Pseudotyping Lentiviral Vectors: When the Clothes Make the Virus


1
Université de Strasbourg, CNRS, Architecture et Réactivité de l’ARN, UPR9002, 67000 Strasbourg, France
2
Interdisciplinary Thematic Institute (ITI) InnoVec, Université de Strasbourg, 67000 Strasbourg, France
*
Author to whom correspondence should be addressed.
Viruses 2020, 12(11), 1311; https://doi.org/10.3390/v12111311
Submission received: 28 August 2020
/
Revised: 10 November 2020
/
Accepted: 11 November 2020
/
Published: 16 November 2020
(This article belongs to the Special Issue Lentiviral Vectors)
Abstract
:Delivering transgenes to human cells through transduction with viral vectors constitutes one of the most encouraging approaches in gene therapy. Lentivirus-derived vectors are among the most promising vectors for these approaches. When the genetic modification of the cell must be performed in vivo, efficient specific transduction of the cell targets of the therapy in the absence of off-targeting constitutes the Holy Grail of gene therapy. For viral therapy, this is largely determined by the characteristics of the surface proteins carried by the vector. In this regard, an important property of lentiviral vectors is the possibility of being pseudotyped by envelopes of other viruses, widening the panel of proteins with which they can be armed. Here, we discuss how this is achieved at the molecular level and what the properties and the potentialities of the different envelope proteins that can be used for pseudotyping these vectors are.

1. Gene Therapy Using Viral Vectors
According to the definition provided by the NIH Genetics Home Reference, gene therapy is an experimental technique aimed at treating or preventing a disease by using genes [1]. This can be achieved by various means. When the disease is of genetic origin and, particularly, when it is caused by a single defective gene, the ultimate goal is replacing the defective gene with a wild-type one. This has been possible only recently with the development of powerful genome editing techniques [2,3,4]. Although, these are not applicable routinely and alternative approaches are followed, the most common of which is the introduction of a gene conferring a dominant wild-type phenotype to the modified cell [5]. Whatever the approach followed, gene therapy relies on the use of vectors that allow the efficient genetic modification of cells, or tissues, combined with a high specificity for the target cells to reduce adverse effects [6]. Introducing exogenous genetic material in cells is efficiently performed by cellular “parasites”—phages for bacteria or viruses for eukaryotic cells. In particular, the vast range of human viruses provides a large panel of promising tools for vectorization (by transduction) in sight of intervention on human cells. How to reprogram human viruses for the purposes mentioned above is a major challenge in molecular medicine.
A main watershed in gene therapy is whether the genetic modification of the cell must be carried out ex vivo or in vivo. If the cells’ target for the therapy can be isolated from the patient, modified ex vivo, and reinfused in the patient, essentially no specific tropism is required for the vector since the cells to modify are the only ones it comes into contact with [7,8,9,10]. In this case, the vectors can therefore carry pan-tropic envelope proteins such as, for example, the vescicular stomatitis virus (VSV) envelope protein G (see below). If, in contrast, the modification of the cells must be carried out in vivo, a high specificity for the target cells is required to avoid off-target transduction. The nature of the envelope proteins carried by the viral vector is the major determinant for the specificity of transduction.
Most gene therapy clinical trials carried out to date have relied on the use of adeno-associated vectors (AAVs) or retroviral vectors, which might be derived from γ-retroviruses or lentiviruses [11]. Modification of cells in vivo (liver, muscles, central nervous system and retina) has been restricted to the use of AAV-derived vectors, while ex vivo approaches (for the genetic modification of T cells and of human hematopoietic stem and progenitor cells) have relied on the use of vectors derived from murine γ-retroviruses and human lentiviruses. The neat division between clinical trials where AAV vectors have been used and those involving retroviral vectors is in part explained by the natural tropism of the viruses from which these vectors have been constructed.
AAV are non-enveloped non-integrative single-stranded DNA viruses of the Parvoviridae family. They require coinfection by adenoviruses to replicate and are non-pathogenic for humans. They infect replicating as well as quiescent cells and enter into the target cells by interaction with sialic acid, heparan sulfate, or galactose present on their surface, and therefore possess a large tropism. Differences in the capsid protein of AAV determine cell type-specific preferences and define the existence of the eleven serotypes of this virus. For gene therapy, according to the type of target tissue, serotypes that naturally target that type of tissue, when such serotypes exist, are the preferred choice for building a viral vector. To date, in gene therapy, eight serotypes (1–2 and 4–9) have been used to orient viral transduction toward the tissue of interest [12].
In sharp contrast to AAV, γ-retroviruses and lentiviruses do not present different serotypes and no variation in tissue specificity is found for these viruses, which both target blood cells. For example, in human immunodeficiency virus (HIV), despite its impressive genetic diversity, which is particularly high at the level of its envelope proteins, infection remains essentially restricted either to CD4+/CCR5+ or CD4+/CXCR4+ cells. However, an interest of retroviral-derived vectors (and therefore of lentiviral-derived vectors as well) comes from the possibility of replacing the original envelope proteins with those of other viruses, a process called pseudotyping. In this review article, we focus on the perspectives on which pseudotyping lentiviral-derived vectors (LV vectors) open and how this is achieved.
