
Abstract
Ferric chloride (FeCl3) has widespread use as an etchant in the PCB industry and for photochemical machining. Although the corrosion process of copper in FeCl3 is well known, the use of organic inhibitors of copper corrosion in FeCl3 solutions has not been reported. Such inhibition may allow microfabrication of special PCB designs and electrochemical sensors. Here, we investigate the inhibition of copper corrosion by FeCl3 (0.10 or 2.46 M) solutions with organic inhibitors. The most promising inhibitors are identified, investigated in 0.01 M FeCl3 solution using electrochemical techniques at different temperatures and exposure times, and the modified surfaces of copper are characterized by a variety of techniques in order to determine the inhibition mechanism. 5-methyl-1H-benzotriazole (MBTA, 65 mM) and 1H-benzotriazole (BTA, 65 mM) are found to be the most attractive inhibitors, with inhibition efficiencies between 96.5% and 99.5% at room temperature, depending on the exposure time and the measurement technique.
Similar content being viewed by others
Introduction
The electro-dissolution of pure copper in chloride-containing aqueous solutions is often described by the following reactions1,2:
These reactions are generally believed to be reversible1. High-chloride concentrations stabilize the Cu(I) ion in the form of the complex anions \({\mathrm{CuCl}}_2^ -\) and \({\mathrm{CuCl}}_3^{2 - }\)3,4, and the Cu(II) ion in a \({\mathrm{CuCl}}_4^{2 - }\) complex4. The uncomplexed Cu(I) cuprous ion is colorless, while the uncomplexed Cu(II) cupric ion is light blue5. Complexation often results in change in the color of the solution. Insoluble CuCl is an intermediate that can deactivate the electrode surface4. In contrast, complexes of Cu(I) may be soluble. Soluble hydrolyzed complexes may also form. Hydrolysis is pH dependent, so are the solubility and stability of surface films of copper oxide and copper chloride1. At sufficiently low pH, copper dissolution is accompanied by hydrogen evolution, and with increasing chloride concentration—the stability of the solid phase CuCl2·3Cu(OH)2 increases compared to Cu2O and CuO3. At potentials close to the corrosion potential, the kinetics of the anodic reaction is typically under mixed charge transfer and mass transport control, where the rate-determining step is the transport of a cuprous chloride complex away from the metal surface to the bulk electrolyte1,2. The formation of surface films generally reduces the rate of both anodic and cathodic reactions1. Pourbaix diagrams of copper in aqueous chloride solutions at different temperatures have been drawn1,6,7. For 0.01 M FeCl3 at pH = 2.5 and T = 25 °C (the conditions of the electrochemical tests in this work), solid Cu, \({\mathrm{CuCl}}_2^ -\), or Cu2+ can be expected, depending on the potential3. However, it should be borne in mind that these diagrams only reflect thermodynamics, and that they do not account for the effect of soluble copper complexes (or ligands), which might affect equilibria5,8.
Printed circuit boards (PCBs) are an integral part of the microelectronics industry and are used in a wide range of electronic devices. The conductors on PCBs are usually made of copper due to its excellent electrical and thermal conductivities9. The layout of conductors on the board is formed by a chemical etching process in which a concentrated ferric chloride (FeCl3) solution is utilized for the complete etching of the unprotected Cu film, which was pre-deposited on the board. High concentration of FeCl3 (often, either 4.11 M or 2.46 M) is used to achieve high manufacturing rates of industrial processes; for a fresh solution heated to 40 °C, the etching time is typically 10–15 min9.
The dissolution of FeCl3 in water follows the reaction:
which is accompanied by increase in acidity. The oxidation of Cu in FeCl3 solutions10,11 is attributed to the redox reaction between the ferric ion (Fe3+), which acts as a strong oxidizer, and Cu—the reducing agent, following Eqs. (7) and (8)1:
This redox reaction, however, is very fast and cannot be controlled easily. Once all areas not protected by resist have been cleared of Cu, the board should be immediately removed from solution, otherwise “undercutting” of the resist and consequent thinning of tracks and pads will occur9. FeCl3-based solutions are also used as etchants of Cu, Al, Ni, and Fe in industrial photochemical machining10.
In special cases where only thinning of Cu on board is required, the reaction rate must be reduced significantly. For example, in cheap disposable PCBs there is economic advantage to fabricate trace fuses directly on the board using the originally deposited Cu. The electrical characteristics of the trace fuse are determined by the cross-section of the Cu line. A very sensitive trace fuse can be produced by developing a slow, controllable etching process, which will allow manipulation of the line thickness12,13. Another potential application for organic inhibitors of Cu corrosion in FeCl3 solutions is in electrochemical time sensors, which are based on increase in the resistance of the reduced Cu line cross-section.
There are two possible ways to decrease the corrosion rate of Cu in FeCl3 solutions: (i) decrease the solution concentration, and (ii) use corrosion inhibitors8,14,15,16,17,18,19. To the best of our knowledge, the second approach has not been reported before (besides a minor note by Walker20 that 0.12 g L–1 benzotriazole (BTA) in a FeCl3 solution promoted Cu corrosion, while pretreatment of Cu in a hot solution of 5 g L–1 BTA gave 40% inhibition efficiency in solution of FeCl3).
Some organic compounds are known to be good corrosion inhibitors of Cu and its alloys. They are often non-toxic, eco-friendly, and cost-effective. A major group of organic corrosion inhibitors consists of compounds containing heterocyclic aromatic rings with polar atoms such as nitrogen, oxygen and sulfur, which have a lone pair of electrons that establish chemical bonds with the metal surface. This process allows the adsorption of a hydrophobic protective layer that blocks the corrosion sites21,22,23. Commonly studied Cu corrosion inhibitors that rely on an adsorption mechanism include amino acids24, triazoles17,25,26,27,28, imidazoles29,30,31, and surfactants17,32. A wide variety of amino acids have been tested as corrosion inhibitors under acidic conditions17,24.
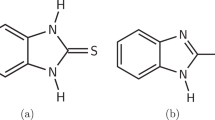
Different studies have demonstrated the ability of triazoles to inhibit corrosion with high efficacy under acidic conditions8. The most common triazole compounds for corrosion inhibition are BTA and its derivatives23,33. These inhibitors have heterocyclic structure that contains three nitrogen atoms. While the inhibition ability of these materials is based on the adsorption of a protective layer over the metal surface, the precise adsorption mechanism is still debated21,23. 5-methyl-1H-benzotriazole (MBTA) is a mixed-type inhibitor, although its anodic reaction inhibition has been claimed more dominant21. Another organic compound that can inhibit corrosion of metals in acidic media is 2-mercaptobenzimidazole (MBI)17. MBI is heterocyclic organic compound that contains a mercapto group with sulfur atom. Some mechanisms suggest that the sulfur atom is strongly adsorbed on the metal surface, while others claim that compounds containing mercapto groups can form insoluble protective complexes, which protect the metal from corrosion29,34,35. Several surfactants inhibit corrosion via aggregation at metal/solution interfaces27. Cetyltrimethylammonium bromide (CTAB) is a known cationic surfactant that provides corrosion inhibition to certain metals in acidic environments8. One of the most investigated amino acids is Tryptophan (Trp), which acts as a mixed-type inhibitor17,36. The mixture of CTAB and Trp has been claimed to mainly inhibit the anodic reaction37.
Although the corrosion reaction between Cu and FeCl3 is well known, the use of organic inhibitors in FeCl3-based solutions has not been reported. The objective of this study is thus to study the effect of organic inhibitors on the corrosion of Cu in FeCl3 solutions by gravimetric and electrochemical measurements. After screening out some organic and inorganic inhibitors (see Supplementary Table 1), four corrosion inhibitors are evaluated: BTA, MBTA, MBI, CTAB + Trp. The inhibition efficiency of these inhibitors is assessed using common, reliable techniques31. Successful inhibition of the Cu dissolution reaction may pave the way to microfabrication of special PCB designs where partial Cu removal is required.
Results
Mass loss measurements
Mass loss measurements were made for inhibitor screening and corrosion rate assessment in FeCl3 solution. Measurements were acquired for the 2.46 M FeCl3 solution, both with and without organic inhibitors. For comparison, mass loss measurements were also made for 0.10 M FeCl3-based solutions.
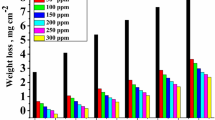
Figure 1 shows the normalized mass loss of copper coupons as a function of immersion time. As expected, the addition of organic inhibitors resulted in a decrease in mass loss for all exposure times. For further analysis, both the corrosion rate and the inhibition efficiency (η%) were calculated. The values obtained this way are listed in Table 1.

a 2.46 M and b 0.10 M. Note the different ranges of the abscissae. Error bars represent the standard deviation of three measurements.
Copper complexes and kinetics of electrode reactions
Different solution colors may indicate different oxidation states of copper (or other) ions in solution (e.g., due to splitting of the 3d subshell and absorption of visible light photons) or formation of complexes with either organic or inorganic ligands. Changes in the coordination number or shape of the complex may also be noticeable this way7,38. In this work it was decided not to use coloring reagents in order to determine the oxidation state/s of the copper ion/s in solution because such reagents would need to compete with other copper-binding ligands that exist in the studied solutions. Spectrophotometric study by other means was not conducted either. Yet, based on visual examination it can be reported that the color of solution changed when adding the inhibitors to copper-free FeCl3 solutions, from orange-brown of the blank solution, gradually darker orange-brown of the BTA- and MBTA-containing solutions, dark brown of the CTAB + Trp solution, and translucent-white of the MBI-containing solution. This indicated on the formation of a distinct iron-based complex in the MBI-containing solution.
Within the potential range of –0.4 to +0.9 V vs. sat’d Ag/AgCl, cyclic voltammetry (CV) revealed only one, reduction peak on glassy carbon working electrode in an uninhibited 0.01 M FeCl3, at –0.045 V (ν = 30 mV s–1), see Fig. 2a. Since in acidic conditions the inert potential region for glassy carbon is ca. –0.77 to +1.41 V vs. sat’d Ag/AgCl39, this peak may be associated with the Fe3+/Fe2+ redox reaction. This was further supported by a characteristic increase in peak height when the concentration of FeCl3 was increased. There was no difference in the shape of the voltammograms on glassy carbon electrode when a fresh blank solution was used compared to when a solution aged for 4 days was used.

a On glassy carbon working electrode. b On copper working electrode.
