study has targeted PI3K as a way to treat ASD, but it is considered a plausible new pharmacologic target for ASD [55, 138].
ERK/MAPK Pathway
The ERK/MAPK (extracellular signal-regulated kinase/mitogen-activated protein kinase) pathway is another pathway that has been tied to ASD [121]. It is involved in the control of many intracellular processes that regulate proteins, control cell growth, and trigger apoptosis [121, 139]. RASopathies are a collection of syndromes with abnormalities in this pathway. This includes genetic syndromes such as neurofibromatosis type 1 and Noonan syndrome [140]. There is an increased prevalence in autistic traits and ASD in children with RASopathies [141, 142].
Drugs Targeting ERK/MAPK Pathway
Similar to the PI3K/AKT/mTOR pathway, researchers are interested in targeting ERK/MAPK for cancer treatment [143]. Ulixertinib is a novel ERK1/2 inhibitor which is being studied as an oncologic treatment [144]. The impact of targeting the ERK/MAPK pathway for neurodevelopment has been studied in animal models. Mice with 16p11.2 microdeletion, which is a deletion associated with ASD in humans, had increased ERK activity and behavioral deficits. Treatment with an ERK pathway inhibitor rescued some of the behavioral deficit [145]. The effect of ERK upregulation on neurodevelopment is also thought to occur during a critical period. Transient blockage of phosphorylation of ERK at postnatal day 6 in mice resulted in apoptosis in the forebrain, social deficit, and impaired memory. When this was done at postnatal day 14, the same effects were not seen [146].
PI3K/AKT/mTOR and ERK/MAPK Pathways in FXS
In FXS, both the PI3K/AKT/mTOR and ERK/MAPK pathways are dysregulated. Normally, FMRP is thought to suppress the mTOR and ERK/MAPK pathways. As a result, in FXS, signaling through these pathways are elevated [147]. In both mice model and human tissue, studies have found elevated levels of mTOR [148, 149] and ERK [150]. Inhibition of these pathways has been shown to reverse some of the deficits in the Fmr1 knockout (KO) mouse [151, 152]. Medications targeting these pathways are starting to be studied in patients with FXS.
Lovastatin, a 3-hydroxy-3 methylglutaryl-CoA reductase inhibitor, prevents cholesterol biosynthesis. It is a common treatment for hyperlipidemia and hypercholesteremia. Lovastatin has been found to inhibit ERK/MAPK pathway [153]. In the Fmr1 KO mouse, lovastatin corrects excessive hippocampal protein synthesis and decreases seizures [152]. Lovastatin has also been studied as part of a 12-week open-label trial in individuals with FXS. Most patients had behavioral improvement with the treatment [154]. The researchers also found that ERK activity normalized after the treatment and that the change correlated with the clinical response [155]. However, a more recent double-blind, randomized control trial with lovastatin combined with an open-label treatment of parent implemented language intervention (PILI) did not find significant differences between the group treated with PILI without lovastatin and the group treated with PILI with lovastatin, although both groups showed improvement from baseline scores. This study also did not find changes in the ERK level with treatment. Ultimately, this study showed that behavioral interventions with PILI are just as effective as lovastatin treatment. [156].
Metformin is a medication that acts through the AMP-activated protein kinase (AMPK) pathway [157], which then impacts both the mTOR and ERK/MAPK pathways [157, 158]. Metformin is thought to inhibit mTOR through multiple mechanisms. This includes AMPK directly phosphorylating a component of mTOR and AMPK activating the TSC2 gene which inhibits the mTOR activator [159] (see Fig. 2). In the mouse model, metformin has been shown to strongly inhibit mTOR through AMPK [160]. There are also proposed AMPK-independent mechanisms for metformin’s regulation of mTOR [159]. Metformin also impairs the ERK/MAPK signaling pathway, but mechanisms are not well understood [161]. In the Fmr1 KO mice, metformin has been shown to rescue core phenotype and normalized ERK signaling [162].

Metformin and the PI3K/AKT/mTOR pathway. Proposed mechanisms for metformin’s regulation of the PI3K/AKT/mTOR pathway. Black arrows represent activation pathways whereas red lines represent inhibitory pathways. IGF-1 = insulin-like growth factor-1; IGF-1R = insulin-like growth factor-1 receptor; IRS = insulin receptor substrate; PI3K = phosphoinositide 3-kinase; PIP2 = phosphatidylinositol-4,5-bisphosphate; PIP3 = phosphatidylinositol-4,5-triphosphate; PTEN = phosphatase and tensin homolog; PDK1 = phosphoinositide-dependent kinase 1; PKB = protein kinase B or Akt; TSC1 = tuberous sclerosis complex 1 (hamartin); TSC2 = tuberous sclerosis complex 2 (tuberin); Rheb = Ras homolog enriched in brain; mTOR = mammalian target of rapamycin; 4E-BP1 = eukaryotic initiation factor 4E binding protein 1; eIF4E = eukaryotic translation initiation factor 4E; p70S6K = p70 ribosomal protein S6 kinase; S6K = S6 kinase; AMP = adenosine monophosphate; AMPK = AMP-activated protein kinase; AAs = amino acids; Rags = Rag GTPases
In humans, metformin has been approved by the Food and Drug Administration for patients with type 2 diabetes due to its ability to lower blood glucose levels [163]. In a case series following 7 patients with FXS ages 4.5 to 60 years who were treated clinically with metformin, there was noted to be improvement in behavior and language [164]. Additionally, another case series of nine children with FXS, ages 2 to 7, treated off-label with metformin for at least 3 months also showed improvement in behavior and language [165]. Currently, there is a multisite double-blind randomized controlled trial evaluating the safety along with the efficacy of metformin in the treatment of language deficit and behavioral issues in individuals with FXS (NCT03862950, NCT03479476) [166, 167].
Endocannabinoid System
The endocannabinoid system has been found to be dysregulated in FXS. Endocannabinoids can target this deficiency and treat common symptoms in patients with FXS and ASD [168, 169]. For instance, cannabidiol (CBD), the primary nonpsychoactive phytocannabinoid found in cannabis, has therapeutic potential for the treatment of anxiety, sleep disorders, and cognitive deficits [170]. FMRP has significant control over the endocannabinoid system because it regulates the translation of hundreds of messages in the CNS. In the absence of FMRP, there is significant dysregulation of the endocannabinoid system. Anxiety, which is one of the most severe problems in FXS, is also correlated with ASD so that the more severe the anxiety, the more severe the ASD [171, 172]. The presence of symptoms such as anxiety, poor sleep, and seizures in individuals with FXS and communication issues in individuals with ASD could be benefitted by the pharmacological potential of CBD [173]. CBD is generally safe to use and tolerated well [174]. Research indicates that CBD acts in multiple ways such as N-arachidonoylethanolamine (anandamide; AEA) and 2-arachidonoylglycerol (2-AG) binding to two G-protein-coupled receptors, cannabinoid 1 (CB1) and cannabinoid 2 (CB2), and thus interacting with the endocannabinoid system [170]. CBD may have positive effects on behavior issues through gamma-aminobutyric acid (GABA) signaling by acting as a positive allosteric modulator and anxiolytic effects through modulation of serotonin-1A receptor (5-HT1A) [175,176,177].
A phase 1/2 open-label multicenter trial of Zygel transdermal CBD gel in patients with FXS found anxiety and behavioral improvements [178, 179]. A total of 20 patients, mean age of 10.4 years, were treated during a 12-week period (6-week titration period and 6-week maintenance period) at 3 possible doses—50 mg/day, 100 mg/day, or 250 mg/day—based on tolerability and clinical response. The study’s primary outcome measure was the Anxiety, Depression, and Mood Scale (ADAMS). Investigators reported a statistically significant reduction in the mean ADAMS total score (t = − 5.74, p < 0.001), as well as mean reductions on the manic/hyperactive behavior, general anxiety, compulsive behavior, and social avoidance subscales. Secondary outcome measures, including the ABC-Community version modified for FXS (ABC-CFXS), Pediatric Anxiety Rating Scale (PARS-R), and the Pediatric Quality of Life Inventory (PedsQL) also showed significant improvement [178]. A double-blind randomized multicenter controlled trial of Zygel recently completed showed a significant improvement in behavior only in patients with full methylation (over 90% methylation) of the FMR1 gene [180]. This group represents roughly 70% of the population of individuals with FXS and they demonstrated a mean improvement of 40% of their social avoidance subscale scores of the ABC-CFXS [180]. Meanwhile, during an open-label phase 2 trial of Zygel in patients with ASD, it was observed that after 6 months of treatment, patients experienced greater autonomy, especially for activities like showering and dressing independently [181]. They had a mean improvement of 42.5% in the use of inappropriate speech, 39.1% in irritability, 36.4% in social withdrawal, and 35.6% in hyperactivity as shown by the results of their ABC-C scores at week 14 from baseline [181]. These results are further represented in the mean improvement of 46% from a baseline score of 40.8 measured by the Parent Rated Anxiety Scale - Autism Spectrum Disorder (PRAS-ASD) and a 57% increase in Clinical Global Impression-Improvement (CGI-I) scores [181]. These results are impressive for both those with FXS and ASD, but it is unclear at the time of this writing whether the FDA will approve the use of this topical CBD preparation for either disorder.
Glutamatergic–GABAergic Neurotransmitter System
Excitatory–Inhibitory Imbalance in ASD
In ASD, the synaptic excitatory–inhibitory balance is altered with a decrease in inhibitory GABAergic signaling. Although GABA is normally known for being the major inhibitory neurotransmitter, it causes chloride (Cl–) efflux during early development and leads to the excitation of immature neurons [182]. The balance in Cl− levels and the shift from excitatory to inhibitory neurotransmission depends on the action of the sodium–potassium–chloride (Na-K-2Cl) cotransporters (NKCC1), which mediates Cl− influx, and potassium–chloride (K+–2Cl−) cotransporter 2 (KCC2) that mediates efflux. In early development, GABA exerts an excitatory action in postsynaptic neurons. The expression of KCC2 is low; therefore, intracellular Cl− levels are high, and GABA causes Cl− efflux and postsynaptic depolarization. As neurons mature, there is increased expression of KCC2, and intracellular Cl− levels diminish. This is known as the excitatory-to-inhibitory GABA “switch.” As KCC2 expression increases, NKCC1 activity decreases, which leads to lower intracellular Cl− levels and the change from GABA-induced depolarization to hyperpolarization [183, 184]. In humans, the increase in KCC2 and decreased in NKCC1 activity begins at 40 weeks after birth [182, 183]. It has been hypothesized that in individuals with ASD, this GABA switch is unsuccessful and leads to the well-described excitatory–inhibitory imbalance [185, 186].
Evidence from genetic studies, postmortem brain tissues, and in vivo studies in patients with ASD and animal models have shown different alterations of GABAergic signaling [187], such as a reduction in the density of GABAA and GABAB receptors in different brain regions [188,189,190], decreased number of GABAergic Purkinje cells [191], and reduction of GABA levels [192, 193]. Abnormalities in the GABAergic signaling is also implicated in the pathophysiology of FXS [194].
Aberrant glutamate signaling has been implicated in the excitatory–inhibitory imbalance in ASD and FXS. Increased excitatory transmission is mediated by higher expression of amino-hydroxy-methyl-isoxazole propionic acid (AMPA) and N-methyl-d-aspartate (NMDA) has also been identified in ASD [195]. In FXS, there is excitatory–inhibitory imbalance with increased neuronal cell excitability [196]. Fmr1 KO mice models have upregulation of metabotropic glutamate receptors (mGluR) and subsequent aberrant excitatory signaling [197]. FXS hyperexcitability is also mediated by reduced GABAA inhibition [196]. Therefore, targeting the glutamate and GABA signaling pathway has been of interest for the treatment of ASD and FXS.
Targeting the GABAergic System in ASD and FXS
Bumetanide in ASD and FXS
Bumetanide is a loop diuretic that works by inhibiting the Na-K-2Cl cotransporters NKCC1 and NKCC2 in the thick ascending limb of Henle’s loop. NKCC1 is also expressed in the central nervous system and modulates neuronal transmembrane chloride gradient by contributing to Cl− accumulation inside the cell [198]. Due to the NKCC1-mediated excitatory actions in the CNS, bumetanide was proposed to treat neonatal seizures [198]. Bumetanide has also been studied to treat young patients with ASD because, if administered early, it could lead to restoration of the neuronal excitatory–inhibitory balance. It has been demonstrated in animal models of ASD, such as rats with intrauterine exposure to valproic acid or Fmr1 KO mice, that the maternal administration of bumetanide before delivery restored the excitatory–inhibitory imbalance [199, 200].
Early administration of bumetanide can potentially restore the excitatory–inhibitory imbalance in ASD by promoting the GABA switch. Zhang and colleagues [186] assessed the effects of bumetanide in the reduction of symptom severity in patients with ASD. They randomized 83 patients aged 3 to 6 years old to bumetanide or control group (no treatment). They assessed the change in score from baseline to after 3 months of intervention in the CARS and CGI. CARS is a 15-item clinical symptom scale, each item rated on a 7-point scale [201]. They compared the change in total score and the number of items with a score ≥ 3. They also assessed the GABA/glutamate neurotransmitter concentrations ratio in the insular cortex and visual cortex through magnetic resonance spectroscopy (MRS) to evaluate whether the clinical improvement was related to normalization in the excitatory–inhibitory imbalance. Baseline CARS were similar between both groups. After the 3-month period, patients assigned to bumetanide had lower total CARS scores (p = 0.0012) and fewer items with a score of 3 or higher (p = 0.0053). Improvement in CGI was also confirmed using the improvement scale (CGI-I) (p = 0.000036) and efficacy index (p = 0.00056) [186]. The researchers also found that the decrease in the number of items with a CARS score 3 or over was associated with the change in GABA/glutamate ratio in the insular cortex (Spearman’s r = 0.42, p = 0.00194) [186]. Although this study had a small sample size, the results are promising and suggest that bumetanide acts by restoring the excitatory–inhibitory balance. Other clinical trials have found that bumetanide administration reduces clinical symptom severity [202,203,204] and shows better results when used as an add-on therapy for applied behavior analysis (ABA) than ABA alone [205].
Arbaclofen in ASD and FXS
Enhancing GABA inhibitory signaling has been studied in clinical trials in FXS with mixed results. For instance, ganaxolone, a GABAA-positive allosteric modulator, found promising results in participants with more severe anxiety and cognitive involvement in an open-label trial [206, 207]. However, in a randomized, double-blind controlled trial, ganaxolone did not demonstrate efficacy overall, although in those with severe anxiety it was helpful [208]. In contrast, arbaclofen, a GABAB agonist, failed to show an improvement in the primary outcome of two phase 3 clinical trials [209]. Erickson et al. conducted an open-label trial of arbaclofen in 32 patients with ASD, with 25 patients completing the study [210]. After 8 weeks of arbaclofen administration, they found significant improvement in the ABC-Irritability subscale. These results are limited due to lack of placebo control, possible expectancy bias, and the trial’s relatively short duration. Arbaclofen is currently being studied in a subgroup of children with ASD (NCT03682978, NCT03887676).
Acamprosate in ASD and FXS
Acamprosate is a GABA analog used to treat alcohol dependence. It activates both GABAA and GABAB receptors and has been studied in patients with FXS and patients with idiopathic ASD [211,212,213]. Erickson et al. conducted a 10-week open-label trial in 12 patients with FXS and ASD and found improvement in 9 of the subjects on the CGI-I. They also saw improvement in social behavior on the ABC social withdrawal subscale (ABC-SW) and on the ABC social avoidance scale (ABC-SA) [212]. The same group studied acamprosate in individuals with idiopathic ASD. They conducted an open-label study in six patients [213] and later a single-blinded trial in which they enrolled 12 patients, but only six subjects completed the trial [211]. In both, they found improvement in social behavior and reduced inattention/hyperactivity. These findings are limited due to the small sample size and lack of placebo control in both and missing data with the last observation carried forward (LOCF) analysis in the most recent one.
Allopregnanolone
Allopregnanolone is a metabolite of pregnenolone, a natural neurosteroid produced in the CNS. Allopregnanolone has been shown to positively modulate GABAA receptors [214]. Administration of oral pregnenolone has been studied in FXS and ASD. Fung et al. conducted an open-label 12-week trial of oral pregnenolone in 12 adults with ASD, of which 10 completed the trial, with changes in ABC-Irritability scale (ABC-I) as their primary outcome [215]. They found statistically significant improvement in ABC-I, which must be taken with caution due to the study’s small sample size and open-label nature. Ayatollahi et al. conducted a randomized, double-blind, and placebo-controlled trial in adolescents with ASD in which they evaluated the effectiveness of pregnenolone as add-on therapy to risperidone [216]. They enrolled 64 patients, and 59 completed the trial. Their results suggest improvement in irritability, stereotypical movements, and hyperactivity based on ABC, which indicates a possible beneficial add-on effect.
Targeting Glutamate-Mediated Hyperexcitability
Despite the fact that the upregulation of mGluR is implicated in FXS pathogenesis, clinical trials assessing the effects of mGluR antagonists, Mavoglurant or Basimglurant, have not shown positive results in adolescents and adults [217,218,219]. Mavoglurant is currently being studied at multiple centers in young children 3 to 6 years old with FXS and combined with PILI in all children (NCT02920892), but the results are not yet known.
Memantine is an NMDA antagonist that is approved for the treatment of Alzheimer’s disease. Memantine was studied in three phase 2 trials in which 765 participants with ASD completed the study. The initial open-label phase found a clinically significant improvement in mean SRS scores. Despite this, in the double-blind, placebo-controlled trial, they find no statistically significant efficacy parameters [220].
Ketamine is a widely used anesthetic agent that works as an NMDAR antagonist [221]. Interest in this drug as a treatment for ASD arises from its effect on multiple mechanisms linked to ASD pathogenesis. Ketamine not only acts upon glutamate receptors to restore their normal functioning, but preclinical data has also shown that it acts upon the mTOR and BDNF/TrkB signaling pathway [222,223,224]. Wink et al. conducted a randomized, placebo-controlled, and crossover study of intranasal ketamine in 21 adolescents and young adults with ASD [225]. Intranasal ketamine was relatively well tolerated, but researchers found no significant effects on ABC-SW or CGI-I. These results must be viewed in light of the small sample size and the use of clinician and caregiver-reported outcome measures.
Oxytocin
Oxytocin is a neuropeptide involved in social functioning and relationship bonding formation. Animal models have also revealed that similar to bumetanide, oxytocin mediates GABA inhibition during delivery and may promote the GABA excitatory–inhibitory shift [184, 199]. Oxytocin levels are commonly found to be low in patients with ASD [226, 227]. Clinical trials have assessed intranasal oxytocin’s effects on autistic behaviors [228,229,230,231]. Studying the effects of oxytocin is challenging due to the differential effects between acute and repeated dosing and the interactions between oxytocin and other signaling pathways such as glutamatergic neurotransmission, arginine vasopressin (AVP), and serotoninergic pathway [231,232,233,234]. Yamasue et al. conducted a large-scale, randomized, placebo-controlled, double-blind, and clinical trial to assess the effects of intranasal oxytocin on the core social symptoms of patients with ASD [235]. They enrolled 103 male participants aged 18–48 years old. Their primary endpoint was a change in social reciprocity scores on the ADOS module 4 after 6 weeks of intervention. There was a reduction in the ADOS reciprocity scores in both oxytocin and placebo groups with no significant group difference. They found significant improvement in secondary outcomes such as repetitive behavior and increased gaze fixation duration on socially relevant regions in the treatment group. They concluded that there was a high placebo effect in their trial. Crossover designs assessing intranasal oxytocin can account for a placebo effect but risk a carryover effect. Carryover studies have detected significant effects of oxytocin improving social symptoms in ASD, such as social reciprocity [236, 237]. Therefore, the results of trials assessing oxytocin for social symptoms in ASD are conflicting, but this can be explained by differences in trial designs, the placebo effect, and the type of oxytocin dosing administration (acute vs repeated dosing) [238,239,240].
Vasopressin
Similar to the oxytocin mechanism, AVP affects social behavior in both animals and humans [241]. There is a suggested link between the neuropeptide pathway and incidence of ASD [242]. Social behavior regulatory receptors that vasopressin binds to include arginine vasopressin receptor 1A (V1aR), where V1aR specifically mediates behavioral function, and knockout mice demonstrated a decrease in social interaction [241]. Vasopressin concentrations within cerebrospinal fluid (CSF) were found to be stable measures that could predict lower social interaction for rhesus monkeys; thus, it promoted confidence in using vasopressin as a biomarker for ASD through blood testing and CSF collection [243].
The continued study of the effects of vasopressin led to the phase 2 VANILLA clinical trial, in which researchers assessed the effects and safety of balovaptan, a selective vasopressin V1a receptor antagonist, in men with ASD. This was a randomized, multi center, double-blind, and placebo-controlled trial, in which patients received the intervention daily for 12 weeks. Two hundred twenty-three men were randomized to either placebo or one of three balovaptan dose arms (1.5 mg, 4 mg, and 10 mg). They found no statistically significant effect in the primary endpoint which was a change from baseline in the SRS. They found improvements on Vineland-II Adaptive Behavior Scales composite scores, mainly in socialization and communication scores for those assigned to the higher doses of balovaptan. There were no safety concerns identified. This trial supports the potential for targeting the vasopressin pathway for the treatment of social symptoms in ASD [244].
Challenges in the Search for a Targeted Treatment
Despite the large number of clinical trials in ASD, there is still no evidence for the approval of routine use of a new drug to manage the core social symptoms of ASD or for targeted treatment. The only FDA-approved medications for symptomatic management in ASD are risperidone and aripiprazole [245]. What have been the challenges in the conduction of clinical trials in ASD and FXS? Firstly, both ASD and FXS are very heterogeneous with different clinical manifestations and levels of involvement [245, 246]. There are also important comorbidities such as epilepsy and ADHD, meaning many patients participating in trials are receiving other medications [247,248,249]. Aside from the clinical heterogeneity, the pathophysiology is also very complex with multiple pathways being involved. Searching for targeted treatments is further complicated due to the cross talk between pathways such as the Wnt and the mTOR pathways [53, 250] or the multiple interactions of oxytocin with other molecular pathways [251]. The conduct of clinical trials faces challenges such as the high placebo response in ASD [252, 253], observer and rater biases, suboptimal metrics for primary and secondary outcomes [245], adherence, and dropouts rates. It has also been shown that the recruitment strategy can impact expectations and play a crucial role in the placebo response [220, 252]. Lack of efficacy and conflicting outcomes between trials is impacted by the different trial designs, small sample sizes leading to underpowered studies, the use of subjective outcome measures that are susceptible to observer/rater bias, and the lack of molecular biomarkers. Electronic or digital outcome measures such as gaze tracking or event-related potentials (ERPs) can be a more objective outcomes with reduced observer/rater bias [254, 255]. For example, Yamasue et al. found in their study that there was a higher placebo effect on ADOS measurements compared to gaze tracking measurements explaining the lack of correlation between improvements on both measures. The placebo effect may have accounted for this difference, highlighting that more objective measurements such as eye gaze tracking measurements are less susceptible to the placebo effect and might be more robust in assessing the efficacy of interventions [235]. ASD and FXS treatment is an active research field with many challenges to address and many promising results.