2. Lentiviruses and Gene Therapy
Retroviruses are enveloped viruses that integrate in the infected cell. This property has made of these viruses the preferred choice for developing vectors when the expression of the transgene must be stable or when the transgene must be inherited by the progeny of the transduced cell. For these reasons, retroviral vectors have been chosen for the expression of transgenes in hematopoietic stem and progenitor cells (HSPCs) and, more recently, they have been used for the transduction of peripheral blood cells for the generation of CAR-T cells [13]. Gammaretroviral vectors derived from Moloney murine leukemia were used for the earliest gene therapy assays using retroviral vectors. They have been successful in the treatment of several primary immunodeficiencies, such as the X-linked severe combined immunodeficiency (SCID) or the adenosine deaminase deficiency-induced SCID [14,15,16], and they have been employed in the treatment of the Wiskott–Aldrich syndrome and of X-linked chronic granulomatous disease [17,18,19]. However, γ-retroviral vectors have been progressively replaced by the lentiviral vectors (LV vectors), mostly due to the lower levels of induction of the innate immune response they trigger [20,21] and, in particular, for biosafety reasons. Indeed, LV vectors predominantly integrate in transcription units [22], rather than in regions controlling gene expression as promoters and enhancers that are, instead, the preferential sites of integration for gammaretroviral vectors [23,24]. This difference has been shown to lead to a lower probability for lentiviruses to cause insertional oncogenesis [25,26]. LV vectors have thus been used in most recent trials, always for the treatment of blood diseases. Besides treating the same diseases with these new vectors as are treated with γ-retroviral vectors mentioned above [27,28,29,30,31], β-thalassemia [32], Fanconi anemia [33], metachromatic leukodystrophy [34,35], mucopolysaccharidosis type I [36], adrenoleukodystrophy [37] and sickle cell disease [38] have also been made the object of clinical trials using LV vectors.
3. Molecular Biology of Lentiviruses
Lentiviruses belong to the subfamily Lentivirinae of retroviruses [39]. They are considered as “complex” retroviruses, due to the presence of additional genes, compared to other retroviruses. As all retroviruses, they are enveloped integrative viruses. The viral particle is constituted by a spheric matrix shell that lies immediately underneath the lipid bilayer, which consists of a patch of the cell membrane that is carried over during viral budding from the infected cell [40]. More internally, a fullerene-shaped core [41] contains the genomic RNA that is constituted by a single-stranded positive-sense molecule, present in two copies in the viral particle, in a dimeric form. Upon infection of the host cell (that occurs after recognition of a specific receptor on the surface of the cell) the viral capsid enters the cytoplasm. The availability of the nucleotides, to which the capsid is permeable, allows the initiation of reverse transcription. This results in the conversion of the genomic RNA into double-stranded DNA which is then integrated in the cell genome [42,43,44,45].
Where and when this conversion occurs and is achieved remains a matter of debate. The traditional view according to which reverse transcription was completed in the cytoplasm or at the nuclear pore, followed by the dismantling of the capsid core and the import of the preintegration nucleoprotein complex [46,47,48,49], has recently been challenged by the observation of intact or almost-intact cores, as well as the detection of ongoing reverse transcription in the nucleus [50]. However, irrespective of the form under which the genetic material is imported into the nucleus, the import occurs in an active manner, through the interaction of the viral capsid protein p24 with the cellular protein cyclophillin A and the cellular splicing factor CSPF6 [51,52,53]. This interaction leads to the use of the nuclear import pathway relying on the pair of nuclear pore proteins Nup153/Nup358 and transportin 3 (TNPO3) [54]. This complex system allows lentiviruses (in the specific case detailed above, human immunodeficiency virus type 1 (HIV-1)) to infect non-replicating cells. This not only allows LV vectors to deliver transgenes to cells that naturally do not replicate, but also can be exploited for transducing cells, such as HSPCs, that must be kept in a quiescent state to avoid their differentiation and loss of pluripotency. Retroviruses as γ-retroviruses are instead unable to enter the nucleus of the infected cell and require the disassembly of the nuclear membrane at mitosis for reaching the genome of the infected cell for integration.
Integration is carried out by the viral enzyme integrase with poor sequence specificity for the selection of the integration sites, although preferential types of genomic regions (as, for example, regions where actively transcribed genes are located, or the proximity with respect to transcription start sites) can be defined for the different types of retroviruses [55,56]. The reverse transcription product, integrated in the genomic RNA of the infected cell, is called a provirus. The provirus is flanked by the terminal repeated regions (LTRs) that contain the viral promoter sequence (see below). Transcription from the LTR in 5’ will lead to the synthesis of the new genomic RNA as well as the viral proteins required for infection to be continued. At the moment of assembly of the viral particle, the dimers of viral genomic RNA will be packaged in the budding particle [57]. The particle will also incorporate the envelope proteins at their surface, as detailed below, and be released in the extracellular space as an immature particle. Activation of the viral protease in the immature particle, will then lead to viral maturation and the production of an infectious virus [40].
4. Molecular Bases for the Making of LV Vectors
4.1. Structure of the Genomic RNA
LV vectors are generally derived from the best characterized lentivirus—human immunodeficiency virus type 1 (HIV-1). Lentiviral infection, detailed above, is conceptually composed of two phases. Entry and the conversion of the genomic RNA (gRNA) into DNA that will be integrated in the cell’s chromosomes are considered as the “early phase” of the infectious cycle. With the exception of entry, which depends on the nature of the envelope employed in the viral vector, all the steps of the early phase of HIV-1 infection are carried out essentially in the same manner during LV vector-mediated transduction. The late phase, constituted by the production of the gRNA and of the viral proteins, is instead absent in the case of LV vector transduction.
The viral gRNA of HIV-1 is characterized, proceeding from 5’ to 3’, by the terminal repeated sequence R, the unique sequence in 5’ U5; then, contiguous to U5, are found the 18 nucleotides that constitute the sequence to which the tRNA Lys3 anneals for priming reverse transcription (primer binding sequence (PBS)) [58] followed by an untranslated 5’ region that is responsible for the dimerization of the gRNA and its packaging in the viral particle [57]. Then, the main three genes (gag, pol and env) follow, overlapping the sequences for the auxiliary proteins Vif, Vpr and Vpu, as well as the proteins Tat and Rev (Figure 1). Finally, partially overlapping with the 3’ portion of env, the Nef coding sequence is found, followed by the unique sequence in 3’ U3, the repeated sequence R and the polyA tail [59]. The sequences required for priming the synthesis of the second strand of DNA (3’ and central polypurine tracts, -3’ PPT and cPPT, respectively) are located immediately upstream of the U3 sequence and in the 3’ end portion of pol, respectively [60,61]. The Rev Responsive Element (RRE) sequence that, when bound by the Rev protein allows the export of partially unspliced RNAs from the nucleus, is located in the portion of env encoding the gp41 protein [62].
