
Abstract
Aquatic birds represent a vast reservoir from which new pandemic influenza A viruses can emerge1. Influenza viruses contain a negative-sense segmented RNA genome that is transcribed and replicated by the viral heterotrimeric RNA polymerase (FluPol) in the context of viral ribonucleoprotein complexes2,3. RNA polymerases of avian influenza A viruses (FluPolA) replicate viral RNA inefficiently in human cells because of species-specific differences in acidic nuclear phosphoprotein 32 (ANP32), a family of essential host proteins for FluPol activity4. Host-adaptive mutations, particularly a glutamic-acid-to-lysine mutation at amino acid residue 627 (E627K) in the 627 domain of the PB2 subunit, enable avian FluPolA to overcome this restriction and efficiently replicate viral RNA in the presence of human ANP32 proteins. However, the molecular mechanisms of genome replication and the interplay with ANP32 proteins remain largely unknown. Here we report cryo-electron microscopy structures of influenza C virus polymerase (FluPolC) in complex with human and chicken ANP32A. In both structures, two FluPolC molecules form an asymmetric dimer bridged by the N-terminal leucine-rich repeat domain of ANP32A. The C-terminal low-complexity acidic region of ANP32A inserts between the two juxtaposed PB2 627 domains of the asymmetric FluPolA dimer, suggesting a mechanism for how the adaptive PB2(E627K) mutation enables the replication of viral RNA in mammalian hosts. We propose that this complex represents a replication platform for the viral RNA genome, in which one of the FluPol molecules acts as a replicase while the other initiates the assembly of the nascent replication product into a viral ribonucleoprotein complex.
Similar content being viewed by others


Main
The influenza virus genome is composed of negative-sense single-stranded RNA segments, which are assembled into separate viral ribonucleoprotein (vRNP) complexes with FluPol, a heterotrimeric complex of PB1, PB2 and PA (P3 in influenza C virus) proteins, and the viral nucleoprotein (NP)2,3. Following virus entry, vRNPs are trafficked to the cell nucleus where FluPol, in complex with various host factors, directs the transcription and replication of the viral genome5. For replication, FluPol first generates complementary RNA (cRNA), which then serves as template for viral RNA (vRNA) synthesis. The molecular details of the replicase complex and, in particular, the role of the ANP32 family of host proteins, known to be essential for genome replication6, remain unknown. Here we investigate the role of ANP32A in promoting FluPol function and uncover a complex of ANP32A with a FluPol dimer, which we propose acts as a replication platform for the viral genome.
ANP32A forms a complex with a FluPol dimer
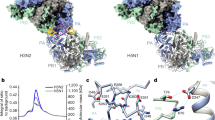
Host ANP32 proteins are essential for the activity of FluPol of both influenza A and influenza B viruses7,8. To address whether FluPol from influenza C virus also requires ANP32 proteins, we performed minigenome assays in human cells lacking ANP32A and ANP32B (Extended Data Fig. 1). No detectable FluPol activity was observed in the double-knockout cells, but expression of human ANP32A (huANP32A), human ANP32B (huANP32B) or chicken ANP32A (chANP32A) restored FluPolC activity. We then used cryo-electron microscopy (cryo-EM) to solve the structures of FluPolC in complex with huANP32A and chANP32A at a resolution ranging from 3.0 Å to 3.6 Å (Fig. 1a, b, Extended Data Figs. 2, 3, Extended Data Table 1, Supplementary Video 1). In these structures, two heterotrimeric FluPolC molecules assemble into an asymmetric dimer with the N-terminal leucine-rich repeat domain of ANP32A (ANP32ALRR)9 bridging the two FluPolC molecules. The C-terminal low-complexity acidic region of ANP32A (ANP32ALCAR) could not be fully resolved in the structures. The structures of FluPolC dimers with huANP32A and chANP32A are largely identical, exhibiting the same FluPolC–FluPolC and FluPolC–ANP32ALRR interaction interfaces. About 22% of particles in the FluPolC–chANP32A dataset lack density for ANP32A and were used to reconstruct a 3.4 Å resolution polymerase-only structure, which shows the same arrangement of FluPolC dimer as the ANP32A-containing complexes (Fig. 1c, Extended Data Figs. 2 and 3, Extended Data Table 1).

a–c, Cryo-EM structures of dimers of FluPolC heterotrimers with huANP32A (a) or chANP32A (b), and without ANP32A (c).
FluPol forms an asymmetric dimer
The first FluPol molecule (hereafter designated as the FluPol replicase, FluPolR) is fully resolved in the density map and shows a configuration of the peripheral flexible domains that is distinct from the one in the cap-snatching competent transcriptase, but similar to the configuration previously observed for RNA-free FluPol and FluPol bound to cRNA10,11 (Fig. 1, Extended Data Fig. 4, Supplementary Video 1). Specifically, the cap-binding domain of PB2 (PB2cap) is immobilized through extensive contacts with the palm subdomain of PB1, while the nuclear localization signal domain of PB2 (PB2NLS) intimately associates with the N-terminal endonuclease domain of P3 (P3endo). The 627 domain of PB2 (PB2627), named after the host-specific amino acid residue 627, makes relatively few contacts with the rest of FluPolR. FluPolR is bound to a 47-nucleotide long vRNA, whose 5′ terminus is clearly resolved and shows the typical hook structure bound in a pocket formed by P3 and PB1, as observed previously12,13 (Fig. 1, Extended Data Fig. 5). In the majority of particles, the 3′ vRNA terminus cannot be resolved, but the density observed around the template entry channel suggests that it has entered the active site. In about 10% of FluPolC–chANP32A and about 7% of FluPolC–huANP32A particles, the 3′ vRNA terminus is bound at the interface of the C-terminal domain of P3, the thumb subdomain of PB1 and the N1 subdomain of PB2, as recently described for FluPolA and influenza D FluPol12,14,15. The second FluPol molecule (hereafter designated as the encapsidating FluPol, FluPolE) shows a markedly different configuration of the flexible domains. The C-terminal flexible domains of PB2 (PB2cap, PB2 mid-link domain (PB2mid-link), PB2627 and PB2NLS) flip over to pack against the C-terminal domain of the P3 subunit (P3CTD) (Extended Data Fig. 4, Supplementary Video 1). P3endo, several N-terminal domains of PB2 and the C-terminal 18 amino acid residues of PB2NLS are disordered in FluPolE. Furthermore, approximately 52% of FluPolC–chANP32A and all FluPolC–huANP32A particles lack density for PB2cap and PB2mid-link in FluPolE, suggesting that these domains are flexible. No RNA was found associated with FluPolE. The two FluPolC molecules make extensive interactions, mainly between their respective PB2 and P3 subunits, to form a large polymerase dimer interface burying a total of ~1,500 Å2 (Fig. 2a). The PB2627 domain of FluPolR interacts with the P3CTD, PB2627 and PB2NLS domains of FluPolE (Fig. 2b), and the P3CTD domain of FluPolE stacks against the PB2 N2 (PB2N2) and PB2mid-link domains of FluPolR (Fig. 2c). In addition, the P3 arch and PB1 β-hairpin of FluPolE contact the P3CTD and PB2N2 domains of FluPolR (Fig. 2d). This dimer interface is distinct from the one observed previously in FluPolA, which promotes template realignment and is required for replication initiation on a cRNA12,16. To address the functional relevance of this dimer interface in influenza A virus, we introduced alanine mutations to several clusters of amino acid residues in FluPolA located at or close to the dimer interface on the basis of the structure of the FluPolC heterotrimeric dimer. These include the residues PA K324/H326/E327, PA K339/Q340 and PB2 P132, which are structurally equivalent to P3 R299/K301/D302, P3 N318/Q319 and PB2 E139 in FluPolC, respectively (Supplementary Fig. 1). All of these mutations resulted in a reduction of viral RNA levels in a minigenome assay analysing FluPol function (Extended Data Fig. 6a). These data indicate that amino acid residues at the asymmetric FluPol dimer interface we have identified in the FluPolC–ANP32A complexes are important for FluPol activity.