References
Zablotsky B, Black LI, Maenner MJ, Schieve LA, Danielson ML, Bitsko RH, et al. Prevalence and trends of developmental disabilities among children in the United States: 2009–2017.J Pediatr. 2019;144(4):e20190811. https://doi.org/10.1542/peds.2019-0811%.
Maenner M, Shaw K, Baio J, et al. Prevalence of autism spectrum disorder among children aged 8 years — Autism and Developmental Disabilities Monitoring Network, 11 sites, United States. MMWR Surveill Summ. 2020. 2016;69:1–12. https://doi.org/10.15585/mmwr.ss6904a1.
American Psychiatric Association. DSM-5 Task Force. Diagnostic and statistical manual of mental disorders : DSM-5. 5th ed. Washington, DC.: American Psychiatric Association; 2013.
Anderson DK, Oti RS, Lord C, Welch K. Patterns of growth in adaptive social abilities among children with autism spectrum disorders. J Abnorm Child Psychol. 2009;37(7):1019–34. https://doi.org/10.1007/s10802-009-9326-0.
Brignell A, Chenausky KV, Song H, Zhu J, Suo C, Morgan AT. Communication interventions for autism spectrum disorder in minimally verbal children. Cochrane Database Syst Rev. 2018;11(11):CD012324-CD. https://doi.org/10.1002/14651858.CD012324.pub2.
Anderson DK, Lord C, Risi S, DiLavore PS, Shulman C, Thurm A, et al. Patterns of growth in verbal abilities among children with autism spectrum disorder. J Consult Clin Psychol. 2007;75(4):594–604. https://doi.org/10.1037/0022-006x.75.4.594.
Hollocks MJ, Lerh JW, Magiati I, Meiser-Stedman R, Brugha TS. Anxiety and depression in adults with autism spectrum disorder: a systematic review and meta-analysis. Psychol Med. 2019;49(4):559–72. https://doi.org/10.1017/s0033291718002283.
Goldman SE, Alder ML, Burgess HJ, Corbett BA, Hundley R, Wofford D, et al. Characterizing sleep in adolescents and adults with autism spectrum disorders. J Autism Dev Disord. 2017;47(6):1682–95. https://doi.org/10.1007/s10803-017-3089-1.
Cheroni C, Caporale N, Testa G. Autism spectrum disorder at the crossroad between genes and environment: contributions, convergences, and interactions in ASD developmental pathophysiology. Mol Autism. 2020;11(1):69. https://doi.org/10.1186/s13229-020-00370-1.
Werling DM, Brand H, An J-Y, Stone MR, Zhu L, Glessner JT, et al. An analytical framework for whole-genome sequence association studies and its implications for autism spectrum disorder. Nat Genet. 2018;50(5):727–36. https://doi.org/10.1038/s41588-018-0107-y.
Wang W, Corominas R, Lin GN. De novo mutations from whole exome sequencing in neurodevelopmental and psychiatric disorders: from discovery to application. Front Genet. 2019;10:258. https://doi.org/10.3389/fgene.2019.00258.
Bae SM, Hong JY. The Wnt signaling pathway and related therapeutic drugs in autism spectrum disorder. Clin Psychopharmacol Neurosci. 2018;16(2):129–35. https://doi.org/10.9758/cpn.2018.16.2.129.
Kalkman HO. A review of the evidence for the canonical Wnt pathway in autism spectrum disorders. Mol Autism. 2012;3(1):10–. https://doi.org/10.1186/2040-2392-3-10.
Casanova MF, Casanova EL, Frye RE, Baeza-Velasco C, LaSalle JM, Hagerman RJ, et al. Editorial: secondary vs. idiopathic autism. Front Psychiatry. 2020;11:297–. https://doi.org/10.3389/fpsyt.2020.00297.
Qin M, Kang J, Burlin TV, Jiang C, Smith CB. Postadolescent changes in regional cerebral protein synthesis: an in vivo study in the FMR1 null mouse. J Neurosci. 2005;25(20):5087–95. https://doi.org/10.1523/JNEUROSCI.0093-05.2005.
Li Z, Zhang Y, Ku L, Wilkinson KD, Warren ST, Feng Y. The fragile X mental retardation protein inhibits translation via interacting with mRNA. Nucleic Acids Res. 2001;29(11):2276–83. https://doi.org/10.1093/nar/29.11.2276.
Tassone F, Hagerman RJ, Iklé DN, Dyer PN, Lampe M, Willemsen R, et al. FMRP expression as a potential prognostic indicator in fragile X syndrome. Am J Med Genet. 1999;84(3):250–61.
Gothelf D, Furfaro JA, Hoeft F, Eckert MA, Hall SS, O'Hara R, et al. Neuroanatomy of fragile X syndrome is associated with aberrant behavior and the fragile X mental retardation protein (FMRP). Ann Neurol. 2008;63(1):40–51. https://doi.org/10.1002/ana.21243.
Harris SW, Hessl D, Goodlin-Jones B, Ferranti J, Bacalman S, Barbato I, et al. Autism profiles of males with fragile X syndrome. Am J Ment Retard. 2008;113(6):427–38. https://doi.org/10.1352/2008.113:427-438.
Hatton DD, Sideris J, Skinner M, Mankowski J, Bailey DB, Jr., Roberts J, et al. Autistic behavior in children with fragile X syndrome: prevalence, stability, and the impact of FMRP. Am J Med Genet A. 2006;140A(17):1804–13. https://doi.org/10.1002/ajmg.a.31286.
Roberts JE, Weisenfeld LA, Hatton DD, Heath M, Kaufmann WE. Social approach and autistic behavior in children with fragile X syndrome. J Autism Dev Disord. 2007;37(9):1748–60. https://doi.org/10.1007/s10803-006-0305-9.
Li J, Wang CY. TBL1-TBLR1 and beta-catenin recruit each other to Wnt target-gene promoter for transcription activation and oncogenesis. Nat Cell Biol. 2008;10(2):160–9. https://doi.org/10.1038/ncb1684.
Niehrs C. The complex world of WNT receptor signalling. Nat Rev Mol Cell Biol 2012;13(12):767–79. https://doi.org/10.1038/nrm3470.
Willert K, Nusse R. Wnt proteins. Cold Spring Harb Perspect Biol. 2012;4(9):a007864. https://doi.org/10.1101/cshperspect.a007864.
Komiya Y, Habas R. Wnt signal transduction pathways. Organogenesis. 2008;4(2):68–75. https://doi.org/10.4161/org.4.2.5851.
Hirabayashi Y, Itoh Y, Tabata H, Nakajima K, Akiyama T, Masuyama N, et al. The Wnt/beta-catenin pathway directs neuronal differentiation of cortical neural precursor cells. Development. 2004;131(12):2791–801. https://doi.org/10.1242/dev.01165.
Munji RN, Choe Y, Li G, Siegenthaler JA, Pleasure SJ. Wnt signaling regulates neuronal differentiation of cortical intermediate progenitors. J Neurosci. 2011;31(5):1676–87. https://doi.org/10.1523/JNEUROSCI.5404-10.2011.
Breen MS, Browne A, Hoffman GE, Stathopoulos S, Brennand K, Buxbaum JD, et al. Transcriptional signatures of participant-derived neural progenitor cells and neurons implicate altered Wnt signaling in Phelan-McDermid syndrome and autism. Mol Autism. 2020;11(1):53. https://doi.org/10.1186/s13229-020-00355-0.
Quesnel-Vallieres M, Weatheritt RJ, Cordes SP, Blencowe BJ. Autism spectrum disorder: insights into convergent mechanisms from transcriptomics. Nat Rev Genet. 2019;20(1):51–63. https://doi.org/10.1038/s41576-018-0066-2.
Satterstrom FK, Kosmicki JA, Wang J, Breen MS, De Rubeis S, An JY, et al. Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell. 2020;180(3):568–84 e23. https://doi.org/10.1016/j.cell.2019.12.036.
Dong F, Jiang J, McSweeney C, Zou D, Liu L, Mao Y. Deletion of CTNNB1 in inhibitory circuitry contributes to autism-associated behavioral defects. Hum Mol Genet. 2016;25(13):2738–51. https://doi.org/10.1093/hmg/ddw131.
Medina MA, Andrade VM, Caracci MO, Avila ME, Verdugo DA, Vargas MF, et al. Wnt/beta-catenin signaling stimulates the expression and synaptic clustering of the autism-associated Neuroligin 3 gene. Transl Psychiatry. 2018;8(1):45. https://doi.org/10.1038/s41398-018-0093-y.
Bernier R, Golzio C, Xiong B, Stessman HA, Coe BP, Penn O, et al. Disruptive CHD8 mutations define a subtype of autism early in development. Cell. 2014;158(2):263–76. https://doi.org/10.1016/j.cell.2014.06.017.
Kawamura A, Katayama Y, Nishiyama M, Shoji H, Tokuoka K, Ueta Y, et al. Oligodendrocyte dysfunction due to Chd8 mutation gives rise to behavioral deficits in mice. Hum Mol Genet. 2020;29(8):1274–91. https://doi.org/10.1093/hmg/ddaa036.
Katayama Y, Nishiyama M, Shoji H, Ohkawa Y, Kawamura A, Sato T, et al. CHD8 haploinsufficiency results in autistic-like phenotypes in mice. Nature. 2016;537(7622):675–9. https://doi.org/10.1038/nature19357.
Platt RJ, Zhou Y, Slaymaker IM, Shetty AS, Weisbach NR, Kim JA, et al. Chd8 mutation leads to autistic-like behaviors and impaired striatal circuits. Cell Rep. 2017;19(2):335–50. https://doi.org/10.1016/j.celrep.2017.03.052.
Durak O, Gao F, Kaeser-Woo YJ, Rueda R, Martorell AJ, Nott A, et al. Chd8 mediates cortical neurogenesis via transcriptional regulation of cell cycle and Wnt signaling. Nat Neurosci. 2016;19(11):1477–88. https://doi.org/10.1038/nn.4400.
Suetterlin P, Hurley S, Mohan C, Riegman KLH, Pagani M, Caruso A, et al. Altered neocortical gene expression, brain overgrowth and functional over-connectivity in Chd8 haploinsufficient mice. Cereb Cortex. 2018;28(6):2192–206. https://doi.org/10.1093/cercor/bhy058.
Stolerman ES, Smith B, Chaubey A, Jones JR. CHD8 intragenic deletion associated with autism spectrum disorder. Eur J Med. Genet 2016;59(4):189–94. https://doi.org/10.1016/j.ejmg.2016.02.010.
Stessman HA, Xiong B, Coe BP, Wang T, Hoekzema K, Fenckova M, et al. Targeted sequencing identifies 91 neurodevelopmental-disorder risk genes with autism and developmental-disability biases. Nat Genet. 2017;49(4):515–26. https://doi.org/10.1038/ng.3792.
Kwan V, Unda BK, Singh KK. Wnt signaling networks in autism spectrum disorder and intellectual disability. J Neurodev Disord. 2016;8:45. https://doi.org/10.1186/s11689-016-9176-3.
Beurel E, Grieco SF, Jope RS. Glycogen synthase kinase-3 (GSK3): regulation, actions, and diseases. Pharmacol Ther. 2015;148:114–31. https://doi.org/10.1016/j.pharmthera.2014.11.016.
Kim WY, Snider WD. Functions of GSK-3 signaling in development of the nervous system. Front Mol Neurosci. 2011;4:44. https://doi.org/10.3389/fnmol.2011.00044.
Franklin AV, King MK, Palomo V, Martinez A, McMahon LL, Jope RS. Glycogen synthase kinase-3 inhibitors reverse deficits in long-term potentiation and cognition in fragile X mice. Biol Psychiatry. 2014;75(3):198–206. https://doi.org/10.1016/j.biopsych.2013.08.003.