To generate the gRNA of the LV vector, the viral gRNA is modified by removing all the coding sequences for the viral proteins and leaving the elements required in cis for genomic RNA packaging, reverse transcription and integration. Specifically, the gRNA of the vector must contain: the PBS sequence; the 3’ PPT and cPPT sequences; the region (located in the 5’ untranslated portion of the genome) responsible for the packaging and dimerization of the genomic RNA; the RRE sequence; the repeated terminal sequence R and the sequence U5, which are required for achieving reverse transcription and integration [63]. The U3 sequence, instead, is only partially preserved, since a large deletion (approximately half of its total length) is made in this sequence [64]. The deletion is essential for inactivating, in LV vectors, the promoter activity of U3, generating what are known as self-inactivating (SIN) vectors [64]. In natural infections, the U3 sequence is located inside the LTR sequences, present at both ends of the proviral DNA (Figure 2A). The U3 sequence located in the 5’ LTR contains the promoter that is used to drive the transcription of the genomic RNA. The genomic RNA contains only the U3 sequence of the 3’ LTR (Figure 2A). After reverse transcription of this genomic RNA, the LTRs are again generated (Figure 2A). In the case of SIN LV vectors, the U3 sequence of the 3’ LTR carries the deletion (in black in Figure 2B) and it will be this sequence that will be present in the genomic RNA. After reverse transcription, this deleted version of U3 will be present in both LTR, the 5’ and the 3’ regions (Figure 2B). Transcription is thereby no longer possible from this proviral DNA, since the promoter in the U3 sequence in the 5’ LTR is not functional. Taking into account these requirements, the gRNA of the LV vector can accommodate up to 8 kb of exogenous sequences.
For the generation of the LV vector particle, the plasmid leading to the synthesis of the gRNA is cotransfected with transcomplementation plasmids leading to the synthesis of the viral proteins. Depending on which generation of LV vectors is considered, the structure of the plasmids varies as well as which viral proteins are provided (Figure 3). In this setting, in order to change the tropism of the viral vector through pseudotyping, the plasmid encoding the envelope proteins will be chosen to carry the desired, non-HIV, envelope protein coding sequences.
4.2. Mechanism of Entry in HIV-1
The need for pseudotyping LV vector particles comes, as mentioned above, from the difficulty of modifying the mechanism of viral entry of the natural HIV envelope proteins. HIV-1 encodes two envelope proteins—gp41 and the gp120. The gp41 is a transmembrane protein that, associating in a non-covalent manner to the gp120 (that is located on the external side of the virus), forms an unstable heterodimer [65]. Three of these heterodimers associate to form a trimer of dimers that constitute the viral spike [65]. Viral entry occurs by fusion of the cell and viral membranes, carried out by the viral envelope proteins. For this, the spike interacts, through the gp120 component, with the natural HIV-1 receptor, the CD4 molecule [66,67]. This triggers a conformational change that leads to the generation in the gp120 of a binding site for the HIV coreceptor, generally the transmembrane protein CCR5, or CXCR4 [68,69,70,71,72]. This second interaction is responsible for another structural rearrangement of the gp120/gp41 dimer that releases the gp41 from the interaction with the gp120 [73]. The gp41 that was maintained in a metastable state by the interaction with the gp120 inserts its highly hydrophobic N-terminal portion, called “fusion peptide”, in the internal portion of the spike, in the membrane of the target cell [74]. Once this has occurred, the gp41 folds back on itself to reach the most stable conformation possible. This brings the fusion peptide (still inserted in the cell membrane) in proximity of the viral membrane leading to the fusion of the membranes [75,76,77,78] and to the creation of a pore that, once enlarged, allows the entry of the viral core into the cytoplasm. Because of the complex series of conformational transitions required for the functionality of the envelope, the interactions between the two Env proteins must be based on highly unstable equilibria that are extremely difficult to retain if one wishes to modify this system, in order to redesign the tropism of the virus. Therefore, changing the tropism of a HIV-derived LV vectors is a fairly difficult goal to achieve through the modification of the natural HIV envelope proteins. However, the relative ease with which a LV vector can be efficiently pseudotyped by exogenous viral envelope proteins provides alternative solutions to bypass these difficulties.
4.3. Mechanism of Recruitment of HIV-1 Envelope Proteins on the Surface of the Virus: Bases for Pseudotyping
The HIV-1 particle is enveloped by the plasma membrane of the cell from which the virus has budded. Consequently, the lipid and protein compositions of the viral membrane reflect that of the infected cell at the site of budding. The peculiar composition of lipids and proteins of the viral particle with respect to that of the cell, suggests that viral budding occurs in specific regions of the membrane with a particular lipid composition. To be incorporated in the budding viral particle a protein must be addressed to the cell compartment where viral budding occurs [79]. These observations set the bases to conceive the possibility of pseudotyping lentiviral particles.
HIV-1 assembles at lipid rafts, areas of the membrane enriched in cholesterol and sphingolipids. This lipid composition tends to be enriched in glycosylphosphatidylinositol-anchored proteins at the site of budding [80]. Potentially, any protein with a high “affinity” for lipid rafts has a higher probability than the average protein to be found on the viral particle. This strategy allows the virus to reduce immunogenicity during natural infections and, consequently, also leads to the generation of poorly immunogenic HIV-derived LV vector particles. Assembling at lipid rafts indeed provides a lipid membrane of the viral particle enriched in proteins such as CD46, CD55 and CD59 [81,82,83,84] which are known to inhibit complement activation [85,86,87]. Accordingly, when the virus is produced in glycosylphosphatidylinositol-anchors deficient cells, it becomes sensitive to degradation by the immune system [88].
In HIV-1, the envelope proteins are recruited at lipid rafts through the interaction between the cytoplasmic tail of the gp41 and the precursor polyprotein Pr55 Gag, which is localized at the lipid rafts thanks to the myristoyl group that is present at its N-terminus [89]. It also has been shown that the acylation of the transmembrane domains of proteins was sufficient to address these proteins to the lipids raft [90,91]. Acylated proteins potentially prevail in viral particles that bud from lipid raft rich areas. This characteristic of acylated proteins provides a “tool” to induce pseudotyping in LV vectors. Accordingly, VSV glycoproteins are acylated [92], as well as the E2 envelope glycoprotein of alphaviruses such as Semliki forest virus and Sindbis Virus [93,94]. All these envelope proteins efficiently pseudotype LV vectors. For other viral envelope proteins, such as the rabies ones for instance, the molecular mechanism leading to pseudotyping LV vectors is known in much less detail, but it is logical to expect that, also in this case, pseudotyping occurs through addressing these proteins to lipid rafts.
5. Dressing LV Vectors (Pseudotyping)
Pseudotyping LV vectors with envelope proteins of different viruses allows combining the properties of lentiviruses with those of viral entry of other viruses. The envelope proteins of several types of viruses have been shown to be able to pseudotype LV vectors. Among these viruses, some possess a large tropism and therefore the use of their proteins for treatments in vivo cannot be envisaged. However, some of these proteins can be used as starting platforms for engineering variants that specifically target a desired cell population. Envelope proteins from other viruses, instead, present a tropism restricted to certain types of cells (neurons, for example), and can be employed “opportunistically” when these cell types constitute the target of the intervention strategy. Envelope proteins from several viruses have been described to successfully pseudotype LV vectors. Those for which the molecular mechanism has been elucidated in more detail fall into three viral families and are presented here (Figure 4 and Table 1).
5.1. Rhabdoviruses: Clathrin-Dependent Endocytosis
5.1.1. Vescicular Stomatitis Virus
Pseudotyping LV vectors by the vescicular stomatitis virus envelope glycoprotein G is the most common approach for creating cell lines [117]. VSV is an enveloped virus from the Rhabdoviridae family. It expresses the G glycoprotein on the envelope surface. The first hypothesis to explain the large tropism of the virus suggests that it might use not only a specific, widespread, receptor but that it possibly also uses alternative receptors. The first receptor described for VSV-G were the phosphatidylserines, phospholipids that are a main components of plasma membranes [118,119] present at the surface of almost all cell types [119]. However, more recent work showed that treatment with annexin V, a specific ligand for phosphatidylserines, did not inhibit infection by VSV [120]. In addition, the same work showed an absence of correlation between the content of phosphatidylserines in the plasma membrane and the efficacy of infection by VSV [120]. In 2013 it was demonstrated that the main receptor promoting VSV entry are members of the LDL-Receptor (LDL-R) family [95]. These receptors are involved in the regulation of the homeostasis of cholesterol in mammalian cells and are ubiquitously expressed [121,122]. Once bound to the cell membrane, the VSV envelope protein VSV-G triggers clathrin-dependent endocytosis [123], typical of the Rhabdoviridae family, followed by pH-dependent fusion of endosomal and viral membranes [124,125], leading to the release of the capsid in the cell, although it is still debated whether membrane fusion occurs in the early endosome or in the late endosomes/multivesicular bodies [126,127]. To begin fusion, a conformational change of the G protein is required first to anchor the virus into the cell membrane and then to operate a physical connection between the two lipid bilayers that ultimately allows membrane fusion [128].
The quasi-universal tropism provided by the glycoproteins G of VSV makes it difficult to conceive a safe manner for the systemic inoculation of these vectors in patients, because of obvious problems related to off-target delivery. When the natural biodistribution that follows the systemic administration of vectors is favorable, as for example when the liver is the target of the therapy, LV vectors pseudotyped by the VSV envelope protein G have proved to be successful for in vivo treatment, as for the case of the induction of the expression of the coagulation factor IX (FIX) in mice and hemophilic dog models [129,130]. Alternatively, their local administration can be considered in vivo, as it has been shown for colorectal administration in mouse models [131]. However, to date, pseudotyping LV vectors by the VSV envelope protein G for gene therapy is employed, in the majority of the cases, for transduction ex vivo [132,133].
5.1.2. Rabies Virus
Rabies viruses (RVs) are negatively stranded RNA Rhabdoviruses, with a natural tropism for neurons. Accordingly, the use of their envelope proteins for pseudotyping LV vectors is strictly related to intervention on these cells. RVs infect neurons through their terminal axons and spread through the synapses in a retrograde direction, a feature that is maintained when RV-pseudotyped LV vectors are used [134]. Recognition of the receptor is ensured by the G protein, which interacts with a panel of different receptors, all expressed on neurons. After receptor recognition, RV particles are endocytosed following a clathrin-dependent uptake [135]. The internalized vesicles then fuse with the early endosomes, as a consequence of the acidification of the endosome, with a VSV-like mechanism [123] (Figure 3).
The first receptor described for RV was the nicotinic acetylcholine receptor (nAChR) [97]. This receptor is present at a high density in neuromuscular junctions [136]. Another receptor used by RV is CD56 (or NCAM) [98], involved in the adhesion of neural cells, the development of neurites and the synapses’ plasticity. However, CD56 is also abundant on natural killer cells, raising a concern about the specificity of its use. RVs have also been shown to interact with the nerve growth factor receptor (NGFR) superfamily, the p75NTR (Low-affinity Nerve Growth Factor Receptor: LNGFR) [99]. However, despite this interaction, infection is limited to around 20% of neurons, while more than 80% are p75NTR positive [137]. These results tend to show that the LNGFR is not the most important receptor for RV uptake. Finally, mGluR2, abundant in the central nervous system [100], has also been described as a receptor for RV. Indeed, it has been observed that RV and mGluR2 are internalized into cells and transported to early and late endosomes in close association, suggesting their functional interaction.
In conclusion, the efficient transduction by the RV G glycoprotein involves a wide panel of receptors, but is strictly limited to neurons. This characteristic can be exploited to transduce specifically neurons. Indeed, it has been shown that an injection of RV-pseudotyped LV vectors directly in the muscle can lead to gene transfer in the spinal cord motoneurons while, under the same conditions, a vector pseudotyped with VSV-G have transduced muscle cells around the injection site, without any expression in the spinal cord neurons [134]. However, crossing the hemato-encephalic barrier by LV vectors in adults is mostly restricted to some more permissive areas as the median eminence (hypothalamus), pituitary, choroids plexus or pineal gland [138], limiting the use of RV-pseudotyped LV vectors.
5.2. Paramyxoviruses: Splitting Binding and Fusion in H and F Proteins
5.2.1. Measles Virus
Measles virus (MV) is an enveloped, single-stranded RNA virus of negative polarity belonging to the family of Paramixoviridae. It is responsible for the measles disease in humans, against which extensive vaccinal strategies were developed over decades. Its envelope glycoproteins confer a wide cell tropism. Recognition of the receptors is ensured by the H protein (hemagglutinin) present at the surface of the virus. Once the virus is bound to the target cell, membrane fusion is ensured by the F protein (where F stands for fusion). For this, F undergoes a first conformational change that allows it to anchor the cell membrane and, subsequently, start the fusion [139], which is achieved by bringing the viral and cell membranes in close proximity [140]. As for all Paramixoviridae, the mechanism of membrane fusion is pH independent and it occurs directly at the plasma membrane [141] (Figure 3).
The first receptor to be identified for measles was CD46, a molecule present on the surface of most human cells except erythrocytes [142,143,144]. Consequently, the use of these envelope proteins would result in an extremely large tropism, conferring only a little advantage with respect to the use of VSV-G. Furthermore, it appears that CD46 is not the only receptor for measles virus, since passages in cell culture of the MV on lymphoblastoid cell lines led to the selection of a strain capable of infecting cells not expressing CD46 [101,102,103]. This laboratory strain had the ability to infect lymphocyte cell lines through the use of the receptor SLAM (also named CDw150) [104,105,106]. However, SLAM is also expressed on some subsets of B and T human cells. Therefore, the problem of the lack of specificity encountered with CD46 is still present, although considerably reduced, if pseudotyping is performed with an envelope issued from a SLAM-tropic strain. Finally, for the wild-type MV and for the strain used in vaccinal approaches (the Edmonston laboratory strain), another receptor can be used—nectin-4 [145,146]. Nectins are responsible for calcium-dependent cell adhesion and are therefore found on epithelial cells. Nectin-4 is found overexpressed on the surface of tumor cells in some ovarian [147] or lung [148] cancers and could therefore be used for treatment of cancers expressing this receptor. Therefore, three types of receptors have been identified for this virus. While for two of them the tropism is too large for their use, for the third one it is possible to envisage its use if the cells to target match the natural tropism of this viral variant.
The H protein can also be used as a basis for redirecting viral tropism. In this sense, mutations that abolished binding to CD46 and SLAM were identified, an important issue for reducing off-target delivery [149]. The first relevant case of complete MV retargeting consisted of the modification of the H glycoprotein, by inserting a single-chain antibody directed against CD38 or EGFR to be used as an oncolytic vector [150]. The engineered viruses mediated efficient infection through their respective receptors targeted, but not through, CD46 or SLAM. This work showed the possibility of a complete specificity change of the H protein. Single-chain antibodies are not the only way to retarget the specificity of the MV envelopes, though. It has been shown that the insertion of designed ankyrin repeat proteins (DARPins), consisting of at least three ankyrin repeats, in H protein can allow the retargeting of a MV-pseudotyped LV vectors [151]. In addition to an efficient retargeting, the H protein fused to a DARPin has a higher level of surface expression than the H proteins fused to a single-chain antibody [151].
The major obstacle to the development of a pseudotyping approach based on the use of MV envelope proteins is constituted by the neutralization of the vectors, due to the large vaccinal coverage against measles virus present in the human population. Since H protein seems to be the main target of the immune response [152], a promising approach is constituted by the introduction of mutations in this protein that allow escape, to a certain extent, from immune recognition while preserving its functional activity [153].
5.2.2. Nipah Virus
Another Paramyxovirus, the Nipah virus (NiV), a virus that can cause severe flu-like disease in human with potentially fatal issues, constitutes a promising option as source of envelope proteins for pseudotyping LV vector particles [154]. Infections by NiV are very rare, making the existence of a pre-existing humoral immunity that would interfere with gene transmission unlikely [109]. Nipahs have two envelope proteins—F and G glycoproteins (NiV-F and Niv-G, respectively). NiV-G is responsible for binding to the viral receptor, while F is the protein that carries out membrane fusion with the same mechanism than MV, as previously described [139] (Figure 3).
The NiV-G glycoprotein consists of a stem domain and a globular head. It forms dimers linked by disulfide bonds which combine in pairs to generate tetramers [155]. Functional pseudotyping of LV vectors by these proteins is possible but it requires the truncation of the cytoplasmic tail of the NiV-F protein. Recently, indeed, a truncated variant mutated in four residues of the cytoplasmic tail (FcΔ22) also shows a ten-fold increased efficiency of pseudotyping [110,156]. Concerning the NiV-G protein, it has been reported that the full-length form can be used, although two truncated forms of the protein, GcΔ33 and GcΔ34, in which have been deleted respectively the 33 and 34 N-ter amino acids, provide optimized pseudotyping [109,156]. It has already been described that shortened cytoplasmic tails of the attachment proteins increase the titers of Paramyxoviridae-pseudotyped LV vectors [157,158]. It is therefore not unexpected that this strategy is transposable to pseudotyping LV vectors by Nipah envelope proteins [109]. The advantages conferred by the truncation can be explained by the location of the truncated forms of the NiV-G proteins in plasma membrane regions far from ephrin-B2, which is normally located at the junction of neighboring cells. For the truncated forms, this limits, at the same time, the cytopathic effect exerted at these junctions and their sequestration by ephrin-B2 molecules expressed in producer cells. This enhances their incorporation into LV vector particles [109].
Attachment of Nipah on the target cell occurs through binding to Ephrin-B2 [159] as a primary receptor, although Ephrin-B3 can also be recognized for attachment albeit with an affinity ten times lower [160]. Ephrin receptors are membrane-associated tyrosine kinase (RTK) receptors with different roles in several biological processes such as neurogenesis and angiogenesis. The main receptor, Ephrin-B2, is highly expressed in the arterial endothelium and, to a lower extent, in pericytes and vascular smooth muscle cells [161,162]. Usually expressed at the cell–cell junctions, Ephrin-B2/Ephrin-B4 interactions are strongly involved in angiogenesis, cell migration and tumor invasion. Pseudotyping LV vectors with wt Nipah envelope proteins would therefore allow to address viral vectors to these areas, potentially interfering with the setting up of the angiogenic processes that are associated with tumor expansion.
Besides these potential applications of LV vectors pseudotyped by the wild-type Nipah envelope proteins, the use of engineered proteins can also be envisaged. Namely, to increase the specificity of the vector pseudotyped by NiV-G, NiV-GcΔ34 has been modified by point mutations in order to abolish binding to Ephrin-B2 [156]. This study showed that combining some mutations can reduce binding to a level below detection, providing a basis for the development of variants with an entirely new tropism. By the insertion of new ligands in the mutated H protein [156], it has been possible to transduce efficiently model cells expressing either CD20 (a marker of B cells), CD8 (a marker of cytotoxic T cells), or EPCAM (a putative marker of early tumor cells [163]) with the same selectivity as reference LV vectors pseudotyped with receptor-targeted measles virus envelope proteins. Thanks to the lower exposure of the population to Nipah virus, engineered NiV envelope proteins could therefore provide an alternative for LV vector pseudotyping with a reduced risk of antibody neutralization of the vector with respect to MV.
5.3. Togaviridae: The Pair E1-E2 Dissociates to Trigger Membrane Fusion
5.3.1. Chikungunya Virus
Chikungunya virus (ChikV) is an alphavirus from the Togaviridae family. Akin to all togaviruses, its spikes are composed by the E1, E2 and E3 envelope glycoproteins, associated non-covalently. E1 is responsible for pH-dependent viral and cell membrane fusion [164] after the clathrin-dependent endocytosis [165,166] of the virus. E2 carries out the association with cellular receptors [167] while E3 is responsible for the translocation of the spikes to the endoplasmic reticulum. E3 also displays chaperone activity to assist the correct folding of the E2 precursor and, by acting as a clamp, stabilizes the association between E1 and E2 [168,169]. E3 is ultimately cleaved from E2 in the trans-Golgi network. This cleavage is necessary to remove the “clamp” since, upon binding to the target cell at low pH conditions, dissociation of the E2-E1 heterodimer becomes necessary in order to mediate fusion between the viral and host cell membranes during viral entry [170]. Indeed, for entry, once E2 and the target receptor have interacted, dissociation of the E1/E2 complex, probably induced by the acidification of the endosome, is required to trigger fusion [171]. The dissociation exposes a non-polar domain at the E1 apex, which is able to start the fusion by anchoring to the membrane of the endosome. Nevertheless, some studies revealed the existence of alternative entry pathways such as macropinocytosis [172], probably coexisting with the main clathrin-mediated, one (Figure 3).
The tropism conferred by this envelope is not fully defined, although heparan sulfate, integrins and the matrix remodeling-associated protein 8 (Mxra8, a protein expressed on a large panel of cell types) have been shown to be involved in binding of the virus to the cell surface. The widespread expression of these proteins can explain the large variety of symptoms observed during infection with wild-type ChikV: encephalitis and neurological complications [173], arthralgia/myalgia [174] and ophthalmic disorders [175]. The neurological disorders associated with this virus have suggested the use of ChikV-pseudotyped LV vectors for transducing cells of the central nervous system. A study has indeed shown that ChikV-pseudotyped LV vectors efficiently transduce neurons and astrocytes both in vitro and in vivo [176]. Despite these results, it has not been shown that this type of pseudotyping allows specific targeting in vivo. Another receptor, Prohibitin (PHB), has also been described as a target for ChickV [111]. PHB is an evolutionarily conserved and ubiquitously expressed protein. It has a broad range of functions: it has a structural role at the level of the cell membrane, it is involved in transcription, in mitochondrial morphogenesis and apoptosis [177,178]. Even if PHB is overexpressed in some cancers, such as diffuse large B-cell lymphomas [178], also in this case, its widespread expression does not allow it to be used as a target for specific interventions in vivo. Therefore, at least until variants with a more restricted tropism will be developed, the most straightforward therapeutic application for the use of ChickV envelope proteins seems to be the reprogramming of target cells in vitro [176]. Furthermore, the frequent recognition of the ChikV envelope by the immune system, having oriented the research to focus preferentially on another less immunogenic but just as promising as ChikV for genetic manipulations, alphavirus envelope: the one of the Sindbis virus.
5.3.2. Sindbis Virus
The Sindbis virus (SV), similar to ChikV, is an alphavirus from the Togaviridae family. Since these viruses are genetically related [179], their envelope proteins have similar structures, with a step of dissociation between E1 and E2 essential for triggering membrane fusion in both cases. Due to the similarity between alphaviruses, the entry mechanism of Sindbis virus is described as being strictly similar to that of ChikV.
Despite their relatedness, the receptors used differ between these two viruses. The most well documented receptor capable of interactions with SV E2 is the 67 kDa non-integrin high affinity laminin receptor, 67LR [115]. 67LR is found in lipid rafts of a wide variety of normal cell types such as intestinal epithelium cells [180], neurons, hematopoietic cells [181] and in some cancer cells as carcinomas [182]. The natural resistance-associated macrophage protein 2 receptor (NRAMP2, also known as DMT1, for divalent metal transporter 1), expressed in almost all cell types [183,184,185], has also been described as being an E2 interactant [116]. As for ChikV, viral entry into the cell follows a clathrin-dependent pathway with the subsequent release of the capsid into the cytoplasm (Figure 3). It is noteworthy that some studies support the idea that Sindbis fusion could occur at the plasma membrane with no need for clathrin-dependent endocytosis [186,187] (Figure 3).
The use of the wt Sindbis virus envelope in pseudotyping for the purposes of viral vectorization in vivo is limited because of its wide tropism. However, the main interest of its envelope proteins comes from their versatility in genetic modifications. Numerous studies have described variants of SV envelope proteins that have been modified by genetic engineering and used to pseudotype LV vectors. In particular, a loop of the E2 protein allows insertion of exogenous motifs without affecting the ability of the SV envelopes to promote viral entry in the target cell. The most successful results obtained so far concern the insertion of the ZZ domain derived from the IgG-binding domain of protein A of Staphylococcus aureus between amino acids 71 and 74 of E2 [188]. This strategy made it possible to bind the envelope protein to the Fc fragment of an IgG directed against specific receptors in order to reorient the vector to enter a defined type of cells thanks to the interaction between the ZZ domain and the IgG. Other works aimed at reducing non-specific targeting through abolishing the natural tropism of the virus. The most successful one resulted in the creation of a variant called m168 [189] which was fusion competent but deficient for binding to the natural receptors of SV. The mutations present in the m168 variant are, in addition to the insertion of the ZZ motif in E2, a deletion of the residues 61-64 of E3 and the insertion of four point mutations in E2 (K159A, E160A, E216A and T218A) [190]. Therefore E1, responsible for the fusion of the membranes, was not mutated. The m168 mutations were then combined to coupling an IgG directed against the P glycoprotein that is expressed in the lungs [191]. When the vector was administered by intravenous injection in model mice, the pseudotyped lentiviruses were addressed to the lungs with a low dispersion of the vectors in the rest of the organism. However, this strategy requires a step of non-covalent coupling between the viral particle and the antibodies, which, being a labile interaction, constitutes the critical point of the strategy.
In conclusion, alphaviruses’ wt envelope proteins cannot be used for in vivo approaches, due to their natural broad tropism. However, the marked separation of the binding and of the fusion functions, carried out by E2 and by E1, respectively, allows envisaging modifications to the E2 protein so as to allow the most specific interactions possible with the desired targets without affecting the fusion step. The main concern is the preservation of the association between E1 and E2 at the surface of the viral particle.
6. Concluding Remarks
Viral vectorization for gene therapy is a cutting-edge technique whose development has not yet shown its full potential. Even if several virus-based vectors have been identified as being potentially useful for these approaches, so far, the choice of preference regularly falls on a handful of vectors, among which are lentiviral vectors. A feature of LV vectors that makes them particularly promising is their possibility to uptake envelope proteins from other viruses. These exogenous envelopes can either possess a limited natural tropism that can match the tropism required for the intervention or can have a widespread tropism. In this latter case, they cannot be employed the way they are but, if they have a high plasticity with respect to genetic engineering, they can be used as starting points for the elaboration of artificial envelopes with a specific tropism of interest for the approach followed. LV vectors play the role of mannequins for these approaches for both the “prêt à porter” envelopes and for the “tailor-made” ones, being extremely useful for the development of these therapeutic approaches. Furthermore, emerging viruses may provide interesting alternatives to the envelopes already studied. In this sense, viruses responsible for today’s illnesses may become part of tomorrow’s therapies.
Author Contributions
Both authors outlined, wrote and revised the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
Work on lentiviral vectors in the laboratory of Matteo Negroni is funded by the French “Ligue contre le cancer”. A.D. is supported by a grant from the French “Ministère de l’Enseignement Supérieur et de la Recherche”.
Acknowledgments
The authors are grateful to Daniela Lener for helpful discussion and critical reading of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; nor in the writing of the manuscript.
References
- U.S. National Library of Medicine. What is Gene Therapy? Genetics Home Reference—NIH. Available online: https://ghr.nlm.nih.gov/primer/therapy/genetherapy (accessed on 27 August 2020).
- Reyon, D.; Tsai, S.Q.; Khayter, C.; Foden, J.A.; Sander, J.D.; Joung, J.K. FLASH assembly of TALENs for high-throughput genome editing. Nat. Biotechnol. 2012, 30, 460–465. [Google Scholar] [CrossRef] [PubMed]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; Dicarlo, J.E.; Norville, J.E.; Church, G.M. RNA-Guided Human Genome Engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wirth, T.; Parker, N.; Ylä-Herttuala, S. History of gene therapy. Gene 2013, 525, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, K.B.; Büning, H.; Galy, A.; Schambach, A.; Grez, M. Gene therapy on the move. EMBO Mol. Med. 2013, 5, 1642–1661. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.A.; Walters, M.C.; Kwiatkowski, J.; Rasko, J.E.; Ribeil, J.-A.; Hongeng, S.; Magrin, E.; Schiller, G.J.; Payen, E.; Semeraro, M.; et al. Gene Therapy in Patients with Transfusion-Dependent β-Thalassemia. N. Engl. J. Med. 2018, 378, 1479–1493. [Google Scholar] [CrossRef] [PubMed]
- Zonari, E.; DeSantis, G.; Petrillo, C.; Boccalatte, F.E.; Lidonnici, M.R.; Kajaste-Rudnitski, A.; Aiuti, A.; Ferrari, G.; Naldini, L.; Gentner, B. Efficient Ex Vivo Engineering and Expansion of Highly Purified Human Hematopoietic Stem and Progenitor Cell Populations for Gene Therapy. Stem Cell Rep. 2017, 8, 977–990. [Google Scholar] [CrossRef] [Green Version]
- Gowing, G.; Svendsen, S.; Svendsen, C.N. Ex Vivo gene therapy for the treatment of neurological disorders. In Progress in Brain Research; Elsevier BV: Amsterdam, The Netherlands, 2017; Volume 230, pp. 99–132. [Google Scholar]
- Rosenberg, S.A. Gene Therapy for Cancer. JAMA 1992, 268, 2416–2419. [Google Scholar] [CrossRef]
- Dunbar, C.E.; High, K.A.; Joung, J.K.; Kohn, D.B.; Ozawa, K.; Sadelain, M. Gene therapy comes of age. Science 2018, 359, eaan4672. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Asokan, A.; Samulski, R.J. Adeno-associated Virus Serotypes: Vector Toolkit for Human Gene Therapy. Mol. Ther. 2006, 14, 316–327. [Google Scholar] [CrossRef]
- Wilkins, O.; Keeler, A.M.; Flotte, T.R. CAR T-Cell Therapy: Progress and Prospects. Hum. Gene Ther. Methods 2017, 28, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, H.B.; Cooray, S.; Gilmour, K.C.; Parsley, K.L.; Adams, S.; Howe, S.J.; Al Ghonaium, A.; Bayford, J.; Brown, L.; Davies, E.G.; et al. Long-Term Persistence of a Polyclonal T Cell Repertoire After Gene Therapy for X-Linked Severe Combined Immunodeficiency. Sci. Transl. Med. 2011, 3, 97ra79. [Google Scholar] [CrossRef] [PubMed]
- Cavazzana-Calvo, M.; Hacein-Bey, S.; Basile, G.D.S.; Gross, F.; Yvon, E.; Nusbaum, P.; Selz, F.; Hue, C.; Certain, S.; Casanova, J.-L.; et al. Gene Therapy of Human Severe Combined Immunodeficiency (SCID)-X1 Disease. Science 2000, 288, 669–672. [Google Scholar] [CrossRef]
- Aiuti, A.; Vai, S.; Mortellaro, A.; Casorati, G.; Ficara, F.; Andolfi, G.; Ferrari, G.; Tabucchi, A.; Carlucci, F.; Ochs, H.D.; et al. Immune reconstitution in ADA-SCID after PBL gene therapy and discontinuation of enzyme replacement. Nat. Med. 2002, 8, 423–425. [Google Scholar] [CrossRef] [PubMed]
- Boztug, K.; Schmidt, M.; Schwarzer, A.; Banerjee, P.P.; Díez, I.A.; Dewey, R.A.; Böhm, M.; Nowrouzi, A.; Ball, C.R.; Glimm, H.; et al. Stem-Cell Gene Therapy for the Wiskott–Aldrich Syndrome. N. Engl. J. Med. 2010, 363, 1918–1927. [Google Scholar] [CrossRef] [Green Version]
- Braun, C.J.; Boztug, K.; Paruzynski, A.; Witzel, M.; Schwarzer, A.; Rothe, M.; Modlich, U.; Beier, R.; Göhring, G.; Steinemann, D.; et al. Gene Therapy for Wiskott-Aldrich Syndrome--Long-Term Efficacy and Genotoxicity. Sci. Transl. Med. 2014, 6, 227ra33. [Google Scholar] [CrossRef]
- Ott, M.G.; Schmidt, M.; Schwarzwaelder, K.; Stein, S.; Siler, U.; Koehl, U.; Glimm, H.; Kühlcke, K.; Schilz, A.; Kunkel, H.; et al. Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1. Nat. Med. 2006, 12, 401–409. [Google Scholar] [CrossRef]
- Piras, F.; Riba, M.; Petrillo, C.; Lazarevič, D.; Cuccovillo, I.; Bartolaccini, S.; Stupka, E.; Gentner, B.; Cittaro, D.; Naldini, L.; et al. Lentiviral vectors escape innate sensing but trigger p53 in human hematopoietic stem and progenitor cells. EMBO Mol. Med. 2017, 9, 1198–1211. [Google Scholar] [CrossRef] [Green Version]
- Gao, D.; Wu, J.; Wu, Y.-T.; Du, F.; Aroh, C.; Yan, N.; Sun, L.; Chen, Z.J. Cyclic GMP-AMP Synthase Is an Innate Immune Sensor of HIV and Other Retroviruses. Science 2013, 341, 903–906. [Google Scholar] [CrossRef] [Green Version]
- Schröder, A.R.; Shinn, P.; Chen, H.; Berry, C.; Ecker, J.R.; Bushman, F.D. HIV-1 Integration in the Human Genome Favors Active Genes and Local Hotspots. Cell 2002, 110, 521–529. [Google Scholar] [CrossRef] [Green Version]
- Cattoglio, C.; Pellin, D.; Rizzi, E.; Maruggi, G.; Corti, G.; Miselli, F.; Sartori, D.; Guffanti, A.; Di Serio, C.; Ambrosi, A.; et al. High-definition mapping of retroviral integration sites identifies active regulatory elements in human multipotent hematopoietic progenitors. Blood 2010, 116, 5507–5517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romano, O.; Peano, C.; Tagliazucchi, G.M.; Petiti, L.; Poletti, V.; Cocchiarella, F.; Rizzi, E.; Severgnini, M.; Cavazza, A.; Rossi, C.; et al. Transcriptional, epigenetic and retroviral signatures identify regulatory regions involved in hematopoietic lineage commitment. Sci. Rep. 2016, 6, 24724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montini, E.; Cesana, D.; Schmidt, M.; Sanvito, F.; