
Abstract
Recently, we demonstrated that the oligosaccharide portion of ganglioside GM1 is responsible, via direct interaction and activation of the TrkA pathway, for the ability of GM1 to promote neuritogenesis and to confer neuroprotection in Neuro2a mouse neuroblastoma cells. Recalling the knowledge that ganglioside GM1 modulates calcium channels activity, thus regulating the cytosolic calcium concentration necessary for neuronal functions, we investigated if the GM1-oligosaccharide would be able to overlap the GM1 properties in the regulation of calcium signaling, excluding a specific role played by the ceramide moiety inserted into the external layer of plasma membrane. We observed, by calcium imaging, that GM1-oligosaccharide administration to undifferentiated Neuro2a cells resulted in an increased calcium influx, which turned out to be mediated by the activation of TrkA receptor. The biochemical analysis demonstrated that PLCγ and PKC activation follows the TrkA stimulation by GM1-oligosaccharide, leading to the opening of calcium channels both on the plasma membrane and on intracellular storages, as confirmed by calcium imaging experiments performed with IP3 receptor inhibitor. Subsequently, we found that neurite elongation in Neuro2a cells was blocked by subtoxic administration of extracellular and intracellular calcium chelators, suggesting that the increase of intracellular calcium is responsible of GM1-oligosaccharide mediated differentiation. These results suggest that GM1-oligosaccharide is responsible for the regulation of calcium signaling and homeostasis at the base of the neuronal functions mediated by plasma membrane GM1.
Similar content being viewed by others
Introduction
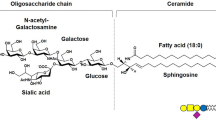
Among gangliosides, a particular class of sialic acid-containing glycosphingolipids enriched in neuronal membranes, particular attention is given to GM1, β-Gal-(1–3)-β-GalNAc-(1–4)-[α-Neu5Ac-(2–3)]-β-Gal-(1–4)-β-Glc-(1–1)-Cer (II3Neu5Ac-Gg4Cer) by virtue of its relevant implication in neuronal differentiation and in neuronal recovery and protection [1,2,3,4,5,6]. Component of all mammalian brains, GM1 is inserted into the outer layer of the plasma membrane (PM) with the hydrophobic moiety, the ceramide, while the saccharide portion protrudes into the extracellular milieu interacting with a wide range of membrane-associated proteins, including receptors and enzymes [1,2,3,4,5,6]. It has been extensively studied how GM1 induction of neurite outgrowth and neuroprotective phenomena are accomplished through GM1 specific interaction with neurotrophin tyrosine kinase receptors (Trk) and via its ability to modulate cellular calcium (Ca2+) levels, acting on Ca2+ influx channels, Ca2+ exchangers, and various Ca2+-utilizing enzymes [3, 7, 8]. Elevation of endogenous PM GM1 or exogenous applied GM1 in mouse neuroblastoma cells Neuro2a (N2a) and in other neuroblastoma cells leads to the increase of intracellular Ca2+, which is accompanied by neurite outgrowth and extension [7, 9, 10]. Additionally, cultured neurons from the B4galnt1 knock out mouse, characterized by deficiency of GM1 and its oligosialo derivatives, showed impaired Ca2+ regulatory capability, which translates in significantly retarded outgrowth [11, 12] and in increased vulnerability to KCl and glutamate excitotoxicity, which is rescued after GM1 application [12].
Although the numerous properties of GM1 have been extensively studied over the years, its mechanism of action is still being explored. It has been proved that GM1-oligosaccharide, β-Gal-(1–3)-β-GalNAc-(1–4)-[α-Neu5Ac-(2–3)]-β-Gal-(1–4)-Glc (OligoGM1; II3Neu5Ac-Gg4), exogenously added to the culture medium of N2a neuroblastoma cells, was able alone to induce the neuritogenesis process by directly interacting with nerve growth factor (NGF)-specific receptor TrkA at the PM, reserving to the ceramide an exclusively anchor and structural role [13]. Subsequently we found that OligoGM1 administered to cerebellar granule neurons enhanced neuron clustering, neurite sprouting and networking, thus confirming the specific role of the oligosaccharide chain in the processes of neuronal differentiation and maturation, known to be regulated by the entire GM1 [14]. Additionally, the OligoGM1 was found to mediate also the neuroprotective phenomena attributed to ganglioside GM1, being able to induce protection from 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine hydrochloride (MPTP) neurotoxicity in N2a neuroblastoma cells [15] acting on mitochondria [16], and to rescue the physical and biochemical defects due to the partial lack of GM1 content in a mouse model of Parkinson’s disease [17].
The present work aims to further investigate the mechanism of action of OligoGM1, as the bioactive component of ganglioside GM1, focusing on its ability to modulate the cell Ca2+ flow, mechanism at the base of GM1-mediated neuronal differentiation. We describe that the addition of OligoGM1 to the culture medium of N2a cells is able to activate the TrkA-phospholipase C gamma (PLCγ) pathway at the cell surface, leading to an increase of cytosolic Ca2+ necessary for OligoGM1-induced cell differentiation. These data contribute to explain the molecular mechanism underlying the neurotrophic action of OligoGM1, shedding new light on the mechanism of action of ganglioside GM1.
Methods
Materials
Commercial chemicals were of the highest purity available, common solvents were distilled before use and water was doubly distilled in a glass apparatus.
Phosphate buffered saline (PBS), sodium orthovanadate (Na3VO4), phenylmethanesulfonyl fluoride (PMSF), aprotinin, protease inhibitor cocktail (IP), ethylenediamine tetraacetic acid (EDTA), bovine serum albumin (BSA), Xestspongin C, calcium ionophore A23187, mouse anti-alpha-tubulin (RRID: AB_477579) antibody and mouse neuroblastoma N2a cells (RRID: CVCL_0470) were from Sigma-Aldrich (St. Louis, MO, USA). TrkA-inhibitor (CAS 388626-12-8) was from Merk Millipore (Billerica, MA, USA). Fluo-4 acetoxymethyl (AM), 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid AM (BAPTA-AM), Hank’s Balanced Salt Solution containing calcium and magnesium (HBSS+) and Sodium pyruvate were from Thermo Fisher Scientific (Waltham, MA, USA). Rabbit anti-TrkA (RRID: AB_10695253), rabbit anti-phospho-TrkA (Tyr 490) (RRID: AB_10235585), rabbit anti-flotillin-1 (RRID: AB_2773040), and goat anti-rabbit IgG (RRID: AB_2099233) antibodies were from Cell Signaling Technology (Danvers, MA, USA). Mouse anti-PLCγ1 (RRID: AB_628119), anti-phospho-PLCγ1 (Tyr 783) (RRID: AB_2163561), Protein kinase C (PKC) (RRID: AB_628139), anti-phospho-PKCα (Ser 657) (RRID not found) antibodies were from Santa Cruz Biotechnology (Dallas, TX, USA). Mouse anti-calnexin (RRID: AB_397884) antibody was from BD Biosciences. Chemiluminescent kit for western blot was from Cyanagen (Bologna, Italy). 4 − 20% Mini-PROTEAN® TGX™ Precast Protein Gels, Turbo Polyvinylidene difluoride (PVDF) Mini -Midi membrane and DC™ protein assay kit were from BioRad (Hercules, CA, USA). Triton X -100 were from Merk Millipore (Frankfurten, Germany). Cell culture flasks, dishes, and plates were purchased from Corning (Corning, NY, USA). Dulbecco’s modified Eagle’s high glucose medium (DMEM HG), fetal bovine serum (FBS), L-glutamine (L-Glut), Penicillin/streptomycin (10.000 Units/ml) (10.000 U/mL), Streptomycin (10 mg/mL), and acrylamide were purchased from EuroClone (Paignton, UK).