Mines MA, Yuskaitis CJ, King MK, Beurel E, Jope RS. GSK3 influences social preference and anxiety-related behaviors during social interaction in a mouse model of fragile X syndrome and autism. PLoS One. 2010;5(3):e9706. https://doi.org/10.1371/journal.pone.0009706.
Jope RS, Johnson GV. The glamour and gloom of glycogen synthase kinase-3. Trends Biochem Sci. 2004;29(2):95–102. https://doi.org/10.1016/j.tibs.2003.12.004.
Min WW, Yuskaitis CJ, Yan Q, Sikorski C, Chen S, Jope RS, et al. Elevated glycogen synthase kinase-3 activity in fragile X mice: key metabolic regulator with evidence for treatment potential. Neuropharmacology. 2009;56(2):463–72. https://doi.org/10.1016/j.neuropharm.2008.09.017.
Wang L, Liu Y, Li S, Long ZY, Wu YM. Wnt signaling pathway participates in valproic acid-induced neuronal differentiation of neural stem cells. Int J Clin Exp Pathol. 2015;8(1):578–85.
Wiltse J. Mode of action: inhibition of histone deacetylase, altering WNT-dependent gene expression, and regulation of beta-catenin--developmental effects of valproic acid. Crit Rev Toxicol. 2005;35(8-9):727–38. https://doi.org/10.1080/10408440591007403.
Go HS, Kim KC, Choi CS, Jeon SJ, Kwon KJ, Han SH, et al. Prenatal exposure to valproic acid increases the neural progenitor cell pool and induces macrocephaly in rat brain via a mechanism involving the GSK-3beta/beta-catenin pathway. Neuropharmacology. 2012;63(6):1028–41. https://doi.org/10.1016/j.neuropharm.2012.07.028.
Schneider T, Przewlocki R. Behavioral alterations in rats prenatally exposed to valproic acid: animal model of autism. Neuropsychopharmacology. 2005;30(1):80–9. https://doi.org/10.1038/sj.npp.1300518.
Qin L, Dai X. [Effect of sulindac on improving autistic behaviors in rats]. Nan Fang Yi Ke Da Xue Xue Bao. 2015;35(8):1162-5.
Kumar S, Reynolds K, Ji Y, Gu R, Rai S, Zhou CJ. Impaired neurodevelopmental pathways in autism spectrum disorder: a review of signaling mechanisms and crosstalk. J Neurodev Disord 2019;11(1):10. https://doi.org/10.1186/s11689-019-9268-y.
Zhang Y, Yang C, Yuan G, Wang Z, Cui W, Li R. Sulindac attenuates valproic acid-induced oxidative stress levels in primary cultured cortical neurons and ameliorates repetitive/stereotypic-like movement disorders in Wistar rats prenatally exposed to valproic acid. Int J Mol Med. 2015;35(1):263–70. https://doi.org/10.3892/ijmm.2014.1996.
Baranova J, Dragunas G, Botellho MCS, Ayub ALP, Bueno-Alves R, Alencar RR, et al. Autism spectrum disorder: signaling pathways and prospective therapeutic targets. Cell Mol Neurobiol. 2020. https://doi.org/10.1007/s10571-020-00882-7.
Warkus ELL, Marikawa Y. Fluoxetine inhibits canonical Wnt signaling to impair embryoid body morphogenesis: potential teratogenic mechanisms of a commonly used antidepressant. Toxicol Sci. 2018;165(2):372–88. https://doi.org/10.1093/toxsci/kfy143.
Xia MY, Zhao XY, Huang QL, Sun HY, Sun C, Yuan J, et al. Activation of Wnt/beta-catenin signaling by lithium chloride attenuates d-galactose-induced neurodegeneration in the auditory cortex of a rat model of aging. FEBS Open Bio. 2017;7(6):759–76. https://doi.org/10.1002/2211-5463.12220.
Berry-Kravis E, Sumis A, Hervey C, Nelson M, Porges SW, Weng N, et al. Open-label treatment trial of lithium to target the underlying defect in fragile X syndrome. J Dev Behav Pediatr. 2008;29(4):293–302. https://doi.org/10.1097/DBP.0b013e31817dc447.
Chugani DC, Muzik O, Rothermel R, Behen M, Chakraborty P, Mangner T, et al. Altered serotonin synthesis in the dentatothalamocortical pathway in autistic boys. Ann Neurol. 1997;42(4):666–9. https://doi.org/10.1002/ana.410420420.
Chugani DC, Muzik O, Behen M, Rothermel R, Janisse JJ, Lee J, et al. Developmental changes in brain serotonin synthesis capacity in autistic and nonautistic children. Ann Neurol. 1999;45(3):287–95. https://doi.org/10.1002/1531-8249(199903)45:3<287::aid-ana3>3.0.co;2-9.
Chugani DC, Chugani HT, Wiznitzer M, Parikh S, Evans PA, Hansen RL, et al. Efficacy of low-dose buspirone for restricted and repetitive behavior in young children with autism spectrum disorder: a randomized trial. J Pediatr. 2016;170:45-53 e1–4. https://doi.org/10.1016/j.jpeds.2015.11.033.
Greiss Hess L, Fitzpatrick SE, Nguyen DV, Chen Y, Gaul KN, Schneider A, et al. A randomized, double-blind, placebo-controlled trial of low-dose sertraline in young children with fragile X syndrome. J Dev Behav Pediatr. 2016;37(8):619–28. https://doi.org/10.1097/DBP.0000000000000334.
Winarni TI, Schneider A, Borodyanskara M, Hagerman RJ. Early intervention combined with targeted treatment promotes cognitive and behavioral improvements in young children with fragile X syndrome. Case Rep Genet. 2012;2012:280813. https://doi.org/10.1155/2012/280813.
Potter LA, Scholze DA, Biag HMB, Schneider A, Chen Y, Nguyen DV, et al. A randomized controlled trial of sertraline in young children with autism spectrum disorder. Front Psychiatry. 2019;10:810. https://doi.org/10.3389/fpsyt.2019.00810.
Rajaratnam A, Potter LA, Biag HMB, Schneider A, Petrasic IC, Hagerman RJ. Review of autism profiles and response to sertraline in fragile X syndrome-associated autism vs. non-syndromic autism; next steps for targeted treatment. Front Psychiatry. 2020;11:581429. https://doi.org/10.3389/fneur.2020.581429.
Roh MS, Seo MS, Kim Y, Kim SH, Jeon WJ, Ahn YM, et al. Haloperidol and clozapine differentially regulate signals upstream of glycogen synthase kinase 3 in the rat frontal cortex. Exp Mol Med. 2007;39(3):353–60. https://doi.org/10.1038/emm.2007.39.
Sutton LP, Honardoust D, Mouyal J, Rajakumar N, Rushlow WJ. Activation of the canonical Wnt pathway by the antipsychotics haloperidol and clozapine involves dishevelled-3. J Neurochem. 2007;102(1):153–69. https://doi.org/10.1111/j.1471-4159.2007.04527.x.
Alimohamad H, Sutton L, Mouyal J, Rajakumar N, Rushlow WJ. The effects of antipsychotics on beta-catenin, glycogen synthase kinase-3 and dishevelled in the ventral midbrain of rats. J Neurochem. 2005;95(2):513–25. https://doi.org/10.1111/j.1471-4159.2005.03388.x.
Mines MA, Jope RS. Brain region differences in regulation of Akt and GSK3 by chronic stimulant administration in mice. Cell Signal. 2012;24(7):1398–405. https://doi.org/10.1016/j.cellsig.2012.03.001.
Hagerman RJ, Murphy MA, Wittenberger MD. A controlled trial of stimulant medication in children with the fragile X syndrome. Am J Med Genet 1988;30(1-2):377–92. https://doi.org/10.1002/ajmg.1320300138.
Hagerman RJ, Berry-Kravis E, Kaufmann WE, Ono MY, Tartaglia N, Lachiewicz A, et al. Advances in the treatment of fragile X syndrome. Pediatrics. 2009;123(1):378–90. https://doi.org/10.1542/peds.2008-0317.
Tapia-Rojas C, Inestrosa NC. Loss of canonical Wnt signaling is involved in the pathogenesis of Alzheimer’s disease. Neural Regen Res. 2018;13(10):1705–10. https://doi.org/10.4103/1673-5374.238606.
Varela-Nallar L, Ramirez VT, Gonzalez-Billault C, Inestrosa NC. Frizzled receptors in neurons: from growth cones to the synapse. Cytoskeleton (Hoboken). 2012;69(7):528–34. https://doi.org/10.1002/cm.21022.
Klaus A, Birchmeier W. Wnt signalling and its impact on development and cancer. Nat Rev Cancer. 2008;8(5):387–98. https://doi.org/10.1038/nrc2389.
Vu TH, Hoffman AR. Imprinting of the Angelman syndrome gene, UBE3A, is restricted to brain. Nat Genet. 1997;17(1):12–3. https://doi.org/10.1038/ng0997-12.
Rougeulle C, Cardoso C, Fontés M, Colleaux L, Lalande M. An imprinted antisense RNA overlaps UBE3A and a second maternally expressed transcript. Nat Genet. 1998;19(1):15–6. https://doi.org/10.1038/ng0598-15.
Hochstrasser M. Ubiquitin-dependent protein degradation. Annu Rev Genet. 1996;30:405–39. https://doi.org/10.1146/annurev.genet.30.1.405.
de Bie P, Ciechanover A. Ubiquitination of E3 ligases: self-regulation of the ubiquitin system via proteolytic and non-proteolytic mechanisms. Cell Death Differ. 2011;18(9):1393–402. https://doi.org/10.1038/cdd.2011.16.
Yi JJ, Berrios J, Newbern JM, Snider WD, Philpot BD, Hahn KM, et al. An autism-linked mutation disables phosphorylation control of UBE3A. Cell. 2015;162(4):795–807. https://doi.org/10.1016/j.cell.2015.06.045.
Dindot SV, Antalffy BA, Bhattacharjee MB, Beaudet AL. The Angelman syndrome ubiquitin ligase localizes to the synapse and nucleus, and maternal deficiency results in abnormal dendritic spine morphology. Hum Mol Genet. 2008;17(1):111–8. https://doi.org/10.1093/hmg/ddm288.
Xu X, Li C, Gao X, Xia K, Guo H, Li Y, et al. Excessive UBE3A dosage impairs retinoic acid signaling and synaptic plasticity in autism spectrum disorders. Cell Res. 2018;28(1):48–68. https://doi.org/10.1038/cr.2017.132.
LaSalle JM, Reiter LT, Chamberlain SJ. Epigenetic regulation of UBE3A and roles in human neurodevelopmental disorders. Epigenomics. 2015;7(7):1213–28. https://doi.org/10.2217/epi.15.70.
Glessner JT, Wang K, Cai G, Korvatska O, Kim CE, Wood S, et al. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature. 2009;459(7246):569–73. https://doi.org/10.1038/nature07953.
Baron CA, Tepper CG, Liu SY, Davis RR, Wang NJ, Schanen NC, et al. Genomic and functional profiling of duplicated chromosome 15 cell lines reveal regulatory alterations in UBE3A-associated ubiquitin–proteasome pathway processes. Hum Mol Genet. 2006;15(6):853–69. https://doi.org/10.1093/hmg/ddl004%JHumanMolecularGenetics.
Chamberlain SJ, Lalande M. Angelman syndrome, a genomic imprinting disorder of the brain. J Neurosci. 2010;30(30):9958–63. https://doi.org/10.1523/jneurosci.1728-10.2010.
Peters SU, Beaudet AL, Madduri N, Bacino CA. Autism in Angelman syndrome: implications for autism research. Clin Genet. 2004;66(6):530–6. https://doi.org/10.1111/j.1399-0004.2004.00362.x.
