
Abstract
Innate lymphoid cells (ILCs) are a heterogeneous family of immune regulators that protect against mucosal pathogens but can also promote intestinal pathology. Although the plasticity between ILCs populations has been described, the role of mucosal pathogens in inducing ILC conversion leading to intestinal pathology remains unclear. Here we demonstrate that IFNγ-producing ILCs are responsible for promoting intestinal pathology in a mouse model of enterocolitis caused by Campylobacter jejuni, a common human enteric pathogen. Phenotypic analysis revealed a distinct population of IFNγ-producing NK1.1−T-bet+ILCs that accumulated in the intestine of C. jejuni-infected mice. Adoptive transfer experiments demonstrated their capacity to promote intestinal pathology. Inactivation of T-bet in NKp46+ ILCs ameliorated disease. Transcriptome analysis and cell-fate mapping experiments revealed that IFNγ-producing NK1.1−ILCs correspond to ILC1 profile and develop from RORγt+ progenitors. Collectively, we identified a distinct population of NK1.1−ex-ILC3s that promotes intestinal pathology through IFNγ production in response to C. jejuni infection.
Similar content being viewed by others


Introduction
Innate lymphoid cells (ILCs) are a recently identified family of immune cells that have emerged as important regulators of immune homeostasis at mucosal surfaces1,2,3. In the intestine ILCs contribute to immune defense by producing cytokines that support epithelial barrier integrity and regulate the initiation, maintenance, and resolution of inflammation1,2,3. Although the protective roles of ILCs during infection or dissemination of commensal bacteria are well documented4,5,6,7, mounting evidence from clinical studies, supported by animal studies, suggest that dysregulated ILC responses can promote intestinal pathology8,9,10. IFNγ-producing ILC1s accumulate in the inflamed tissue of Crohn’s disease patients while ILC3s were diminished, suggesting that ILC1s play a pathogenic role in inflammatory bowel disease (IBD)8,10,11. Experimental evidence in mice lacking adaptive lymphocytes further defined the pathogenic potential of ILC1s8,9. Furthermore, cell fate-mapping experiments in mice provided evidence that IFNγ-producing ILC1s can develop from RORγt-expressing ILC3 progenitors, named as “ex-ILC3s” because of their cellular ontogeny3,4. However, the conditions that direct the in vivo conversion of ILCs, particularly the role of mucosal pathogens in this process has not been fully analyzed.
Campylobacter is a major human pathogen that infects an estimated 2.5 million people each year resulting in a $1.9 billion economic loss in the U.S.12,13. Among the Campylobacter species, C. jejuni is the primary human pathogen that causes gastroenteritis, which manifests as cramping and diarrhea12,13. In addition to these acute symptoms, mounting epidemiological evidence implicates Campylobacter infection as a cause of long-term intestinal dysfunction such as post-infectious irritable bowel syndrome, which appears to be immune mediated12,13. Experiments in mice support this hypothesis since infection of wild-type mice with C. jejuni causes persistent colonization but does not produce overt symptoms of disease. In contrast, mice lacking IL-10, a key anti-inflammatory cytokine, develop symptoms and pathology that resemble human campylobacteriosis14. Since IL-10 is known to suppress inflammation, and since C. jejuni infection causes disease in IL-10-deficient mice, it is thought that an overly aggressive host response by T cells promotes disease in this model of colitis; however, the role of ILCs in promoting inflammation remains controversial15,16.
In the present study, we investigated the role of ILCs in intestinal inflammation caused by C. jejuni. We found that a population of IFNγ-producing ILCs promoted colitis independently of T cells. These pathogenic ILCs lack NK1.1 and exhibited ILC3 > ILC1 lineage plasticity and, in accordance with the established nomenclature, we designate these cells as NK1.1−ex-ILC3s.
Results
Innate lymphoid cells promote C. jejuni-induced colitis
Prior studies established that IL-10−/− mice, but not wild type mice, develop severe colitis when infected by C. jejuni14,17. The commensal microbiota strongly influences the development of colitis in IL-10−/− mice18. To exclude the variability associated with the microbiota and to facilitate colonization, IL-10−/− and heterozygous littermates were pre-treated with an antibiotic cocktail in the drinking water prior to oral inoculation with C. jejuni and evaluated for weight loss and diarrhea (Fig. 1a). Whereas infected IL-10 heterozygotes were asymptomatic, IL-10−/− mice lost significant weight and began to succumb to C. jejuni infection after 10 days (Fig. 1b). Gross examination of the intestine at day 10 revealed marked thickening and inflammation of the cecum and colon (data not shown). Histologically, inflammatory lesions consisted of mixed leukocytic mucosal and submucosal infiltrates with distention of the submucosa. Associated with the infiltrates was mucosal hyperplasia with prominent mitotic figures in the crypts adjacent to regions of inflammation. In the most severely affected sections, many of the crypts contained necrotic cellular debris and mucus (Fig. 1c). Histological scoring of the colon and colonic mass-to-length measurements, an indicator of tissue pathology, confirmed that IL-10−/− mice develop C. jejuni-induced colitis whereas IL-10 heterozygous littermates were colonized but did not develop severe pathology (Fig. 1d, e, f). WT mice treated with IL-10Rα blocking mAb developed similar pathology to IL-10−/− mice when infected with C. jejuni (Fig. 1d–e).

Antibiotic pre treated mice were inoculated by gavage with vehicle (Naive) or C. jejuni (Cj). Ten days later, disease severity and bacterial colonization were evaluated. a C. jejuni-induced colitis model in mice. Arrows indicate administration of monoclonal antibodies (mAb) for depletion of specific cells or neutralization of cytokines/cytokine receptors. Disease severity in IL-10-deficient (IL-10 KO) and heterozygous (IL-10 Het) mice: b loss of body weight, c, d histological examination of H&E stained colon sections, and e colonic mass-to-length ratio. f C. jejuni colonization of colons. g, h RAG-deficient (RAG−/−) and RAG/IL-2Rγ double deficient (RAG−/−γc−/−) mice were treated with IL-10Rα blocking antibody and infected with C. jejuni. g Colon mass-to-length ratio. h C. jejuni colonization. i–j TCRβ/δ−/−IL-10−/− and TCRβ/δ−/−IL-10+/− mice were infected with C. jejuni and treated with either Thy1.2-depleting (αThy1.2) or isotype control mAb (Ctrl Ig). i Colon mass-to-length ratio. j C. jejuni colonization. Data represent individual mice with horizontal lines and error bars depicting means and SEM, respectively. Data is representative of two to three independent experiments. P values were calculated by two-way ANOVA (b) with Bonferroni’s multiple hypothesis corrections or unpaired Student’s t-test with Welch’s correction when warranted (d–j). *p < 0.05, **p < 0.01, ***p < 0.001.
To delineate the contribution of ILCs and lymphocytes to C. jejuni-induced pathology in absence of adaptive immunity, we blocked IL-10Rα in RAG−/− and RAG−/−γc−/− mice prior to infection with C. jejuni. Infected RAG−/− mice lost weight, although at a lower rate than IL-10−/− mice and developed similar pathology to IL-10−/− mice (Fig. 1g and data not shown). Surprisingly, RAG−/−γc−/− mice, which lack Thy1.2+ILCs in addition to T and B cells (Fig. S1a) showed significantly less inflammation and weight loss, and harbored fewer C. jejuni in the colon compared to RAG−/− mice (Fig. 1g, h). These results suggest that Thy1.2+ILCs promote C. jejuni-induced intestinal pathology.
To further examine the role of Thy1.2+ILCs in promoting C. jejuni-induced colitis independently of T cells, we intercrossed T cell-deficient TCRβ/δ−/− mice (Fig. S1b, c) and IL-10-deficient mice. We treated these mice with either Thy1.2 depleting or isotype control mAb prior to inoculation with C. jejuni. Infected TCRβ/δ−/−IL-10−/− mice developed pathology that was significantly ameliorated by depletion of Thy1.2+ cells (Fig. 1i). Depletion of Thy1.2+ ILCs cells also significantly reduced the numbers of C. jejuni in the colon (Fig. 1j). Collectively, these data suggest that ILCs promote C. jejuni-induced colitis independently of T cells.