GM1-oligosaccharide preparation
GM1 ganglioside was purified from the total ganglioside mixture extracted from fresh pig brains collected at the slaughterhouse of the Galbani Company (Melzo, Italy), according to the procedure developed previously [18]. Briefly, high amount of GM1 was obtained by the sialidase treatment of the total pig brain ganglioside mixture. This simplified the purification process as the major part of polysialogangliosides were transformed into GM1 [19]. The ganglioside mixture, 5 g as sialic acid, was dissolved in pre-warmed (36 °C) 500 mL of 0.05 M sodium acetate, 1 mM CaCl2 buffer pH 5.5. Vibrio cholerae sialidase (1 unit) was added to the solution every 12 h. Incubation at 36 °C and magnetic stirring was maintained for two days, and the solution dialyzed at 23 °C for 4 days against 10 L of water changed 5 times a day. The sialidase treated ganglioside mixture was subjected to 150 cm x 2 cm silica gel 100 column chromatography equilibrated and eluted with chloroform/methanol/water, 60:35:5 by vol. The fractions containing GM1, identified by TLC, were pooled, dried and submitted to a further column chromatographic purification using the above experimental conditions. Fractions containing pure GM1 were collected and dried. The residue was dissolved in chloroform/methanol (2:1 v/v) and precipitated by adding 4 volumes of cold acetone. After centrifugation (15,000 x g) the GM1 pellet was separated from the acetone, dried, dissolved in 50 mL of deionized water and lyophilized giving 1,350 mg of white powder which was stored at -20 °C.
The OligoGM1 was prepared by ozonolysis of GM1 followed by alkaline degradation [20] (Supplementary Fig. S1). Briefly, GM1 was dissolved in methanol and slowly saturated with ozone at 23 °C. Triethylamine / water (5:1) was added to the mixture bringing the pH to 10.5–11.0 and the reaction continued for three days. Then, solvent was evaporated and OligoGM1 was purified by flash chromatography using chloroform/methanol/2-propanol/water 60:35:5:5 by vol as eluent. The oligosaccharide was dissolved in methanol and stored at 4 °C.
The NMR spectrum showed a correct ratio between the main peaks and contained no unclear signals, mass spectrometry indicated the correct molecular mass, and HPTLC analyzes performed showed a single band. Overall, and considering that the OligoGM1 was prepared from homogeneous GM1, these results suggest homogeneity for the prepared oligosaccharide (Supplementary Figure S2).
N2a cell cultures
Murine neuroblastoma cells N2a were cultured and propagated as monolayer on 75 cm2 flasks in DMEM HG medium supplemented with 10% heat inactivated FBS, 1% L-glutamine, 1% Penicillin/Streptomycin and 1 mM sodium pyruvate, at 37 °C in a humidified atmosphere of 95% air / 5% CO2. Cells were sub-cultured to a fresh culture when growth reached the 80–90% confluence (i.e. every 3–4 days). In sub-cultures passages cells were washed once with PBS and detached by 0.02% EDTA − 0.6% glucose in PBS (w/v).
Cell treatments
N2a cells were plated at 5 × 103 / cm2 on 6-well plates in complete DMEM HG medium for 24 h to allow cells attachment and recovery in complete medium before treatments.
OligoGM1 treatment
24 h after plating, growth medium was removed and N2a cells were pre-incubated in pre-warmed (37 °C) DMEM HG medium containing 2% FBS, 1% L-glutamine, and 1% penicillin/streptomycin, for 30 min at 37 °C.
Subsequently, OligoGM1 was solubilized in water at 2 mM concentration by vortex agitation and sonication in water bath 3 times for 30 sec. Solubilized OligoGM1 was administered to cells at the final concentration of 50 µM. This dose condition has been previously found to promote neurodifferentiation and neuroprotection in N2a cells via TrkA-ERK1/2 signaling pathway activation [13, 15]. Control cells were incubated under the same experimental conditions but omitting any addition of OligoGM1.
Inhibition of TrkA receptor
To block TrkA activity in N2a cells, TrkA inhibitor (120 nM) was added to the incubation medium 1 h before the addition of OligoGM1 [13, 21].
EGTA and BAPTA-AM treatment
To chelate Ca2+ ions in N2a cells, the extracellular Ca2+ chelating agent EGTA (100 µM) or the intracellular Ca2+ chelating agent BAPTA-AM (1 µM) were added to the incubation medium together with OligoGM1 or alone as control condition [22].
Morphological analysis and neurite outgrowth evaluation
N2a cells untreated (control) or treated with 50 µM OligoGM1 in presence or absence of EGTA/BAPTA-AM for 24 h were observed by phase contrast microscopy (Olympus BX50 microscope; Olympus, Tokyo, Japan).
The neurite-like processes length was measured after treatment with OligoGM1 on bidimensional images acquired with 200X magnification with phase contrast microscopy and expressed as the ratio between neurite length and cell body diameter [13, 23, 24]. Five random fields were examined from each well, giving a total cell count of at least 200 cells per well.
Isolation of detergent-resistant membrane (DRM) fractions
N2a cells were incubated in the absence (control) or in the presence of 50 µM OligoGM1 for 3 h at 37 °C. Detergent-resistant membrane (DRM) were prepared by ultracentrifugation on discontinuous sucrose gradient of cells subjected to homogenization with 1% Triton X-100, as previously described [25, 26]. Briefly, cells were mechanically harvested in PBS 1X and centrifuged at 270 x g for 10 min at 4 °C. Cell pellet was lysed in 1.2 mL of 1% Triton X-100 in TNEV buffer (10 mM TrisHCl pH 10, 150 mM NaCl, 5 mM EDTA pH 7.5) in the presence of 1 mM Na3VO4, 1 mM PMSF, and 75 mU/mL aprotinin and homogenized for 11-folds with tight Dounce. Cell lysate (2 mg of cell protein/mL) was centrifuged for 5 min at 1,300 x g at 4 °C to remove nuclei and cellular debris and obtain a post nuclear supernatant (PNS). A volume of 1 mL of PNS was mixed with an equal volume of 85% sucrose (w/v) in TNEV buffer containing 1 mM Na3VO4, placed at the bottom of a discontinuous sucrose gradient (30–5%), and centrifuged for 17 h at 200,000 x g at 4 °C. After ultracentrifugation, 12 fractions were collected starting from the top of the tube. The light scattering band, corresponding to the DRM fraction, was located at the interface between 5 and 30% sucrose corresponding to fractions 4–6. The entire procedure was performed at 0–4° C on ice immersion. Equal amounts from each fraction were diluted with Laemmli sample buffer (0.15 M DTT, 94 mM Tris-HCl, 3% SDS w/v, 0.015% blue bromophenol, v/v) without glycerol and used for protein analysis as reported below.