a, The FluPolC–FluPolC dimer interface with interacting regions in FluPolR and FluPolE highlighted on the molecular surface; the two molecules are shown peeled apart by 30° to highlight the surface. Letters denote the close-up views shown in b–d. b–d, Close-up views of the FluPolC–FluPolC-interaction interface indicated in a. Dashed lines indicate hydrogen bonds. e, ANP32A–FluPolC interface with interacting regions in FluPolC highlighted. Letters denote the close-up views shown in f–h. f–h, Close-up views of the ANP32A–FluPolC-interaction interface indicated in e. Dashed lines indicate hydrogen bonds.
ANP32ALRR bridges the asymmetric FluPol dimer
In both the FluPolC–huANP32A and FluPolC–chANP32A structures, the ANP32ALRR domain binds in a depression formed by the FluPolC dimer interface (Figs. 1a, b, 2e, Supplementary Video 1). The N-terminal region of ANP32ALRR interacts with FluPolR, involving multiple regions of PB2, including the lid domain (PB2lid), PB2627 and PB2NLS (Fig. 2e, f). These interactions bury a total of about 600 Å2 at the interface between ANP32A and FluPolR. The C-terminal region of ANP32ALRR interacts with the P3CTD domain and the N terminus of PB1 of FluPolE, and buries a total of about 490 Å2 at the interface (Fig. 2e, g, h). ANP32A N129 and D130, previously identified as important for FluPolA binding and activity8,17, are directly involved in the interaction with FluPolE. Specifically, N129 is sandwiched between M387 and K391 of P3 and interacts with K391 directly through hydrogen bonding, whereas D130 interacts with K608 of P3 (Fig. 2g). To address the functional relevance of this interaction, we mutated PA K413 in FluPolA—equivalent to P3 K391 in FluPolC (Fig. 2g, Supplementary Fig. 1)—to alanine. This mutant FluPolA showed reduced binding to huANP32A and loss of activity in a minigenome assay (Extended Data Fig. 7). Further mutations in FluPolA at PA D529 and PB2 T609, corresponding to P3 E513 and PB2 H631 in FluPolC (Fig. 2f, g, Supplementary Fig. 1), had similar inhibitory effects on both ANP32A binding and FluPol activity. Together, these data confirm the functional relevance of the identified ANP32A–FluPol interaction and provide an explanation for why chicken ANP32B (chANP32B) with I129 and N130 is unable to support FluPol activity8,17.
ANP32ALCAR interacts with PB2627
Species-specific differences in ANP32 proteins underpin the low activity of avian FluPolA in mammalian cells4. Amino acid differences important for FluPol activity have been identified in both ANP32ALRR and ANP32ALCAR, but a 33-amino-acid insertion in the avian ANP32ALCAR relative to mammalian ANP32ALCAR was found to be the most critical for the ability of avian ANP32A to support the activity of avian FluPolA4,18,19 (Fig. 3a). Although the ANP32ALCAR could not be assigned unambiguously in the map, continuous density shows that it contacts PB2627 of FluPolR (Fig. 3b, c, Supplementary Video 2). These data are in agreement with previous reports of a direct interaction between ANP32A and the PB2627 domain using biochemical methods and NMR20,21,22. In the FluPolC–chANP32A structure, the negatively charged chANP32ALCAR binds in a basic groove formed by the PB2627 domains of FluPolR and FluPolE, with the avian-specific 33-amino-acid insertion directly contacting the PB2627 domain of FluPolR. This interaction positions a previously identified hydrophobic SUMO interaction motif-like sequence23 (Extended Data Fig. 1c) on top of PB2627, as well as a downstream region of the ANP32LCAR which contains a mixture of basic and acidic amino acid residues (176-VLSLVKDR-183) (Fig. 3a–c). Specifically, this motif is located next to PB2 K649 (equivalent to E627 in the PB2 of avian FluPolA) and V614. PB2 V614 is equivalent to K591 in the 2009 H1N1 pandemic influenza A virus, which was of swine origin, and retained E627 in its avian PB2 but used a Q591K adaptation to increase polymerase activity24,25 (Extended Data Fig. 8, Supplementary Fig. 1). Interaction of this region of ANP32A with PB2627 could be critical for stabilization of the interaction between ANP32A and FluPol and, consequently, for the ability of ANP32A to support FluPol activity. The presence of a mixture of basic and acidic amino acid residues in this region could explain why avian ANP32A is able to support FluPol with either avian-specific 627E or mammalian-specific 627K in PB2. The corresponding region in huANP32A (176-EEEYDEDA-183), as well as in the huANP32B isoform (176-DEEDEDDE-183), is entirely acidic (Fig. 3a), potentially explaining the need to eliminate the acidic residue 627 in avian FluPolA upon viral transmission to a mammalian host. To test this hypothesis, we replaced the acidic 176–183 region of huANP32A with the corresponding region from chANP32A and found that this mutant form of huANP32A was able to fully support the activity of FluPolA with 627E in mammalian cells (Fig. 3d). These data are also in agreement with a previous study reporting that FluPolA can tolerate a range of non-acidic amino acid residues at position 627 and activity is positively correlated with the pI value of the amino acid26. Thus, our data provide an explanation for adaptive mutations observed in FluPolA upon transmission of influenza A virus from an avian to a human host.