IFNγ from ILCs is critical to promote C. jejuni-induced colitis
Several pro- and anti-inflammatory cytokines produced by ILCs and known to regulate intestinal inflammation8,13,19,20,21. Therefore, we analyzed cytokine expression in colons during C. jejuni infection. IL-17A, IFNγ, TNF, and IL-22 were upregulated in colons of IL-10−/− mice after infection (Fig S2). Interestingly, IFNγ, TNF, and IL-22 but not IL-17A were upregulated in the colons of infected TCRβ/δ−/−IL-10−/− mice (Fig. 2a) suggesting that mainly T cells were responsible for IL17A production. We also found that C. jejuni infection drove colonic expression of IL-12 and IL-23 (Figs. 2b and S2b), known regulators of IFNγ, TNF, IL-22, and IL-17A production during inflammation22. Together, these results suggest that increased innate production of pro-inflammatory cytokines, such as IFNγ is associated with C. jejuni-induced intestinal pathology.

Antibiotic pre treated TCRβ/δ−/−IL-10−/− mice were infected with C. jejuni. Ten days after infection colon and MLN were analyzed. a, b Expression of proinflammatory cytokines were measured in colon by real-time PCR. Data combined from two independent experiments with similar results (n = 7–9 mice per group). c Intracellular staining of IFNγ and IL-17A in live, Thy1.2+ gated cells from MLN and cecum after restimulation with PMA and ionomycin. Open black histograms: FMO control; filled histograms: stained panel, red open histograms: stained panel samples without PMA and ionomycin restimulation, blue open histogram: samples after restimulation with PMA and ionomycin. Histograms are a concatenation of four samples. d Intracellular staining of IFNγ in naïve (red histogram) and infected (blue histogram) mice after PMA and ionomycin stimulation. e IFNγ-reporter mice were treated with IL-10Rα-blocking and CD4 and CD8 depleting mAbs prior to infection with C. jejuni. IFNγ expression (EYFP) was analyzed in cecal lymphocytes directly ex vivo (without restimulation) 10 days after infection. Flow cytometry plots from four concatenated samples show EYFP+ cells stained for Thy1.2 and CD4 and CD8. f–i TCRβ/δ−/−IL-10−/− mice were treated with neutralizing mAb or isotype control (Ctrl Ig). Ten days later, disease severity and C. jejuni colonization was evaluated. f Histological examination, g pathology disease scores, h colonic mass-to-length ratio, i enumeration of C. jejuni in colon. Data shown in g–i are pooled from two independent experiments (n = 7–8 mice per group). Bars depict means and SEM. Data represents an individual mouse with the horizontal lines depicting means. Real-time PCR data were normalized to hprt expression. P values were calculated by Mann–Whitney test (a, b) or unpaired Student’s t-test with Welch’s correction when warranted (g–i). *p < 0.05, **p < 0.01, ***p < 0.001.
Both IFNγ and IL-17A have been shown to contribute to C. jejuni infection15. T cells, ILCs and NK cells are the major producers of IFNγ during C. jejuni infection15 (see also Fig. S3a, b). To identify contribution of ILCs in IFNγ and IL-17A production during C. jejuni –induced colitis, we infected TCRβ/δ−/−IL-10−/− mice with C. jejuni and isolated cells from the mesenteric lymph nodes (MLN) and cecum. Innate Thy1.2+ cells from both MLN and cecum were robust producers of IFNγ but not IL-17A (Fig. 2c). Furthermore, C. jejuni infection induced high levels of IFNγ production by innate Thy1.2+ cells compared to naïve controls (Fig. 2d), indicating that infection is a potent inducer of cytokine production by these cells. To confirm that our findings were not biased by in vitro stimulation and to evaluate the expression of IFNγ directly ex vivo without stimulation, we analyzed cells from IFNγ reporter mice23 after T-cell depletion. Flow cytometry analysis showed that >80% of the cells that express IFNγ during C. jejuni infection were innate Thy1.2+ cells (Fig. 2e) and there were no production of IFNγ in naïve mice by these cells (Fig. S3c).
Next, to test whether IFNγ was required to promote pathology during C. jejuni infection, we treated TCRβ/δ−/−IL-10−/− mice with IFNγ neutralizing mAb prior to C. jejuni infection. Neutralization of IFNγ significantly ameliorated colitis and reduced C. jejuni numbers in the colon (Fig. 2f–i). These results indicate that Thy1.2+ILCs are a prominent source of IFNγ that contributes to C. jejuni induced intestinal disease.
Distinct population of IFNγ+NK1.1− ILCs accumulates in the inflamed intestine
IFNγ-producing ILCs are a heterogeneous population of cells that express the transcription factor T-bet1,3. This group of ILCs includes cytotoxic NK cells and helper-like ILC1s that produce IFNγ and TNF1,3. To characterize the IFNγ-producing ILCs during C. jejuni-induced colitis, we analyzed ILCs (IFNγ+CD3−Lin−Thy1.2+) isolated from MLN and colon lamina propria (cLP) of infected WT mice with disrupted IL-10R signaling. Flow cytometry analysis revealed two distinct populations of IFNγ-producing ILCs: NK1.1+ILCs and NK1.1−ILCs (Fig. 3a, b). Murine ILC1s are reported to express NKp46 and NK1.1 markers3,4,9. Interestingly, we found a marked increase of colonic IFNγ-producing NK1.1−ILCs after infection, whereas the absolute number of IFNγ+NK1.1+ILCs was unchanged (Fig. 3b) indicating that C. jejuni infection drives the accumulation of NK1.1−IFNγ+ILCs in the colon. Moreover, C. jejuni infection rather than antibiotic treatment or IL-10R blockade induced the accumulation of NK1.1−IFNγ+ILCs, since antibiotic-treated or aIL-10Rα blocked uninfected mice did not display increased number of colonic ILCs (Fig. S4e), in agreement with previous study showing no inflammation in aIL-10Rα blocked uninfected mice16. Further characterization of IFNγ-producing ILCs revealed that these cells express IL-7Rα (CD127) (Fig. 3c), NKp46 (Fig. 3d), but do not express c-kit, IL-17, IL-22 (Fig. S4b) and KLRG1 (Fig. S4h). The absence of KLRG1 expression in IFNγ-producing ILCs indicates that these cells represent ILC1s or ILC3s but not ILC2s. Moreover, IFNγ+ILCs also lacked CCR6, the marker associated with LTi-like ILC3s (Fig. 3c).

a–f WT mice were treated with IL-10Rα-blocking antibody and infected with C. jejuni (Cj). Ten days after infection, colon LP leukocytes were isolated. Cells were restimulated with PMA and ionomycin and stained for flow cytometry. a NK1.1 levels in IFNγ+ ILCs cells. b Frequency (left panel) and absolute number (right panel) of IFNγ+ ILCs. c Histograms show expression of indicated markers in IFNγ+CD3−Lin−Thy1+ ILCs (red) relative to FMO control (black). d T-bet, RORγt, and NKp46 expression in IFNγ+ ILCs. Black line: FMO control. e Frequency (left panel) and absolute number (right panel) of IFNγ+NK1.1+ ILCs. f Frequency (left panel) and absolute number (right panel) of IFNγ+NK1.1− ILCs. Data shown in (d) and (e–f) are pooled from two independent experiments. Lineage: CD11c, B220, Gr1, Ter119. g–h IFNγ-reporter mice were depleted of T cells by αCD4 and αCD8 depleting mAbs. Absolute numbers of (g) IFNγ+NK1.1+ ILCs and (h) IFNγ+NK1.1− ILCs directly ex vivo (without restimulation). Bars depict means and SEM. Data represent an individual mouse with the horizontal lines depicting means. P values were calculated by unpaired Student’s t-test with Welch’s correction when warranted (b, e–h). ns not significant, **p < 0.01, ***p < 0.001.
To define the role of IFNγ+-producing ILCs independently of T cells, we next analyzed NK1.1− and NK1.1+ILCs in TCRβ/δ−/−IL-10−/− mice. Consistent with the data obtained in T-cell sufficient mice (Figs. 3b, e and S4d), the frequency of NK1.1+IFNγ+ILCs in colon and MLN of TCRβ/δ−/−IL-10−/− mice was decreased during C. jejuni infection, whereas the frequency of NK1.1−IFNγ+ILCs was increased (Fig. S5a–d). To confirm our findings without in vitro stimulation, we infected IFNγ reporter mice with C. jejuni and analyzed colonic ILCs by flow cytometry (Fig S4h). C. jejuni infection of IFNγ reporter mice did not impact the number of NK1.1+ILC1s but enhanced the accumulation of NK1.1−ILCs to the colon, compared to uninfected mice (Figs. 3g, h and S4i). Furthermore, depletion of T cells by mAb treatment did not influence the number of NK1.1−ILCs or NK1.1+ILCs (Figs. 3g, h and S4i), suggesting that the presence of T cells has no impact on accumulation of IFNγ-producing NK1.1−ILCs to the colon. Accordingly, the number of IFNγ+ NK1.1−ILCs in the colon was similar between WT and TCRβ/δ−/− mice (Fig. S5e–g) indicating that the lack of T cells does not influence IFNγ production by NK1.1−ILCs. Additionally, confocal microscopy detected IFNγ-producing NK1.1−ILCs clusters in colon of IFNγ-reporter mice treated with CD4, CD8, NK1.1 depleting antibodies (Fig. S5h).
T-bet is a key regulator of IFNγ production by ILC11,2,4. In addition to ILC1 subset, T-bet can be co-expressed with RORγt, the signature transcriptional factor for ILC3 subset19. Both NK1.1+ and NK1.1−ILCs expressed T-bet and were the major producers of IFNγ (Figs. 3e, f and S4c). However, in contrast to NK1.1+ILCs, NK1.1−ILCs did not express Eomes, indicating their distinct phenotype from NK cells (Figs. 3c and S4c). In agreement with the reduced frequency of NK1.1+IFNγ+ cells in the colon during C. jejuni infection, we observed the significant reduction of the frequency of T-bet+NK1.1+ cells, although the absolute number of these cells was not affected (Figs. 3e and S4g). Conversely, T-bet+NK1.1−ILCs were significantly increased in cLP of C. jejuni-infected mice compared to naïve mice (Fig. 3f), but not in MLN (Fig. S4f). Interestingly, the absolute number of NK1.1+ and NK1.1− IFNγ-producing ILCs that express both T-bet and RORγt was increased in the colon (Fig. 3e, f), indicating their intermediate ILC3-ILC1 phenotype.
Together, these results suggest that population of IFNγ-producing Lin−NK1.1−ILCs accumulates in the intestine during C. jejuni infection and that these cells are phenotypically distinct from NK1.1+ILC1s.
NK1.1− IFNγ-producing ILCs can promote C. jejuni-induced colitis
Our immunophenotyping data identified two populations of IFNγ-producing ILCs that reside in the inflamed intestine during C. jejuni infection, NK1.1+ILC1s and NK1.1−ILCs (Fig. 3). Prior studies implicated NK1.1+ILC1s in promoting colitis in mice8,9. To further define the role of NK1.1+ILC1s in C. jejuni-induced colitis, we treated TCRβ/δ−/−IL-10−/− mice with either NK1.1 depleting or isotype control antibody prior to C. jejuni infection. This strategy has been used successfully to ameliorate ILC1-dependent αCD40-driven colitis8,9. Despite effective depletion of NK1.1+ cells (Figs. 4a and S6), this treatment did not affect colitis or altered C. jejuni numbers in the colon (Fig. 4b–f). Furthermore, depletion of NK1.1+ILCs did not impact IFNγ secretion by MLN cells when stimulated in vitro with heat-killed C. jejuni (Fig. 4g). Together these results suggest that NK1.1+ILC1s are not critical mediators of C. jejuni-induced colitis.

a–f NK1.1+ILCs are dispensable for C. jejuni-induced colitis. TCRβ/δ−/−IL-10−/− mice were treated with NK depleting (αNK1.1) or isotype control (Ctrl) mAbs. Disease severity, intestinal lymphocytes and IFNγ expression were analyzed 10 days after infection. a The efficacy of in vivo NK1.1 depletion. Histograms depict fully stained panels (filled histograms) relative to FMO controls (outlined) on live, Thy1.2+ cells. Each flow cytometry plot is a concatenation of three (Naïve) or four (Cj) samples. b Loss of body weight (c) H&E stained colon sections. d Pathology disease scores of colons. e Colonic mass-to-length ratio. f Enumeration of C. jejuni in the colon of infected mice. All data represent an individual mouse from three independent experiments with horizontal lines depicting means. Histopathology score bar graphs depict the mean and SEM of nine (Naïve) or 12 (Cj) mice from three independent experiments. g The capacity for IFNγ secretion by MLN cells after NK1.1 depletion. 1 × 106 mesenteric lymph node cells were restimulated with control (medium alone) or heat-killed C. jejuni for 48 h. After restimulation, IFNγ in culture supernatants was measured by ELISA. Bars represent the mean and SEM of six (naïve) or eight (Cj) mice from two independent experiments. h–n Cells from the MLN, cecum and colon were isolated from C. jejuni-infected TCRβ/δ−/−IL-10−/− mice. ILCs were sorted based on Thy1.2+Lin−NK1.1− phenotype and 2 × 104 purified ILCs were adoptively transferred by i.v. injection to RAG−/−γc−/− recipients treated with IL-10Rα-blocking mAb. ILCs were transferred to recipient mice one day prior to C. jejuni infection, compared to sham mice injected with PBS. h Sorting scheme and (i) purity of Thy1.2+Lin−NK1.1− ILCs. Numbers in the plots show the frequency of cells within the gate. Ten days after infection, disease severity in recipient mice was measured by (j–k) histological examination of H&E stained colon sections (25× magnification) (l) colonic mass-to-length ratio and (m) C. jejuni colonization. Data represent two independent combined experiments (n = 2–3 mice per group). Data represent an individual mouse with bars depicting the mean. P values were calculated by two-way ANOVA (b and g) with Bonferroni’s multiple hypothesis corrections or unpaired Student’s t-test with Welch’s correction when warranted (d–f, k–n). *p < 0.05, **p < 0.01, ***p < 0.001.
Since depletion of NK1.1+ILC1s did not impact C. jejuni-induced colitis, we hypothesized that IFNγ-producing NK1.1−ILCs that accumulate in the inflamed intestine during C. jejuni infection could promote intestinal pathology. To determine the pathogenic potential of these ILCs, we sorted Thy1.2+Lin−NK1.1− cells (purity > 95%, as confirmed by flow cytometry, Fig. 4h–i) from the MLN, ceca, and colons of C. jejuni-infected TCRβ/δ−/−IL-10−/− mice. The purified cell population was transferred to RAG−/−γc−/− recipient mice which lack both adaptive and innate lymphocytes and do not show symptoms or pathology after C. jejuni infection (Fig. 1g). One day after receiving either ILCs or vehicle, recipient mice were infected with C. jejuni and disease severity and bacteria burden were evaluated 10 days later. Mice receiving NK1.1−ILCs showed increased histological scores, colonic mass-to-length ratio and C. jejuni colonization of the colon (Fig. 4j–m). These findings suggest that Lin–NK1.1−IFNγ-producing ILCs can promote C. jejuni-induced intestinal pathology.
IFNγ-producing ILCs predominantly express ILC1 signature genes
Next, to further characterize IFNγ-producing ILCs using an unbiased approach, we sort-purified CD3–Thy1.2+Lin−IFNγ+ cells (Fig. 5a) from cLP and MLN of C. jejuni infected mice treated with aIL-10Rα antibodies. Transcriptome profiles were analyzed by RNA-seq. Principal component analysis (PCA) was used to simplify the complexity of high-dimensional data. The first Principal component, PC1, which represents 29.9% of variation, distinguished between colon and MLN cells, whereas PC2, which accounted for 21.1% of variation, separated ILCs and T cells (Fig. 5b). Hierarchical clustering of 25 previously reported signature genes of distinct ILC subsets from siLP24 revealed that the major transcripts expressed at higher level in IFNγ-producing ILCs from cLP shown to be characteristic genes for ILC1, such as Ifng, Tbx21, Xcl1, Il21r, Ccl3, Ccl4, and Ccl5 (Fig. 5c)24,25,26,27. Although most upregulated genes were ILC1s signature genes, we also detected upregulation of Il22, Tcf7, Areg, and Ccl1 in cLP ILCs that are characteristic genes for ILC3 and ILC2 subtypes, respectively (Fig. 5c). In contrast, IFNγ-producing ILCs from MLN upregulated predominantly ILC1-characteristic transcripts, but had much lower Il22, Areg, Ccl1 transcript levels compared to cLP ILCs, whereas the expression of Tcf7 gene was the same between MLN and cLP ILCs (Fig. 5c). Thus, the transcriptional identity of IFNγ-producing ILCs that accumulate in the MLN and colon during C. jejuni infection represent predominantly ILC1 subtype.

IFNγ-reporter mice were treated with IL-10Rα-blocking mAb and infected with C. jejuni. Ten days after infection, leukocytes from colon LP and MLN were isolated and sorted. Transcriptome profile was analyzed by RNAseq. a Gating strategy for sorting of IFNγ-producing ILCs and T-cells. b Principal component analysis of the ILCs (ln_ilc or lp_ilc) and T-cells (ln_t or lp_t) using the top 9525 most variable genes. Numbers along the axes indicate relative scaling of the principal variables. c Heat map represents the relative expression of key signature genes of ILCs subsets from Gury-BenAri et al. and our MLN and LP expression data. d Estimated proportions of distinct ILC subsets, ILC1, ILC2, and ILC3) from siLP in MLN and cLP. The results were determined by a custom signature matrix from Gury-BenAri et al. and generated by the CIBERSORT website. In the table, all results are reported as relative fractions (normalized to 1 across all cell subsets); P value is the statistical significance of the deconvolution result across all cell subsets; Pearson’s correlation coefficient is estimated from comparing the original mixture with the estimated mixture; RMSE (root mean squared error) is the error between the original mixture and the imputed mixture. Both measurements of correlation and RSEM were restricted to genes in the signature gene file.
To define the transcriptional identity of IFNγ-producing ILCs from cLP and MLN, we compared transcriptional profiles of these population with known ILC signature genes described by Gury-BenAri et al.24 using CIBERSORT method28. This statistical deconvolution method provides a useful tool to evaluate the abundances of different cell types in a mixed cell population utilizing gene expression data. To estimate the relative proportion of each ILC subset within IFNγ-producing ILCs in C. jejuni-induced colitis, an input matrix of reference gene expression signatures was made based on RNA-seq data generated by Gury-BenAri et al.24. Our analysis revealed that IFNγ-producing ILCs in colon had mainly ILC1 and ILC3 gene expression profile whereas ILCs from MLN were represented exclusively by ILC1 (Fig. 5c, d). Together, these findings indicate that pathogenic IFNγ-producing ILCs in colon represent predominantly ILC1 subtype with distinct ILC3 gene signatures.
T-bet+ ILCs are required for C. jejuni-induced colitis
Our data demonstrate that IFNγ-producing ILCs in the inflamed intestine express T-bet (Fig. 3b–d). T-bet is a key regulator of IFNγ production in both ILCs and T cells1,2,4. To further define the pathogenic role of T-bet+ ILCs in a T-cell sufficient environment, we generated NKp46-Cre x Tbx21fl/fl (NKp46-Tbet) mice which are harboring genetic deletion of T-bet in ILC1s, NKp46+ ILC3s, NK cells and some minor populations of T cells29. Following C. jejuni infection, NKp46-Tbet mice exhibited reduced intestinal pathology compared to WT mice (Fig. 6a–d), although there was no difference in bacterial burden compared to the control group (Fig. 6e). These data indicate that NKp46+T-bet+ cells had no impact on C. jejuni colonization, yet promoted inflammation.

NKp46-Tbet mice were treated with IL-10Rα-blocking antibody, infected orally with C. jejuni and evaluated at day 10. a Changes in body weight. b, c Histological examination of H&E stained colon sections. d Colonic mass-to-length ratio. e Bacterial burdens of C. jejuni. f Expression of IFNγ in colon measured by real-time PCR. Data were normalized to hprt expression. Data represent two independent combined experiments (n = 6–9 mice per group). g–h, j–k IFNγ-producing ILCs and NK cells from colon were analyzed by flow cytometry. cLP lymphocytes were restimulated with PMA/ionomycin in vitro for 4 h before intracellular staining of IFNγ. g Absolute cell numbers of IFNγ-producing ILCs and NK cells (h) and percentage of IFNγ+ ILCs and NK cells as a frequency of live cells. i Representative plots of IFNγ-producing NK1.1+ and NK1.1− ILCs. j Absolute cell numbers of IFNγ-producing ILCs. k Percentage of IFNγ+ ILCs and NK cells as a frequency of live cells. ILCs: CD3−Lin−Thy1+ Eomes−IFNγ+, NK cells: CD3-NK1.1+T-bet+Eomes+IFNγ+. Lineage: CD11c, B220, Gr1, Ter119, CD5. Data show results of three independent experiments (n = 7–9 mice per group). Data represent an individual mouse with bars depicting the mean. P values were calculated by two-way ANOVA with Bonferroni’s multiple hypothesis corrections (a) or Mann–Whitney test (c–h, j–k). ns not significant, *p < 0.05, **p < 0.01, ***p < 0.001.
Given that IFNγ is critical for C. jejuni-induced colitis15, we next analyzed IFNγ expression in NKp46-Tbet and WT mice. As shown in Fig. 6f, IFNγ expression was significantly reduced in colon of NKp46-Tbet mice on day 10 after C. jejuni infection. Flow cytometry analysis revealed marked reduction of IFNγ by ILCs, whereas IFNγ levels by NK cells were similar to control mice (Fig. 6g, h). NK1.1− and NK1.1+ ILCs isolated from NKp46-Tbet mice showed significantly reduced IFNγ levels compared to WT mice (Fig. 6i–k), consistent with T-bet expression in these cell populations (Fig. 3d). Together, these data indicate that T-bet+ ILCs contribute to C. jejuni-induced intestinal disease in T-cell sufficient environment.
IFNγ+Lin−NK1.1− T-bet-expressing ILCs develop from RORγt-expressing ILC progenitors
Our adoptive transfer experiments suggested an important role of Lin−NK1.1− IFNγ-producing ILCs in promoting colitis. RNAseq data revealed that ILCs from the colon mostly represent ILC1 subtype, although several ILC3 characteristics transcripts have been revealed. Next, we examined the developmental origin of these cells. In vitro studies with human ILCs and cell fate-mapping studies in mice revealed that RORγt-expressing ILC3s can convert to an ILC1-like phenotype that express T-bet and IFNγ9,25,30,31. Therefore, we hypothesized that C. jejuni infection drives the conversion of RORγt+ ILC3s to pathogenic IFNγ+T-bet+Lin−NK1.1−ILCs.
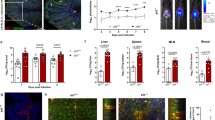
To test whether RORγt plays a role in the development of IFNγ+Lin−NK1.1−ILCs, we infected RORγt−/− and heterozygous littermates on the RAG1−/− background with C. jejuni and evaluated Lin−NK1.1−ILCs and intestinal pathology. We found that whereas RORγt heterozygous mice developed severe colitis, RORγt−/− littermates developed only mild inflammation despite being colonized with pathogen at comparable levels (Fig. 7a–e). Flow cytometry analysis revealed that RORγt−/− mice showed reduced frequency and absolute number of IFNγ+Lin−NK1.1−ILCs in the cecum compared to heterozygous littermates (Fig. 7f–g). These results indicate that RORγt can participate in development and maintenance of IFNγ+Lin−NK1.1−ILCs. However, these results do not exclude a possibility that lack of lymph nodes or gut-associated lymphoid tissues in RORγt−/− mice may affect the course of disease.

RORγt-deficient (RORγt KO) and heterozygous littermates (RORγt Het) on the RAG1−/− background were treated with IL-10Rα-blocking mAb and infected with C. jejuni. a Loss of body weight (b) H&E stained colons (25x magnification). c Pathology disease scores of colon sections. d Colonic mass-to-length ratio. e Enumeration of C. jejuni from the colons of infected mice. Flow cytometry analysis of (f) frequency and (g) absolute number of IFNγ+Lin−NK1.1− ILCs in the cecum. Data is representative of two independent experiments. h–l The history of RORγt expression (current or prior RORγt expression) was determined using a cell fate-mapping approach with RORγtTg-creRosa26stop-EYFP mice treated with IL-10Rα-blocking mAb and either infected with C. jejuni (Cj) or sham infected with vehicle alone (Naïve). h NK1.1− ILCs (Thy1.2+Lin−IFNγ+) from the intestine or (i) MLN were analyzed for their history of RORγt expression. Open histogram: Rosa26stop-EYFP (Cre-) control mice. Filled histogram: RORγtTg-creRosa26stop-EYFP (Cre+) cell fate-map mice. The frequency of EYFP+ cells in (j) cecum and (k) MLN. l T-bet and RORγt expression by ILCs and T cells. CD4, CD8: mAb against CD4 and CD8 were both in the same channel. Lineage: CD11b, CD11c, B220, Gr1, Ter119, CD3, CD5. Data are representative of two independent experiments. Data represent individual mice with bars depicting means. P values were calculated by two-way ANOVA (a) with Bonferroni’s multiple hypothesis corrections or unpaired Student’s t-test with Welch’s correction when warranted (c–g, j–k). **p < 0.01, ***p < 0.001.
To further address the role of RORγt in NKp46+ ILC3s in C. jejuni-induced colitis, we generated NKp46-Cre x RORγt fl/fl (NKp46-RORγt) mice in which RORγt deficiency is selectively restricted to NKp46+ ILC3 subsets32,33. We did not find significant difference in disease severity (Fig. S7a–c) or IFNγ production by ILCs (Fig. S7d) in NKp46-RORγt mice compared to controls. The number of colonic NKp46+ILC3s in NKp46-RORγt mice was slightly reduced, whereas the number of NKp46−ILC3s was unchanged (Fig. S7e). These data indicate that NKp46+ILC3s are not sufficient to drive C. jejuni-induced pathology.
Our data suggest that NK1.1−ILCs are the major producers of IFNγ during C. jejuni infection. Next, we determined the developmental origin of these IFNγ+Lin−NK1.1−ILCs using RORγt cell fate map mice (RORγtfm). Mice expressing EYFP that is preceded by a loxP-flanked STOP sequence in the Rosa26 locus34 were crossed to mice expressing Cre recombinase under the RORγt promoter35 to generate RORγtfm mice. In these mice, cells that express RORγt are permanently marked by EYFP expression, allowing the identification of the cells that have unstable or transient expression of RORγt. We found that the majority of NK1.1−ILCs (Thy1.2+Lin−IFNγ+) from the intestine and MLN were EYFP+ compared to uninfected controls (Fig. 7h, i), indicating that C. jejuni infection promotes ILC3 plasticity. Moreover, T cells, which express RORγt in the thymus36, displayed efficient recombinase activity in RORγtfm mice since >94% of T cells in the cecum and MLN were EYFP+ (Figs. 7j, k and S8). We found that 90% of Lin−NK1.1−ILCs from the MLN were EYFP+ as well as 89% of cecal IFNγ+Lin−NK1.1−ILCs. Whereas EYFP+ T cells (Thy1.2+Lin−CD4+ or CD8+) predominantly expressed T-bet, EYFP+ ILCs (Thy1.2+Lin−CD4−CD8−) were heterogeneous in their RORγt and T-bet expression (Fig. 7l), suggesting their plasticity. Together, these findings indicate that IFNγ+Lin−NK1.1−ILCs develop from RORγt-expressing progenitors during C. jejuni infection and represent NK1.1−ex-ILCs.
Discussion
The discovery of ILCs has greatly expanded our understanding of effector immune cells that contribute to host defense against pathogens and promote tissue repair after injury. Nevertheless, mounting clinical evidence suggests that dysregulated ILCs responses can promote chronic inflammatory pathologies such as IBD. How ILCs become dysregulated and contribute to disease remains unclear. Using a mouse model of campylobacteriosis, we demonstrate that C. jejuni infection induces a population of IFNγ+Lin−NK1.1−ILCs that exhibits ILC3 > ILC1 phenotypic plasticity and promotes IFNγ-dependent intestinal inflammation.
C. jejuni is a major foodborne pathogen and a significant cause of immune-mediated post-infectious sequelae12,37. IL-10-deficient mice develop severe colitis with an accumulation of lymphocytes in the intestine when associated with members of the bacterial order Campylobacterales, particularly Helicobacter hepaticus and C. jejuni14,15,38. Depletion of neutrophils16 or Thy1+ lymphocytes15 significantly ameliorated colonic inflammation in IL-10−/− mice infected with C. jejuni, revealing the pathogenic role of dysregulated host responses during infection by this pathogen. In our study, depletion of Thy1+ cells in T-cell deficient TCRβ/δ−/−IL-10−/− mice markedly reduced pathology after C. jejuni infection, suggesting a role for dysregulated ILCs in promoting colitis. Consistent with this, RAG−/−γc−/− mice that are deficient for both adaptive immune cells and all ILCs were better protected from C. jejuni-induced colitis compared to RAG−/− mice. Therefore, we conclude that ILCs can promote C. jejuni-induced intestinal disease.
IFNγ is a pro-inflammatory cytokine that has important functions in immune defense against intracellular pathogens including the upregulation of antigen presenting MHC molecules, the induction of anti-microbial immunity and the production of chemokines that facilitate leukocyte trafficking39. In clinical trials, IFNγ levels correlated with disease resistance when people were rechallenged with C. jejuni40, suggesting that IFNγ plays a protective role during repeated exposure to this pathogen. Despite IFNγ role in combating infection, its excessive production is implicated in the etiology of IBD20 and the number of IFNγ-producing ILCs are increased in the colon of Crohn’s disease patients compared to healthy cohorts8,10. IFNγ-producing lymphocytes also accumulate in intestine during experimental infection with H. hepaticus or C. jejuni and neutralization of IFNγ ameliorated disease in these models of colitis15. In our experiments, neutralization of IFNγ in TCRβ/δ−/−IL-10−/− mice significantly ameliorated C. jejuni-induced colitis and reduced bacterial counts in the colon, suggesting that IFNγ-producing ILCs contribute to intestinal pathology.
IFNγ-producing ILCs include cytotoxic NK cells and helper-like ILC1s that produce cytokines1. To date, murine ILC1s are defined by their expression of the transcription factor T-bet and surface markers NK1.1 and NKp463,4. Although IFNγ production by NK1.1+ILC1s has been implicated in protective responses to intracellular pathogens, dysregulated production of IFNγ by ILCs could also promote immune-mediated pathology4,19. In addition to ILC1, NKp46+ and NKp46−CCR6−ILC3 can also produce IFNγ in response to environmental cues or bacterial infection4,19. Anti-NK1.1 antibody cell depletion studies demonstrated the pathogenic potential of murine ILC1s in αCD40 experimental innate colitis model8,9. In our study we revealed two populations of IFNγ-producing ILCs in the colons of C. jejuni infected mice, one that expresses markers consistent with previously described murine NK1.1+ILC1s and another one that does not express NK1.1 (NK1.1− ILC1s). Both of these ILCs populations expressed T-bet and RORγt. Surprisingly, depletion of NK1.1+ILCs did not impact C. jejuni-induced intestinal pathology, suggesting that NK1.1+ILC1s are dispensable for C. jejuni-induced intestinal disease. We next therefore focused on the role of IFNγ-producing NK1.1−ILCs which are greatly increased in the inflamed mouse intestine during C. jejuni infection. Using an adoptive transfer strategy, we confirmed that Lin−NK1.1−ILCs can facilitate C. jejuni-induced pathology. Our data demonstrate the importance of Lin−NK1.1−ILCs for the development of C. jejuni-induced colitis. In line with our study, recent report demonstrated that Lin−NK1.1−ILCs are the major producer of IFNγ during Yersinia enterocolitica infection and can contribute to protection32.
Infection or inflammation can affect local ILCs populations. In fact, the number of ILC1s is increased in the intestinal inflamed mucosa of CD patients8,30. Previous studies showed that IFNγ-producing ILC1s can arise from CCR6−RORγt+ILCs population that upregulates T-bet expression in response to signals from the environmental cues9,19. Our data suggest that colonic IFNγ-producing ILCs mostly have transcriptional signatures of ILC1s with distinct signature genes of ILC3s during C. jejuni infection, whereas in MLN IFNγ-producing ILCs exclusively display transcriptional signatures of ILC1s. This difference in transcriptional profiles can depend on the local cytokine microenvironment and the ability of ILCs to migrate within the organs. A recent report indicates that ILCs can migrate between the MLN and intestine41.
The contribution of ILCs to pathology during bacterial infection in immune sufficient host can be easily overlooked due to contribution of T cells. Importantly, our data demonstrate that T-bet deficiency in NKp46+ cells (mainly ILC1, NKp46+ILC3, and NK cells) reduced pathology and IFNγ production in the colon highlighting the role of IFNγ production by ILCs in the early stages of infection in a T-cell sufficient environment and intact development of gut-associated lymphoid tissues. Although impaired IFNγ production was observed in both NK1.1−ILCs and NK1.1+ILCs, our NK1.1 depletion experiments indicate that NK1.1−ILCs rather then NK1.1+ cells have a major impact in pathogenesis of C.jejuni-induced colitis. Dissecting the relative contribution of ILCs and NK cells in mice with selective NK cell deficiency will be important for further studies of campylobacteriosis.
Transcriptional analyses revealed extensive heterogeneity and plasticity among ILC subsets24,26,31. Although the in vivo conditions that promote ILC conversion are still poorly understood, in vitro experiments showed that ILC3 > ILC1 transition can be stimulated by IL-15, IL-18, and IL-128,19,30. Although our results with NKp46-RORγt mice suggest that NKp46+ ILC3s are not sufficient to drive C. jejuni-induced pathology, cell fate mapping experiments indicate that majority of IFNγ-producing Lin−NK1.1−ILCs have a history of RORγt expression, and therefore represent “ex-ILC3s”. Surprisingly, these cells lack NK1.1 and only partially express NKp46 despite increased T-bet and reduced RORγt expression. In addition to ex-ILC3s, we also do not exclude the role of IFNγ-producing ILC1s in C. jejuni-induced intestinal pathology. In line with our results, recent studies demonstrate that NKp46 expression in RORγt+ILC3s is unstable and can be lost in adult mice during antibiotic treatment suggesting that signals from commensal microbiota may regulate this cell population31,42,43. Additionally, previous study described that both NKp46+RORγt+ and NKp46−RORγt+ ILCs can downregulate RORγt to induce T-bet-dependent IFNγ production in response to Salmonella enterica infection19.
In conclusion, we have described the plasticity between RORγt+ILC3 and NK1.1−ILC1 populations induced by C. jejuni. We propose that induction of IL-12, IL-18, and IL-15 cytokines by C. jeuni infection can drive the conversion of RORγt+ILC3s towards IFNγ-producing NK1.1−T-bet+ILC3s (“ex-ILC3s”) by upregulating T-bet expression and downregulating RORγt expression. Thus, ex-ILC3s produce IFNγ thereby promoting intestinal inflammation. It is tempting to speculate that C. jejuni-induced dysregulation of ILCs is beneficial for the pathogen to induce inflammation, tissue damage, and diarrhea, thereby facilitating dissemination and spread to new hosts.
Stimulation of RORγt+ILC3-facilitated protective responses mediated by IL-22 and IL-2 may provide a therapeutic potential to ameliorate the intestinal inflammation and promote mucosal healing in IBD21,44. On the other side, ILC3 plasticity induced in response to mucosal bacterial infection can promote ILC3 to ILC1 conversion leading to IFNγ-dependent intestinal pathology3,19. Therefore, context-dependent strategies are required for optimal targeting of ILCs responses in intestinal disease.
Thus, our findings demonstrate that Campylobacter infection induces a population of IFNγ+NK1.1−ILCs that exhibits ILC3 to ILC1 phenotypic plasticity and promotes IFNγ-dependent intestinal pathology.
Materials and methods
Mice
All animal studies were conducted in accordance with the Trudeau Institute and the University of Texas Health Science Center at San Antonio Animal Care and Use guidelines. Six to twelve-week-old male and female mice were used for experiments. The following mice were obtained from The Jackson Laboratory (Bar Harbor, ME) and housed under specific pathogen-free conditions: C57Black/6 wild-type (WT), B6.129S7-Rag1tm1Mom/J23,45, B6.129S4-IFNγtm3.1Lky/J)23,45, B6.129P2-Tcrbtm1MomTcrdtm1Mom/J45, B6.129P2-IL10tm1Cgn/J46, RORγt-Cre35, ROSA26-YFP34, Tbx21tm2Srnr47, Rorctm3Litt48. RAG−/−γc−/− mice (B10; B6-Rag2tm1FwaIl2rgtm1Wjl) were from Taconic (Germantown, NY). NKp46-Cre mice were previously described49.
In vivo antibody treatments
In vivo antibodies (BioXCell, West Lebanon, NH) are listed in Supplementary Table 1 and were administered by i.p. injections. To antagonize IL-10Rα in vivo, mice were treated with 350 μg of clone 1B1.3 A 12–16 h prior to infection and again 1, 4, and 7 days after infection. To deplete NK cells, mice were given 250 μg of clone PK136 using the same schedule as IL-10Rα blockade. T cells were depleted by administering 500 μg of clone GK1.5 and clone 2.43 one day prior to infection and again one day after infection. To deplete ILCs in TCRβ/δ−/−IL-10−/− mice, 250 μg of clone 30H12 was given starting the day of infection and again on days 2, 4, 6, and 8 post infection. IFNγ was neutralized by administering 500 μg clone XMG1.2 one day prior to infection and again on days 3 and 7 post infection.
Bacterial growth and infections
Frozen stock cultures of C. jejuni NCTC 11168 were streaked onto Mueller Hinton (MH) agar and grown at 37 °C microaerobically (85% N2, 10% CO2, 5% O2) for 48 h. Several colonies were picked from the initial growth and subcultured on MH agar for 24–28 h. Mice were pre treated with an antibiotic cocktail for 7 days in the drinking water as previously described50. One day after the antibiotics water was removed, mice were inoculated with either MH broth or 1−5 × 109 CFU C. jejuni by gavage in a 200 μl volume. The infection dose was confirmed for each experiment by plating serial dilutions onto MH agar.
Evaluation of colitis and enumeration of C. jejuni
Ten days after initiating infection, mice were sacrificed, and the severity of colitis was assessed by histology and colonic mass-to-length ratio. Resected colons were cut longitudinally, the feces were removed, and the colon’s length and mass recorded. Swiss rolls were prepared from the proximal half of the colons and fixed in 10% neutral buffered formalin (Fisher Scientific). Colons were then embedded in paraffin, sectioned and stained with hematoxylin and eosin (Richard Allan Scientific). Stained sections were evaluated for characteristics of inflammation and images were taken with a Zeiss Axiophot2 (Thornwood, NY). For histopathologic scoring, evaluated parameters were: crypt architecture, mucosal thickening, cellular infiltrates, hemorrhage, goblet cell depletion, and tissue damage. Each parameter was scored within a range from 0, indicating no marked change from normal, to 3, indicating severe change. The overall histological score was calculated as the sum of the scores of individual parameters. Colon-associated C. jejuni was enumerated by plating serial dilutions onto MH agar as previously described51. Briefly, the distal half of the colon was cut longitudinally and washed three times in sterile PBS. The washed tissue was finely chopped and incubated in 0.5 ml of MH broth containing 375U Type XI collagenase and 15 ug Type IV DNase for 1 h microaerobically. After incubation, the digested tissue was homogenized by passing through a large pipette tip, serially diluted in PBS and plated. Plates were incubated at 42 °C for 48 h before counting.
Isolation of lamina propria leukocytes
The cecum and colon were cut open and rinsed twice in PBS to remove feces. The tissue was finely chopped and incubated in complete medium (DMEM supplemented with 10% FCS, 1 mM sodium pyruvate, 0.1 mM nonessential amino acids, 1 mM penicillin-streptomycin, 55 μM 2-mercaptoethanol) containing 2 mM EDTA for 30 min at 37 °C. After vortexing, the remaining tissue was collected and digested in serum-free medium containing 375U Type XI collagenase for 40 min at 37 °C. The digested tissue was passed through a mesh screen, washed with PBS containing 2% FCS and separated by a 60/40% Percoll gradient. Cells were collected at the interphase, washed, and viable cells counted by Trypan blue exclusion.
Flow cytometry
Antibodies used for flow cytometry are listed in Supplementary Table 1. A lineage cocktail included antibodies against CD5, CD3, CD11b, CD11c, B220, GR1, Ter119, unless specified differently. Single-cell suspensions (1 × 106 cells) were stimulated with 100 ng/ml PMA and 1.5 μg/ml ionomycin with 5 μg/ml brefeldin A (all from Sigma-Aldrich) in complete medium for 4 h. After stimulation, cells were stained for viability with LIVE/DEAD® (Invitrogen, Carlsbad, CA) for 20 min on ice. After washing, the FcR was blocked with 1 μg of mAb clone 2.4G2 (BioXCell) and stained for surface antigens for 30 min on ice. For intracellular cytokine staining, cells were fixed in 4% paraformaldehyde for 20 min on ice, washed, then stained with cytokine antibodies in permeabilization buffer (0.1% NP-40, 2% heat-inactivated FCS in PBS) for 30 min on ice. For nuclear staining, washed cells were fixed and permeabilized with Foxp3/Transcription Factor Permeabilization buffer (eBioscience) for 1 h at 4 °C. Data was acquired on either a FACSCanto II or LSRII (BD Biosciences) and analyzed with FlowJo (FlowJo LLC).
Adoptive transfer of ILCs
Isolated ILCs from cecum and MLN from C. jejuni-infected TCRβ/δ−/−IL-10−/− mice were sorted and 1×104 purified ILCs were adoptively transferred by i.v. injection into RAG−/−γc−/− recipients treated with IL-10Rα-blocking mAb one day prior to C. jejuni infection.
RNA-seq analysis
cLP and MLN cells were isolated from IFNγ-reporter mice as described above. ILCs, defined as CD45+Lin−CD3−CD90.2+IFNγ+ (Lineage: CD11c, B220, GR1, Ter119) were sort purified using FACSAria (BD Biosciences) and RNAs from the sorted cells were isolated using RNeasy Micro kit (Qiagen), following the manufacturer’s instructions. RNA integrity was determined using Fragment Analyzer (Agilent, Santa Clara, CA) prior to library preparation. RNA-seq libraries were prepared according to SMART-seq2 protocol52, with the following modifications: PCR preamplification to 15 cycles, two rounds beads cleanup with 1:1 ratio after cDNA synthesis, and 0.6-0.8 dual beads cleanup for Nextera XT DNA-seq library purification. RNA-seq libraries were sequenced using Illumina HiSeq 3000 system (Illumina, San Diego, CA) with 54 bp single-read sequencing module. Upon sequencing completion, short-read sequences from RNAseq were first aligned to UCSC mm9 genome build using TopHat2 aligner53 and then quantified for gene expression by HTSeq54 to obtain raw read counts per gene, and then converted to RPKM (Read Per Kilobase of gene length per Million reads of the library). The transcriptional identity of IFNγ+ILCs was analyzed in comparison with available RNAseq data (GSE85154)24. Signature genes of different ILC subsets from intestine24 were analyzed using CIBERSORT method28.
Real-time reverse transcriptase PCR analysis
RNA from colon was isolated using E.Z.N.A. Total RNA kit I (Omega Bio-tek). cDNA synthesis and real-time RT-PCR were performed as described previously55. PCR primers are listed in Supplementary Table 2.
Quantification of secreted cytokines
MLNs were collected and processed into single-cell suspensions by passing through a fine gauge screen. Antigens for restimulation were prepared by harvesting C. jejuni bacterial lawns in PBS and heat inactivated by incubation at 65 °C for 1.5 h. Protein concentration was determined by bicinchoninic acid assay (Thermo Scientific, Rockford IL). Bacteria were confirmed to be non-culturable by plating on agar. 1 × 106 test cells were cultured with an equal number of heat-killed bacteria (10 μg/ml of protein). Cytokines secreted into the supernatant were assayed by capture ELISA after 48 h (IFNγ) using commercially available antibodies (IFNγ: BD Biosciences, San Jose CA).
Statistical analysis
Results are expressed as mean±S.E.M. Differences between groups were evaluated by two-tailed t test after assessing assumptions of normality and variance by D’Agostino Omnibus and F tests, and visualization of residual and Q-Q plots. Student’s t-test or two-way analysis of variance (ANOVA) were performed when the dataset met statistical assumptions. Nonparametric analyses (Mann–Whitney and Welch’s correction) were performed when statistical assumptions were not valid. All statistical computations were performed using GraphPad Prism 8 program. ns – not significant, *p < 0.05, **p < 0.01, ***p < 0.001.
Change history
30 August 2021
A Correction to this paper has been published: https://doi.org/10.1038/s41385-021-00449-9
References
Vivier, E. et al. Innate lymphoid. Cells.: 10 Years. Cell 174, 1054–1066 (2018).
Artis, D. & Spits, H. The biology of innate lymphoid cells. Nature. 517, 293–301 (2015).
Colonna, M. Innate lymphoid cells: diversity, plasticity, and unique functions in immunity. Immunity. 48, 1104–1117 (2018).
Klose, C. S. N. et al. Differentiation of type 1 ILCs from a common progenitor to all helper-like innate lymphoid cell lineages. Cell 157, 340–356 (2014).
Tumanov, A. V. et al. Lymphotoxin controls the IL-22 protection pathway in gut innate lymphoid cells during mucosal pathogen challenge. Cell Host Microbe. 10, 44–53 (2011).
Sonnenberg, G. F. et al. Innate lymphoid cells promote anatomical containment of lymphoid-resident commensal bacteria. Science. 336, 1321–1325 (2012).
Abt, M. C. et al. Innate immune defenses mediated by two ilc subsets are critical for protection against acute clostridium difficile infection. Cell Host Microbe. 18, 27–37 (2015).
Fuchs, A. et al. Intraepithelial type 1 innate lymphoid cells are a unique subset of IL-12- and IL-15-responsive IFN-gamma-producing cells. Immunity. 38, 769–781 (2013).
Vonarbourg, C. et al. Regulated expression of nuclear receptor RORgammat confers distinct functional fates to NK cell receptor-expressing RORgammat(+) innate lymphocytes. Immunity. 33, 736–751 (2010).
Bernink, J. H. et al. Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nat. Immunol. 14, 221–229 (2013).
Li, J., Doty, A. L., Iqbal, A. & Glover, S. C. The differential frequency of Lineage(-)CRTH2(-)CD45(+)NKp44(-)CD117(-)CD127(+)ILC subset in the inflamed terminal ileum of patients with Crohn’s disease. Cell Immunol. 304-305, 63–68 (2016).
O’Brien, S. J. The consequences of Campylobacter infection. Curr. Opin. Gastroenterol. 33, 14–20 (2017).
Al-Banna, N. A., Cyprian, F. & Albert, M. J. Cytokine responses in campylobacteriosis: linking pathogenesis to immunity. Cytokine Growth Factor Rev. 41, 75–87 (2018).
Mansfield, L. S. et al. C57BL/6 and congenic interleukin-10-deficient mice can serve as models of Campylobacter jejuni colonization and enteritis. Infect. Immun. 75, 1099–1115 (2007).
Malik, A., Sharma, D., St Charles, J., Dybas, L. A. & Mansfield, L. S. Contrasting immune responses mediate Campylobacter jejuni-induced colitis and autoimmunity. Mucosal. Immunol. 7, 802–817 (2014).
Sun, X., Liu, B., Sartor, R. B. & Jobin, C. Phosphatidylinositol 3-kinase-gamma signaling promotes Campylobacter jejuni-induced colitis through neutrophil recruitment in mice. J. Immunol. 190, 357–365 (2013).
Mansfield, L. S. et al. Genetic background of IL-10(-/-) mice alters host-pathogen interactions with Campylobacter jejuni and influences disease phenotype. Micro. Pathog. 45, 241–257 (2008).
Keubler, L. M., Buettner, M., Hager, C. & Bleich, A. A multihit model: colitis lessons from the interleukin-10-deficient mouse. Inflamm. Bowel Dis. 21, 1967–1975 (2015).
Klose, C. S. et al. A T-bet gradient controls the fate and function of CCR6-RORgammat+ innate lymphoid cells. Nature 494, 261–265 (2013).
Neurath, M. F. Cytokines in inflammatory bowel disease. Nat. Rev. Immunol. 14, 329–342 (2014).
Goldberg, R., Prescott, N., Lord, G. M., MacDonald, T. T. & Powell, N. The unusual suspects-innate lymphoid cells as novel therapeutic targets in IBD. Nat. Rev. Gastroenterol. Hepatol. 12, 271–283 (2015).
Tait Wojno, E. D., Hunter, C. A. & Stumhofer, J. S. The immunobiology of the interleukin-12 family: room for discovery. Immunity 50, 851–870 (2019).
Reinhardt, R. L., Liang, H. E. & Locksley, R. M. Cytokine-secreting follicular T cells shape the antibody repertoire. Nat. Immunol. 10, 385–393 (2009).
Gury-BenAri, M. et al. The spectrum and regulatory landscape of intestinal innate lymphoid. Cells Are Shaped Microbiome. Cell 166, 1231–1246 e1213 (2016).
Cella, M. et al. Subsets of ILC3-ILC1-like cells generate a diversity spectrum of innate lymphoid cells in human mucosal tissues. Nat. Immunol. 20, 980–991 (2019).
Robinette, M. L. et al. Transcriptional programs define molecular characteristics of innate lymphoid cell classes and subsets. Nat. Immunol. 16, 306–317 (2015).
Pokrovskii, M. et al. Characterization of transcriptional regulatory networks that promote and restrict identities and functions of intestinal innate lymphoid cells. Immunity 51, 185–197 e186 (2019).
Newman, A. M. et al. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 12, 453–457 (2015).
Kwong, B. et al. T-bet-dependent NKp46(+) innate lymphoid cells regulate the onset of TH17-induced neuroinflammation. Nat. Immunol. 18, 1117–1127 (2017).
Bernink, J. H. et al. Interleukin-12 and -23 control plasticity of CD127(+) Group 1 and Group 3 innate lymphoid cells in the intestinal lamina propria. Immunity 43, 146–160 (2015).
Melo-Gonzalez, F. & Hepworth, M. R. Functional and phenotypic heterogeneity of group 3 innate lymphoid cells. Immunology 150, 265–275 (2017).
Seo, G. Y. et al. LIGHT-HVEM signaling in innate lymphoid cell subsets protects against enteric bacterial infection. Cell Host Microbe 24, 249–260 e244 (2018).
Song, C. et al. Unique and redundant functions of NKp46+ ILC3s in models of intestinal inflammation. J. Exp. Med 212, 1869–1882 (2015).
Srinivas, S. et al. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev. Biol. 1, 4 (2001).
Eberl, G. et al. An essential function for the nuclear receptor RORgamma(t) in the generation of fetal lymphoid tissue inducer cells. Nat. Immunol. 5, 64–73 (2004).
Sun, Z. et al. Requirement for RORgamma in thymocyte survival and lymphoid organ development. Science 288, 2369–2373 (2000).
Crim, S. M. et al. Preliminary incidence and trends of infection with pathogens transmitted commonly through food - Foodborne Diseases Active Surveillance Network, 10 U.S. sites, 2006-2014. MMWR Morbidity Mortal. Wkly. Rep. 64, 495–499 (2015).
Kullberg, M. C. et al. Helicobacter hepaticus triggers colitis in specific-pathogen-free interleukin-10 (IL-10)-deficient mice through an IL-12- and gamma interferon-dependent mechanism. Infect. Immun. 66, 5157–5166 (1998).
Schroder, K., Hertzog, P. J., Ravasi, T. & Hume, D. A. Interferon-gamma: an overview of signals, mechanisms and functions. J. Leukoc. Biol. 75, 163–189 (2004).
Tribble, D. R. et al. Assessment of the duration of protection in Campylobacter jejuni experimental infection in humans. Infect. Immun. 78, 1750–1759 (2010).
Mackley, E. C. et al. CCR7-dependent trafficking of RORgamma(+) ILCs creates a unique microenvironment within mucosal draining lymph nodes. Nat. Commun. 6, 5862 (2015).
Viant, C. et al. Transforming growth factor-beta and Notch ligands act as opposing environmental cues in regulating the plasticity of type 3 innate lymphoid cells. Sci. Signal 9, ra46 (2016).
Verrier, T. et al. Phenotypic and functional plasticity of murine intestinal NKp46+ group 3 innate lymphoid cells. J. Immunol. 196, 4731–4738 (2016).
Zhou, L. et al. Innate lymphoid cells support regulatory T cells in the intestine through interleukin-2. Nature 568, 405–409 (2019).
Mombaerts, P. et al. RAG-1-deficient mice have no mature B and T lymphocytes. Cell 68, 869–877 (1992).
Kuhn, R., Lohler, J., Rennick, D., Rajewsky, K. & Muller, W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell 75, 263–274 (1993).
Intlekofer, A. M. et al. Anomalous type 17 response to viral infection by CD8+ T cells lacking T-bet and eomesodermin. Science 321, 408–411 (2008).
Eberl, G. & Littman, D. R. Thymic origin of intestinal alphabeta T cells revealed by fate mapping of RORgammat+ cells. Science 305, 248–251 (2004).
Narni-Mancinelli, E. et al. Fate mapping analysis of lymphoid cells expressing the NKp46 cell surface receptor. Proc. Natl Acad. Sci. USA 108, 18324–18329 (2011).
Sun, X., Threadgill, D. & Jobin, C. Campylobacter jejuni induces colitis through activation of mammalian target of rapamycin signaling. Gastroenterology 142, 86–95 e85 (2012).
Muraoka, W. T. & Zhang, Q. Phenotypic and genotypic evidence for L-fucose utilization by Campylobacter jejuni. J. Bacteriol. 193, 1065–1075 (2011).
Picelli, S. et al. Full-length RNA-seq from single cells using Smart-seq2. Nat. Protoc. 9, 171–181 (2014).
Kim, D. et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36 (2013).
Anders, S., Pyl, P. T. & Huber, W. HTSeq-a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015).
Wang, Y. et al. Lymphotoxin beta receptor signaling in intestinal epithelial cells orchestrates innate immune responses against mucosal bacterial infection. Immunity 32, 403–413 (2010).
Acknowledgements
This research was supported by grants from NIH (AI135574, AI111000), Pfizer (WI215053), and Trudeau Institute. WTM was supported by USDA NIFA (2014-67012-22276). AVT was supported by the Crohn’s and Colitis Foundation (SRA#294083), the Max and Minnie Tomerlin Voelcker Fund, and by the William and Ella Owens Medical Research Foundation. We thank Dr. Joseph Sun (MSKCC) for providing NKp46-Cre mice. Genome Sequencing Facility at the Greehey Children’s Cancer Research Institute is supported with funding from NIH (NCI P30 CA54174, CTSA 1UL1 RR025767-01, and 1S10 OD021805-01), and from Cancer Prevention and Research Institute of Texas (RP160732) with support to YC, and ZL. The E.V. laboratory at CIML and Assistance-Publique des Hôpitaux de Marseille is supported by funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (TILC, grant agreement No. 694502), the Agence Nationale de la Recherche including the PIONEER Project (ANR-17-RHUS-0007), MSDAvenir, Innate Pharma and institutional grants to the CIML (INSERM, CNRS, and Aix-Marseille University) and to Marseille Immunopole. Data was generated in the Flow Cytometry Shared Resource Facility which is supported by UT Health San Antonio, NIH-NCI P30 CA054174-20 (CTRC at UTHSCSA) and UL1 TR001120 (CTSA grant).
Author information
Authors and Affiliations
Contributions
Study concept and design: WTM and AVT. Designed and performed experiments, analyzed data, wrote manuscript: WTM, AAK, EPK, and AVT. Performed experiments: QX, SAS, XJ, and LWK. Provided important experimental materials: MM and EV. Performed RNA seq and bioinformatics analysis: ZL, KW, L-JW, and YC.
Corresponding author
Ethics declarations
Competing interests
Eric Vivier is an employee of Innate Pharma.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Muraoka, W.T., Korchagina, A.A., Xia, Q. et al. Campylobacter infection promotes IFNγ-dependent intestinal pathology via ILC3 to ILC1 conversion. Mucosal Immunol 14, 703–716 (2021). https://doi.org/10.1038/s41385-020-00353-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41385-020-00353-8