Protein determination
Protein concentration of samples was assessed using a DC™ protein assay kit according to manufacturer’s instructions, using BSA as standard.
Protein analysis
N2a cells were washed with cold PBS containing 1 mM Na3VO4 and lysed by hot Laemmli sample buffer (0.15 M DTT, 94 mM Tris-HCl, 15% glycerol, v/v, 3% SDS w/v, 0.015% blue bromophenol, v/v). After the probe sonication (50 W, 30 kHz Vibra-Cell™ Ultrasonic VXC130) and the boiling of the lysed samples for 5 min at 99 °C, equal amounts of denatured proteins derived from OligoGM1 treated and untreated cells were separated on 4–20% precast polyacrylamide gels, and transferred to PVDF membranes using the Trans -Blot® Turbo™ Transfer System (Bio -Rad).
PVDF membranes were blocked with 5% milk (w/v) in TBS-0.1% tween-20 (v/v) at 23 °C for 1 h under gentle shaking. The presence of TrkA and p-TrkA was determined by using specific rabbit primary antibodies, both diluted 1:1,000 in 5% BSA (w/v) in TBS-0.1% tween-20. PLCγ1, P-PLCγ1, PKCα, P-PKCα were detected by the specific mouse primary antibodies diluted 1:500 in 5% milk (w/v) in TBS-0.1% tween-20 (v/v). Flotillin was detected by the specific rabbit primary antibody diluted 1:1,000 in 5% milk (w/v) in TBS-0.1% tween-20 (v/v). Calnexin, used as loading controls, weas detected by the specific mouse primary antibodies diluted 1:40,000 in 5% milk (w/v) in TBS-0.1% tween-20 (v/v) and 1:1,000 in 5% BSA (w/v) in TBS-0.1% tween-20, respectively. The incubation was performed overnight (i.e. 16 h) at 4 °C under gentle shaking. Following, PVDF membranes were washed three times with TBS-0.1% tween-20. The reaction with secondary horseradish peroxidase-conjugated antibodies was following performed at 23 °C 1 h in gentle agitation. The data acquisition and analysis were performed using Alliance Uvitec (Cleaver Scientific Ltd, UK).
Calcium-imaging
N2a cells were plated at 7.5 × 103 / cm2 on a 24 mm coverglass in complete DMEM HG medium. 48 h after plating medium was removed and cells were rinsed three times with HBSS containing calcium and magnesium (HBSS+). After washing, cells were incubated with 2.5 µM Fluo-4 AM (494/506 nm) in HBSS+ for 30 min at 23 °C in the dark. Subsequently cells were washed three times with HBSS+ to remove any dye that is not specifically associated with the cell surface and then incubated in HBSS+. Fluorescent emission was examined by live cell analysis using Axio Observer (Zeiss Axio Observer.Z1 with Hamamatsu EMCCD 9100–02) with 400X magnification. The frames were acquired every 5 sec for 20 min (Supplementary Fig. S3). After 3 min of acquisition, 50 µM OligoGM1 solubilized in HBSS+ was administered to the cells and after 15 min the Ca2+ ionophore A23187 (2 µM) in HBSS+ was added to the cells. Control cells were subjected to the same experimental conditions but HBSS+ alone without OligoGM1 was administered. Only ionophore responsive cells were analysed. At least 6 cells for field were quantified. The fluorescence for each acquisition (F) was related to the basal fluorescence (Fmin) according to the following formula:
To evaluate the involvement of TrkA receptor in the induction of Ca2+ influx by the OligoGM1, the TrkA inhibitor (120 nM) was added to the growth medium 30 min before the incubation with Fluo-4. After 30 min, cells were washed three times with HBSS+ and then incubated with Fluo-4 AM in HBSS+ containing 120 nM TrkA inhibitor for 30 min at 23 °C in the dark. Subsequently, cells were washed three times with HBSS+ and incubated in HBSS+ with 120 nM TrkA inhibitor.
To inhibit the inositol trisphosphate (IP3)-receptors, the selective membrane-permeable inhibitor of IP3 receptor Xestospongin C (2.5 µM) [27, 28] was administered together with Fluo-4 for 30 min. After washing with HBSS+, Xestospongin C was added again to the working solution and left for the entire duration of the experiment.
Statistical analysis
Data are expressed as mean ± SEM. The analysis was performed with Prism software (GraphPad Software, Inc. La Jolla, CA, USA). The normality distribution was verified using Kolmogorov–Smirnov, D’ Agostino & Pearson and Shapiro-Wilk tests; in case of a non-Gaussian distribution of data, non-parametric tests were used as indicated in the legend of the figures. A p-value < 0.05 was considered significant.
Other analytical methods
NMR spectra were recorded with a Bruker AVANCE-500 spectrometer at a sample temperature of 298 K. NMR spectra were recorded in CDCl3 or CD3OD and calibrate using the TMS signal as internal reference. Mass spectrometric analysis were performed in positive or negative ESI-MS. Mass spectra were recorded on a Thermo Quest Finningan LCQTM DECA ion trap mass spectrometer, equipped with a Finnigan ESI interface; data were processed by Finnigan Xcalibur software system. All reactions were monitored by TLC on silica gel 60 plates (Merck).
Results
OligoGM1 neuritogenic effect depends on calcium levels modulation
Exogenously administered GM1, or its endogenous increase, induces the differentiation of murine neuroblastoma cells as well as the maturation state of primary neurons [3, 29]. Importantly, the GM1 mediated neurite outgrowth is known to be strictly dependent on Ca2+ influx [30,31,32].
It has recently been observed that the administration of the GM1-oligosaccharide component alone induces the same neuritogenic action in N2a cells, demonstrating that GM1 pentasaccharide is responsible for this effect [13, 14].
Additionally, proteomic analysis of N2a cells exposed to OligoGM1 identified the ex-novo expression of several proteins [15] involved in the regulation of Ca2+ homeostasis and in Ca2+-dependent differentiative and neuroprotective pathways (Table 1), suggesting a possible modulation of Ca2+ signaling by OligoGM1.
Thus, to study the influence of Ca2+ on OligoGM1 enhanced neuritogenesis, 50 µM OligoGM1 was administered to N2a cells in the presence or absence of subtoxic concentrations of extracellular (EGTA, 100 µM) or intracellular (BAPTA-AM, 1 µM) Ca2+ chelators for 24 h. None EGTA or BAPTA-AM at indicated concentration affected the N2a cells morphology in our experimental conditions with respect to control N2a cells (Fig. 1).

Morphological outcomes of N2a cells treated with 50 µM OligoGM1 in presence or absence of 100 µM EGTA or 1 µM BAPTA-AM for 24 h. a Cells were observed by phase contrast microscopy with 200X magnification. Images are representative of three independent experiments (n = 3); b Evaluation of neurite sprouting and elongation in N2a cells. Neurite extensions were evaluated as the ratio between process length and cell body diameter. The bars show the mean values ± SEM from three different experiments (n = 3, OligoGM1 ****p < 0.0001 vs. CTRL, OligoGM1 + EGTA, OligoGM1 + BAPTA, non-parametric one-way ANOVA)
As highlighted in the images reported in Fig. 1a, the presence of both extracellular and intracellular Ca2+ chelators abolished the neurite sprouting induced by OligoGM1 after 24 h. As it emerges from the graph in Fig. 1b, the length of the neuritogenesis in cells incubated with OligoGM1 resulted at least two-fold higher compared to the control cells but equalized the control condition when Ca2+ chelators were administered in combination with OligoGM1. This result suggests that, as already reported for GM1 [3], the increase of cytoplasmic Ca2+ is fundamental for OligoGM1-mediated neuronal differentiation.