a, Schematic of huANP32A and chANP32A, highlighting the 33-amino-acid insertion (33 aa) (yellow) in chANP32A and sequence differences. huANP32A(ch176–183) is a huANP32A construct in which residues 176–183 are replaced by those from chANP32A. Acidic and basic amino acid residues are highlighted in red and blue, respectively. b, Close-up view of the cryo-EM density attributed to chANP32A (grey, threshold 0.934) with the chANP32ALRR domain represented in cartoon (orange) and the positions of PB2627 of FluPolR (blue) and FluPolE (green). Regions potentially corresponding to the N-terminal part of ANP32ALCAR and part of the avian 33-amino-acid insertion are highlighted in cyan and yellow, respectively. V614 and K649 in FluPolC correspond to K591 (Q) and K627 (E) in FluPolA (residues in parentheses are avian species-specific), respectively. c, Same view as shown in b, with PB2627 of FluPolR and FluPolE shown in surface representation. d, Effect of wild-type chANP32A and huANP32A, and huANP32A(ch176–183) on the activity of FluPolA with mammalian-adapted PB2K627 or avian-signature PB2E627 in a vRNP-reconstitution assay in mammalian HEK 293T cells. Top, primer-extension analysis of viral RNA levels with quantification. Bottom, western blot analysis of ANP32A levels with molecular weight markers. Data are mean ± s.e.m., n = 3 biologically independent samples from 3 independent experiments. Ordinary one-way ANOVA with Dunnett’s post hoc test for multiple comparisons. P < 0.05 is considered significant to reject the null hypothesis. Gel source data are presented in Supplementary Fig. 2.
Implications for genome replication
The product exit channel of vRNA-bound FluPolR, located along the PB1 finger domains and PB2cap, points towards the 5′-cRNA binding site of FluPolE, which is located next to the PA arch (residues 343–376) and the PB1 β-hairpin (residues 353–370)13,15. A path lined with basic amino acid residues connects this exit channel with the 5′-cRNA binding site of FluPolE (Fig. 4a, Supplementary Video 3) and FluPolE is thus ideally positioned to capture the 5′ end of a nascent cRNA product emerging from the active site of FluPolR. This suggests that in the FluPolC–ANP32A complex, FluPolR functions as the replicase, whereas FluPolE acts as an ‘encapsidating’ polymerase, initiating the co-replicative assembly of the nascent cRNA with NP into a complementary ribonucleoprotein (cRNP) complex. To test this hypothesis, we performed a cRNA-stabilization assay27 to examine the ability of FluPol to encapsidate nascent cRNA product. Mutations in FluPolE at PA K324/H326/E327 and PA K339/Q340 caused a substantial reduction in cRNA accumulation. Note that PB2 P132 is located on the FluPolR side of the interface and therefore this mutation could not be tested using this assay. These data confirm that the FluPolR–FluPolE interface is important for encapsidation of a nascent cRNA product (Extended Data Fig. 6b). The PB2627 domain of FluPolE has been implicated in cRNA encapsidation using this assay, which is consistent with our model28. We propose that a similar mechanism is likely to apply for cRNA-bound FluPolR, owing to intrinsic similarities between cRNA synthesis and vRNA synthesis, which both require co-replicative encapsidation of nascent RNA.

a, Relative positions of the RNA-product exit channel in FluPolR and the 5′-RNA binding site in FluPolE in the chANP32A–FluPolC complex. The positions of the RNA-product exit channel in FluPolR and the 5′-RNA binding site in FluPolE were determined by superposing FluPolR and FluPolE with the structure of FluPolA bound to capped RNA and vRNA template (Protein Data Bank (PDB): 6RR7). b, Model for the role of FluPol–ANP32A complex in the replication of the influenza virus RNA genome and its assembly into vRNPs. FluPol in the context of vRNP or cRNP is flexible (1) but is stabilized in a replicase FluPolR conformation upon binding of a newly synthesized FluPol in the presence of ANP32A (2). FluPolR initiates replication in a primer-independent manner (3), with a trans-activating FluPol involved in cRNA to vRNA replication by promoting cRNA template realignment (3′). As the 5′ end of the nascent replication product is released from the polymerization active site of FluPolR, it is captured in the 5′-RNA binding pocket of the encapsidating FluPolE bound to FluPolR (4), initiating the encapsidation of the nascent RNA with NP (5). Nascent vRNA or cRNA assemble into vRNPs or cRNPs, respectively (6), and are released upon FluPolR termination. FluPolE becomes the resident polymerase of the newly produced vRNP or cRNP.
Such a role for the FluPol–ANP32A complex in viral genome replication is consistent with previous observations; in particular, ANP32A and the adaptive PB2 E627K mutation have been specifically linked to viral genome replication rather than transcription7,29,30 providing strong support for a role of the FluPol–ANP32A complex in replication. We were unable to fully resolve ANP32ALCAR in our structures, indicating that this part of ANP32A is highly dynamic. Replication involves viral NP, which co-replicatively coats viral RNA along its length, but it is currently unknown how NP is recruited onto the growing nascent chain of RNA. Non-segmented negative-strand RNA viruses encode an acidic phosphoprotein (P) that is an essential component of their RNA polymerase complex and is involved in recruiting NP to nascent replication product31. Segmented negative-strand RNA viruses do not encode an equivalent protein and it is tempting to speculate that its function is performed by a cellular protein such as ANP32A. Specifically, the unstructured region of ANP32ALCAR, downstream of the region interacting with FluPolR, could act as a molecular whip recruiting NP in a manner analogous to that proposed for the P phosphoprotein.
Understanding the structural basis for why ANP32A is fundamental to influenza virus replication opens a new perspective on the role of ANP32A and host-adaptive mutations in FluPol function. Specifically, our data suggest that the FluPol–ANP32A complex represents a replication platform for the influenza virus RNA genome. The mechanistic insights gained from this study have enabled us to build a more complete picture of the structural basis for the viral replication cycle. During infection, incoming vRNPs bind to the C-terminal domain of host RNA polymerase II through interactions primarily mediated by the C-terminal domain of FluPol PA32,33. This interaction stabilizes FluPol in a cap-snatching-competent conformation, enabling the cleavage of host capped RNAs that serve as primers for the initiation of viral mRNA synthesis33. Accumulation of newly synthesized FluPol favours genome replication over transcription, consistent with a transition from transcription to replication as infection proceeds, through forming an asymmetric dimer with the vRNP-associated resident FluPol and stabilizing it in a replication-competent conformation, enabling primer-independent initiation of replication (Fig. 4). ANP32A contributes to the stabilization of this dimer by bridging the two FluPol molecules. Several lines of evidence suggest that ANP32A is involved in cRNA-to-vRNA replication, although there is also evidence in favour of a role during vRNA-to-cRNA replication7,18,29,30. In particular, avian polymerase in human cells is incapable of generating replication-competent cRNPs, suggesting that cRNA encapsidation—and hence the correct assembly of cRNPs—might be affected29; this is fully consistent with our data showing that mutations at the FluPolR–FluPolE dimer interface interfere with cRNA encapsidation. ANP32A may have a particularly important role in stabilizing the interaction of a free FluPol with a cRNP-associated FluPol, as cRNPs need to make at least two different interactions with free FluPol molecules using two different interaction interfaces, to promote template realignment and nascent-product encapsidation. These interactions need to be fine-tuned in a host-dependent manner, in agreement with the observation of adaptive mutations at the dimer interfaces12,34. Dependence on free FluPol to ensure template realignment during replication initiation on a cRNA template12 and co-replicative genome assembly into a vRNP both provide the virus with a safety mechanism to avoid the generation of naked viral RNA in the absence of viral protein synthesis, which could otherwise be a trigger for activating innate immune responses35. The identification of amino acid residues involved in mediating ANP32A–FluPol interactions will facilitate both the development of novel small-molecule inhibitors that disrupt the interaction interface and the design of genetically engineered animals resilient to influenza virus.
Methods
No statistical methods were used to predetermine sample size. The experiments were not randomized and investigators were not blinded to allocation during experiments and outcome assessment.
Cells
Human embryonic kidney 293T (HEK 293T) and Sf9 insect cells were sourced from the Cell Bank of the Sir William Dunn School of Pathology, University of Oxford. HEK 293T cells were maintained in Dulbecco’s Modified Eagle Medium (DMEM) and Sf9 cells were maintained in Sf-900 II serum free medium (Gibco). Human eHAP cells (Horizon Discovery), control or lacking ANP32A and ANP32B proteins (dKO), were cultured in Iscove’s modified Dulbecco’s medium (IMDM; Sigma) supplemented with 10% fetal bovine serum (FBS; Sigma), 1% nonessential amino acids (NEAA; Gibco), and 1% penicillin/streptomycin (Gibco) and have been described7. Cell lines have not been authenticated but tested negative for mycoplasma contamination.
Protein expression and purification
The three subunits of influenza C/Johannesburg/1/1966 virus polymerase, together with human or chicken ANP32A, were co-expressed in Sf9 cells from codon-optimized genes (GeneArt) cloned into a single baculovirus using the MultiBac system36. Expression and purification of the co-expressed complex were performed as previously described10,12 with minor modifications: the salt concentration in all buffers was set to 150 mM to maintain FluPolC–ANP32A complex formation. Size-exclusion chromatography was performed using 25 mM HEPES–NaOH, pH 7.5, 150 mM NaCl and 5% (v/v) glycerol on a Superdex 200 Increase 10/300 GL column (GE Healthcare).
Cryo-EM sample preparation
Protein surface charges of the purified complex were neutralized by adding 0.001% glutaraldehyde for 20 min on ice to minimize preferential orientation of particles. After quenching the reaction by adding Tris-HCl, pH 8.0 to a final concentration of 100 mM, the sample was injected onto a Superdex 200 Increase 10/300 GL column (GE Healthcare) together with a 47 nt vRNA (5′-AGUAGAAACAAGGGUAUUUUUCUUUACUAGUCUACCCUGCUUUUGCU-3′) using 25 mM HEPES–NaOH, pH 7.5, 150 mM NaCl as the running buffer. This 47-nucleotide (nt) vRNA contains the 5′ and 3′ promoter elements recognized and bound by FluPol. The fractions of interest were concentrated to 0.23–0.28 mg ml−1 and RNA binding was confirmed by measuring the A260/A280 ratio. A final concentration of 0.005% (v/v) Tween 20 was added before grid preparation. A volume of 3–4 μl of sample was placed on glow-discharged Quantifoil Holey Carbon grids (R 2/1, with 2 μm holes and 1 μm spacing and applied on biocompatible 200 gold mesh), before blotting for 3.0–3.5 s and flash-freezing in liquid ethane. All grids were prepared using a Vitrobot mark IV (FEI) at 95–100% humidity.
Cryo-EM image collection and processing
Cryo-EM data for the FluPolC–chANP32A sample were collected on a 300 kV Titan Krios microscope (Thermo Fisher Scientific) at the Division of Structural Biology (Strubi). Automated data collection was setup in EPU 2.1 using a K2 Summit (Gatan) direct electron detector at 1.37 Å per pixel operating in counting mode and a GIF Quantum energy filter (Gatan) with 20 eV slit. Sample was collected with a tilt angle of 30° at a dose of ~38.8 e− Å−2 across 44 frames for a dose rate of ~3.527 e− Å−2 s−1, using a defocus range of −2 μm to −3.5 μm. Cryo-EM data for the FluPolC–huANP32A sample were collected on a 300 kV Titan Krios microscope (Thermo Fisher Scientific) at Electron Bio-Imaging Centre (eBIC). Automated data collection was setup in SerialEM 3.7 using a K3 (Gatan) direct electron detector operating in super-resolution mode at 0.5425 Å per pixel and a GIF Quantum energy filter (Gatan) with 20 eV slit. Sample was collected with a tilt angle of 20° at a dose of ~32.1 e− Å−2 across 34 frames for a dose rate of ~16 e− Å−2 s−1, using a defocus range of −2 μm to −3.5 μm. Sample-specific data collection parameters are summarized in Extended Data Table 1. Data processing pipelines are shown in Extended Data Fig. 1. In brief, raw movies were processed using MotionCor2-1.1.037, with a five-by-five patch-based alignment, keeping all of the frames and dose-weighting up to the total exposure. For the K3 super-resolution, the data have been binned 2 times at the motion correction step, giving a final pixel size of 1.085 Å per pixel. The contrast transfer function (CTF) of full-dose, non-weighted micrographs was initially estimated using Gctf-v.1.0638 or cryoSPARC v.2.12.039 Patch-CTF. Poor-quality images were discarded after manual inspection. Particles were blob picked in cryoSPARC v.2.12.039 and the 2D classes were inspected and classes of interest were selected to generate templates for complete particle picking. For the FluPolC–chANP32A dataset, a total number of 2,534,332 particles were picked and a final number of 772,989 particles were exported to RELION 3.140 after 2D and 3D class selection. The consensus map was refined to 3.50 Å. Bayesian polishing and per particle CTF refinement and B-factor estimation with beam tilt correction were performed in RELION 3.1 which improved map resolution to 3.2 Å. High order aberration and magnification anisotropy refinement improved map resolution to 3.1 Å. Global and focus 3D classification were performed within RELION (Extended Data Fig. 2a). For the 3D variability analysis, polished particles and model were imported into cryoSPARC v.2.12.0 and refined using the non-uniform refinement job. A mask was then designed around the density corresponding to the PB2627 domains of both polymerases and chANP32A. The 3D variability analysis was then performed using this mask, asking for solving the three main modes on a structure low pass filtered to 6 Å. The results of this principal component analysis were clustered in twenty sub-populations and models were reconstructed for each of the individual clusters. One cluster was showing a clear density for the chANP32ALCAR region and was refined using the non-uniform refinement job to 3.6 Å (Subclass 2). In parallel, all of the particles having a strong density for chANP32A were selected and refined using the non-uniform refinement job to 3.1 Å. The same 3D variability analysis was performed, asking for solving the three main modes on a structure low pass filtered to 6 Å. Mode 3 is displayed as Supplementary Video 2. For the FluPolC–huANP32A dataset, a total number of 2,312,045 particles were picked and a final number of 835,198 particles were exported to RELION 3.1 after 2D and 3D class selection. The consensus map was refined to 3.2 Å. Bayesian polishing and per particle CTF refinement and B-factor estimation with beam tilt correction were performed in RELION 3.1 which improved map resolution to 2.8 Å. Global and focus 3D classification were performed within RELION (Extended Data Fig. 2b). CryoEF41 and 3D Fourier shell correlation (FSC)42 sphericity scores were used to confirm that the final maps were not suffering from distortion bias due to preferred specimen orientation. The structures were modelled by first fitting ANP32ALRR (PDB ID: 4XOS), FluPolC (PDB ID: 5D98) and 5′ and 3′ vRNA termini (PDB ID: 6RR7) using UCSF Chimera43. One cycle of rigid body real space refinement followed by manual adjustment in Coot44 was performed to correctly position the Cα chain into the density. Locally sharpened maps were generated using LocScale45 in the CCP-EM46 package. Finally, cycles of PHENIX47 real space refinement and manual building in Coot44 were used to improve model geometry. Map-to-model comparison in PHENIX mtriage validated that no over-fitting was present in the structures. Model geometry was validated for all models using MolProbity48. All map and model statistics are detailed in Extended Data Table 1. A homology model of FluPolA–ANP32A complex structure was generated using SWISS-MODEL49.
Plasmids
Plasmids pHMG-PB1, pHMG-PB2, pHMG-P3 and pHMG-NP expressing the subunits of FluPolC and NP of influenza C/Johannesburg/1/66 virus and pPolI-Luci-CP3-RT expressing a luciferase reporter have been described50,51. pcDNA-PB1, pcDNA-PB1a (catalytically inactive; D445A/D446A), pcDNA-PB2, pcDNA-PB2 K627E, pcDNA-PA, pcDNA-NP and pPOLI-NA, which encodes an NA vRNA segment, are derived from influenza A/WSN/33 virus and have been described previously27,52,53,54. pCAGGS-chANP32A has been described previously4. pcDNA-huANP32A and pCAGGS-huANP32A-Strep were produced from pCAGGS-huANP32A4 by restriction-ligation cloning. pcDNA-huANP32A(ch176-183), pcDNA-PB2 T609A, pcDNA-PB2 P132A, pcDNA-PA D529A/R531A/E533A, pcDNA-PA K413A, pcDNA-PA K324A/H326A/E327A and pcDNA-PA K339A/Q340A were generated from pcDNA-huANP32A, pcDNA-PB2 and pcDNA-PA by site-directed PCR mutagenesis and validated by Sanger sequencing.
Influenza C virus vRNP-reconstitution and luciferase-reporter assays
Approximately 0.2 × 106 eHAP cells, control or dKO, were transfected with plasmids pHMG-PB1, pHMG-PB2, pHMG-P3, pHMG-NP, pPolI-Luci-CP3-RT and pcDNA-Renilla using Lipofectamine 2000 according to the manufacturer’s instructions. Where indicated, plasmids pCAGGS-huANP32A, pCAGGS-huANP32B or pCAGGS-chANP32A were included. Cells were lysed 24 h after transfection using Passive lysis buffer (Promega). Firefly and Renilla luciferase activities were measured using a Promega dual luciferase kit and firefly activity levels were normalized to the Renilla control.
Influenza A virus vRNP-reconstitution assay and primer extension analysis
Approximately 0.2 × 106 HEK 293T cells were transfected with plasmids pcDNA-PB1, pcDNA-PB2, pcDNA-PA, pcDNA-NP and pPOLI-NA using Lipofectamine 2000 according to the manufacturer’s instructions. Where indicated, plasmids encoding ANP32A proteins or mutant polymerase subunits were included. Total cellular RNA was extracted 20 h after transfection using TRI reagent (Sigma) according to the manufacturer’s instructions. NA segment RNA levels were assessed using primer extension55. In brief, primers complementary to the positive (5′-TGGACTAGTGGGAGCATCAT-3′) and negative (5′-TCCAGTATGGTTTTGATTTCCG-3′) sense NA segment viral RNA species were labelled with 32P and total cellular RNA was reverse transcribed. A primer complementary to 5S rRNA (5′-TCCCAGGCGGTCTCCCATCC-3′) was included as a loading control. Products were resolved by 6% denaturing PAGE and visualized by phosphorimaging. Product bands were identified by comparison with previously published data56,57. Analysis was carried out using ImageJ58 and Prism 8 (GraphPad), and viral RNA signals were normalized to the 5S rRNA loading control.
Western blotting
Western blotting of all proteins was carried out using specific rabbit polyclonal antibodies. PB1 and PB2 were blotted using commercially available antibodies (Genetex), while PA was blotted using a custom-made antibody59. ANP32A and actin were blotted using commercially available antibodies (Sigma). In all cases the secondary antibody used was goat anti-rabbit conjugated to HRP, and Amersham ECL Western Blotting Detection Reagents (GE Healthcare) were used for detection.
Affinity purification assays
Approximately 2 × 106 HEK 293T cells were transfected with pcDNA-PB1, pcDNA-PB2, pcDNA-PA and pCAGGS-huANP32A-Strep. 48 h after transfection, cells were lysed in 500 μl of lysis buffer (50 mM Tris-HCl pH 8.0, 200 mM NaCl, 25% glycerol, 0.5% Igepal CA-630, 1 mM DTT, 1 mM PMSF, 1 × complete EDTA-free protease inhibitor cocktail tablet (Roche)) for 1 h at 4 °C and cellular debris was cleared by centrifugation for 5 min at 17,000g. The supernatant was diluted in 2 ml 150 mM NaCl and incubated with Strep-Tactin beads (IBA) for 2 h at 4 °C. Beads were washed three times in wash buffer (10 mM Tris-HCl pH 8.0, 150 mM NaCl, 10% glycerol, 0.1% Igepal CA-630, 1 mM PMSF) and complexes were eluted in 1× buffer E (IBA) for 16 h at 4 °C. Eluted complexes were analysed by 12% SDS PAGE and western blotting.
cRNA-encapsidation assays
cRNA-encapsidation assays were performed as described previously27. In brief, approximately 1 × 106 HEK 293T cells were transfected with pcDNA-PB1a, pcDNA-PB2, pcDNA-PA and pcDNA-NP using Lipofectamine 2000 according to the manufacturer’s instructions. Forty-eight hours after transfection, culture medium was replaced with DMEM containing 0.5% FBS and 5 μg ml−1 actinomycin D and influenza A/WSN/33 virus at multiplicity of infection 5.0. Total cellular RNA was extracted 6 h after infection using Trizol (Sigma) according to the manufacturer’s instructions, and viral RNA levels were determined by primer extension analysis as described above.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
All data are available from the corresponding authors and/or included in the manuscript or Supplementary Information. Cryo-EM density maps with the corresponding atomic coordinates have been deposited in the Electron Microscopy Data Bank with accession codes EMD-10665 (FluPolC–huANP32A subclass 1), EMD-10667 (FluPolC–huANP32A subclass 2), EMD-10666 (FluPolC–chANP32A subclass 1), EMD-10659 (FluPolC–chANP32A subclass 2), EMD-10662 (FluPolC–chANP32A subclass 3) and EMD-10664 (FluPolC–chANP32A subclass 4), and the Protein Data Bank with accession codes 6XZQ (FluPolC–huANP32A subclass 1), 6Y0C (FluPolC-huANP32A subclass 2), 6XZR (FluPolC–chANP32A subclass 1), 6XZD (FluPolC–chANP32A subclass 2), 6XZG (FluPolC–chANP32A subclass 3) and 6XZP (FluPolC–chANP32A subclass 4).
References
Krammer, F. et al. Influenza. Nat. Rev. Dis. Primers 4, 3 (2018).
Fodor, E. & Te Velthuis, A. J. W. Structure and function of the influenza virus transcription and replication machinery. Cold Spring Harb. Perspect. Med. 10, a038398 (2019).
Wandzik, J. M., Kouba, T. & Cusack, S. Structure and function of influenza polymerase. Cold Spring Harb. Perspect. Med. 10, a038372 (2020).
Long, J. S. et al. Species difference in ANP32A underlies influenza A virus polymerase host restriction. Nature 529, 101–104 (2016).
Walker, A. P. & Fodor, E. Interplay between influenza virus and the host RNA polymerase II transcriptional machinery. Trends Microbiol. 27, 398–407 (2019).
Peacock, T. P., Sheppard, C. M., Staller, E. & Barclay, W. S. Host determinants of influenza RNA synthesis. Annu. Rev. Virol. 6, 215–233 (2019).
Staller, E. et al. ANP32 proteins are essential for influenza virus replication in human cells. J. Virol. 93, e00217-19 (2019).
Zhang, H. et al. Fundamental contribution and host range determination of ANP32A and ANP32B in influenza A virus polymerase activity. J. Virol. 93, e00174-19 (2019).
Huyton, T. & Wolberger, C. The crystal structure of the tumor suppressor protein pp32 (Anp32a): structural insights into Anp32 family of proteins. Protein Sci. 16, 1308–1315 (2007).
Hengrung, N. et al. Crystal structure of the RNA-dependent RNA polymerase from influenza C virus. Nature 527, 114–117 (2015).
Thierry, E. et al. Influenza polymerase can adopt an alternative configuration involving a radical repacking of PB2 domains. Mol. Cell 61, 125–137 (2016).
Fan, H. et al. Structures of influenza A virus RNA polymerase offer insight into viral genome replication. Nature 573, 287–290 (2019).
Pflug, A., Guilligay, D., Reich, S. & Cusack, S. Structure of influenza A polymerase bound to the viral RNA promoter. Nature 516, 355–360 (2014).
Peng, Q. et al. Structural insight into RNA synthesis by influenza D polymerase. Nat. Microbiol. 4, 1750–1759 (2019).
Wandzik, J.M. et al. A structure-based model for the complete transcription cycle of influenza polymerase. Cell 181, 877–893 (2020).
Chang, S. et al. Cryo-EM structure of influenza virus RNA polymerase complex at 4.3 Å resolution. Mol. Cell 57, 925–935 (2015).
Long, J. S. et al. Species specific differences in use of ANP32 proteins by influenza A virus. eLife 8, e45066 (2019).
Bi, Z. et al. Insights into species-specific regulation of ANP32A on the mammalian-restricted influenza virus polymerase activity. Emerg. Microbes Infect. 8, 1465–1478 (2019).
Zhang, H. et al. A unique feature of swine ANP32A provides susceptibility to avian influenza virus infection in pigs. PLoS Pathog. 16, e1008330 (2020).
Baker, S.F., Ledwith, M.P. & Mehle, A. Differential splicing of ANP32A in birds alters its ability to stimulate rna synthesis by restricted influenza polymerase. Cell Rep. 24, 2581–2588 (2018).
Camacho-Zarco, A. R. et al. Molecular basis of host-adaptation interactions between influenza virus polymerase PB2 subunit and ANP32A. Nat. Commun. 11, 3656 (2020).
Mistry, B. et al. Elucidating the interactions between influenza virus polymerase and host factor ANP32A. J. Virol. 94, e01353-19 (2020).
Domingues, P. & Hale, B. G. Functional insights into ANP32A-dependent influenza A virus polymerase host restriction. Cell Rep. 20, 2538–2546 (2017).
Mehle, A. & Doudna, J. A. Adaptive strategies of the influenza virus polymerase for replication in humans. Proc. Natl Acad. Sci. USA 106, 21312–21316 (2009).
Yamada, S. et al. Biological and structural characterization of a host-adapting amino acid in influenza virus. PLoS Pathog. 6, e1001034 (2010).
Chin, A. W. H. et al. Influenza A viruses with different amino acid residues at PB2-627 display distinct replication properties in vitro and in vivo: revealing the sequence plasticity of PB2-627 position. Virology 468–470, 545–555 (2014).
Vreede, F. T., Jung, T. E. & Brownlee, G. G. Model suggesting that replication of influenza virus is regulated by stabilization of replicative intermediates. J. Virol. 78, 9568–9572 (2004).
Nilsson, B. E., Te Velthuis, A. J. W. & Fodor, E. Role of the PB2 627 domain in influenza A virus polymerase function. J. Virol. 91, e02467-16 (2017).
Mänz, B., Brunotte, L., Reuther, P. & Schwemmle, M. Adaptive mutations in NEP compensate for defective H5N1 RNA replication in cultured human cells. Nat. Commun. 3, 802 (2012).
Sugiyama, K., Kawaguchi, A., Okuwaki, M. & Nagata, K. pp32 and APRIL are host cell-derived regulators of influenza virus RNA synthesis from cRNA. eLife 4, e08939 (2015).
Pan, J. et al. Structure of the human metapneumovirus polymerase phosphoprotein complex. Nature 577, 275–279 (2020).
Lukarska, M. et al. Structural basis of an essential interaction between influenza polymerase and Pol II CTD. Nature 541, 117–121 (2017).
Serna Martin, I. et al. A mechanism for the activation of the influenza virus transcriptase. Mol. Cell 70, 1101–1110 (2018).
Chen, K. Y., Santos Afonso, E. D., Enouf, V., Isel, C. & Naffakh, N. Influenza virus polymerase subunits co-evolve to ensure proper levels of dimerization of the heterotrimer. PLoS Pathog. 15, e1008034 (2019).
Killip, M. J., Fodor, E. & Randall, R. E. Influenza virus activation of the interferon system. Virus Res. 209, 11–22 (2015).
Bieniossek, C., Imasaki, T., Takagi, Y. & Berger, I. MultiBac: expanding the research toolbox for multiprotein complexes. Trends Biochem. Sci. 37, 49–57 (2012).
Zheng, S. Q. et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017).
Zhang, K. Gctf: Real-time CTF determination and correction. J. Struct. Biol. 193, 1–12 (2016).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Scheres, S. H. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 180, 519–530 (2012).
Naydenova, K. & Russo, C. J. Measuring the effects of particle orientation to improve the efficiency of electron cryomicroscopy. Nat. Commun. 8, 629 (2017).
Tan, Y. Z. et al. Addressing preferred specimen orientation in single-particle cryo-EM through tilting. Nat. Methods 14, 793–796 (2017).
Pettersen, E. F. et al. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D 66, 486–501 (2010).
Jakobi, A. J., Wilmanns, M. & Sachse, C. Model-based local density sharpening of cryo-EM maps. eLife 6, e27131 (2017).
Burnley, T. Introducing the Proceedings of the CCP-EM Spring Symposium. Acta Crystallogr. D 73, 467–468 (2017).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D 66, 213–221 (2010).
Davis, I. W. et al. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 35, W375–W383 (2007).
Waterhouse, A. et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 46 (W1), W296–W303 (2018).
Crescenzo-Chaigne, B., Naffakh, N. & van der Werf, S. Comparative analysis of the ability of the polymerase complexes of influenza viruses type A, B and C to assemble into functional RNPs that allow expression and replication of heterotypic model RNA templates in vivo. Virology 265, 342–353 (1999).
Tang, Y. S., Lo, C. Y., Mok, C. K., Chan, P. K. & Shaw, P. C. The extended C-terminal region of influenza C virus nucleoprotein is important for nuclear import and ribonucleoprotein activity. J. Virol. 93, e02048-18 (2019).
Fodor, E. et al. A single amino acid mutation in the PA subunit of the influenza virus RNA polymerase inhibits endonucleolytic cleavage of capped RNAs. J. Virol. 76, 8989–9001 (2002).
Fodor, E. et al. Rescue of influenza A virus from recombinant DNA. J. Virol. 73, 9679–9682 (1999).
Paterson, D., te Velthuis, A. J., Vreede, F. T. & Fodor, E. Host restriction of influenza virus polymerase activity by PB2 627E is diminished on short viral templates in a nucleoprotein-independent manner. J. Virol. 88, 339–344 (2014).
Robb, N. C., Smith, M., Vreede, F. T. & Fodor, E. NS2/NEP protein regulates transcription and replication of the influenza virus RNA genome. J. Gen. Virol. 90, 1398–1407 (2009).
Deng, T. et al. Role of ran binding protein 5 in nuclear import and assembly of the influenza virus RNA polymerase complex. J. Virol. 80, 11911–11919 (2006).
Vreede, F. T. & Brownlee, G. G. Influenza virion-derived viral ribonucleoproteins synthesize both mRNA and cRNA in vitro. J. Virol. 81, 2196–2204 (2007).
Schneider, C. A., Rasband, W. S. & Eliceiri, K. W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 (2012).
Engelhardt, O. G., Smith, M. & Fodor, E. Association of the influenza A virus RNA-dependent RNA polymerase with cellular RNA polymerase II. J. Virol. 79, 5812–5818 (2005).
Robert, X. & Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 42, W320–W324 (2014).
Acknowledgements
We thank N. Naffakh, P.-C. Shaw, G. G. Brownlee and F. Vreede for plasmids; I. Berger for the MultiBac system; D. Karia, A. Howe and D. Clare for assistance with cryo-EM; and G. G. Brownlee and members of the Fodor and Grimes laboratories for helpful comments and discussions. This work was supported by Medical Research Council (MRC) programme grant MR/R009945/1 (to E.F.), Wellcome Investigator Awards 200835/Z/16/Z (to J.M.G.) and 205100/Z/16/Z (to W.S.B.), MRC Studentship (to A.P.W.) and Imperial College President’s PhD Scholarship (to E.S.). We thank Diamond Light source for access and support of the cryo-EM facilities at the UK national Electron Bio-Imaging Centre (eBIC) (proposal EM20223), funded by the Wellcome, MRC and BBSRC. Further electron microscopy provision was provided through the OPIC electron microscopy facility, which was founded by a Wellcome JIF award (060208/Z/00/Z) and is supported by a Wellcome equipment grant (093305/Z/10/Z). Computation was performed at the Oxford Biomedical Research Computing (BMRC) facility, a joint development between the Wellcome Centre for Human Genetics and the Big Data Institute supported by Health Data Research UK and the NIHR Oxford Biomedical Research Centre. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health. Part of this work was supported by Wellcome administrative support grant (203141/Z/16/Z).
Author information
Authors and Affiliations
Contributions
L.C., H.F., A.P.W., J.R.K., E.F. and J.M.G. conceived and designed the study. H.F., L.C. and J.R.K. carried out cloning of recombinant baculoviruses and protein purification, collected and processed electron microscopy data and built and refined models. A.P.W. and J.S. performed functional assays and analysed data. E.S. and W.S.B. provided plasmids and cell lines. J.M.G. and E.F. supervised the structural and functional studies, respectively. L.C., H.F., A.P.W., J.R.K., E.F. and J.M.G. wrote the manuscript with input from all co-authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature thanks Julien Lescar, Yi Shi, Xiu-Feng Wan and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 FluPolC activity depends on ANP32A and alignment of ANP32 proteins.
a, b, Luciferase reporter gene activities reflecting FluPolC activity in control (a) and dKO (b) eHAP cells in the presence or absence of overexpressed huANP32A, huANP32B or chANP32A. Data are presented as mean values ± s.e.m. n = 3 biologically independent samples from n = 3 independent experiments. Ordinary one-way ANOVA with Dunnett’s post hoc test for multiple comparisons. P < 0.05 is considered significant to reject the null hypothesis. c, Sequence alignment of huANP32A, huANP32B, chANP32A, chANP32B. Residues involved in hydrogen bonding interactions with FluPolC are indicated in orange. The chANP32A avian-specific 33 amino acid insertion is highlighted in cyan. The SUMO interaction motif (SIM) sequence is indicated by black triangles. The figure was prepared with Espript 3.060.
Extended Data Fig. 2 Data collection, processing and analysis scheme.
a, b, Flowchart for the processing and the classification of the FluPolC-huANP32A complex (a) and FluPolC-chANP32A complex (b).
Extended Data Fig. 3 Single-particle cryo-EM analysis of FluPolC-huANP32A and FluPolC-chANP32A complexes.
a, e, Representative micrograph of FluPolC-huANP32A (a) and FluPolC-chANP32A (e) embedded in vitreous ice. Scale bar, 200 Å. b, f, Representative 2D class averages of FluPolC-huANP32A (b) and FluPolC-chANP32A (f). c, d, Data analysis for FluPolC-huANP32A Subclass1 (c) and Subclass2 (d). 3D reconstruction locally filtered and coloured according to RELION local resolution (left panel). FSC curve indicating overall map resolution and model-to-map FSC (middle panel). Curves are shown for phase randomization, unmasked, masked and phase-randomization-corrected masked maps. Angular distribution of particle projections with the cryo-EM map shown in grey (right panel). g–j, Data analysis for FluPolC-chANP32A Subclass1 (g), Subclass2 (h), Subclass3 (i) and Subclass4 (j). 3D reconstruction locally filtered and coloured according to RELION local resolution (top panel). FSC curve indicating overall map resolution and the model-to-map FSC (middle panel). Curves are shown for phase randomization, unmasked, masked and phase-randomization-corrected masked maps. Angular distribution of particle projections with the cryo-EM map shown in grey (bottom panel).
Extended Data Fig. 4 Comparison of FluPolR and FluPolE structures with the transcriptase and apo conformations of FluPol.
a–d, Comparison of structures of human influenza A/NT/60/68 (H3N2) bound to vRNA and capped RNA in the transcriptase conformation (PDB: 6RR7) (a) and human influenza C/Johannesburg/1/66 in the apo conformation (PDB: 5D98) (b) with structures of FluPolR (c) and FluPolE (d) in the FluPolC-chANP32A complex. e–h, Comparison of the PB2 domain arrangements in the complexes shown in a–d.
Extended Data Fig. 5 Close-up view of the interaction of 5′ and 3′ vRNA termini with FluPolR.
a, c, Close-up view of the 3′ vRNA pointing towards the active site in the FluPolC-huANP32A (a) and FluPolC-chANP32A (c) structures. b, d, Close-up view of the 3′ vRNA binding in a groove located between P3CTD and the PB1thumb and PB2N1 subdomains in the FluPolC-huANP32A (b) and FluPolC-chANP32A (d) structures.
Extended Data Fig. 6 Effect of FluPolR-FluPolE dimer interface mutations on FluPolA activity.
a, b, Effect of mutations at the FluPolR-FluPolE dimer interface on FluPolA activity in viral minigenome assays (a) and cRNA encapsidation by FluPolA (b). Data are presented as mean values ± s.e.m. n = 3 biologically independent samples from n = 3 independent experiments. Ordinary one-way ANOVA with Dunnett’s post hoc test for multiple comparisons. P < 0.05 is considered significant to reject the null hypothesis. Western blot analyses were repeated from n = 3 independent experiments with similar results. For gel source data, see Supplementary Fig. 2.
Extended Data Fig. 7 Effect of FluPolA mutations at the FluPolA-ANP32A interface on FluPolA activity and interaction with huANP32A.
a, b, Effect of FluPolA mutations at the FluPolA-ANP32A interface on FluPolA activity in viral minigenome assays on (a) and FluPolA-ANP32A interaction (b). Data are presented as mean values ± s.e.m. n = 3 biologically independent samples from n = 3 independent experiments. Ordinary one-way ANOVA with Dunnett’s post hoc test for multiple comparisons. P < 0.05 is considered significant to reject the null hypothesis. Western blot analyses were repeated from n = 3 independent experiments with similar results. For gel source data, see Supplementary Fig. 2.
Extended Data Fig. 8 Structural comparison of PB2627 domains of FluPolA and FluPolC.
Structures of the PB2627 domains from crystal structures of FluPol from influenza C/Johannesburg/1/1966 (a, PDB ID: 5D98) and A/NT/60/1968 (H3N2) (b, PDB ID: 6QNW) viruses are aligned and shown in cartoon mode. Residues discussed in this study are highlighted in stick mode and coloured in orange.
Supplementary information
Supplementary Figure
Supplementary Fig. 1 Sequence alignment of FluPolA and FluPolC. a - c, Sequence alignment of FluPol subunits (a, PA/P3; b, PB1; c, PB2) from influenza A/WSN/33 (H1N1) and C/Johannesburg/1/1966 viruses. Residues involved in forming the asymmetric FluPol dimer interface are highlighted in cyan, and residues involved in ANP32A binding are highlighted in orange. The figure was prepared with Espript 3.060.
Supplementary Figure
Supplementary Fig. 2 Source data (gels).
Video 1
Overview of the FluPolC-chANP32A structure. The movie shows how vRNA-bound FluPolC (FluPolR) assembles with an apo FluPolC (FluPolE) to form an asymmetric dimer stabilized by ANP32A.
Video 2
Cryo-EM map of the FluPolC-chANP32A complex. The movie shows the repositioning of the reconstructed densities, corresponding to the PB2627 domains of both polymerases and chANP32A, along the third eigenvector.
Video 3
Cryo-EM map of the FluPolC-chANP32A complex. The movie shows the cryo-EM density of the FluPolC-chANP32A complex, highlighting the proximity of the product exit channel of FluPolR to the 5’ RNA binding site of FluPolE.
Rights and permissions
About this article

Cite this article
Carrique, L., Fan, H., Walker, A.P. et al. Host ANP32A mediates the assembly of the influenza virus replicase. Nature 587, 638–643 (2020). https://doi.org/10.1038/s41586-020-2927-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-020-2927-z
This article is cited by
-
Structural basis for dimerization of a paramyxovirus polymerase complex
Nature Communications (2024)
-
The host RNA polymerase II C-terminal domain is the anchor for replication of the influenza virus genome
Nature Communications (2024)
-
Structural and functional analysis of the minimal orthomyxovirus-like polymerase of Tilapia Lake Virus from the highly diverged Amnoonviridae family
Nature Communications (2023)
-
The ubiquitination landscape of the influenza A virus polymerase
Nature Communications (2023)
-
Creating resistance to avian influenza infection through genome editing of the ANP32 gene family
Nature Communications (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