After immersion of copper sheet in the solution for one day and its removal shortly before the CV experiment began, the voltammogram also shown in Fig. 2a was constructed. Here, two reduction peaks (at –0.227 and +0.007 V) and one large oxidation peak (at +0.101 V) were evident. Similar results were obtained when using platinum working electrode. Under potentiostatic conditions of –0.2 V for 10 min, Cu was deposited on the Pt electrode. It can thus be concluded that the reduction peak at –0.227 V vs. sat’d Ag/AgCl is associated with the reduction of Cu+ to Cu0. When the immersion period of the copper sheet in solution was increased from one day to four days, two reduction peaks (at –0.151 and +0.014 V) and two oxidation peaks at (+0.111 and +0.162 V) were observed, see Fig. 2a. Similar results were obtained when repeating the experiment on a gold working electrode. Based on the results above, the reduction peak at –0.151 V should be associated with the reduction of Cu+ to Cu0, while the reduction peak at +0.014 V may be associated with both Fe3+/Fe2+ and Cu2+/Cu+ reduction reactions40. The Cl− anion in solution stabilizes the Cu(I) ion, which may originate from oxidation of Cu. Accordingly, the oxidation peaks at +0.111 V and +0.162 V may be associated with the Cu0/Cu+ and Cu+/Cu2+ oxidation reactions, respectively40.
In an uninhibited solution, the voltammogram of a copper working electrode (Fig. 2b) appeared different than the voltammograms of inert working electrodes in copper ions-containing uninhibited solutions. At a scan rate of 30 mV s–1, only one reduction peak is evident, at –0.080 V. This peak may be associated with either Fe3+/Fe2+ or Cu2+/Cu+ reduction reactions. Addition of 0.065 M BTA to the solution resulted in a significant decrease in the background currents and the appearance of only one oxidation peak, at around +0.03 V.
The Randles–Ševčík equation describes the dependence of the peak current (Ip) on scan rate8. In general, as the scan rate is increased, the size of the diffusion layer decreases, thus higher currents are monitored. Drawing the Ip values in the voltammograms of glassy carbon in an uninhibited solution in which a copper sheet was first immersed for one day both versus ν and versus ν1/2, and fitting linearly with pre-fix intercept equals zero, higher coefficient of determination (R2) values were obtained in the latter case, namely R2 = 0.97773, 0.99114, and 0.99837 for the oxidation peak, less-negative reduction peak, and more-negative reduction peak, respectively. Such a proportionality of the peak current to the square root of the potential scan rate in the absence of stirring can indicate: (1) a Nernstian (reversible) diffusion-controlled process; (2) a totally irreversible process. The reduction peak current in the case of an uninhibited solution and a copper working electrode was also found to be better fitted linearly versus ν1/2 (R2 = 0.98584). In contrast, the addition of 0.065 M BTA to the solution yielded an oxidation peak with a current that was neither linearly dependent on ν nor on ν1/2 (R2 = 0.00598 and 0.11046, respectively).
Polarization measurements
The effect of different organic inhibitors on the corrosion rate of Cu in 0.01 M FeCl3 solution was investigated using potentiodynamic polarization tests (Fig. 3a). Tafel extrapolation was used to determine the corrosion current density (jcorr) and corrosion potential (Ecorr). The resulting values are listed in Table 2.

a Polarization curves of Cu in 0.01 M FeCl3, with and without organic inhibitors. b–d Langmuir adsorption isotherms based on Tafel extrapolation data: b BTA-inhibited, c MBTA-inhibited, d MBI-inhibited.
The adsorption of MBTA, BTA, and MBI on copper surface was found to follow the Langmuir adsorption isotherm:
where c is the concentration of the inhibitor, θ is the fractional surface coverage, and Kads is the adsorption equilibrium constant.
As evident from Fig. 3b–d, the surface coverage increases with increase in the concentration of the three inhibitors, indicating that inhibitor films were forming on the Cu surface. The coefficients of determination of the linear regression are R2 = 0.99973, 0.99958, and 0.99038 for BTA, MBTA, and MBI, respectively, supporting the excellent fit of the Langmuir adsorption mode to the experimental data. The values of the adsorption equilibrium constant can be deduced from the intercept of the straight lines with the ordinate. Thus, one obtains Kads = 340, 400, and 1337 L mol–1 for BTA, MBTA, and MBI, respectively. The standard free energy of adsorption \(\Delta G_{{\mathrm{ads}}}^ \circ\) (J mol–1) can be calculated by
where R is the molar gas constant (8.31447 J mol–1 K–1), T is the thermodynamic temperature (K), and 55.5 is the molar concentration of water in the solution (mol L–1). Thus, one obtains \(\Delta G_{{\mathrm{ads}}}^ \circ\) = –24.4, –24.8, and –27.8 kJ mol–1 for BTA, MBTA, and MBI, respectively. The slopes of the Langmuir isotherms are 0.99178 ± 0.01154, 0.99447 ± 0.01447, and 0.89817 ± 0.06245 for BTA, MBTA, and MBI, respectively. Deviation of the slope from unity may reflect interactions between adsorbed molecules on the surface25. Based on the values above, such interactions, if exist, are more significant in the case of MBI.
The effect of temperature on the corrosion rate of Cu in 0.01 M FeCl3 solution with and without organic inhibitors was studied. The corrosion rate was calculated based on the jcorr values deduced from Tafel extrapolation on potentiodynamic polarization curves constructed at 25, 40, 50, and 60 °C. In Fig. 4a, the dependence of corrosion rate on temperature is shown. To calculate the activation energy, Ea, of the corrosion process, which is required to better understand the differences between inhibition mechanisms, Arrhenius plots were calculated, as shown in Fig. 4b. The linearity of the curves for the blank, BTA-, MBTA-, and MBI-containing solutions (coefficients of determination of the linear regression R2 = 0.93, 0.96, 0.92, and 0.98, respectively) imply that they all follow an Arrhenius behavior. In contrast, the mixture of Trp and CTAB does not follow an Arrhenius behavior. The activation energies and the pre-exponential coefficient, A, deduced from Fig. 4b are tabulated in Table 3.

a Corrosion rate values deduced from potentiodynamic polarization measurements. b Arrhenius plots. Error bars represent the standard deviation of three measurements.
Electrochemical impedance spectroscopy (EIS) measurements were conducted both in uninhibited and in inhibited 0.01 M FeCl3 solutions. Typical results are shown in Fig. 5a in the form of Nyquist plots. All five Nyquist plots reveal only one, slightly depressed, semicircle. Such non-perfect semicircles have been described by a semi-ellipse model and have been attributed, among others, to a non-ideal electrochemical behavior at the electrode/solution interphase due to heterogeneities at the metal surface41,42,43. No capacitive loops are apparent in the Nyquist plots. In addition to the depressed semicircle, a Warburg impedance component is evident at low frequencies in the case of MBI and Trp + CTAB solutions, indicating a diffusion limitation8,44, possibly of soluble copper species from the electrode surface to the bulk solution. In these two cases, a second time constant appeared at low frequencies in the modulus and phase shift Bode plots (not shown herein). It cannot be ruled out that the Warburg component is screened in the cases of BTA and MBTA by some other Faradaic process with the same time constant (namely, the diffusion limitation is always there, but does not show in the EIS spectra).

a Nyquist plots acquired immediately after reaching OCP (t = 0). b, c Electrical equivalent circuits for the electrochemical system without (b) and with (c) Warburg impedance. d Nyquist plots acquired after 3 immersion days.
The Nyquist plots, with and without a Warburg impedance (ZW), can be well simulated using the simple Randles equivalent circuit shown in Fig. 5b. Similar equivalent circuits have been proposed to describe the corrosion of copper-based alloys and steels in sulfide- and chloride-containing environments, with and without inhibitors41,42,43,45. Table 4 summarizes the data extracted from the Nyquist plots and fitted to these equivalent circuits. The solution resistance, RS, is determined based on the displacement of the semicircle from the origin of coordinates (i.e., its intercept with the ReZ axis at high frequency of 100 Hz)42,46. In Fig. 5b, the polarization resistance (Rp) is used, rather than the charge-transfer resistance (Rct). While the latter is related to a single, kinetically controlled electrochemical reaction at equilibrium, without any mixed potential and at constant surface concentrations, the former is measured between the working electrode and a reference electrode in solution, and therefore it may encompass various resistive elements beyond Rct, e.g., the diffuse layer resistance (Rd), adsorbed species resistance (Ra) attributed to adsorbed inhibitor molecules and corrosion products, surface film resistance (Rf), etc.8,41,42,46. Under simplified conditions, where Rd is the only additional contribution, Rd defines the deviation of the measured polarization resistance Rp from the pure Rct, by41:
According to the semi-ellipse model, Rp is the long axis of the ellipse (Rp = 2a) while the maximum complex of the depressed semicircle (\(Z{\prime\prime}_{\max }\)) is half the short axis of the ellipse (i.e., b). As in an ellipse, where the foci are defined by a2 – b2 = c2, Rd and Rct can be calculated according to41:
Their values are given in Table 4. In the presence of redox processes, Rp, and not Rct, is the most closely correlated with the corrosion rate47. In addition, the Rp values obtained from EIS are often higher than those measured by other electrochemical techniques41.
Figure 5b includes a constant phase element (CPE), which represents a non-ideal capacitance and replaces the double-layer capacitance (Cdl) with the aim of better matching the measurement results and the equivalent circuits. The use of CPE takes into consideration surface characteristics such as surface roughness, discontinuity of the inhibitor layer adsorbed on the metal surface, and inhomogeneity in the conductance or dielectric constant, which cause a non-uniform distribution in Cdl32,46,48. Poor surface uniformity may be related, for example, to a pore structure through which electrolyte, ions and neutral molecules could permeate48. The presence of depressed semicircles supports the use of CPE. In the case of MBI and Trp + CTAB solutions, the straight line at low frequencies actually has a slope slightly lower than unity (or, a phase shift of 45°): 0.911 ± 0.011 (R2 = 0.998) and 0.913 ± 0.010 (R2 = 0.999), respectively. This behavior is usually attributed to either microscopic surface roughness or to slow adsorption of ions and chemical inhomogeneity of the surface, thus further supporting the use of CPE.
The robustness of the different inhibitors and the development of the layer that they may form on the surface was evaluated from EIS measurements, up to 72 h. Comparing Fig. 5d to Fig. 5a, it is apparent that the polarization resistance of all samples increased from t = 0 to t = 72 h, although in the case of MBTA and Trp + CTAB it was not monotonic. From Table 4 it is evident that for Cu in an uninhibited solution, Rp increased by a factor of ~1.9. For an activation-controlled process, this would mean a decrease in the corrosion current density by this factor. This may be due to the formation of either an oxide layer or an insoluble CuCl layer on the surface. In the blank solution, Rp and Rd increased monotonically while RS and Rct decreased monotonically over time. In both MBTA- and BTA-inhibited solutions, Rp increased by a higher factor of ~2.5. The maximal inhibition efficiency was achieved, in both cases, after 24 h. No degradation in the inhibition efficiency occurred during 72 h. All of these indicate on the formation of an effective, protective surface layer. However, while in the case of MBTA the changes in Rp, Rct, RS, and Rd were not monotonic over time, in the case of BTA, Rp, Rct and, Rd all increased monotonically over time. In the case of an MBI-inhibited solution, Rp increased by a slightly lower factor of ~2.3. The maximal inhibition efficiency was also achieved after 24 h. However, between 48 and 72 h, a minor decrease in inhibition efficiency was evident. While Rp increased monotonically over time, the changes in RS, Rct, and Rd were not monotonic. The lack of Warburg impedance in Fig. 5c (both MBI and Trp + CTAB inhibited solutions) is related to its shift to lower frequencies. In a measurement not shown herein it was observed when extending the low frequency range from 0.01 Hz to 0.001 Hz. The behavior over time of Cu in Trp + CTAB inhibited solution was much different. In this case, Rp increased by a factor of only ~1.1, and mainly in the first 24 h, and the inhibition efficiency consistently decreased over time (~21% decrease in 72 h). However, the changes in Rp, RS, Rct, and Rd were not monotonic over time.
Surface characterization
The protective layers formed on the Cu surface due to exposure to inhibited FeCl3 solutions were identified by Fourier-transform infrared (FTIR) spectroscopy. Figure 6a shows a typical FTIR mapping (after exposure to BTA-inhibited solution, in this case) of the Cu surface. The different colors in this figure represent the different magnitude of the same compound characteristic peaks in the FTIR spectrum—the red color representing the highest amount of inhibitor, the blue color representing no inhibitor. The FTIR spectra of the different samples are shown in Fig. 6c–f. Three different zones are revealed on the surface in Fig. 6a,b: (A) is the area that was in direct contact with the inhibited solution, (B) is the boundary between the exposed area and the 3 M tape-masked area, and (C) is the area protected by the 3 M tape from the solution. Zone A is covered with a homogenous layer of organic compound. This compound is identified as BTA in the FTIR spectrum in Fig. 6c. In Zone B, less organic compound is present due to limited penetration of the inhibitor through the interphase boundary. In Zone C, no organic compound is found.

a FTIR mapping of the area shown in b after exposure to BTA-inhibited solution. Scale bar, 100 μm. c–f FTIR spectra following immersion in c BTA, d MBTA, e MBI, and f Trp + CTAB solutions. g, i, k, m, o Topography and h, j, l, n, p Volta potential difference scans obtained using SKP measurements on Cu surfaces after 2-week immersion in FeCl3 solutions either uninhibited (g, h), or BTA- (i, j), MBTA- (k, l), Trp + CTAB (m, n), and MBI- (o, p) inhibited solutions. In all scans, the etched zone of the sample is on the left-hand side (lower x) while the non-etched zone is on the right-hand side. The grid lines on the color maps depict the actual tip scanning step size.
The spectrum of BTA, Fig. 6c, includes absorbance bands at 1210 and 740 cm–1, which represent the C–H in-plane and out-of-plane bending of the aromatic benzene ring. The small peaks at 1550–1650 cm–1 represent the N–H covalent bonds. The spectrum of MBTA, Fig. 6d, is similar to that of BTA due to their identical chemical structure, except the methyl group located on the benzene ring in the MBTA molecule. The spectrum of MBI, Fig. 6e, includes a wide peak at 1320 cm–1, that represents the C–S single bond. In addition, stronger peaks within 1480–1550 cm–1 are observed due to the C=C and C=N bonds. The spectrum following exposure to Trp + CTAB mixture, Fig. 6f, shows a strong absorbance band at 3400 cm–1, which represents the O–H and N–H bonds in tryptophan. The peaks at 2800–3000 cm–1 represent the C–H bonds in both materials. At 1720 cm–1, C=O bond stretching in the tryptophan molecule is noticed.
In order to gain some insight on the nature of the protective coatings built up during the etching of Cu in the presence of different inhibitors, scanning Kelvin probe (SKP) measurements were performed. The topography and the Volta potential (or electron work function) scan results are summarized in Fig. 6g–p. All scans in this figure are across the interface between etched (on the left-hand side) and non-etched (on the right-hand side) zones on the sample. The non-etched zone of each sample was masked with an adhesive 3 M tape prior to exposure to the etching solution. It can be seen that the etching in uninhibited FeCl3 solution led to loss of material (Fig. 6g). With all inhibitors other than BTA (Fig. 6i), exposure to the etching solution clearly resulted in the formation of a film. In particular, a very thick film was detected on the area exposed to MBI-inhibited solution (Fig. 6o and Supplementary Table 3); this film was non-uniform and fragmented, exposing pits in the underlying Cu. The higher film thickness of this film is supported by the distinct surface morphology evident from scanning electron microscope (SEM) images (see Supplementary Fig. 1). Exposure to BTA-inhibited solution presumably led to loss of material, albeit less compared to the uninhibited solution (Supplementary Table S3). It should be borne in mind that all samples were immersed in the etching solution for 2 weeks prior to the SKP measurements. In addition, the Volta potential difference distribution on the sample exposed to BTA-inhibited solution is more similar to that on the sample exposed to MBTA-inhibited solution than to that exposed to uninhibited solution. Thus, it may be concluded that either the initial corrosion rate was higher than the film growth rate on this sample, or the BTA-induced film started degrading during the 2-week immersion.
SKP measurements also revealed that etching in uninhibited solution led to a very inhomogeneous surface, with Volta potential difference variations of ±0.10 V (see Supplementary Table 3). On all other surfaces exposed to inhibited solutions, except MBI, the variations were somewhat smaller (±0.05 V), indicating more uniformity. In the absence of inhibitor, the Volta potential difference is close to the true Volta potential difference of the exposed metal, and the variability is probably due to the existence of distinct active cathodic (more positive) and anodic (more negative) sites. With inhibitors that form films on the metal surface, the metal Volta potential difference is partially masked, and the uniformity indicates the uniformity of the chemical nature of both the metal surface and film49. With MBI-inhibited solution, the film is evidently non-uniform (±0.15 V). With all inhibitors except MBI, the Volta potential difference at the etched surface was lower than that of Cu, suggesting that the films were positively charged50. In contrast, after exposure to an MBI-inhibited solution, the Volta potential difference was on average higher than that of Cu, suggesting it was negatively charged, in agreement with the energy-dispersive X-ray spectroscopy (EDS) results showing that after exposure to MBI-inhibited solution, the film contained significantly more chloride (see Supplementary Table 4). Supplementary Table 3 summarizes the surface topography and Volta potential difference measured at the etched surfaces relative to those measured at bare, unexposed Cu.
Supplementary Fig. 1 shows typical SEM images revealing the surface morphologies of copper electrodes before (a) and after (b–f) immersion for 3 days in either uninhibited or inhibited 0.01 M FeCl3 solutions. Supplementary Table 4 tabulates typical chemical compositions of these surfaces, as analyzed by EDS. Before immersion (Supplementary Fig. 1a), scratches and some other mechanical damage due to grinding on SiC paper (to expose fresh copper) are evident. Chemical analysis reveals Si and C from the grinding paper, a small concentration of O, and mainly Cu, with no Cl. After immersion in a blank FeCl3 solution (Supplementary Fig. 1b), the original morphology is no longer evident. Instead, a corrosion product is evident, with some local inhomogeneities. Si is no longer apparent, and the concentration of C is lower, possibly due to masking by the corrosion product, which may contain CuO and CuCl. After exposure to MBTA-containing solution (Supplementary Fig. 1c), the surface looks more uniform, yet with a surface morphology different from the non-immersed sample. The relatively high concentration of C may be related to an organic film that formed on the surface. After exposure to BTA-containing solution (Supplementary Fig. 1d), the surface morphology looks similar to that of the non-immersed sample, although the chemical composition is similar to that of the MBTA-exposed sample. This implies that the film formed on the surface due to BTA is thinner than that due to MBTA. After exposure to MBI-containing solution (Supplementary Fig. 1e), the surface morphology is more nodular and different than that of the other samples. The surface layer is not fully dense and contains some pores and grooves, which could indicate it as being thicker than the other surface layers. The absence of C and Si in the EDS spectrum further supports masking of the Cu surface. The surface layer is richer in Cl and O compared to all other samples. Finally, after exposure to Trp + CTAB, the surface morphology is significantly different from all other samples. The absence of Si in the EDS spectrum may indicate that this organic layer is thick, which could also explain the absence of Cl.
Contact angle (CA) measurements were conducted on copper samples pre-immersed in either uninhibited or inhibited 0.01 M FeCl3 solutions. The results are summarized in Supplementary Table 5. Cu surface pretreated in an uninhibited FeCl3 solution had CA ≅ 89°. Pretreatment in MBI, MBTA and BTA inhibited solutions slightly increased the CA, thus reflecting a more hydrophobic surface. In contrast, pretreatment in Trp + CTAB inhibited solution made the surface significantly more hydrophilic. This may be one of the reasons for the significantly lower inhibition efficiency of this inhibitors mixture and its degradation over immersion time.
Auger electron spectroscopy (AES) analysis of a Cu sample exposed for 1 h to uninhibited 0.01 M FeCl3 solution revealed the elements C, O, Cu, N (5.4 at.%), Cl (3.7 at.%), Si, Fe (0.45 at.%) and Zn. The origin of nitrogen is mainly from storage and transportation (see the Methods section). The presence of chloride indicates the formation of a copper chloride (and possibly also iron chloride) layer at the surface. A large quantity of organic contaminations (and possibly also residues from grinding on SiC papers) that can be involved in Cu bonding made the conclusions possibly drawn from this sample limited. X-ray photoelectron spectroscopy (XPS) revealed a Cu 2p3/2 peak at a binding energy, Eb, of 932.5 eV, which may be related to either Cu2O or CuCl (both typically at Eb = 932.5 eV)51. Cu(II), either as CuO (Eb = 933.6)51 or CuCl2 (Eb = 935.1 eV)51 is not likely. The Cu LMM spectrum showed a strong peak at Eb = 568.0 eV (kinetic energy, Ek = 918.65 eV), which may be related to Cu0 (Ek = 918.6 eV)51. A smaller broad peak, between Ek = 914.6 and 915.9 eV, which seems to be composed of two convolved peaks, may be associated with CuCl2 (Ek = 915.3 eV)51, but also with CuCl (Ek = 915.0 eV)51. Cu2O (Ek = 916.2 eV)51 is less likely. A strong N 1s peak appeared at Eb = 400.2 eV (Supplementary Fig. 2 (a)).
AES analysis of a Cu sample exposed for 1 h to BTA-inhibited 0.01 M FeCl3 solution revealed the elements C, O, N (10.6 at.%), Si, Cu, Cl (1.4 at.%), Fe, and Zn. The increase in the concentration of N is related to the nitrogen-containing BTA molecule. The formation of an organic layer on the surface could also explain the decrease in the Cl concentration if its origin was mainly a corrosion product layer on top of Cu. XPS revealed a Cu 2p large peak at Eb = 932.95 eV and a smaller peak at 935.45 eV. The former peak may be related to Cu0 (Eb = 932.7 eV)51, Cu2O or CuCl. The latter peak is related to CuCl2. The Cu LMM spectrum showed a strong peak at Eb = 572.45 eV (Ek = 914.15 eV), which may be related to either Cu–NC or Cu–C(CN)3 bond (both at Ek = 914.5 eV)51. The Cu–N bonding indicates a Cu/inhibitor film interface. A small peak at Ek = 916.3 eV can be ascribed to Cu2O (Ek = 916.2 eV)51.
AES analysis of a Cu sample exposed for 1 h to MBTA-inhibited 0.01 M FeCl3 solution revealed the elements C, O, N (5.8 at.%), Cu, Cl (3.7 at.%), Br, Fe, and Zn. The origin of the Br contamination is unclear. Also, the N and Cl concentrations are similar to those on Cu exposed to uninhibited solution. Angle-resolved XPS (ARXPS) spectra were acquired at take-off angles of 30°, 45° and 75°, in an attempt to describe the arrangement of surface layers. At 30°, XPS revealed a Cu 2p large peak at Eb = 932.75 eV that may be related to Cu0, Cu2O, or CuCl. The Cu LMM spectrum showed a peak at Eb = 568.25 eV (Ek = 918.4 eV), which may be related to Cu0. A broad peak at Ek ~ 914.25–915.6 eV can be related to Cu–NC or Cu–C(CN)3 bonding, CuCl2 and CuCl. The Cu–N bonding indicates a Cu/inhibitor film interface. N 1s peaks appeared at Eb = 400.1, 402.7, ~407.25, and ~398.6 eV. At 45°, XPS revealed a Cu 2p large peak at Eb = 932.65 eV (Supplementary Fig. 2 (b1)), which can be related to Cu0, CuCl or Cu2O. A “riding” peak is ascribed to CuClx (see Supplementary Fig. 2 (b1)). The typical Cu 2p3/2 and Cu 2p1/2 peaks reflect significant split spin-orbit components (Δmetal ≅ 19.75 eV). The Cu LMM spectrum showed a peak at Ek = 918.4 eV, which can be related to Cu0. Another, small peak at Ek = 915.8 eV can be related to Cu2O or CuCl2, while a broad peak at Ek = 914.35 eV can be related to Cu–NC or Cu–C(CN)3 bonding. N 1 s peaks appeared at Eb = ~399, 400.2, 402.75, and ~407.5 eV (Supplementary Fig. 2 (b2)). The 1st peak can be related to Cu–N–C bonding, as in Cu1.9C77.7N19.3 (Eb = 399.5 eV)52, while the 3rd peak can be related to N–O bonding (~402.9 eV)53. The 4th peak can be ascribed to NO3 (~408.15 eV), or in general to N–Ox bonding where x ≥ 253. At 75°, XPS revealed a Cu 2p large peak at Eb = 932.75 eV, which can be related to Cu0, but also to CuCl and Cu2O. The Cu LMM spectrum (Supplementary Fig. 2 (b3)) showed a peak at Ek = 918.4 eV, which can be related to Cu0. At Eb ~ 570.6 eV, a spectral feature related to Cu0, which usually appears when the Cu0 content increases, appeared. Another peak at Ek = 915.4 eV can be related to CuCl2, but also to CuCl, Cu2O, or Cu(NO3)2. A peak at Ek = 914.35 eV can be related to Cu–NC or Cu–C(CN)3 bonding. Two strong N 1s peaks appeared at Eb = 400.1 and 402.7 eV. For the MBTA-inhibited sample it can thus be concluded that an inorganic inhibitor film was bonded to Cu via Cu–NC or Cu–C(CN)3 chemical bonding, and that a CuClx layer co-existed. The shift of the CuClx peak in the Auger structure in ARXPS was minor: Ek = 915.6 eV at 30°, 915.8 eV at 45°, and 915.4 eV at 75°. The signal-to-noise in the vicinity of this peak was rather poor and did not allow a very precise peak maximum identification. In addition, this peak was “riding” upon another peak related to the metallic Cu (Cu0) Auger structure; when metallic Cu was sensed more (at 75°), the overlap of the two structures was more pronounced. Furthermore, since the layers were not chemically uniform laterally, each take-off angle resulted in the beam falling on a somewhat different point (chemically). The CuClx and Cu-inhibitor layers may thus have only partial coverage, and are in competition with one another on the Cu surface. Because of the sample lateral and depth inhomogeneity, it was difficult to define schematically the exact order of (hypothetically uniform) layers. This conclusion is in agreement with our EIS data.
AES analysis of a Cu sample exposed for 1 h to MBI-inhibited 0.01 M FeCl3 solution revealed the elements C, O, Cu, N (5.3 at.%), Cl (2.4 at.%), Fe (1.4 at.%), S (1.3 at.%), Si and Zn. XPS revealed a Cu 2p large peak at Eb = 932.7 eV, which may be related to Cu0, Cu2O or CuCl. The Cu LMM spectrum showed a strong peak at Ek = 918.4 eV, which may be related to Cu0. Another strong, but broad, peak appeared at Ek = 915.55 eV. This peak may be ascribed to Cu(NO3)2 or CuCl2, although CuCl cannot be excluded. A small “riding” peak at Ek = 916.45 eV can be related to Cu2O (Ek = 916.2 eV)51. In this sample, the chemical bonding between the inhibitor film and Cu was not only via Cu–N bond, but also via Cu–S bond (see the S 2p fitting in Supplementary Fig. 2 (c)).
AES analysis of a Cu sample exposed for 1 h to Trp + CTAB inhibited 0.01 M FeCl3 solution revealed the elements C, O, Cu, N (4.1 at.%), Br (4.0 at.%), Cl (3.3 at.%), Si, Fe, and Zn. The CTAB molecule is the origin of Br. The identification of Br on the surface supports the adsorption of inhibitor molecules on the surface and the possible formation of a surface film. XPS revealed a Cu 2p large peak at Eb = 932.6 eV, which may be related to Cu0, Cu2O, or CuCl. The Cu LMM spectrum showed a peak at Ek = 918.4 eV, which can be related to Cu0. At Eb ~ 570.6 eV, the spectral feature related to Cu0 appeared. Another peak at Ek = 915.4 eV can be related to CuCl2, but also to CuCl, Cu2O, or Cu(NO3)2. A peak at Ek = 914.35 eV can be related to Cu–NC or Cu–C(CN)3 bonding. N 1s peaks appeared at Eb = 400.2 and 402.6 eV.
Discussion
Weight loss measurements showed that the corrosion rate of Cu in 2.46 M FeCl3 was significantly higher than that in 0.10 M FeCl3 (see Table 1). While the concentration of the former solution is 24.6 times higher, the corrosion rate of Cu in it was only 5.3 times higher, possibly due to reactants diffusion control. In both FeCl3 solutions, all inhibitors decreased the corrosion rate and showed an inhibition efficiency of the same order of magnitude. The inhibition efficiencies of all inhibitors were higher in the 0.10 M FeCl3 solution, probably due to the faster metal oxidation rate in the concentrated solution, which prevented sufficiently rapid adsorption of the inhibitor on the metal surface.
Potentiodynamic polarization measurements showed that all inhibitors shifted the corrosion potential to less-negative values (see Fig. 3a and Table 2). Along with the decrease in the corrosion current density, this shift indicates that all tested compounds behave like anodic inhibitors8, i.e., they adsorb onto the surface and suppress the anodic reaction. Since the shift in Ecorr is higher than 85 mV, these compounds cannot be regarded as mixed-type inhibitors54. At room temperature, MBTA, BTA, and MBI exhibited inhibition efficiency higher than 97%, while that of Trp + CTAB was significantly lower.
From potentiodynamic polarization measurements it was also evident that the adsorption of MBTA, BTA, and MBI on copper surface followed the Langmuir adsorption isotherm8,17. This isotherm implies that the inhibitor molecules are adsorbed as a unimolecular layer with minimal molecular interaction. The inhibition efficiency of organic molecules mainly depends on their adsorption ability at the metal/solution interphase, which takes place by the replacement of water molecules by organic molecules25. The values of the adsorption equilibrium constant, Kads, deduced from these isotherms reflect the adsorption ability of the inhibitor (higher values reflecting higher ability, thus MBI seems to have the highest ability while BTA has the lowest). The standard free energy of adsorption, \(\Delta G_{{\mathrm{ads}}}^ \circ\), is another important thermodynamic property. It is commonly argued that values lower than ~20 kJ mol–1 (absolute value) indicate electrostatic interaction between the charged molecules of the adsorbed inhibitor and the metal surface (i.e., physisorption), while those higher than ~40 kJ mol–1 involve charge sharing or charge transfer from the inhibitor molecules to the metal surface to form coordinate covalent bonds25,33,42. Here, the values of \(- \Delta G_{{\mathrm{ads}}}^ \circ\) for BTA, MBTA, and MBI were between 24 and 28 kJ mol–1, the value for MBI being most negative. The negative values indicate spontaneous adsorption of the inhibitor molecules on the Cu surface, namely the interaction between the inhibitor molecules and the metallic surface is stronger than the interaction between the metallic surface and water molecules55. The values themselves suggest that the adsorption of the three inhibitors took place neither via pure physisorption nor via pure chemisorption, but via a combination of both (i.e., physiochemisorption55).
The effect of temperature on the dissolution rate of Cu was investigated in different solutions at 25, 40, 50, and 60 °C (see Fig. 4 and Table 3). The corrosion rate of Cu increased significantly with the increase of solution temperature, following an Arrhenius behavior. Several studies have shown that the corrosion rate of Cu and other metals in acidic media increases exponentially with temperature and may be described by an Arrhenius law10,42,56. The dissolution of Cu in acidic solutions of FeCl3 was reported10 to be a heterogeneous reaction with an activation energy of 24.9 kJ mol–1. A similar value of 26.8 kJ mol–1 is found in the current study for the uninhibited solution (see Table 3). It is evident from Fig. 4a that the corrosion rate increased more moderately in the presence of inhibitors, probably due to blocking of active surface sites. The inhibition efficiencies decreased as the solution temperature was increased, possibly due to desorption of some adsorbed inhibitor molecules.
The change in inhibition efficiency of the Trp + CTAB mixture solution was more moderate than that of the BTA, MBTA, and MBI solutions. The activation energy of the corrosion reaction increased significantly in the presence of BTA, MBTA, and MBI. In contrast, the mixture of Trp + CTAB cannot be described by an Arrhenius law, the effect of temperature on its inhibition ability was minor compared to the other three solutions, and the activation energy of copper corrosion was lower in this solution than in the blank solution (see Table 3). The latter has been claimed to indicate chemisorption of the organic molecules on the copper surface56.
The Nyquist plots for Cu in 0.01 M FeCl3 blank, MBTA-, and BTA-containing solutions showed only a slightly depressed semicircle (see Fig. 5a) and one time constant. A Warburg impedance (and a second time constant) was evident in the case of MBI and Trp + CTAB solutions. It indicates that concentration polarization prevails, due to either depletion of the reactant concentration in the vicinity of the Cu surface, diffusion of soluble copper species from the electrode surface to the bulk solution, or formation of a surface layer (i.e., passivation or film)8,23,32,57. The depressed semicircles along with a Warburg impedance slope lower than unity indicate surface roughness and inhomogeneity42,58. The polarization resistance was higher in all inhibited solutions than in the uninhibited solution. All resistive components (Rp, Rct, Rd, and RS) in MBTA- and BTA-inhibited solutions were higher than in the other solutions, reflecting the highest corrosion resistance of Cu (or, highest inhibition efficiencies) in these solutions, probably due to strong adsorption of the organic inhibitors onto the Cu surface. It is evident from Table 4 that both the polarization resistance and charge-transfer resistance were strongly influenced by the inhibitor type. In addition, the Rct values are only 14–49% the counterpart Rp values, implying that the process was not under pure activation control. The CPE values of solutions/metal interphases in the presence of inhibitors were lower than the value in the uninhibited solution. The decrease in CPE values, most noticeably in MBTA- and BTA-inhibited solutions, may result from a decrease in the local dielectric constant and/or an increase in the thickness of the electrical double-layer (EDL), suggesting either adsorption of inhibitor molecules on the Cu surface or formation of surface film59. The inhibition efficiencies based on EIS measurements exhibit a similar trend as those deduced from potentiodynamic polarization measurements.
The efficiency of corrosion inhibitors may change during immersion31,42,45,47,60,61. In this study, we evaluated the inhibition efficiency of the different inhibitors until 72 h of immersion, using EIS. The inhibition efficiency of BTA, MBTA and MBI slightly increased during the first 24 h, indicating on the continuous growth of a surface film on Cu during this time, which resulted in enhanced corrosion resistance of Cu. While the inhibition efficiency of BTA and MBTA remained constant (99.5% in both cases) during the rest of the exposure time, the inhibition efficiency of MBI slightly decreased during 48 to 72 h, probably due to formation of defects in the surface film, which enabled aggressive ions to reach the metal/inhibitor interface.
The contact angle of water on Cu pretreated in blank FeCl3 solution was ∼89°. After exposure to inhibited solutions, it slightly increased, up to ∼103°. Fresh Cu surfaces have been reported to have CA of either 65°62 or 74°25. Higher CA values have been attributed to either oxidized or smoother Cu surfaces25. Etching Cu in FeCl3 solutions has been reported to increase the CA significantly, even to the formation of superhydrophobic Cu surfaces62. An increase in the CA due to inhibitors was reported before17. This has been attributed to the adsorption of inhibitor molecules and formation of a more hydrophobic surface film25. The CA depends among others on the structure of the adsorbed molecule. For example, benzene rings arranged towards the solution were found to result in a more hydrophobic surface25.
The Volta potential is of electrostatic nature, does not account for chemical interactions, and therefore does not always correlate with the corrosion potential, in particular when the measurements are conducted in “dry” air50,63. Yet, it does reflect the tendency of a metal to corrode, and has therefore been used to study localized corrosion, micro-galvanic couples in the microstructure, coating/paint delamination, and effectiveness of conversion coatings. With respect to its use in the study of corrosion inhibitors, Ferreira et al.64,65 were probably the first to apply scanning Kelvin probe force microscopy (SKPFM) for the study of organic inhibitors. Here, Fig. 6g–p and Supplementary Table 3 summarize our SKP results. It can be seen that MBI is differentiated from the other inhibitors, with a significantly thicker surface film and having a Volta potential difference higher than that of bare Cu. Decreased Volta potential difference may indicate that the inhibitor-induced films act by inhibiting the cathodic reaction (in a manner similar to that observed with chromate conversion coatings on steel66), creating a barrier towards oxygen in air and possibly Fe(III) in solution.
This interpretation is, however, contradictory to what the potentiodynamic curves indicate and to the reported dominant anodic inhibition action of Trp + CTAB37, MBTA (though classified as a mixed-type inhibitor)21, and BTA67. We believe that the trend seen in the SKP results, namely the lowering of the Volta potential difference, is due to the chemical nature of the coatings, in particular oxygen vacancies and defects in the oxide films50,68. The chemical nature of the MBI-induced film is probably more complex. In addition to charge distribution effects in the film, the Volta potential difference may be affected by dipoles induced by hydration of the film69. We believe, however, that the Volta potential difference of the MBI-inhibitor films is pushed to higher values due to enrichment of the copper oxide with copper in the oxidation state of Cu(I)68.
The organic inhibitors act to stabilize these films and act as surfactants or wetting agents to smooth out the formed films. This is true upon exposure to BTA, MBTA, and Trp + CTAB, where the established films are more uniform. With MBI the situation is much different. The porosity and inhomogeneity of the MBI-induced film, observed by SKP and supported by SEM and EIS, may suggest that MBI is less efficient as a wetting agent. We do not, however, understand why this is so and why MBI-induced films are much thicker.
The XPS data supports chemisorption of BTA, MBTA, and MBI on the Cu surface followed by the formation of a polymeric complex, which can be represented, for example, by70:
Cu–N bond was identified by XPS after exposure to all four inhibited solutions. Nitrogen is known to be electron pair donor while copper being an electron pair acceptor71. Only in the case of Cu exposed to BTA-inhibited solution were satellite peaks between Cu 2p3/2 and Cu 2p1/2 detected. Such satellite peaks have been related to Cu(II), for example in CuO72. Therefore, according to our XPS results only in the case of BTA it is possible to conclude that both Cu(I)- and Cu(II)-containing corrosion products and complexes co-exist.
To summarize, four organic inhibitors—BTA, MBTA, MBI, and a mixture of Trp + CTAB—were studied for their efficiency and stability in inhibiting the corrosion of copper in FeCl3 solutions. The corrosion rate of Cu in both 0.10 M and 2.46 M FeCl3 solutions was reduced by all four inhibitors.
In the uninhibited solution and in the absence of copper ions in solution, Fe3+/Fe2+ reduction was monitored. In the presence of copper ions in solution, Cu+/Cu0 reduction was also monitored, and with a greater copper concentration—reversible Cu2+/Cu+ reaction was monitored in solution too, and insoluble CuCl was detected on the surface. The corrosion process in this solution was characterized by diffusion control, Arrhenius behavior, and surface heterogeneity.
BTA provided inhibition efficiencies of 79.9%, 98.9%, 96.5%, 99.2% and 99.5% in 2.46 M FeCl3 (gravimetric), 0.10 M FeCl3 (gravimetric), 0.01 M FeCl3 (potentiodynamic), 0.01 M FeCl3 (EIS, 0 days exposure), and 0.01 M FeCl3 (EIS, 3 days exposure), respectively. BTA was found to have the lowest adsorption ability and to form a relatively thin polymeric complex surface layer by spontaneous physiochemisorption, following Langmuir adsorption mode. The corrosion process in this solution was characterized by Arrhenius behavior and surface heterogeneity. The inhibitor made the surface slightly more hydrophobic and positively charged compared to that of copper exposed to uninhibited solution. The inhibition efficiency was not degraded within 72 h of immersion (at least). BTA acted as both a wetting agent and an anodic inhibitor, forming a passive surface layer. This was the only inhibitor for which Cu(II), either in CuO or CuCl2, was detected in the surface layer. Cu2O was detected in addition to a distinct inhibitor film, bonded to the copper surface via Cu–NC chemical bond where the lone pair of electrons of a nitrogen atom in the triazole ring are shared with Cu on the surface. Physisorption by the alkyl group likely precedes the chemical bonding. The orientation of the molecule relative to the surface can have its own effect, as described elsewhere67. The role of heteroatoms such as N and S in the interaction between organic inhibitor compounds and metallic surfaces has also been described in detail elsewhere55.
MBTA provided inhibition efficiencies of 80.6%, 99.1%, 97.3%, 99.3%, and 99.5%, respectively, under the conditions aforementioned for BTA. MBTA behaved similar to BTA, except having higher adsorption ability and creating a thicker polymeric complex surface layer, with CuClx rich regions in it.
MBI provided inhibition efficiencies of 78.4%, 97.4%, 97.2%, 98.4%, and 98.7%, respectively, under the conditions aforementioned for BTA. MBI behaved differently from BTA and MBTA in certain aspects. First, it seemed to form distinct complexes with iron ions. It also formed the thickest layers, albeit non-uniform, rougher, with pits, and charge opposite to that of the BTA and MBTA layers (possibly due to high-chloride incorporation in the film). Concentration polarization prevailed, and a minor decrease in inhibition efficiency was observed between 48 h and 72 h. MBI was less efficient as a wetting agent compared to BTA and MBTA. The surface layers included Cu2O, and the polymeric complex surface layer was bonded to the copper surface both via Cu–N bond and Cu–S bond.
The Trp + CTAB mixture was the least efficient inhibitor in this study. It provided inhibition efficiencies of 51.2%, 89.5%, 61.3%, 77.9%, and 61.4%, respectively, under the conditions aforementioned for BTA. It did not follow Arrhenius-like dependence on temperature, and its inhibition efficiency decreased significantly in 72 h, among others possibly due to the hydrophilic surface that was formed. The inhibitor film was bonded to the copper surface via Cu–NC bond.
Methods
Materials
The samples used in this study were rectangular copper coupons (99.95% pure, 1 mm thick, Scope Metals Group, Bne Ayish, Israel). The coupons were wrapped with a 3 M Kapton 5413 polyimide amber insulating tape (St. Paul, MN). The tape was cut to maintain a constant exposed surface area of 8 × 8 mm. The coupons were ground mechanically with SiC papers (Buehler, Lake Bluff, IL), from 120 to 800 grit, and washed with distilled water and MEK (ACS, Merck). Ferric chloride solutions were prepared from FeCl3 anhydrous powder (≥99.9% purity, trace metals basis, Sigma Aldrich). After screening out some organic and inorganic inhibitors (see Supplementary Table 1), five inhibitors were used: (i) 5-methyl-1H-benzotriazole (MBTA, 98%); (ii) 2-mercaptobenzimidazole (MBI, 98%); (iii) cetyltrimethylammonium bromide (CTAB, ≥ 98%); (iv) tryptophan reagent, grade ≥98%; and (v) 1H-benzotriazole (BTA, 99%). The inhibitors were supplied by Sigma Aldrich in powder form, and were weighed using AG64 balance (Mettler Toledo, Columbus, OH, 0.1 mg readability) for solution preparation.
Mass loss measurements8 were carried out using both 0.10 M and 2.46 M FeCl3 aerated solutions, with and without different inhibitors at various concentrations. The pH of all solutions with 0.10 M FeCl3 was 1.4, while that of solutions with 2.46 M FeCl3 was between –0.46 and –0.50. The pH was measured with a calibrated Metrohm digital pH meter model 914 pH/DO/Conductometer Lab. Preliminary experiments were conducted to find the solubility limit of the inhibitors in the higher-concentration (2.46 M) FeCl3 solution at room temperature. The solubility limit was determined by defining the point when the inhibitor stopped dissolving while stirring with magnetic stirrer. The optimal molar ratio between the FeCl3 reagent and each inhibitor was determined from those experiments. The proportion between the FeCl3 solution and the inhibitor’s concentration was maintained also in experiments with 0.10 M FeCl3 solution.
Electrochemical tests were conducted using both uninhibited and inhibited aerated 0.01 M FeCl3 solutions. While significantly higher concentrations of FeCl3 in mass loss measurements enabled to obtain measurable values while reducing significantly the measurement time, consuming significantly smaller volumes of solutions, yet determining the etchant/inhibitor compatibility, the low concentration of FeCl3 in electrochemical tests enabled to develop a mild process with high sensitivity, which is essential for the applications of interest. It should also be borne in mind that electrochemical measurements are accelerated tests. Concentrations below 0.01 M FeCl3 required high solution volumes (to supply sufficiently large amounts of reactant), making the process not suitable for the applications of interest. The inhibitor concentrations were selected to maintain the maximum ratios relative to the FeCl3 concentration as found in the preliminary tests of the solubility limit. Yet, for BTA and MBTA, concentrations lower than the solubility limit (0.130 M at room temperature) were selected to avoid the formation of Fe–BTA/MBTA complexes in high-temperature experiments73. Indeed, lowering the inhibitor concentration to 0.065 M was found efficient in avoiding both complexation and precipitation while retaining high inhibition efficiency. The pH of all solutions was 2.5 and remained essentially constant throughout the experiment.
Mass loss measurements
Mass loss measurements were carried out in accordance with the requirements of ASTM G174 to evaluate the general corrosion rate of copper in the two FeCl3 solutions, inhibited and uninhibited. The corrosion rate in mm y–1 unit and the inhibition efficiency (η%) were calculated using Eqs. (16) and (17)74:
where k (8.76 × 104 mm y–1) is a unit convert, t is the time of exposure (h), A is the exposed area (cm2), w is the mass loss (g), ρ is the density (g cm–3), CRblank is the corrosion rate of the blank solution, and CRinh is the corrosion rate in the presence of an inhibitor.
Each coupon sample was immersed in a 50 mL solution. Experiments with 0.10 M FeCl3 solutions lasted 24 h, while those with 2.46 M FeCl3 solutions were performed for 4 h due to the high corrosion rate disparity. After immersion, each sample was removed from the solution and was weighed after a cleaning process. The latter consisted of wiping with cloth, acetic acid wash (99.98%, Sigma Aldrich), distilled water rinse, and drying with air pressure.
Electrochemical potentiodynamic polarization tests
Potentiodynamic polarization tests8 were carried out with the same copper coupons having the exposed area of 0.64 cm2, both in uninhibited and in inhibited 0.01 M FeCl3 solutions. A three-electrode cell comprises of copper coupon as working electrode, a saturated calomel electrode (SCE) as reference electrode, and a platinum foil as counter electrode was used, along with an Autolab PGSTAT128N Electrochemical Analyzer Workstation (Metrohm, Herisau, Switzerland). In all measurements, the system was given one hour to reach a steady open-circuit potential (OCP). The cathodic and anodic polarization curves in different solutions and temperatures were recorded at a scan rate of 1 mV s–1.
Parameters such as inhibition efficiencies were calculated based on the Tafel equations, as determined by the Tafel extrapolation method and following ASTM G598,75. Equations (18) and (19) were used to calculate the inhibition efficiency and corrosion rate (assuming uniform corrosion), respectively.
where jcorr is the corrosion current density in the blank solution and \(j{\prime}_{{\mathrm{corr}}}\) is the corrosion current density in solution with inhibitor.
where B is a conversion factor (3.27 × 106 µm g (A cm y)–1), jcorr in units of µA cm–2, and EW is the equivalent weight.
Experiments at elevated temperatures were carried out by placing the electrochemical cell in a beaker with heated water on a heating electronic plate, while continuously monitoring the solution temperature with thermometer. The activation energies and the pre-exponential coefficient, A, were calculated from the best-fitting regression lines, with respect to the relation between jcorr and temperature:
where T is the absolute temperature (K), R is the universal gas constant (kJ (K mol)–1), A is the Arrhenius pre-exponential factor, and Ea in units of kJ mol–1.
As part of the investigation of the adsorption mechanism of the inhibitors on the surfaces of Cu electrodes, adsorption isotherms were constructed by fitting the experimental data of surface coverage as a function of concentration of three inhibitors—MBTA, BTA, and MBI. Several adsorption isotherms, including Langmuir, Frumkin, Freundlich, Flory-Huggins, Dubinin–Radushkevich, and Tempkin isotherms8,17,25,33,42,58,60,70 were considered. The models were matched to the inhibition efficiency deduced from Tafel plots in potentiodynamic polarization measurements. The fractional surface coverage (θ) was obtained from division of Eq. (16) by 100. The measurements were conducted in 0.01 M FeCl3-based solutions at room temperature and a constant pH of 2.5.
Electrochemical impedance spectroscopy (EIS) tests
EIS tests8,76,77,78 were conducted with the same system setup. Each coupon was immersed in the solution for 1 h, until a steady-state OCP was reached. A sine wave excitation voltage (10 mV peak-to-peak) was superimposed at frequencies between 0.1 Hz and 10,000 Hz. The results are reported as Nyquist and Bode plots (only the former are shown herein). The values of RS, Rp, and CPE were extracted from the different plots. CPE admittance and impedance are defined by Eqs. (21) and (22):
where Y0 is the magnitude of the CPE, ω is the angular frequency, and α the exponential term of the CPE32,46. The α values range from 0 to 1, the former representing an ideal resistor whereas the latter representing an ideal capacitor48.
The inhibition efficiency was calculated using Eq. (23)32:
where \({R^{\prime}}_{\mathrm{p}}\) is the polarization resistance in the inhibited solutions and Rp the one in the uninhibited solution.
The influence of immersion time on the inhibition efficiency was evaluated for each inhibitor. Each sample was placed in a special, isolated cell, and was immersed in the same solution throughout measurement. The cell was sealed between tests using paraffin film. The coupon was immersed in the solution for 1 h, until a steady-state OCP was reached. Immediately after reaching the OCP, the first measurement was taken. Subsequently, measurements were taken at 24, 48, and 72 h.
Cyclic voltammetry (CV)
CV measurements were conducted in 0.01 M based FeCl3 solutions at room temperature and pH = 2.5 using a three-electrode cell, a saturated Ag/AgCl reference electrode, and a platinum mesh counter electrode. The forward (cathodic) sweep was from positive to negative potentials, whereas the reverse (anodic) sweep was from negative to positive potentials8,79. All experiments were run in quiescent (i.e., non-stirred) solutions. A multichannel potentiostat/galvanostat model VSP from BioLogic SAS was used. Glassy carbon (GC), gold, copper, and platinum working electrodes were used and compared. Both uninhibited and 0.065 M MBTA-inhibited solutions were studied. Both fresh solutions and solutions aged for 4 days were evaluated. In some cases, a copper electrode was immersed in the solution for either 3 h, 1 day, or 4 days and was then removed before the CV experiment started. The goal of these experiments was to evaluate whether the system behaves differently when the copper source is ions in solution instead of a solid electrode. In order to better understand the nature of electrode reactions, the experiments were repeated at three different potential scan rates (\(\nu\)) of 5, 10, and 30 mV s–1. The peak currents in each experiment were then drawn both versus \(\nu\) and versus \(\nu\)1/2, looking for the best fit by linear regression.
Surface characterization
FTIR spectroscopy tests were carried out on the copper coupons after exposure to the different solutions. The coupons were partially masked with a polyimide tape, leaving 0.64 cm2 unmasked. They were then immersed in each solution for 96 h. FTIR studies were performed using a Nicolet iN10 infrared microscope (Thermo Scientific, Waltham, MA) with an optimized room temperature DTGS detector (spectral range of 7600–450 cm–1). Each sample was characterized using the reflection method (ATR) for thin films on reflective substrates for compound identification. The data obtained was analyzed using Scientific™ OMNIC™ Picta Software. The results are reported both as FTIR spectra and as compound surface mapping.
SEM was used to characterize the surface morphology of copper after exposure to the different solutions. EDS was used for semi-quantitative chemical analysis of relatively large scan areas. For this purpose, a Quanta 200 FEG ESEM combined with an Oxford X-Max SDD EDS detector was used. The conditions for image acquisition were an accelerating voltage of 20 kV and magnification from ×100 to ×40,000. Neither special sample cleaning nor coating with a conductive film was applied before these analyses.
Sessile drop (static) contact angle (CA) measurements were conducted at T = 20 °C on copper samples pre-immersed in either uninhibited or inhibited 0.01 M FeCl3 solutions, in order to determine whether the hydrophobicity changed due to formation of corrosion products or surface films. To this aim, a Krüss DSA25S system with ADVANCE software ver. 1.7.1.0 was used. After placing a 5 μL deionized (DI) water drop on the surface, the CA was measured three times. Average values are reported herein.
Scanning Kelvin probe (SKP) measurements were performed using a M470 instrument from BioLogic Sciences Instruments. The probe tip was a 500 μm diameter tungsten (W) wire surrounded by a brass sheath. The tip was carefully cleaned with acetone and soft tissue prior to every scan. Each SKP scan was preceded by a topography scan. The topography scan was done in capacitive tracking mode (CTM), where the probe height changes throughout the measurement to maintain the same probe-to-sample distance. In both CTM and SKP scans, the probe was vibrating at 80 Hz with an amplitude of 30 μm in the vertical direction. During the CTM topography scan the sample was held at a constant bias of –10 V, while during the SKP scan neither the tip nor the sample were biased. Instead, the tip and sample were shortened to equilibrate the Fermi levels of the two. The up-down vibration of the tip led to change in capacitance and, hence, to current. A backing potential was then set automatically by the instrument’s PID controlled circuitry to null this current. The so-obtained potential is equal to the Volta potential difference (between the tip and the sample) with a minus sign. During the SKP scan, the obtained CTM topography map was used to keep the probe tip at a constant distance from the surface, of ca. 100 μm. All scans were performed on a 2000 × 2000 μm area with 100 μm steps in the x and y directions. The scan speed was 200 μm s–1, and each step was an average of 100 samples taken at a rate of 1000 Hz. All measurements were performed in air immediately after taking the sample out of the etching solution and after a short rinse with DI water and drying with pressurized argon. All samples were immersed for 2 weeks in the etching solution prior to SKP measurement.
Auger electron spectroscopy (AES) and X-ray photoelectron spectroscopy (XPS) measurements were conducted for compositional and chemical bonding analysis. Five samples, each with dimension of 0.8 × 0.8 cm2, were analyzed—Cu samples exposed for 1 h to either uninhibited solution or BTA, MBTA, MBI, and Trp + CTAB inhibited solutions. It has been claimed that 1 h of immersion of Cu in an inhibited solution is sufficiently to study molecules bonding to the metallic surface, while longer-term immersion might make the XPS analysis less useful80. The samples were mechanically polished prior to immersion. Their storage and transportation was in nitrogen atmosphere, to minimize their oxidation.
XPS measurements were performed in UHV (2.5 × 10–10 Torr base pressure) using 5600 Multi-Technique System (PHI, USA). The sample was irradiated with an Al Kα monochromated source (1486.6 eV), and the outcome electrons were analyzed by a Spherical Capacitor Analyzer using a slit aperture of 0.8 mm. The samples were analyzed at the surface only; sputtering was not performed to avoid artificial reduction of the Cu ions that can occur due to preferential sputtering, and skew the results towards lower oxidation states (e.g., zero oxygen sputter reduction in Cu2O vs. 5.8–10% oxygen reduction in CuO81). Moreover, the rate of sputtering of different elements is not the same, and ions with charge opposite to that of the incident beam (i.e., anions in our case) would be selectively removed, thus making representative conclusions difficult. This issue is sometimes overlooked in the literature. The samples were charged during measurements; the charge was compensated with a charge neutralizer. The C 1s adventitious carbon peak at a binding energy of 285.0 eV was used to correct the binding energy scale and possible charging effect. First, survey spectra were acquired in a wide energy range (0–1400 eV). Next, utility multiplex spectra were taken for different peaks of all the elements considered for atomic concentration, in a low energy range window at an intermediate resolution. Finally, high-resolution multiplex spectra were taken for different peaks in a pass energy of 11.75 eV at steps of 0.05 eV. These measurements allow precise energy position and peak shape determination, which are necessary for chemical bonding analysis. Various spectra were acquired from each sample, including both the pure core-level transitions (Cu 2p, C 1s, O 1s, and N 1s) and the X-ray excited Auger Cu L3M4,5M4,5 spectrum. In the case of Cu exposed to MBTA-inhibited solution, an attempt was made to describe the arrangement of surface layers based on ARXPS spectra80 acquired at take-off angles of 30°, 45°, and 75°. The dependence of the analyzed depth on the take-off angle θ is expressed as 3·IMFP·sin θ, where IMFP is the inelastic mean free path80. For IMFP = 2.0–4.0 nm80 we estimate the analyzed depth in the range of 3.0–11.6 nm. By decreasing θ, one can detect the topmost species and distinguish them from the species present in the deeper subsurface regions80.
Data availability
The datasets generated during and/or analyzed during the current study are not publicly available due to an ongoing technology development.
References
Kear, G., Barker, B. D. & Walsh, F. C. Electrochemical corrosion of unalloyed copper in chloride media–a critical review. Corros. Sci. 46, 109–135 (2004).
Liao, X. et al. Corrosion behaviour of copper under chloride-containing thin electrolyte layer. Corros. Sci. 53, 3289–3298 (2011).
Lilja, C. Influence of High Chloride Concentration on Copper Corrosion. Document ID 1398014 (Swedish Nuclear Fuel and Waste Management Co, Stockholm, Sweden, June 2013).
Zhao, H., Chang, J., Boika, A. & Bard, A. J. Electrochemistry of high concentration copper chloride complexes. Anal. Chem. 85, 7696–7703 (2013).
Arjmand, F. & Adriaens, A. Influence of pH and chloride concentration on the corrosion behavior of unalloyed copper in NaCl solution: A comparative study between the micro and macro scales. Materials 5, 2439–2464 (2012).
King, F. Critical Review of the Literature on the Corrosion of Copper by Water (SKB-TR–10-69, Swedish Nuclear Fuel and Waste Management Co, Stockholm, Sweden, November 2009).
Pourbaix, M. Atlas of Electrochemical Equilibria in Aqueous Solutions, 2nd edn, Section 14.1, pp. 384–392 (NACE, Houston, TX, USA, 1974).
Eliaz, N. & Gileadi, E. Physical Electrochemistry: Fundamentals, Techniques, and Applications, 2nd edn. (Wiley-VCH, Weinheim, 2019).
Tooley, M. Electronic Circuits–Fundamentals & Applications, 5th edn. (Routledge, London, 2019).
Wang, Z., Che, J. & Ye, C. Application of ferric chloride both as oxidant and complexant to enhance the dissolution of metallic copper. Hydrometallurgy 105, 69–74 (2010).
Allen, D. M. & Almond, H. J. A. Characterization of aqueous ferric chloride etchants used in industrial photochemical machining. J. Mater. Process. Technol. 149, 238–245 (2004).
Dimitrov, B., Cruden, A., Sharkh, S. & Determining, P. C. B. Trace capability and fusing, used in switch mode power supplies, based on modeling and simulation with finite. Acta Electrotech. Inform. 18, 3–10 (2018).
Codreanu, N. D., Bunea. R. & Svasta, P. Advanced investigations on PCB traces fusing in high power applications. In 2010 IEEE 16th Inter. Symp. Design and Technology in Electronic Packaging (SIITME), pp. 193–196 (IEEE, 2010).
Raja, P. B. et al. Reviews on corrosion inhibitors: a short view. Chem. Eng. Commun. 203, 1145–1156 (2016).
Dariva, C. G. & Galio, A. F. In Developments in Corrosion Protection, Ch. 16, pp. 365–379 (IntechOpen, Rijeka, 2014).
Brusic, V. et al. Copper corrosion with and without inhibitors. J. Electrochem. Soc. 138, 2253–2259 (1991).
Fateh, A., Aliofkhazraei, M. & Rezvanian, A. R. Review of corrosive environments for copper and its corrosion inhibitors. Arab. J. Chem. 13, 481–544 (2020).
Petrović Mihajlović, M. B. & Antonijević, M. M. Copper corrosion inhibitors. Period 2008– 2014. A Review. Int. J. Electrochem. Sci. 10, 1027–1053 (2015).
Kuznetsov, Y. I. Organic corrosion inhibitors: where are we now? A review. Part III. Passivation and the role of the chemical structure of organophosphates. Int. J. Corros. Scale Inhib. 6, 209–239 (2017).
Walker, R. Benzotriazole as a corrosion inhibitor for immersed copper. Corrosion 29, 290–298 (1973).
Geuli, O. & Mandler, D. The synergistic effect of benzotriazole and trimethylsiloxysilicate towards corrosion protection of printed Cu-based electronics. Corros. Sci. 143, 329–336 (2018).
Rani, B. E. A. & Basu, B. B. J. Green inhibitors for corrosion protection of metals and alloys: An overview. Int. J. Corros. 12, Article 380217 (2012).
Finšgar, M. & Milošev, I. Inhibition of copper corrosion by 1,2,3-benzotriazole: a review. Corros. Sci. 52, 2737–2749 (2010).
Hluchan, V., Wheeler, B. L. & Hackerman, N. Amino acids as corrosion inhibitors in hydrochloric acid solutions. Mater. Corros. 39, 512–517 (1998).
Khan, P. F., Shanthi, V., Babu, R. K., Muralidharan, S. & Barik, R. C. Effect of benzotriazole on corrosion inhibition of copper under flow conditions. J. Env. Chem. Eng. 3, 10–19 (2015).
Mansfeld, F. & Smith, T. Benzotriazole as corrosion inhibitor for copper II. Acid NaCl solutions. Corrosion 29, 105–107 (1973).
Fox, P. G., Lewis, G. & Boden, P. J. Some chemical aspects of the corrosion inhibition of copper by benztriazole. Corros. Sci. 19, 457–467 (1979).
Da Costa, S. L. F. A., Rubim, J. C. & Agostinho, S. M. L. Spectroelectrochemical study of the corrosion of a copper electrode in deaerated 1.0 M HCl solutions containing Fe(III): Effect of the corrosion inhibitor benzotriazole. J. Electroanal. Chem. Interf. Electrochem. 220, 259–268 (1987).
Finšgar, M. 2-Mercaptobenzimidazole as a copper corrosion inhibitor: Part II. Surface analysis using X-ray photoelectron spectroscopy. Corros. Sci. 72, 90–98 (2013).
Petrović Mihajlović, M. B., Radovanović, M. B., Tasić, Z. Z. & Antonijević, M. M. Imidazole based compounds as copper corrosion inhibitors in seawater. J. Mol. Liq. 225, 127–136 (2017).
Taheri, P. et al. On the importance of time-resolved electrochemical evaluation in corrosion inhibitor-screening studies. NPJ Mater. Degrad. 4, Article 12 (2020).
Ma, H., Chen, S., Yin, B., Zhao, S. & Liu, X. Impedance spectroscopic study of corrosion inhibition of copper by surfactants in the acidic solutions. Corros. Sci. 45, 867–882 (2003).
Liu, S. et al. Corrosion inhibition of copper–phosphorus brazing alloys in tetra-n-butylammonium bromide aerated aqueous solution by benzotriazole. Mater. Corros. 6, 932–939 (2013).
Ohsawa, M. & Suëtaka, W. Spectro-electrochemical studies of the corrosion inhibition of copper by mercaptobenzothiazole. Corros. Sci. 19, 709–722 (1979).
Yadav, M., Yadav, S. & Yadav, A. Inhibition of corrosion of copper by substituted triazoles in 3.5% NaCl solution. J. Chem. Pharm. Res. 3, 576–583 (2011).
Moretti, G. & Guidi, F. Tryptophan as copper corrosion inhibitor in 0.5 M aerated sulfuric acid. Corros. Sci. 44, 1995–2011 (2002).
Mobin, M., Parveen, M. & Alam Khan, M. Inhibition of mild steel corrosion using Ltryptophan and synergistic surfactant additives. Port. Electrochim. Acta 29, 391–403 (2011).
Popova, T. V. & Aksenova, N. V. Complexes of copper in unstable oxidation states. Russ. J. Coord. Chem. 29, 743–765 (2003).
Benck, J. D., Pinaud, B. A., Gorlin, Y. & Jaramillo, T. F. Substrate selection for fundamental studies of electrocatalysts and photoelectrodes: inert potential windows in acidic, neutral, and basic electrolyte. PLoS ONE 9, e107942 (2014).
Daniele, S. & Pena, M. J. Cyclic voltammetric investigation of the Cu—histidine system at platinum conventional and microelectrodes. Electrochim. Acta 38, 165–174 (1993).
Dehri, I. & Erbil, M. Application of semi-ellipse model in impedance studies of corrosion of 63–37 brass in neutral aqueous solutions containing sulphate, ammonium, and chloride ions. Br. Corros. J. 34, 299–303 (1999).
Solmaz, R., Kardaş, G., Çulha, M., Yazici, B. & Erbil, M. Investigation of adsorption and inhibitive effect of 2-mercaptothiazoline on corrosion of mild steel in hydrochloric acid media. Electrochim. Acta 53, 5941–5952 (2008).
Kashkovskiy, R., Strelnikova, K. & Fedotova, A. Application of electrochemical impedance spectroscopy to study hydrogen sulphide corrosion of steel and its inhibition: a review. Corros. Eng. Sci. Technol. 54, 493–515 (2019).
Eliaz, N. et al. The use of electrochemical impedance spectroscopy (EIS) and vibrating sample magnetometer (VSM) for measuring the corrosion rate of polymer-coated ferromagnetic metals. Mater. Technol. 16, 90–97 (2001).
Toumiat, K., Guibadj, A. & Taouti, M. B. Copper corrosion inhibition using BTAH inhibitor in sodium chloride medium: experimental and theoretical studies. Eurasian J. Anal. Chem. 12, 275–294 (2017).
Zhang, Q. B. & Hua, Y. X. Corrosion inhibition of mild steel by alkylimidazolium ionic liquids in hydrochloric acid. Electrochim. Acta 54, 1881–1887 (2009).
Qafsaoui, W., Kendig, M. W., Perrot, H. & Takenouti, H. Coupling of electrochemical techniques to study copper corrosion inhibition in 0.5 mol L–1 NaCl by 1-pyrrolidine dithiocarbamate. Electrochim. Acta 87, 348–360 (2013).
Bacelis, A., Veleva, L. & Alpuche-Avilés, M. A. Copper corrosion behavior in simulated concrete-pore solutions. Metals 10, 474 (2020).
Guiñón, J. L., García-Antón J., Pérez-Herranz, V. The effect of benzotriazole on mass transfer in the corrosion of a copper rotating disk electrode. J. Appl. Electrochem. 30, 379–384 (2000).
Örnek, C., Leygraf, C. & Pan, J. On the Volta potential measured by SKPFM–fundamental and practical aspects with relevance to corrosion science. Corros. Eng. Sci. Technol. 54, 185–198 (2019).
Moulder, J. F., Stickle, W. F., Sobol, P. E. & Bomben, K. D. Handbook of X-Ray Photoelectron Spectroscopy (Perkin-Elmer Corp., Eden Prairie, MN, USA, 1992).
Ewen, R. J. & Honeybourne, C. L. X-ray photoelectron spectroscopy of clean and gas-doped films of phthalocyanines. J. Phys. Condens. Matter 3, S303–S310 (1991).
Graf, N. et al. XPS and NEXAFS studies of aliphatic and aromatic amine species on functionalized surfaces. Surf. Sci. 603, 2849–2860 (2009).
Xu, X., Singh, A., Sun, Z., Ansari, K. R. & Lin, Y. Theoretical, thermodynamic and electrochemical analysis of biotin drug as an impending corrosion inhibitor for mild steel in 15% hydrochloric acid. R. Soc. Open Sci. 4, 170933 (2017).
Verma, C., Verma, D. K., Ebenso, E. E. & Quraishi, M. A. Sulfur and phosphorus heteroatom-containing compounds as corrosion inhibitors: An overview. Heteroat. Chem. 29, e21437 (2018).
Popova, A., Sokolova, E., Raicheva, S. & Christov, M. AC and DC study of the temperature effect on mild steel corrosion in acid media in the presence of benzimidazole derivatives. Corros. Sci. 45, 33–58 (2003).
Li, S. et al. Investigation on some Schiff bases as HCl corrosion inhibitors for copper. Corros. Sci. 41, 1273–1287 (1999).
Döner, A., Solmaz, R., Özcan, M. & Kardaş, G. Experimental and theoretical studies of thiazoles as corrosion inhibitors for mild steel in sulphuric acid solution. Corros. Sci. 53, 2902–2913 (2011).
Liu, S., Duan, J. M., Jiang, R. Y., Feng, Z. P. & Xiao, R. Corrosion inhibition of copper in tetra-n-butylammonium bromide aqueous solution by benzotriazole. Mater. Corros. 62, 47–52 (2011).
Marušić, K., Otmačić Ćurković, H. & Takenouti, H. Inhibiting effect of 4-methyl-1-p-tolylimidazole to the corrosion of bronze patinated in sulphate medium. Electrochim. Acta 56, 7491–7502 (2011).
Radovanović, M. B. & Antonijević, M. M. Protection of copper surface in acidic chloride solution by non-toxic thiadiazole derivative. J. Adhes. Sci. Technol. 31, 369–387 (2017).
Talesh Bahrami, H. R., Ahmadi, B. & Saffari, H. Preparing superhydrophobic copper surfaces with rose petal or lotus leaf property using a simple etching approach. Mater. Res. Express 4, 055014 (2017).
Jadhav, N. & Gelling, V. J. Review—The use of localized electrochemical techniques for corrosion studies. J. Electrochem. Soc. 166, C3461–C3476 (2019).
Zheludkevich, M. L., Yasakau, K. A., Poznyak, S. K. & Ferreira, M. G. S. Triazole and thiazole derivatives as corrosion inhibitors for AA2024 aluminium alloy. Corros. Sci. 47, 3368–3383 (2005).
Lamaka, S. V., Zheludkevich, M. L., Yasakau, K. A., Montemor, M. F. & Ferreira, M. G. S. High effective organic corrosion inhibitors for 2024 aluminium alloy. Electrochim. Acta 52, 7231–7247 (2007).
Mekhalif, Z., Forget, L. & Delhalle, J. Investigation of the protective action of chromate coatings on hot-dip galvanized steel: role of wetting agents. Corros. Sci. 47, 547–566 (2005).
Antonijević, M. M. & Petrović, M. B. Copper corrosion inhibitors. A review. Int. J. Electrochem. Sci. 3, 1–28 (2008).
Greiner, M. T., Chai, L., Helander, M. G., Tang, W. M. & Lu, Z. H. Transition metal oxide work function: the influence of cation oxidation state and oxygen vacancies. Adv. Funct. Mater. 22, 4557–4568 (2012).
Guillaumin, V., Schmutz, P. & Frankel, G. S. Characterization of corrosion interfaces by the scanning Kelvin probe force microscopy technique. J. Electrochem. Soc. 148, B163–B173 (2001).
Appa Rao, B. V., Chaitanya Kumar, K. & Nebalkar, N. Y. X-ray photoelectron spectroscopy depth-profiling analysis of surface films formed on Cu–Ni (90/10) alloy in seawater in the absence and presence of 1,2,3-benzotriazole. Thin Solid Films 556, 337–344 (2014).
Huang, H. et al. Water soluble corrosion inhibitors for copper in 3.5 wt.% sodium chloride solution. Corros. Sci. 123, 339–350 (2017).
Wang, Y. B. et al. A robust bilayer nanofilm fabricated on copper foam for oil–water separation with improved performances. J. Mater. Chem. A 26, 10294–10303 (2016).
Anastasiadis, N. C. et al. Iron(III) chloride–benzotriazole adducts with trigonal bipyramidal geometry: Spectroscopic, structural and catalytic studies. Polyhedron 64, 189–202 (2013).
ASTM. G1–03 (2017) e1: Standard Practice for Preparing, Cleaning, and Evaluating Corrosion Test Specimens (American Society for Testing and Materials, West Conshohocken, PA, 2017).
ASTM. G59–97(2014): Standard Test Method for Conducting Potentiodynamic Polarization Resistance Measurements (American Society for Testing and Materials, West Conshohocken, PA, 2014).
Jüttner, K. & Lorenz, W. J. Electrochemical impedance spectroscopy (EIS) of corrosion processes on inhomogeneous surfaces. Mater. Sci. Forum 44-45, 191–204 (1989).
Mitton, D. B. et al. The correlation between substrate mass loss and electrochemical impedance spectroscopy data for a polymer-coated metal. J. Electrochem. Soc. 149, B265–B271 (2002).
Lorenz, W. J. & Mansfeld, F. Determination of corrosion rates by electrochemical DC and AC methods. Corros. Sci. 21, 647–672 (1981).
Elgrishi, N. et al. Practical beginner’s guide to cyclic voltammetry. J. Chem. Educ. 95, 197–206 (2018).
Finšgar, M. The first X-ray photoelectron spectroscopy surface analysis of 4-methyl-2-phenyl-imidazole adsorbed on copper. Anal. Methods 7, 6496–6503 (2015).
Mitchell, D. F., Sproule, G. I. & Graham, M. J. Sputter reduction of oxides by ion bombardment during Auger depth profile analysis. Surf. Interf. Anal. 15, 487–497 (1990).
Acknowledgements
We gratefully acknowledge the financial support from the Israel Ministry of Defense (grant no. 4440932895). We thank Dr. Larisa Burstein and Dr. Pini Shekhter from the Wolfson Applied Materials Research Centre at Tel-Aviv University for AES/XPS measurements and data analysis. We thank Dr. Nabasmita Maity for her peer review and constructive remarks, and Mr. Nadav Genosar for his helpful advice with SKP measurements.
Author information
Authors and Affiliations
Contributions
N.E. and S.D. conceptualized and directed the project and were responsible for funding acquisition. O.S. conducted all experiments except CV, SEM-EDS, CA, SKP, and AES-XPS under the supervision of N.E. and S.D. D.E. performed CV, SEM-EDS, and CA experiments under the supervision of N.E. E.S. conducted SKP measurements. O.S. and N.E. were responsible for data curation. All authors discussed the data interpretation. O.S. wrote the paper first draft, revised significantly by N.E. All coauthors essentially contributed during reviewing of the final manuscript. All authors approved the final version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sisso, O., Dor, S., Eliyahu, D. et al. Corrosion inhibition of copper in ferric chloride solutions with organic inhibitors. npj Mater Degrad 4, 38 (2020). https://doi.org/10.1038/s41529-020-00139-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41529-020-00139-0
This article is cited by
-
Hexane extract of Persea schiedeana Ness as green corrosion inhibitor for the brass immersed in 0.5 M HCl
Scientific Reports (2024)
-
In-operando analysis of the corrosion patterns and rates under magnetic fields using metallic film
npj Materials Degradation (2022)
-
New star-fractal antenna devised as wireless sensor of the permittivity characterization measurement of Na2SO3 solutions
Microsystem Technologies (2022)
-
Molecular scale insights into interaction mechanisms between organic inhibitor film and copper
npj Materials Degradation (2021)