OligoGM1 modulates intracellular calcium levels
To study the OligoGM1 ability to modulate Ca2+ flux required for the neuritogenesis process, we performed calcium-imaging experiments on N2a cells using the non-ratiometric calcium-sensitive Fluo-4 probe. The binding of Fluo-4 to Ca2+ results in increased fluorescence excitation at 488 nm and consequently higher fluorescence signal levels.
We find out that the administration of OligoGM1 to undifferentiated N2a cells induces a significant Ca2+ influx, starting from about 5 min after OligoGM1 administration as shown in Fig. 2. On the contrary, no increase in fluorescence is observed in control cells. This result proved a direct modulation of intracellular Ca2+ flux by the exogenous administration of OligoGM1.

OligoGM1 modulation of intracellular Ca2+ level. Intracellular Ca2+ level of N2a cells treated with 50 µM OligoGM1 was analysed measuring the green fluorescent emission of 2.5 µM Fluo-4. The frames were acquired every 5 sec for 20 min with Widefields Zeiss Axio Observer.Z1 with a 400X magnification. After 3 min of basal acquisition, OligoGM1 was administered to the cells and after 15 min the calcium ionophore A23187 (2 µM) was added. Control cells were loaded with HBSS+ alone. Only ionophore responsive cells were analyzed. The fluorescence (F) of each frame (Fx) was related to the fluorescence of the basal condition (F0) (Fx-F0/F0). Results are expressed as the mean ± SEM of fluorescence intensity of at least three independent experiments (OligoGM1 *p < 0.05 vs. basal, one-way ANOVA, n = 11; OligoGM1 **p < 0.01 vs. CTRL, two-way ANOVA; CTRL no significant (NS) vs. basal, one-way ANOVA, n = 5)
OligoGM1-mediated calcium modulation depends on TrkA activation
In N2a neuroblastoma cell line, as well as in primary neurons, the OligoGM1 is not internalized by the cells, and its effects are due to a direct interaction with TrkA receptor at the PM level [13, 14]. Thus, taking into account the reported effect prompted by GM1 on TrkA mediated neuronal differentiation [33,34,35,36,37,38] and considering the recent result where OligoGM1 was found to increase TrkA phosphorylation [13, 14], we investigated if also Ca2+ flow modulation could be a molecular event downstream of TrkA activation induced by OligoGM1.
To evaluate the involvement of TrkA receptor, calcium-imaging experiments were performed in the presence of a cell-permeable and highly selective TrkA inhibitor [13, 21]. First, we confirmed that the co-administration of TrkA inhibitor is able to prevent the specific TrkA receptor activation induced by OligoGM1 after 5–30 min from its addition to cell culture medium. (Fig. 3a). Following, we repeated the calcium-imaging experiment in the presence of TrkA inhibitor. As shown in Fig. 3b and Supplementary Figure S3, there is no significant increase of the fluorescence signal in the presence of TrkA inhibitor, suggesting that the opening of the cell Ca2+ channels and the Ca2+ influx following OligoGM1 administration is mediated by the activation of TrkA receptor.

OligoGM1-mediated Ca2+ modulation depends on TrkA activation. a The TrkA receptor inhibitor (120 nM) was added to the N2a cells 1 h before the administration of 50 µM OligoGM1. Expression of TrkA and p-TrkA (tyrosine 490, Tyr490) was evaluated 5 and 30 min after OligoGM1 treatment by western blot using specific antibodies and revealed by enhanced chemiluminescence. Top: immunoblotting images are shown. Bottom: semiquantitative analysis of p-TrkA related to total level of TrkA. Data are expressed as fold increase over control of the mean ± SEM from three different experiments (*p < 0.05, **p < 0.01, two-way ANOVA, n = 3); b Intracellular Ca2+ level of N2a cells treated with OligoGM1 was analyzed measuring the green fluorescent emission of 2.5 µM Fluo-4. The frames were acquired every 5 sec for 20 min with Widefields Zeiss Axio Observer.Z1 with a 400X magnification. The TrkA inhibitor (120 nM) was added to the culture medium for 30 min before Fluo-4 administration and left for the duration of the experiment. After 3 min of basal acquisition, 50 µM OligoGM1 was administered to the cells and after 15 min the calcium ionophore A23187 (2 µM) was added. Control cells were loaded with HBSS+ alone. Only ionophore responsive cells were analyzed. The fluorescence of each frame (Fx) was related to the fluorescence of the basal condition (F0) (Fx-F0/F0). Results are expressed as the mean ± SEM of fluorescence intensity of at least three independent experiments (OligoGM1 * p < 0.05 vs. basal, one-way ANOVA, n = 11; OligoGM1 **p < 0.01 vs. CTRL, two-way ANOVA; OligoGM1 + TrkA inhibitor NS vs. basal, one-way ANOVA, n = 5; OligoGM1 + TrkA inhibitor NS vs. CTRL, two-way ANOVA)
Identification of the cellular pathways involved in OligoGM1-mediated calcium influx
To biochemically characterize the effect of OligoGM1 on Ca2+ flux in N2a cells following TrkA activation, we investigated the involvement of TrkA downstream effectors known to modulate of Ca2+ signaling such as the phospholipase C gamma (PLCγ) and the protein kinase C (PKC) [39].
To unveil if OligoGM1 administration could lead to the increase of intracellular Ca2+ through this cellular pathway, an immunoblotting analysis evaluating the phosphorylation status of the TrkA receptor, PLCγ1 and PKCα on N2a cells after 5, 30 and 60 min following OligoGM1 administration was performed.
As shown in Fig. 4 we found that 5 min after OligoGM1 administration there is an activation of TrkA receptor, confirming previous results [13]. Moreover, we observed an enhanced activation of PLCγ1 starting from 5 min from OligoGM1 administration followed, after 1 h, by a hyperphosphorylation of PKCα1, which is a priming event that enables its catalytic activation.

OligoGM1 effect on TrkA-PLCγ1-PKCα pathway. N2a cells were culture in the absence (CTRL) or in the presence of 50 µM OligoGM1. Expression of TrkA, p-TrkA (tyr490), PLCγ1, p-PLCγ1, PKCα and p-PKCα was evaluated 5 min, 30 min and 1 h after OligoGM1 treatment by using specific antibodies and revealed by enhanced chemiluminescence. Top: representative immunoblotting images. Bottom: semi-quantitative analysis of signals of phosphorylated TrkA, PLCγ1 and PKCα related to signals of total TrkA, PLCγ1 and PKCα, respectively. Data are expressed as mean ± SEM of the fold increase over control from at least three experiments (*p < 0.05, **p < 0.01, Student’s t-test)
Although the phosphorylation is a key event for the catalytic activity of PKC, it is known that its activation depends on its translocation to the PM [40, 41]. Thus, to verify whether the administration of OligoGM1 was followed by an enrichment of PKC in lipid rafts, 3 h after treating N2a cells with OligoGM1, lipid rafts were isolated as the DRM, according to the procedure described in "Methods" Section.