McLennan Y, Polussa J, Tassone F, Hagerman R. Fragile X syndrome. Curr Genomics. 2011;12(3):216–24. https://doi.org/10.2174/138920211795677886.
Nowicki ST, Tassone F, Ono MY, Ferranti J, Croquette MF, Goodlin-Jones B, et al. The Prader-Willi phenotype of fragile X syndrome. J Dev Behav Pediatr. 2007;28(2):133–8. https://doi.org/10.1097/01.DBP.0000267563.18952.c9
Noroozi R, Omrani MD, Sayad A, Taheri M, Ghafouri-Fard S. Cytoplasmic FMRP interacting protein 1/2 (CYFIP1/2) expression analysis in autism. Metab Brain Dis. 2018;33(4):1353–8. https://doi.org/10.1007/s11011-018-0249-8.
Han K, Chen H, Gennarino VA, Richman R, Lu H-C, Zoghbi HY. Fragile X-like behaviors and abnormal cortical dendritic spines in cytoplasmic FMR1-interacting protein 2-mutant mice. Hum Mol Genet. 2014;24(7):1813–23. https://doi.org/10.1093/hmg/ddu595%JHumanMolecularGenetics.
Kirkpatrick SL, Goldberg LR, Yazdani N, Babbs RK, Wu J, Reed ER, et al. Cytoplasmic FMR1-interacting protein 2 is a major genetic factor underlying binge eating. Biol Psychiatry 2017;81(9):757–69. https://doi.org/10.1016/j.biopsych.2016.10.021.
Davenport EC, Szulc BR, Drew J, Taylor J, Morgan T, Higgs NF, et al. Autism and schizophrenia-associated CYFIP1 regulates the balance of synaptic excitation and inhibition. Cell Rep. 2019;26(8):2037–51.e6. https://doi.org/10.1016/j.celrep.2019.01.092.
Miller MW, Wolf EJ, Logue MW, Baldwin CT. The retinoid-related orphan receptor alpha (RORA) gene and fear-related psychopathology. J Affect Disord. 2013;151(2):702–8. https://doi.org/10.1016/j.jad.2013.07.022.
Devanna P, Vernes SC. A direct molecular link between the autism candidate gene RORa and the schizophrenia candidate MIR137. Sci Rep. 2014;4:3994. https://doi.org/10.1038/srep03994.
Nguyen A, Rauch TA, Pfeifer GP, Hu VW. Global methylation profiling of lymphoblastoid cell lines reveals epigenetic contributions to autism spectrum disorders and a novel autism candidate gene, RORA, whose protein product is reduced in autistic brain. FASEB J. 2010;24(8):3036–51. https://doi.org/10.1096/fj.10-154484.
Sarachana T, Hu VW. Differential recruitment of coregulators to the RORA promoter adds another layer of complexity to gene (dys) regulation by sex hormones in autism. Mol Autism. 2013;4(1):39. https://doi.org/10.1186/2040-2392-4-39.
Sarachana T, Hu VW. Genome-wide identification of transcriptional targets of RORA reveals direct regulation of multiple genes associated with autism spectrum disorder. Mol Autism. 2013;4(1):14. https://doi.org/10.1186/2040-2392-4-14.
Sarachana T, Xu M, Wu RC, Hu VW. Sex hormones in autism: androgens and estrogens differentially and reciprocally regulate RORA, a novel candidate gene for autism. PLoS One. 2011;6(2):e17116. https://doi.org/10.1371/journal.pone.0017116.
Cook DN, Kang HS, Jetten AM. Retinoic acid-related orphan receptors (RORs): regulatory functions in immunity, development, circadian rhythm, and metabolism. Nucl Recept Res. 2015;2. https://doi.org/10.11131/2015/101185.
Clagett-Dame M, DeLuca HF. The role of vitamin A in mammalian reproduction and embryonic development. Annu Rev Nutr. 2002;22:347–81. https://doi.org/10.1146/annurev.nutr.22.010402.102745E.
Wilson JG, Roth CB, Warkany J. An analysis of the syndrome of malformations induced by maternal vitamin A deficiency. Effects of restoration of vitamin A at various times during gestation. Am J Anat. 1953;92(2):189–217. https://doi.org/10.1002/aja.1000920202.
Liu X, Liu J, Xiong X, Yang T, Hou N, Liang X, et al. Correlation between nutrition and symptoms: nutritional survey of children with autism spectrum disorder in Chongqing, China. Nutrients. 2016;8(5). https://doi.org/10.3390/nu8050294.
Guo M, Zhu J, Yang T, Lai X, Lei Y, Chen J, et al. Vitamin A and vitamin D deficiencies exacerbate symptoms in children with autism spectrum disorders. Nutr Neurosci. 2019;22(9):637–47. https://doi.org/10.1080/1028415X.2017.1423268.
Kumar S, Sandell LL, Trainor PA, Koentgen F, Duester G. Alcohol and aldehyde dehydrogenases: retinoid metabolic effects in mouse knockout models. Biochim Biophys Acta. 2012;1821(1):198–205. https://doi.org/10.1016/j.bbalip.2011.04.004.
Chen L, Lau AG, Sarti F. Synaptic retinoic acid signaling and homeostatic synaptic plasticity. Neuropharmacology. 2014;78:3–12. https://doi.org/10.1016/j.neuropharm.2012.12.004.
Cunningham TJ, Duester G. Mechanisms of retinoic acid signalling and its roles in organ and limb development. Nat Rev Mol Cell Biol. 2015;16(2):110–23. https://doi.org/10.1038/nrm3932.
Zieger E, Schubert M. New Insights into the roles of retinoic acid signaling in nervous system development and the establishment of neurotransmitter systems. Int Rev Cell Mol Biol. 2017;330:1–84. https://doi.org/10.1016/bs.ircmb.2016.09.001.
McCaffery P, Drager UC. High levels of a retinoic acid-generating dehydrogenase in the meso-telencephalic dopamine system. Proc Natl Acad Sci U S A. 1994;91(16):7772–6. https://doi.org/10.1073/pnas.91.16.7772.
Crandall JE, Goodman T, McCarthy DM, Duester G, Bhide PG, Dräger UC, et al. Retinoic acid influences neuronal migration from the ganglionic eminence to the cerebral cortex. J Neurochem. 2011;119(4):723–35. https://doi.org/10.1111/j.1471-4159.2011.07471.x.
Casanova MF, Buxhoeveden D, Gomez J. Disruption in the inhibitory architecture of the cell minicolumn: implications for autism. Neuroscientist. 2003;9(6):496–507. https://doi.org/10.1177/1073858403253552.
Hashemi E, Ariza J, Rogers H, Noctor SC, Martinez-Cerdeno V. The number of parvalbumin-expressing interneurons is decreased in the prefrontal cortex in autism. Cereb Cortex. 2018;28(2):690. https://doi.org/10.1093/cercor/bhx063.
Cawley S, Bekiranov S, Ng HH, Kapranov P, Sekinger EA, Kampa D, et al. Unbiased mapping of transcription factor binding sites along human chromosomes 21 and 22 points to widespread regulation of noncoding RNAs. Cell. 2004;116(4):499–509. https://doi.org/10.1016/s0092-8674(04)00127-8.
Liu J, Liu X, Xiong XQ, Yang T, Cui T, Hou NL, et al. Effect of vitamin A supplementation on gut microbiota in children with autism spectrum disorders - a pilot study. BMC Microbiol. 2017;17(1):204. https://doi.org/10.1186/s12866-017-1096-1.
Sarti F, Zhang Z, Schroeder J, Chen L. Rapid suppression of inhibitory synaptic transmission by retinoic acid. J Neurosci. 2013;33(28):11440–50. https://doi.org/10.1523/JNEUROSCI.1710-13.2013.
Soden ME, Chen L. Fragile X protein FMRP is required for homeostatic plasticity and regulation of synaptic strength by retinoic acid. J Neurosci. 2010;30(50):16910–21. https://doi.org/10.1523/JNEUROSCI.3660-10.2010.
Zhang Z, Marro SG, Zhang Y, Arendt KL, Patzke C, Zhou B, et al. The fragile X mutation impairs homeostatic plasticity in human neurons by blocking synaptic retinoic acid signaling. Sci Transl Med. 2018;10(452). https://doi.org/10.1126/scitranslmed.aar4338.
Gantois I, Popic J, Khoutorsky A, Sonenberg N. Metformin for treatment of fragile X syndrome and other neurological disorders. Annu Rev Med. 2019;70:167–81. https://doi.org/10.1146/annurev-med-081117-041238.
Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18(16):1926–45. https://doi.org/10.1101/gad.1212704.
Lipton JO, Sahin M. The neurology of mTOR. Neuron. 2014;84(2):275–91. https://doi.org/10.1016/j.neuron.2014.09.034.
Levitt P, Campbell DB. The genetic and neurobiologic compass points toward common signaling dysfunctions in autism spectrum disorders. J Clin Invest. 2009;119(4):747–54. https://doi.org/10.1172/JCI37934.
Vithayathil J, Pucilowska J, Landreth GE. ERK/MAPK signaling and autism spectrum disorders. Prog Brain Res. 2018;241:63–112. https://doi.org/10.1016/bs.pbr.2018.09.008.
Winden KD, Ebrahimi-Fakhari D, Sahin M. Abnormal mTOR activation in autism. Annu Rev Neurosci. 2018;41:1–23. https://doi.org/10.1146/annurev-neuro-080317-061747.
Crino P, Nathanson K, Henske E: The tuberous sclerosis complex. Vol 355. (2006) www.nejm.org. Accessed August 16 2020.
Zhou J, Parada LF. PTEN signaling in autism spectrum disorders. Curr Opin Neurobiol. 2012;22(5):873–9. https://doi.org/10.1016/j.conb.2012.05.004.
Samanta D. An updated review of tuberous sclerosis complex-associated autism spectrum disorder. Pediatr Neurol. 2020;109:4–11. https://doi.org/10.1016/j.pediatrneurol.2020.03.008.
Ehninger D, Han S, Shilyansky C, Zhou Y, Li W, Kwiatkowski DJ, et al. Reversal of learning deficits in a Tsc2+/- mouse model of tuberous sclerosis. Nat Med. 2008;14(8):843–8. https://doi.org/10.1038/nm1788.
Tsai PT, Hull C, Chu Y, Greene-Colozzi E, Sadowski AR, Leech JM, et al. Autistic-like behaviour and cerebellar dysfunction in Purkinje cell Tsc1 mutant mice. Nature. 2012;488(7413):647–51. https://doi.org/10.1038/nature11310.
Sato A, Kasai S, Kobayashi T, Takamatsu Y, Hino O, Ikeda K, et al. Rapamycin reverses impaired social interaction in mouse models of tuberous sclerosis complex. Nat Commun. 2012;3:1292. https://doi.org/10.1038/ncomms2295.
Schneider M, de Vries PJ, Schonig K, Rossner V, Waltereit R. mTOR inhibitor reverses autistic-like social deficit behaviours in adult rats with both Tsc2 haploinsufficiency and developmental status epilepticus. Eur Arch Psychiatry Clin Neurosci. 2017;267(5):455–63. https://doi.org/10.1007/s00406-016-0703-8.
MacKeigan JP, Krueger DA. Differentiating the mTOR inhibitors everolimus and sirolimus in the treatment of tuberous sclerosis complex. Neuro-Oncology. 2015;17(12):1550–9. https://doi.org/10.1093/neuonc/nov152.
Krueger DA, Sadhwani A, Byars AW, de Vries PJ, Franz DN, Whittemore VH, et al. Everolimus for treatment of tuberous sclerosis complex-associated neuropsychiatric disorders. Ann Clin Transl Neurol. 2017;4(12):877–87. https://doi.org/10.1002/acn3.494.
Overwater IE, Rietman AB, Mous SE, Bindels-de Heus K, Rizopoulos D, Ten Hoopen LW, et al. A randomized controlled trial with everolimus for IQ and autism in tuberous sclerosis complex. Neurology. 2019;93(2):e200–e9. https://doi.org/10.1212/WNL.0000000000007749.
Mizuguchi M, Ikeda H, Kagitani-Shimono K, Yoshinaga H, Suzuki Y, Aoki M, et al. Everolimus for epilepsy and autism spectrum disorder in tuberous sclerosis complex: EXIST-3 substudy in Japan. Brain and Development. 2019;41(1):1–10. https://doi.org/10.1016/j.braindev.2018.07.003.
Yang Q, Modi P, Newcomb T, Queva C, Gandhi V. Idelalisib: first-in-class PI3K delta inhibitor for the treatment of chronic lymphocytic leukemia, small lymphocytic leukemia, and follicular lymphoma. Clin Cancer Res. 2015;21(7):1537–42. https://doi.org/10.1158/1078-0432.CCR-14-2034.
Markham A. Copanlisib: first global approval. Drugs. 2017;77(18):2057–62. https://doi.org/10.1007/s40265-017-0838-6.
Markham A. Alpelisib: first global approval. Drugs. 2019;79(11):1249–53. https://doi.org/10.1007/s40265-019-01161-6.
Wang Q, Weisberg E, Zhao JJ. The gene dosage of class Ia PI3K dictates the development of PTEN hamartoma tumor syndrome. Cell Cycle. 2013;12(23):3589–93. https://doi.org/10.4161/cc.26812.
Enriquez-Barreto L, Morales M. The PI3K signaling pathway as a pharmacological target in autism related disorders and schizophrenia. Mol Cell Ther. 2016;4:2. https://doi.org/10.1186/s40591-016-0047-9.
Lu Z, Xu S. ERK1/2 MAP kinases in cell survival and apoptosis. IUBMB Life. 2006;58(11):621–31. https://doi.org/10.1080/15216540600957438.
Rauen KA. The RASopathies. Annu Rev Genomics Hum Genet. 2013;14:355–69. https://doi.org/10.1146/annurev-genom-091212-153523.
Garg S, Brooks A, Burns A, Burkitt-Wright E, Kerr B, Huson S, et al. Autism spectrum disorder and other neurobehavioural comorbidities in rare disorders of the Ras/MAPK pathway. Dev Med Child Neurol. 2017;59(5):544–9. https://doi.org/10.1111/dmcn.13394.
Adviento B, Corbin IL, Widjaja F, Desachy G, Enrique N, Rosser T, et al. Autism traits in the RASopathies. J Med Genet. 2014;51(1):10–20. https://doi.org/10.1136/jmedgenet-2013-101951.
Roberts PJ, Der CJ. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene. 2007;26(22):3291–310. https://doi.org/10.1038/sj.onc.1210422.
Germann UA, Furey BF, Markland W, Hoover RR, Aronov AM, Roix JJ, et al. Targeting the MAPK signaling pathway in cancer: promising preclinical activity with the novel selective ERK1/2 inhibitor BVD-523 (ulixertinib). Mol Cancer Ther. 2017;16(11):2351–63. https://doi.org/10.1158/1535-7163.MCT-17-0456.
Pucilowska J, Vithayathil J, Pagani M, Kelly C, Karlo JC, Robol C, et al. Pharmacological inhibition of ERK signaling rescues pathophysiology and behavioral phenotype associated with 16p11.2 chromosomal deletion in mice. J Neurosci. 2018;38(30):6640–52. https://doi.org/10.1523/JNEUROSCI.0515-17.2018.
Yufune S, Satoh Y, Takamatsu I, Ohta H, Kobayashi Y, Takaenoki Y, et al. Transient blockade of ERK phosphorylation in the critical period causes autistic phenotypes as an adult in mice. Sci Rep. 2015;5:10252. https://doi.org/10.1038/srep10252.
Richter JD, Bassell GJ, Klann E. Dysregulation and restoration of translational homeostasis in fragile X syndrome. Nat Rev Neurosci. 2015;16(10):595–605. https://doi.org/10.1038/nrn4001.
Sharma A, Hoeffer CA, Takayasu Y, Miyawaki T, McBride SM, Klann E, et al. Dysregulation of mTOR signaling in fragile X syndrome. J Neurosci. 2010;30(2):694–702. https://doi.org/10.1523/JNEUROSCI.3696-09.2010.
Hoeffer CA, Sanchez E, Hagerman RJ, Mu Y, Nguyen DV, Wong H, et al. Altered mTOR signaling and enhanced CYFIP2 expression levels in subjects with fragile X syndrome. Genes Brain Behav. 2012;11(3):332–41. https://doi.org/10.1111/j.1601-183X.2012.00768.x.
Wang X, Snape M, Klann E, Stone JG, Singh A, Petersen RB, et al. Activation of the extracellular signal-regulated kinase pathway contributes to the behavioral deficit of fragile X-syndrome. J Neurochem. 2012;121(4):672–9. https://doi.org/10.1111/j.1471-4159.2012.07722.x.
Bhattacharya A, Kaphzan H, Alvarez-Dieppa AC, Murphy JP, Pierre P, Klann E. Genetic removal of p70 S6 kinase 1 corrects molecular, synaptic, and behavioral phenotypes in fragile X syndrome mice. Neuron. 2012;76(2):325–37. https://doi.org/10.1016/j.neuron.2012.07.022.
Osterweil EK, Chuang SC, Chubykin AA, Sidorov M, Bianchi R, Wong RK, et al. Lovastatin corrects excess protein synthesis and prevents epileptogenesis in a mouse model of fragile X syndrome. Neuron. 2013;77(2):243–50. https://doi.org/10.1016/j.neuron.2012.01.034.
Xu XQ, McGuire TF, Blaskovich MA, Sebti SM, Romero G. Lovastatin inhibits the stimulation of mitogen-activated protein kinase by insulin in HIRcB fibroblasts. Arch Biochem Biophys. 1996;326(2):233–7. https://doi.org/10.1006/abbi.1996.0070.
Caku A, Pellerin D, Bouvier P, Riou E, Corbin F. Effect of lovastatin on behavior in children and adults with fragile X syndrome: an open-label study. Am J Med Genet A. 2014;164A(11):2834–42. https://doi.org/10.1002/ajmg.a.36750.
Pellerin D, Çaku A, Fradet M, Bouvier P, Dubé J, Corbin F. Lovastatin corrects ERK pathway hyperactivation in fragile X syndrome: potential of platelet’s signaling cascades as new outcome measures in clinical trials. Biomarkers. 2016;21(6):497–508. https://doi.org/10.3109/1354750X.2016.1160289.
Thurman AJ, Potter LA, Kim K, Tassone F, Banasik A, Potter SN, et al. Controlled trial of lovastatin combined with an open-label treatment of a parent-implemented language intervention in youth with fragile X syndrome. J Neurodev Disord. 2020;12(1):12. https://doi.org/10.1186/s11689-020-09315-4.
Rena G, Hardie DG, Pearson ER. The mechanisms of action of metformin. Diabetologia. 2017;60(9):1577–85. https://doi.org/10.1007/s00125-017-4342-z.
Protic D, Salcedo-Arellano MJ, Dy JB, Potter LA, Hagerman RJ. New targeted treatments in fragile X syndrome. Curr Pediatr Rev. 2019. https://doi.org/10.2174/1573396315666190625110748.
Pernicova I, Korbonits M. Metformin--mode of action and clinical implications for diabetes and cancer. Nat Rev Endocrinol. 2014;10(3):143–56. https://doi.org/10.1038/nrendo.2013.256.
Howell JJ, Hellberg K, Turner M, Talbott G, Kolar MJ, Ross DS, et al. Metformin inhibits hepatic mTORC1 signaling via dose-dependent mechanisms involving AMPK and the TSC complex. Cell Metab. 2017;25(2):463–71. https://doi.org/10.1016/j.cmet.2016.12.009.
Gantois I, Popic J, Khoutorsky A, Sonenberg N. Metformin for treatment of fragile X syndrome and other neurological disorders. Annu Rev. Med 2018. https://doi.org/10.1146/annurev-med-081117-041238.
Gantois I, Khoutorsky A, Popic J, Aguilar-Valles A, Freemantle E, Cao R, et al. Metformin ameliorates core deficits in a mouse model of fragile X syndrome. Nat Med. 2017;23(6):674–7. https://doi.org/10.1038/nm.4335.
Metformin information. https://www.fda.gov/drugs/postmarket-drug-safety-information-patients-and-providers/metformin-information (2016). Accessed August 16, 2020.
Dy ABC, Tassone F, Eldeeb M, Salcedo-Arellano MJ, Tartaglia N, Hagerman R. Metformin as targeted treatment in fragile X syndrome. Clin Genet 2018;93(2):216–22. https://doi.org/10.1111/cge.13039.
Biag HMB, Potter LA, Wilkins V, Afzal S, Rosvall A, Salcedo-Arellano MJ, et al. Metformin treatment in young children with fragile X syndrome. Mol Genet Genomic Med. 2019:e956. https://doi.org/10.1002/mgg3.956.
A trial of metformin in individuals with fragile X syndrome (Met) - full text view. https://clinicaltrials.gov/ct2/show/NCT03862950 Accessed 16 August 2020.
A trial of metformin in individuals with fragile X syndrome - full text view. https://clinicaltrials.gov/ct2/show/NCT03479476 Accessed 16 August 2020.
Zhang L, Alger BE. Enhanced endocannabinoid signaling elevates neuronal excitability in fragile X syndrome. J Neurosci. 2010;30(16):5724–9. https://doi.org/10.1523/JNEUROSCI.0795-10.2010.
Jung KM, Sepers M, Henstridge CM, Lassalle O, Neuhofer D, Martin H, et al. Uncoupling of the endocannabinoid signalling complex in a mouse model of fragile X syndrome. Nat Commun. 2012;3:1080. https://doi.org/10.1038/ncomms2045.
Tartaglia N, Bonn-Miller M, Hagerman R. Treatment of fragile X syndrome with cannabidiol: a case series study and brief review of the literaturE. Cannabis Cannabinoid Res. 2019;4(1):3–9. https://doi.org/10.1089/can.2018.0053.
Cordeiro L, Ballinger E, Hagerman R, Hessl D. Clinical assessment of DSM-IV anxiety disorders in fragile X syndrome: prevalence and characterization. J Neurodev Disord. 2011;3(1):57–67. https://doi.org/10.1007/s11689-010-9067-y.
Hong MP, Eckert EM, Pedapati EV, Shaffer RC, Dominick KC, Wink LK, et al. Differentiating social preference and social anxiety phenotypes in fragile X syndrome using an eye gaze analysis: a pilot study. J Neurodev Disord. 2019;11(1):1. https://doi.org/10.1186/s11689-019-9262-4.
Tassanakijpanich N, Cabal-Herrera AM, Salcedo-Arellano MJ, Hagerman RJ. Fragile X syndrome and targeted treatments. J Biomed Transl Res. 2020;6(1):23-33. https://doi.org/10.14710/jbtr.v6i1.7321.
Hurd YL. Leading the next CBD wave-safety and efficacy. JAMA Psychiatry. 2020;77(4):341–2. https://doi.org/10.1001/jamapsychiatry.2019.4157.
Bakas T, van Nieuwenhuijzen PS, Devenish SO, McGregor IS, Arnold JC, Chebib M. The direct actions of cannabidiol and 2-arachidonoyl glycerol at GABAA receptors. Pharmacol Res. 2017;119:358–70. https://doi.org/10.1016/j.phrs.2017.02.022.
Campos AC, Guimaraes FS. Involvement of 5HT1A receptors in the anxiolytic-like effects of cannabidiol injected into the dorsolateral periaqueductal gray of rats. Psychopharmacology. 2008;199(2):223–30. https://doi.org/10.1007/s00213-008-1168-x.
Norris C, Loureiro M, Kramar C, Zunder J, Renard J, Rushlow W, et al. Cannabidiol modulates fear memory formation through interactions with serotonergic transmission in the mesolimbic system. Neuropsychopharmacology. 2016;41(12):2839–50. https://doi.org/10.1038/npp.2016.93.
Heussler H, Cohen J, Silove N, Tich N, Bonn-Miller MO, Du W, et al. A phase 1/2, open-label assessment of the safety, tolerability, and efficacy of transdermal cannabidiol (ZYN002) for the treatment of pediatric fragile X syndrome. J Neurodev Disord. 2019;11(1):16. https://doi.org/10.1186/s11689-019-9277-x.
Heussler H, Cohen J, Silove N, Tich N, Sebree T, Siegel S. Transdermal cannabidiol (CBD) Gel for the treatment of fragile X syndrome (FXS). 57th Annual Meeting of the American College of Neuropsychopharmacology. (ACNP). Hollywood, FL 2018.
Zynerba Pharmaceuticals announces top line results from pivotal CONNECT-FX Trial of Zygel™ (CBD Gel) in fragile X syndrome. 2020. https://zynerba.com/zynerba-pharmaceuticals-announces-top-line-results-from-pivotal-connect-fx-trial-of-zygel-cbd-gel-in-fragile-x-syndrome/. Accessed 30 August 2020.
Zynerba Pharmaceuticals announces positive top line results from exploratory open label phase 2 BRIGHT Trial of Zygel in autism spectrum disorder. 2020. https://zynerba.com/zynerba-pharmaceuticals-announces-positive-top-line-results-from-exploratory-open-label-phase-2-bright-trial-of-zygel-in-autism-spectrum-disorder/. Accessed 30 August 2020.
Hu D, Yu ZL, Zhang Y, Han Y, Zhang W, Lu L, et al. Bumetanide treatment during early development rescues maternal separation-induced susceptibility to stress. Sci Rep. 2017;7(1):11878. https://doi.org/10.1038/s41598-017-12183-z.
Dzhala VI, Talos DM, Sdrulla DA, Brumback AC, Mathews GC, Benke TA, et al. NKCC1 transporter facilitates seizures in the developing brain. Nat Med. 2005;11(11):1205–13. https://doi.org/10.1038/nm1301.
Leonzino M, Busnelli M, Antonucci F, Verderio C, Mazzanti M, Chini B. The timing of the excitatory-to-inhibitory GABA switch is regulated by the oxytocin receptor via KCC2. Cell Rep. 2016;15(1):96–103. https://doi.org/10.1016/j.celrep.2016.03.013.
Represa A, Ben-Ari Y. Trophic actions of GABA on neuronal development. Trends Neurosci. 2005;28(6):278–83. https://doi.org/10.1016/j.tins.2005.03.010.
Zhang L, Huang CC, Dai Y, Luo Q, Ji Y, Wang K, et al. Symptom improvement in children with autism spectrum disorder following bumetanide administration is associated with decreased GABA/glutamate ratios. Transl Psychiatry. 2020;10(1):9. https://doi.org/10.1038/s41398-020-0692-2.
Cellot G, Cherubini E. GABAergic signaling as therapeutic target for autism spectrum disorders. Front Pediatr. 2014;2:70. https://doi.org/10.3389/fped.2014.00070.
Oblak AL, Gibbs TT, Blatt GJ. Decreased GABA(B) receptors in the cingulate cortex and fusiform gyrus in autism. J Neurochem. 2010;114(5):1414–23. https://doi.org/10.1111/j.1471-4159.2010.06858.x.
Oblak A, Gibbs TT, Blatt GJ. Decreased GABAA receptors and benzodiazepine binding sites in the anterior cingulate cortex in autism. Autism Res. 2009;2(4):205–19. https://doi.org/10.1002/aur.88.
Mori T, Mori K, Fujii E, Toda Y, Miyazaki M, Harada M, et al. Evaluation of the GABAergic nervous system in autistic brain: (123)I-iomazenil SPECT study. Brain and Development. 2012;34(8):648–54. https://doi.org/10.1016/j.braindev.2011.10.007.
Whitney ER, Kemper TL, Rosene DL, Bauman ML, Blatt GJ. Density of cerebellar basket and stellate cells in autism: evidence for a late developmental loss of Purkinje cells. J Neurosci Res. 2009;87(10):2245–54. https://doi.org/10.1002/jnr.22056.
Harada M, Taki MM, Nose A, Kubo H, Mori K, Nishitani H, et al. Non-invasive evaluation of the GABAergic/glutamatergic system in autistic patients observed by MEGA-editing proton MR spectroscopy using a clinical 3 Tesla instrument. J Autism Dev Disord. 2011;41(4):447-54. https://doi.org/10.1007/s10803-010-1065-0.
Rojas DC, Singel D, Steinmetz S, Hepburn S, Brown MS. Decreased left perisylvian GABA concentration in children with autism and unaffected siblings. Neuroimage. 2014;86:28–34. https://doi.org/10.1016/j.neuroimage.2013.01.045.
Van der Aa N, Kooy RF. GABAergic abnormalities in the fragile X syndrome. Eur J Paediatr. Neurol 2020;24:100–4. https://doi.org/10.1016/j.ejpn.2019.12.022.
Purcell AE, Jeon OH, Zimmerman AW, Blue ME, Pevsner J. Postmortem brain abnormalities of the glutamate neurotransmitter system in autism. Neurology. 2001;57(9):1618–28. https://doi.org/10.1212/wnl.57.9.1618.
Morin-Parent F, Champigny C, Lacroix A, Corbin F, Lepage JF. Hyperexcitability and impaired intracortical inhibition in patients with fragile-X syndrome. Transl Psychiatry. 2019;9(1):312. https://doi.org/10.1038/s41398-019-0650-z.
Telias M. Molecular mechanisms of synaptic dysregulation in fragile X syndrome and autism spectrum disorders. Front Mol Neurosci 2019;12:51. https://doi.org/10.3389/fnmol.2019.00051.
Kharod SC, Kang SK, Kadam SD. Off-label use of bumetanide for brain disorders: an overview. Front Neurosci. 2019;13:310. https://doi.org/10.3389/fnins.2019.00310.
Tyzio R, Nardou R, Ferrari DC, Tsintsadze T, Shahrokhi A, Eftekhari S, et al. Oxytocin-mediated GABA inhibition during delivery attenuates autism pathogenesis in rodent offspring. Science. 2014;343(6171):675–9. https://doi.org/10.1126/science.1247190.
He Q, Nomura T, Xu J, Contractor A. The developmental switch in GABA polarity is delayed in fragile X mice. J Neurosci. 2014;34(2):446–50. https://doi.org/10.1523/JNEUROSCI.4447-13.2014.
Schopler E, Reichler RJ, DeVellis RF, Daly K. Toward objective classification of childhood autism: Childhood Autism Rating Scale (CARS). J Autism Dev Disord. 1980;10(1):91–103. https://doi.org/10.1007/BF02408436.
Lemonnier E, Villeneuve N, Sonie S, Serret S, Rosier A, Roue M, et al. Effects of bumetanide on neurobehavioral function in children and adolescents with autism spectrum disorders. Transl Psychiatry 2017;7(3):e1056. https://doi.org/10.1038/tp.2017.10.
Lemonnier E, Degrez C, Phelep M, Tyzio R, Josse F, Grandgeorge M, et al. A randomised controlled trial of bumetanide in the treatment of autism in children. Transl Psychiatry. 2012;2:e202. https://doi.org/10.1038/tp.2012.124.
Lemonnier E, Ben-Ari Y. The diuretic bumetanide decreases autistic behaviour in five infants treated during 3 months with no side effects. Acta Paediatr. 2010;99(12):1885–8. https://doi.org/10.1111/j.1651-2227.2010.01933.x.
Du L, Shan L, Wang B, Li H, Xu Z, Staal WG, et al. A pilot study on the combination of applied behavior analysis and bumetanide treatment for children with autism. J Child Adolesc Psychopharmacol. 2015;25(7):585–8. https://doi.org/10.1089/cap.2015.0045.
Braat S, D’Hulst C, Heulens I, De Rubeis S, Mientjes E, Nelson DL, et al. The GABAA receptor is an FMRP target with therapeutic potential in fragile X syndrome. Cell Cycle. 2015;14(18):2985–95. https://doi.org/10.4161/15384101.2014.989114.
Braat S, Kooy RF. Insights into GABAAergic system deficits in fragile X syndrome lead to clinical trials. Neuropharmacology. 2015;88:48–54. https://doi.org/10.1016/j.neuropharm.2014.06.028.
Ligsay A, Van Dijck A, Nguyen DV, Lozano R, Chen Y, Bickel ES, et al. A randomized double-blind, placebo-controlled trial of ganaxolone in children and adolescents with fragile X syndrome. J Neurodev Disord. 2017;9(1):26–. https://doi.org/10.1186/s11689-017-9207-8.
Berry-Kravis E, Hagerman R, Visootsak J, Budimirovic D, Kaufmann WE, Cherubini M, et al. Arbaclofen in fragile X syndrome: results of phase 3 trials. J Neurodev Disord. 2017;9:3. https://doi.org/10.1186/s11689-016-9181-6.
Erickson CA, Veenstra-Vanderweele JM, Melmed RD, McCracken JT, Ginsberg LD, Sikich L, et al. STX209 (arbaclofen) for autism spectrum disorders: an 8-week open-label study. J Autism Dev Disord. 2014;44(4):958–64. https://doi.org/10.1007/s10803-013-1963-z.
Erickson CA, Wink LK, Early MC, Stiegelmeyer E, Mathieu-Frasier L, Patrick V, et al. Brief report: pilot single-blind placebo lead-in study of acamprosate in youth with autistic disorder. J Autism Dev Disord. 2014;44(4):981–7. https://doi.org/10.1007/s10803-013-1943-3.
Erickson CA, Wink LK, Ray B, Early MC, Stiegelmeyer E, Mathieu-Frasier L, et al. Impact of acamprosate on behavior and brain-derived neurotrophic factor: an open-label study in youth with fragile X syndrome. Psychopharmacology. 2013;228(1):75–84. https://doi.org/10.1007/s00213-013-3022-z.
Erickson CA, Early M, Stigler KA, Wink LK, Mullett JE, McDougle CJ. An open-label naturalistic pilot study of acamprosate in youth with autistic disorder. J Child Adolesc Psychopharmacol 2011;21(6):565–9. https://doi.org/10.1089/cap.2011.0034.
Majewska MD, Harrison NL, Schwartz RD, Barker JL, Paul SM. Steroid hormone metabolites are barbiturate-like modulators of the GABA receptor. Science. 1986;232(4753):1004–7. https://doi.org/10.1126/science.2422758.
Fung LK, Libove RA, Phillips J, Haddad F, Hardan AY. Brief report: an open-label study of the neurosteroid pregnenolone in adults with autism spectrum disorder. J Autism Dev Disord 2014;44(11):2971–7. https://doi.org/10.1007/s10803-014-2144-4.
Ayatollahi A, Bagheri S, Ashraf-Ganjouei A, Moradi K, Mohammadi MR, Akhondzadeh S. Does pregnenolone adjunct to risperidone ameliorate irritable behavior in adolescents with autism spectrum disorder: a randomized, double-blind, placebo-controlled clinical trial? Clin Neuropharmacol. 2020. https://doi.org/10.1097/WNF.0000000000000405.
Hagerman R, Jacquemont S, Berry-Kravis E, Des Portes V, Stanfield A, Koumaras B, et al. Mavoglurant in fragile X syndrome: results of two open-label, extension trials in adults and adolescents. Sci Rep. 2018;8(1):16970. https://doi.org/10.1038/s41598-018-34978-4.
Youssef EA, Berry-Kravis E, Czech C, Hagerman RJ, Hessl D, Wong CY, et al. Effect of the mGluR5-NAM Basimglurant on behavior in adolescents and adults with fragile X syndrome in a Randomized, double-blind, placebo-controlled trial: FragXis phase 2 results. Neuropsychopharmacology. 2018;43(3):503–12. https://doi.org/10.1038/npp.2017.177.
Berry-Kravis E, Des Portes V, Hagerman R, Jacquemont S, Charles P, Visootsak J, et al. Mavoglurant in fragile X syndrome: results of two randomized, double-blind, placebo-controlled trials. Sci Transl Med. 2016;8(321):321ra5. https://doi.org/10.1126/scitranslmed.aab4109.
Hardan AY, Hendren RL, Aman MG, Robb A, Melmed RD, Andersen KA, et al. Efficacy and safety of memantine in children with autism spectrum disorder: results from three phase 2 multicenter studies. Autism. 2019;23(8):2096-111. https://doi.org/10.1177/1362361318824103.
Orser BA, Pennefather PS, MacDonald JF. Multiple mechanisms of ketamine blockade of N-methyl-D-aspartate receptors. Anesthesiology. 1997;86(4):903–17. https://doi.org/10.1097/00000542-199704000-00021.
Abdallah CG, Averill LA, Gueorguieva R, Goktas S, Purohit P, Ranganathan M, et al. Modulation of the antidepressant effects of ketamine by the mTORC1 inhibitor rapamycin. Neuropsychopharmacology. 2020;45(6):990-7. https://doi.org/10.1038/s41386-020-0644-9.
Welberg L. Psychiatric disorders: ketamine modifies mood through mTOR. Nat Rev Neurosci. 2010;11(10):666. https://doi.org/10.1038/nrn2916.
Réus G, Quevedo J, Rodrigues A. mTOR signaling in the neuropathophysiology of depression: current evidence. J Recept Ligand Channel Res. 2015;8:65–74. https://doi.org/10.2147/JRLCR.S7035170351.
Wink LK, Reisinger DL, Horn P, Shaffer RC, O'Brien K, Schmitt L, et al. Brief report: intranasal ketamine in adolescents and young adults with autism spectrum disorder-initial results of a randomized, controlled, crossover, pilot study, J Autism Dev Disord. 2020. https://doi.org/10.1007/s10803-020-04542-z.
Husarova VM, Lakatosova S, Pivovarciova A, Babinska K, Bakos J, Durdiakova J, et al. Plasma oxytocin in children with autism and its correlations with behavioral parameters in children and parents. Psychiatry Investig. 2016;13(2):174–83. https://doi.org/10.4306/pi.2016.13.2.174.
Quattrocki E, Friston K. Autism, oxytocin and interoception. Neurosci Biobehav Rev. 2014;47:410–30. https://doi.org/10.1016/j.neubiorev.2014.09.012.
Parker KJ, Oztan O, Libove RA, Sumiyoshi RD, Jackson LP, Karhson DS, et al. Intranasal oxytocin treatment for social deficits and biomarkers of response in children with autism. Proc Natl Acad Sci U S A. 2017;114(30):8119–24. https://doi.org/10.1073/pnas.1705521114.
Anagnostou E, Soorya L, Chaplin W, Bartz J, Halpern D, Wasserman S, et al. Intranasal oxytocin versus placebo in the treatment of adults with autism spectrum disorders: a randomized controlled trial. Mol Autism. 2012;3(1):16. https://doi.org/10.1186/2040-2392-3-16.
Gordon I, Jack A, Pretzsch CM, Vander Wyk B, Leckman JF, Feldman R, et al. Intranasal oxytocin enhances connectivity in the neural circuitry supporting social motivation and social perception in children with autism. Sci Rep 2016;6:35054. https://doi.org/10.1038/srep35054.
Yamasue H, Aran A, Berry-Kravis E. Emerging pharmacological therapies in fragile X syndrome and autism. Curr Opin Neurol. 2019;32(4):635–40. https://doi.org/10.1097/WCO.0000000000000703.
Mottolese R, Redoute J, Costes N, Le Bars D, Sirigu A. Switching brain serotonin with oxytocin. Proc Natl Acad Sci U S A. 2014;111(23):8637–42. https://doi.org/10.1073/pnas.1319810111.
Song Z, Albers HE. Cross-talk among oxytocin and arginine-vasopressin receptors: relevance for basic and clinical studies of the brain and periphery. Front Neuroendocrinol. 2018;51:14–24. https://doi.org/10.1016/j.yfrne.2017.10.004.
Francis SM, Sagar A, Levin-Decanini T, Liu W, Carter CS, Jacob S. Oxytocin and vasopressin systems in genetic syndromes and neurodevelopmental disorders. Brain Res. 2014;1580:199–218. https://doi.org/10.1016/j.brainres.2014.01.021.
Yamasue H, Okada T, Munesue T, Kuroda M, Fujioka T, Uno Y, et al. Effect of intranasal oxytocin on the core social symptoms of autism spectrum disorder: a randomized clinical trial. Mol Psychiatry. 2020;25(8):1849–58. https://doi.org/10.1038/s41380-018-0097-2.
Yatawara CJ, Einfeld SL, Hickie IB, Davenport TA, Guastella AJ. The effect of oxytocin nasal spray on social interaction deficits observed in young children with autism: a randomized clinical crossover trial. Mol Psychiatry. 2016;21(9):1225–31. https://doi.org/10.1038/mp.2015.162.
Watanabe T, Kuroda M, Kuwabara H, Aoki Y, Iwashiro N, Tatsunobu N, et al. Clinical and neural effects of six-week administration of oxytocin on core symptoms of autism. Brain. 2015;138(Pt 11):3400–12. https://doi.org/10.1093/brain/awv249.
Hollander E, Novotny S, Hanratty M, Yaffe R, DeCaria CM, Aronowitz BR, et al. Oxytocin infusion reduces repetitive behaviors in adults with autistic and Asperger’s disorders. Neuropsychopharmacology. 2003;28(1):193–8. https://doi.org/10.1038/sj.npp.1300021.
Kruppa JA, Gossen A, Oberwelland Weiss E, Kohls G, Grossheinrich N, Cholemkery H, et al. Neural modulation of social reinforcement learning by intranasal oxytocin in male adults with high-functioning autism spectrum disorder: a randomized trial. Neuropsychopharmacology. 2019;44(4):749–56. https://doi.org/10.1038/s41386-018-0258-7.
Owada K, Okada T, Munesue T, Kuroda M, Fujioka T, Uno Y, et al. Quantitative facial expression analysis revealed the efficacy and time course of oxytocin in autism. Brain. 2019;142(7):2127–36. https://doi.org/10.1093/brain/awz126.
Lee SW, Kim YB, Kim JS, Kim WB, Kim YS, Han HC, et al. GABAergic inhibition is weakened or converted into excitation in the oxytocin and vasopressin neurons of the lactating rat. Mol Brain. 2015;8:34. https://doi.org/10.1186/s13041-015-0123-0.
Shao Y, Wolpert CM, Raiford KL, Menold MM, Donnelly SL, Ravan SA, et al. Genomic screen and follow-up analysis for autistic disorder. Am J Med Genet. 2002;114(1):99–105. https://doi.org/10.1002/ajmg.10153.
Parker KJ, Garner JP, Oztan O, Tarara ER, Li J, Sclafani V, et al. Arginine vasopressin in cerebrospinal fluid is a marker of sociality in nonhuman primates. Sci Transl Med. 2018;10(439). https://doi.org/10.1126/scitranslmed.aam9100.
Bolognani F, Del Valle Rubido M, Squassante L, Wandel C, Derks M, Murtagh L, et al. A phase 2 clinical trial of a vasopressin V1a receptor antagonist shows improved adaptive behaviors in men with autism spectrum disorder. Sci Transl Med. 2019;11(491). https://doi.org/10.1126/scitranslmed.aat7838.
Anagnostou E. Clinical trials in autism spectrum disorder: evidence, challenges and future directions. Curr Opin Neurol. 2018;31(2):119–25. https://doi.org/10.1097/WCO.0000000000000542.
Jacquemont S, Berry-Kravis E, Hagerman R, von Raison F, Gasparini F, Apostol G, et al. The challenges of clinical trials in fragile X syndrome. Psychopharmacology. 2014;231(6):1237–50. https://doi.org/10.1007/s00213-013-3289-0.
Besag FM. Epilepsy in patients with autism: links, risks and treatment challenges. Neuropsychiatr Dis Treat. 2018;14:1–10. https://doi.org/10.2147/NDT.S120509.
Hagerman PJ, Stafstrom CE. Origins of epilepsy in fragile X syndrome. Epilepsy Curr. 2009;9(4):108–12. https://doi.org/10.1111/j.1535-7511.2009.01309.x.
Leitner Y. The co-occurrence of autism and attention deficit hyperactivity disorder in children - what do we know? Front Hum Neurosci. 2014;8:268. https://doi.org/10.3389/fnhum.2014.00268.
Morris SL, Huang S. Crosstalk of the Wnt/beta-catenin pathway with other pathways in cancer cells. Genes Dis. 2016;3(1):41–7. https://doi.org/10.1016/j.gendis.2015.12.003.
Munesue T, Nakamura H, Kikuchi M, Miura Y, Takeuchi N, Anme T, et al. Oxytocin for male subjects with autism spectrum disorder and comorbid intellectual disabilities: a randomized pilot study. Front Psychiatry. 2016;7:2. https://doi.org/10.3389/fpsyt.2016.00002.
Benedetti F, Carlino E, Piedimonte A. Increasing uncertainty in CNS clinical trials: the role of placebo, nocebo, and Hawthorne effects. Lancet Neurol 2016;15(7):736–47. https://doi.org/10.1016/S1474-4422(16)00066-1.
King BH, Dukes K, Donnelly CL, Sikich L, McCracken JT, Scahill L, et al. Baseline factors predicting placebo response to treatment in children and adolescents with autism spectrum disorders: a multisite randomized clinical trial. JAMA Pediatr. 2013;167(11):1045–52. https://doi.org/10.1001/jamapediatrics.2013.2698.
Fujioka T, Inohara K, Okamoto Y, Masuya Y, Ishitobi M, Saito DN, et al. Gazefinder as a clinical supplementary tool for discriminating between autism spectrum disorder and typical development in male adolescents and adults. Mol Autism. 2016;7:19. https://doi.org/10.1186/s13229-016-0083-y.
Constantino JN, Kennon-McGill S, Weichselbaum C, Marrus N, Haider A, Glowinski AL, et al. Infant viewing of social scenes is under genetic control and is atypical in autism. Nature. 2017;547(7663):340–4. https://doi.org/10.1038/nature22999.
Disclosures
RJ Hagerman has received funding from Zynerba, Ovid, and the Azrieli Foundation for carrying out treatment studies in patients with fragile X syndrome (FXS). She has also consulted with Fulcrum, Ovid, and Zynerba regarding treatment studies in individuals with FXS.
Funding
This research was supported by funds from the National Institute of Child Health and Human Development (NICHD) grant R01 HD036071, the Azrieli Foundation, and the MIND Institute Intellectual and Developmental Disabilities Research Center P50 HD103526.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Salcedo-Arellano, M.J., Cabal-Herrera, A.M., Punatar, R.H. et al. Overlapping Molecular Pathways Leading to Autism Spectrum Disorders, Fragile X Syndrome, and Targeted Treatments. Neurotherapeutics 18, 265–283 (2021). https://doi.org/10.1007/s13311-020-00968-6
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-020-00968-6