Western blotting analysis revealed a PKCα enrichment in DRM fractions in OligoGM1 treated cells, while in control cells PKCα is present in the fluid membrane fraction, solubilized by the detergent (Fig. 5).

PKCα translocation in lipid rafts microdomains. N2a cells were incubated in the absence (CTRL), or in the presence of 50 µM OligoGM1 for 3 h at 37 °C. Cells were subsequently subjected to sucrose gradient ultracentrifugation to prepare PM microdomains. Twelve fractions were collected from the top of the tube, with fractions 4–6 corresponding to the detergent resistant membrane (DRM) fractions and fractions 10–12 corresponding to high density (HD) fractions. Expression of PKCα, Flotillin (DRM marker) and Calnexin (HD marker) was evaluated by western blot using specific antibodies and revealed by enhanced chemiluminescence. Images are representative of three independent cell culture preparations (n = 3)
These events could be responsible for the opening of Ca2+ channels on both PM and intracellular storages (i.e. endoplasmic reticulum), resulting in an increase of intracellular Ca2+.
To disclose if the cytoplasmic Ca2+ increase after OligoGM1 administration derives from the extracellular environment through the PM Ca2+ channels or from the intracellular storages, the calcium-imaging experiment was performed in the presence of Xestospongin C, a selective, reversible and potent inhibitor of IP3 receptors on endoplasmic reticulum [28]. The calcium-imaging representative frames (Supplementary Fig. S3) and the relative graph shown in Fig. 6 demonstrate that in the presence of the IP3 receptors inhibitor, the Ca2+ influx following OligoGM1 administration is reduced but is not completely abolished, suggesting that both PM and intracellular channels may be modulated by OligoGM1 administration.

OligoGM1-modulated Ca2+ derives from both the extracellular environment and the intracellular storages. Intracellular Ca2+ level of N2a cells treated with OligoGM1 was analysed measuring the green fluorescent emission of Fluo-4 (2.5 µM). The frames were acquired every 5 sec for 20 min with Widefields Zeiss Axio Observer.Z1 with a 400X magnification. The IP3 receptor inhibitor, Xestospongin C (2.5 µM) was added to the cells together with Fluo-4 for 30 min before starting the acquisitions and left for the entire duration of the experiment. After 3 min of basal acquisition, OligoGM1 (50 µM) was administered to the cells and after 15 min the calcium ionophore A23187 (2 µM) was added. Control cells were loaded with HBSS+ alone. Only ionophore responsive cells were analysed. The fluorescence of each frame (Fx) was related to the fluorescence of the basal condition (F0) (Fx-F0/F0). Results are expressed as the mean ± SEM of fluorescence intensity of at least three independent experiments (OligoGM1 *p < 0.05 vs. basal, one-way ANOVA, n = 11; OligoGM1 **p < 0.01 vs. CTRL, two-way ANOVA; OligoGM1 + Xestospongin C *p < 0.05 vs. basal, one-way ANOVA, n = 5; OligoGM1 + Xestospongin C NS vs. CTRL, two-way ANOVA)
Discussion
For several years ganglioside GM1 has been widely studied for its essential role in neuronal differentiation, protection and restoration [3, 5, 6, 29], accomplished through the cooperation with several players expressed on the PM.
In particular, the GM1 enrichment in PM allows the dimerization and activation of neurotrophins’ receptors belonging to Trk family [3, 42, 43] and modulates Ca2+ influx channels and Ca2+ exchange proteins causing changes in cellular Ca2+ levels [3, 7, 8]. The increase of cytosolic Ca2+ is essential for the morphological changes accompanying the neurodifferentiative properties prompted by GM1, triggering specific signaling cascades resulting in actin depolymerization, axon protrusion, and elongation [7, 44,45,46,47,48,49].
Despite the long-standing research on the GM1, the fine molecular mechanism at the basis of its functions remained obscure until recent years when in a murine neuroblastoma cell line it has been observed that, within the entire molecule, the oligosaccharide chain (OligoGM1) represents, alone, the bioactive component of GM1 ganglioside [13]. Following its isolation from parental compound, the GM1-oligosaccharide added to the culture medium of N2a neuroblastoma cells was observed to induce the neuritogenesis process as equimolar concentration of GM1 did, acting at the PM level by enhancing the phosphorylation of the TrkA receptor followed by the increase in ERK1/2 phosphorylation [13].
This finding was supported by the revelation that, in N2a cells, TrkA and GM1 belong to separate membrane domains. In fact TrkA does not belong to lipid rafts where GM1 is located, suggesting that its interaction with GM1 and the following stabilization which leads to its autophosphorylation involves only the GM1-oligosaccharide chain and the extracellular portion of TrkA, that may flop down on the PM approaching the GM1 saccharide [50]. These results were subsequently translated in a more physiological context by using primary cultures of murine granule cerebellar neurons [14]. According to this study, OligoGM1 administered to primary neurons enhanced cell clustering, neurite sprouting and networking, thus confirming the specific role of the OligoGM1 in the processes of neuronal differentiation and maturation, known to be regulated by the entire GM1.
Here, to further clarify the mechanism of action of OligoGM1, as the bioactive portion of GM1, we examined its ability to modulate the cellular Ca2+ flow, at the basis of the neuritogenic properties induced by GM1, using mouse neuroblastoma cells N2a as experimental model.
As shown by calcium-imaging experiments, the administration of 50 µM OligoGM1 to undifferentiated N2a is capable of inducing a significant Ca2+ intake starting from 5 min after OligoGM1 application (Fig. 2). This latency time suggests that the entry of Ca2+ is not due to the direct interaction of OligoGM1 with Ca2+ channels on the PM neither with intracellular channels, since it was demonstrated [13, 14] that the OligoGM1 is not internalized by the cells but rather it could be a result of the activation of PM receptors and downstream signaling pathways.
Since OligoGM1 carries out its neurotrophic and neuroprotective activities interacting with the TrkA receptor on PM, we verified a direct participation of TrkA in the OligoGM1-mediated Ca2+ modulation, performing the calcium-imaging experiment in the presence of the TrkA inhibitor. In this case, no Ca2+ influx was observed following OligoGM1 administration, indicating that TrkA receptor activation is the upstream event modulating Ca2+ flux upon OligoGM1 addition (Fig. 3b).
Subsequently, we studied in more detail the involvement of signaling proteins downstream of TrkA receptor, known to be responsible for cellular Ca2+ mobilization, such as PLCγ.
PLCγ is a membrane-associated enzyme that cleaves PIP2 into DAG and IP3 [51]. The two products of the PLC catalysed reaction, DAG and IP3, are important second messengers that propagate and regulate cellular signaling via Ca2+ mobilization and activation of protein kinases, such as PKC, and ion channels [51]. When PIP2 is cleaved, DAG remains bound to the membrane, whereas IP3 is released as a soluble molecule into the cytosol, binding IP3-sensitive intracellular Ca2+ channels predominately located in the membrane of the endoplasmic reticulum, regulating the Ca2+ flux from intracellular stores to the cytosol [52]. The other product, DAG, triggers Ca2+ influx from extracellular environment independently from IP3 activity by directing plasma membrane TRP channels [53] and activating PKC [54]. The function of the PKC is regulated by two mechanisms: its phosphorylation allows the correct alignment of the residues necessary for the catalysis, while the increase in the intracellular Ca2+ concentration triggers the membrane translocation of PKC and its association with DAG at the PM microdomains, stimulating the enzyme activity [40, 41, 54]. Additionally, PKC phosphorylates other molecules, modulating several cellular events: the activation of PKC in the nervous system has been involved in the regulation of ion channels activity, neurotransmitter release, growth, differentiation, and neural plasticity [54].
By immunoblotting analysis we found a hyperphosphorylation of PLCγ occurring 5 min upon OligoGM1 administration (Fig. 4), followed by a hyperphosphorylation of PKC 1 h after OligoGM1 was supplemented to the medium and by its enrichment in lipid rafts, confirming its activation (Fig. 5).
The involvement of IP3 in mobilizing Ca2+ from the intracellular stores was confirmed by calcium-imaging experiments performed by administering OligoGM1 together with the selective IP3 inhibitor, Xestospongin C [27, 28]. Interestingly, while a lower increase in intracellular Ca2+ level was recorded (Fig. 6), the Ca2+ influx was not completely abolished, suggesting the possible involvement of Ca2+ channels of both the intracellular and plasma membranes in the modulation of the cytoplasmic Ca2+ levels, which could be activated by IP3 and DAG respectively.
Thus, we finally proved that GM1 neuritogenic effect is mediated by an increase in intracellular Ca2+ following the OligoGM1-TrkA interaction at the PM level, leading to the recruitment and activation of multiple intracellular players eventually promoting neurite sprouting (Fig. 7). In fact, we observed that OligoGM1 is not able to induce neurite emission in N2a cells if Ca2+ ions, both intracellular and extracellular, are chelated (Fig. 1), suggesting that the modulation of cytosolic Ca2+ levels by OligoGM1 is fundamental for the execution of its neurodifferentiative properties.

Schematic representation of molecular mechanism underling OligoGM1 neurotrophic function. OligoGM1 enhances the activation of TrkA signaling pathway, which could be associated to an increased activation of PLCγ1, leading to the formation of second messengers DAG and IP3. These events bring to the opening of Ca2+ channels on the PM and on endoplasmic reticulum, leading to an increase of cytosolic Ca2+ responsible for N2a differentiation. TrkA, neurotrophin tyrosine kinase receptor A; NGF, nerve growth factor; PLCγ, phospholipase C gamma; PIP2, phosphatidylinositol 4,5-bisphosphate; DAG, diacyl-glycerol; IP3, inositol 1,4,5-trisphosphate; PKC, protein kinase C; TRPC, transient receptor potential channel; RER, rough endoplasmic reticulum. GM1 sugar code is according to Varki et al. 2015 [55]. This image is updated from Chiricozzi et al. [13]
Although here the identity of Ca2+ channels modulated by OligoGM1 has not been identified and will be investigate in a later study, the present work demonstrates that the GM1-oligosaccharide is responsible on its own also for the GM1 modulation of Ca2+ homeostasis and that the regulation of Ca2+ signaling is a fundamental mechanism at the base of OligoGM1 neurogenic action, partially revealing the mechanism of action of the oligosaccharide chain of ganglioside GM1.
Abbreviations
- A.U.:
-
arbitrary units
- BAPTA-AM:
-
1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (acetoxymethyl ester)
- BSA:
-
bovine serum albumin
- Ca2+ :
-
calcium
- CTRL:
-
control
- DMEM HG:
-
Dulbecco’s modified Eagle’s high glucose medium
- DRM:
-
Detergent-resistant membrane
- DTT:
-
1,4-Dithiothreitol
- EDTA:
-
ethylenediamine tetraacetic acid
- EGTA:
-
ethylene glycol tetraacetic acid
- ERK1/2:
-
extracellular signal-regulated protein kinases 1 and 2
- FBS:
-
fetal bovine serum
- F:
-
fluorescence
- GM1:
-
II3Neu5Ac-Gg4Cer, β-Gal-(1–3)-β-GalNAc-(1–4)-[α-Neu5Ac-(2–3)]-β-Gal-(1–4)-β-Glc-Cer
- HBSS+ :
-
Hank’s Balanced Salt Solution containing calcium and magnesium
- HPTLC:
-
High-performance thin-layer chromatography
- IP3 :
-
inositol trisphosphate
- N2a:
-
Neuro2a cells
- NS:
-
no significant
- OligoGM1:
-
GM1-oligosaccharide, II3Neu5Ac-Gg4
- PBS:
-
phosphate-buffered saline
- P-ERK1/2:
-
phosphorylated ERK1/2
- PKC:
-
Protein kinase C
- PLCγ:
-
Phospholipase C γ
- PNS:
-
post nuclear supernatant
- P-TrkA:
-
phosphorylated TrkA
- PM:
-
plasma membrane
- PVDF:
-
polyvinylidene difluoride
- RRID:
-
Research Resource Identifiers
- TBS:
-
Tris-buffered saline
- TLC:
-
thin-layer chromatography
- Trk:
-
neurotrophin tyrosine kinase receptor
- Tyr490:
-
tyrosine 490
References
Schengrund, C.L., Ringler, N.J.: Binding of Vibrio cholera toxin and the heat-labile enterotoxin of Escherichia coli to GM1, derivatives of GM1, and nonlipid oligosaccharide polyvalent ligands. J. Biol. Chem. 264, 13233–13237 (1989)
Schengrund, C.L., Mummert, C.M.: Exogenous gangliosides. How do they cross the blood-brain barrier and how do they inhibit cell proliferation. Ann N Y Acad Sci. 845, 278–284 (1998)
Ledeen, R.W., Wu, G.: The multi-tasked life of GM1 ganglioside, a true factotum of nature. Trends Biochem. Sci. 40, 407–418 (2015)
Zhai, H.W., Gong, Z.K., Sun, J., Chen, W., Zhang, M., Zhou, J.J., Zheng, B.: Ganglioside with nerve growth factor for the recovery of extremity function following spinal cord injury and somatosensory evoked potential. Eur. Rev. Med. Pharmacol. Sci. 19, 2282–2286 (2015)
Aureli, M., Mauri, L., Ciampa, M.G., Prinetti, A., Toffano, G., Secchieri, C., Sonnino, S.: GM1 ganglioside: past studies and future potential. Mol. Neurobiol. 53, 1824–1842 (2016)
Chiricozzi, E., Lunghi, G., Di Biase, E., Fazzari, M., Sonnino, S., Mauri, L.: GM1 Ganglioside is a key factor in maintaining the mammalian neuronal functions avoiding neurodegeneration. Int. J. Mol. Sci. 21, 868 (2020)
Wu, G., Fang, Y., Lu, Z.H., Ledeen, R.W.: Induction of axon-like and dendrite-like processes in neuroblastoma cells. J. Neurocytol. 27, 1–14 (1998)
Wu, G., Xie, X., Lu, Z.H., Ledeen, R.W.: Sodium-calcium exchanger complexed with GM1 ganglioside in nuclear membrane transfers calcium from nucleoplasm to endoplasmic reticulum. Proc. Natl. Acad. Sci. U. S. A. 106, 10829–10834 (2009)
Wu, G., Ledeen, R.W.: Stimulation of neurite outgrowth in neuroblastoma cells by neuraminidase: putative role of GM1 ganglioside in differentiation. J. Neurochem. 56, 95–104 (1991)
Wu, G., Lu, Z.H., Obukhov, A.G., Nowycky, M.C., Ledeen, R.W.: Induction of calcium influx through TRPC5 channels by cross-linking of GM1 ganglioside associated with alpha5beta1 integrin initiates neurite outgrowth. J. Neurosci. 27, 7447–7458 (2007)
Wu, G., Lu, Z.H., Xie, X., Ledeen, R.W.: Susceptibility of cerebellar granule neurons from GM2/GD2 synthase-null mice to apoptosis induced by glutamate excitotoxicity and elevated KCl: rescue by GM1 and LIGA20. Glycoconj. J. 21, 305–313 (2004)
Wu, G., Xie, X., Lu, Z.H., Ledeen, R.W.: Cerebellar neurons lacking complex gangliosides degenerate in the presence of depolarizing levels of potassium. Proc. Natl. Acad. Sci. U. S. A. 98, 307–312 (2001)
Chiricozzi, E., Pome, D.Y., Maggioni, M., Di Biase, E., Parravicini, C., Palazzolo, L., Loberto, N., Eberini, I., Sonnino, S.: Role of the GM1 ganglioside oligosaccharide portion in the TrkA-dependent neurite sprouting in neuroblastoma cells. J. Neurochem. 143, 645–659 (2017)
Di Biase, E., Lunghi, G., Fazzari, M., Maggioni, M., Pome, D.Y., Valsecchi, M., Samarani, M., Fato, P., Ciampa, M.G., Prioni, S., Mauri, L., Sonnino, S., Chiricozzi, E.: Gangliosides in the differentiation process of primary neurons: the specific role of GM1-oligosaccharide. Glycoconj. J. 37, 329–343 (2020)
Chiricozzi, E., Maggioni, M., di Biase, E., Lunghi, G., Fazzari, M., Loberto, N., Elisa, M., Scalvini, F.G., Tedeschi, G., Sonnino, S.: The neuroprotective role of the GM1 oligosaccharide, II(3)Neu5Ac-Gg4, in neuroblastoma cells. Mol. Neurobiol. 56, 6673–6702 (2019)
Fazzari, M., Audano, M., Lunghi, G., Di Biase, E., Loberto, N., Mauri, L., Mitro, N., Sonnino, S., Chiricozzi, E.: The oligosaccharide portion of ganglioside GM1 regulates mitochondrial function in neuroblastoma cells. Glycoconj. J. 37(3), 293–306 (2020)
Chiricozzi, E., Mauri, L., Lunghi, G., Di Biase, E., Fazzari, M., Maggioni, M., Valsecchi, M., Prioni, S., Loberto, N., Pome, D.Y., Ciampa, M.G., Fato, P., Verlengia, G., Cattaneo, S., Assini, R., Wu, G., Alselehdar, S., Ledeen, R.W., Sonnino, S.: Parkinson’s disease recovery by GM1 oligosaccharide treatment in the B4galnt1(+/-) mouse model. Sci. Rep. 9, 19330 (2019)
Tettamanti, G., Bonali, F., Marchesini, S., Zambotti, V.: A new procedure for the extraction, purification and fractionation of brain gangliosides. Biochim. Biophys. Acta 296, 160–170 (1973)
Acquotti, D., Poppe, L., Dabrowski, J., Von der Lieth, C.W., Sonnino, S., Tettamanti, G.: Three-dimensional structure of the oligosaccharide chain of GM1 ganglioside revealed by a distance-mapping procedure: A rotating and laboratory frame nuclear Overhauser enhancement investigation of native glycolipid in dimethyl sulfoxide and in water-dodecylphosphocholine solutions. J. Am. Chem. Soc. 112, 7772–7778 (1990)
Wiegandt, H., Bucking, H.W.: Carbohydrate components of extraneuronal gangliosides from bovine and human spleen, and bovine kidney. Eur. J. Biochem. 15, 287–292 (1970)
Wood, E.R., Kuyper, L., Petrov, K.G., Hunter, R.N. III; Harris, P.A., Lackey, K.: Discovery and in vitro evaluation of potent TrkA kinase inhibitors: oxindole and aza-oxindoles. Bioorg. Med. Chem. Lett. 14, 953–7 (2004)
Chiricozzi, E., Fernandez-Fernandez, S., Nardicchi, V., Almeida, A., Bolanos, J.P., Goracci, G.: Group IIA secretory phospholipase A2 (GIIA) mediates apoptotic death during NMDA receptor activation in rat primary cortical neurons. J. Neurochem. 112, 1574–1583 (2010)
Schengrund, C.L., Prouty, C.: Oligosaccharide portion of GM1 enhances process formation by S20Y neuroblastoma cells. J. Neurochem. 51, 277–282 (1988)
Sato, C., Matsuda, T., Kitajima, K.: Neuronal differentiation-dependent expression of the disialic acid epitope on CD166 and its involvement in neurite formation in Neuro2A cells. J. Biol. Chem. 277, 45299–45305 (2002)
Chiricozzi, E., Ciampa, M.G., Brasile, G., Compostella, F., Prinetti, A., Nakayama, H., Ekyalongo, R.C., Iwabuchi, K., Sonnino, S., Mauri, L.: Direct interaction, instrumental for signaling processes, between LacCer and Lyn in the lipid rafts of neutrophil-like cells. J. Lipid Res. 56, 129–141 (2015)
Schiumarini, D., Loberto, N., Mancini, G., Bassi, R., Giussani, P., Chiricozzi, E., Samarani, M., Munari, S., Tamanini, A., Cabrini, G., Lippi, G., Dechecchi, M.C., Sonnino, S., Aureli, M.: Evidence for the involvement of lipid rafts and plasma membrane sphingolipid hydrolases in pseudomonas aeruginosa infection of cystic fibrosis bronchial epithelial cells. Mediat. Inflamm. 2017, 1730245 (2017)
Miyamoto, S., Izumi, M., Hori, M., Kobayashi, M., Ozaki, H., Karaki, H.: Xestospongin C, a selective and membrane-permeable inhibitor of IP(3) receptor, attenuates the positive inotropic effect of alpha-adrenergic stimulation in guinea-pig papillary muscle. Br. J. Pharmacol. 130, 650–654 (2000)
Gafni, J., Munsch, J.A., Lam, T.H., Catlin, M.C., Costa, L.G., Molinski, T.F., Pessah, I.N.: Xestospongins: potent membrane permeable blockers of the inositol 1,4,5-trisphosphate receptor. Neuron. 19, 723–33 (1997)
Schengrund, C.L.: Gangliosides: glycosphingolipids essential for normal neural development and function. Trends Biochem. Sci. 40, 397–406 (2015)
Masco, D., Van de Walle, M., Spiegel, S.: Interaction of ganglioside GM1 with the B subunit of cholera toxin modulates growth and differentiation of neuroblastoma N18 cells. J. Neurosci. 11, 2443–2452 (1991)
Wu, G., Ledeen, R.W.: Gangliosides as modulators of neuronal calcium. Prog. Brain Res. 101, 101–112 (1994)
Fang, Y., Wu, G., Xie, X., Lu, Z.H., Ledeen, R.W.: Endogenous GM1 ganglioside of the plasma membrane promotes neuritogenesis by two mechanisms. Neurochem. Res. 25, 931–940 (2000)
Farooqui, T., Franklin, T., Pearl, D.K., Yates, A.J.: Ganglioside GM1 enhances induction by nerve growth factor of a putative dimer of TrkA. J. Neurochem. 68, 2348–2355 (1997)
Singleton, D.W., Lu, C.L., Colella, R., Roisen, F.J.: Promotion of neurite outgrowth by protein kinase inhibitors and ganglioside GM1 in neuroblastoma cells involves MAP kinase ERK1/2. Int. J. Dev. Neurosci. 18, 797–805 (2000)
Duchemin, A.M., Ren, Q., Mo, L., Neff, N.H., Hadjiconstantinou, M.: GM1 ganglioside induces phosphorylation and activation of Trk and Erk in brain. J. Neurochem. 81, 696–707 (2002)
Da Silva, J.S., Hasegawa, T., Miyagi, T., Dotti, C.G., Abad-Rodriguez, J.: Asymmetric membrane ganglioside sialidase activity specifies axonal fate. Nat. Neurosci. 8, 606–615 (2005)
Mocchetti, I.: Exogenous gangliosides, neuronal plasticity and repair, and the neurotrophins. Cell. Mol. Life Sci. 62, 2283–2294 (2005)
Zakharova, I.O., Sokolova, T.V., Vlasova, Y.A., Furaev, V.V., Rychkova, M.P., Avrova, N.F.: GM1 ganglioside activates ERK1/2 and Akt downstream of Trk tyrosine kinase and protects PC12 cells against hydrogen peroxide toxicity. Neurochem. Res. 39, 2262–2275 (2014)
Huang, E.J., Reichardt, L.F.: Trk receptors: roles in neuronal signal transduction. Annu. Rev. Biochem. 72, 609–642 (2003)
Freeley, M., Kelleher, D., Long, A.: Regulation of Protein Kinase C function by phosphorylation on conserved and non-conserved sites. Cell. Signal 23, 753–762 (2011)
Igumenova, T.I.: Dynamics and membrane interactions of Protein Kinase C. Biochemistry. 54, 4953–68 (2015)
Mutoh, T., Tokuda, A., Miyadai, T., Hamaguchi, M., Fujiki, N.: Ganglioside GM1 binds to the Trk protein and regulates receptor function. Proc. Natl. Acad. Sci. U. S. A. 92, 5087–5091 (1995)
Rabin, S.J., Mocchetti, I.: GM1 ganglioside activates the high-affinity nerve growth factor receptor trkA. J. Neurochem. 65, 347–354 (1995)
Kappagantula, S., Andrews, M.R., Cheah, M., Abad-Rodriguez, J., Dotti, C.G., Fawcett, J.W.: Neu3 sialidase-mediated ganglioside conversion is necessary for axon regeneration and is blocked in CNS axons. J. Neurosci. 34, 2477–2492 (2014)
Wu, G., Lu, Z.H., Nakamura, K., Spray, D.C., Ledeen, R.W.: Trophic effect of cholera toxin B subunit in cultured cerebellar granule neurons: modulation of intracellular calcium by GM1 ganglioside. J. Neurosci. Res. 44, 243–54 (1996)
Milani, D., Minozzi, M.C., Petrelli, L., Guidolin, D., Skaper, S.D., Spoerri, P.E.: Interaction of ganglioside GM1 with the B subunit of cholera toxin modulates intracellular free calcium in sensory neurons. J. Neurosci. Res. 33, 466–475 (1992)
Bachis, A., Rabin, S.J., Del Fiacco, M., Mocchetti, I.: Gangliosides prevent excitotoxicity through activation of TrkB receptor. Neurotox. Res. 4, 225–234 (2002)
Sokolova, T.V., Rychkova, M.P., Avrova, N.F.: [Protective effect of GM1 ganglioside against toxic action of glutamate on cerebellar granule cells]. Zh. Evol. Biokhim. Fiziol. 50, 399–401 (2014)
Park, D.H., Wang, L., Pittock, P., Lajoie, G., Whitehead, S.N.: Increased expression of GM1 detected by electrospray mass spectrometry in rat primary embryonic cortical neurons exposed to glutamate toxicity. Anal. Chem. 88, 7844–7852 (2016)
Chiricozzi, E., Biase, E.D., Maggioni, M., Lunghi, G., Fazzari, M., Pome, D.Y., Casellato, R., Loberto, N., Mauri, L., Sonnino, S.: GM1 promotes TrkA-mediated neuroblastoma cell differentiation by occupying a plasma membrane domain different from TrkA. J. Neurochem. 149, 231–241 (2019)
Fukami, K., Inanobe, S., Kanemaru, K., Nakamura, Y.: Phospholipase C is a key enzyme regulating intracellular calcium and modulating the phosphoinositide balance. Prog. Lipid Res. 49, 429–437 (2010)
Alzayady, K.J., Wang, L., Chandrasekhar, R., Wagner, L.E. II, Van Petegem, F., Yule, D.I.: Defining the stoichiometry of inositol 1,4,5-trisphosphate binding required to initiate Ca2 + release. Sci. Signal. 9, ra35 (2016)
Hofmann, T., Obukhov, A.G., Schaefer, M., Harteneck, C., Gudermann, T., Schultz, G.: Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature. 397, 259–263 (1999)
Huang, K.P.: The mechanism of protein kinase C activation. Trends Neurosci. 12, 425–432 (1989)
Varki, A., Cummings, R.D., Aebi, M., Packer, N.H., Seeberger, P.H., Esko, J.D., Stanley, P., Hart, G., Darvill, A., Kinoshita, T., Prestegard, J.J., Schnaar, R.L., Freeze, H.H., Marth, J.D., Bertozzi, C.R., Etzler, M.E., Frank, M., Vliegenthart, J.F., Lutteke, T., Perez, S., Bolton, E., Rudd, P., Paulson, J., Kanehisa, M., Toukach, P., Aoki-Kinoshita, K.F., Dell, A., Narimatsu, H., York, W., Taniguchi, N., Kornfeld, S.: Symbol Nomenclature for graphical representations of glycans. Glycobiology. 25, 1323–4 (2015)
Chester, M.A.: IUPAC-IUB Joint Commission on Biochemical Nomenclature (JCBN). Nomenclature of glycolipids–recommendations 1997. Eur. J. Biochem. 257, 293–298 (1998)
Acknowledgements
This work was supported by University of Milano departmental funds RV_TAR16SSONN_M to SS and by Mizutani Foundation for Glycoscience funds to EC. The authors acknowledge Euro-BioImaging (www.eurobioimaging.eu) for providing access to imaging technologies and services via the Italian Node (ALEMBIC, Milano, Italy).
Funding
Open access funding provided by Università degli Studi di Milano within the CRUI-CARE Agreement.
Author information
Authors and Affiliations
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Ganglioside nomenclature is in accordance with IUPAC-IUBB recommendations [56].
Electronic supplementary material
ESM 1
(PDF 271 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lunghi, G., Fazzari, M., Di Biase, E. et al. Modulation of calcium signaling depends on the oligosaccharide of GM1 in Neuro2a mouse neuroblastoma cells. Glycoconj J 37, 713–727 (2020). https://doi.org/10.1007/s10719-020-09963-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10719-020-09963-7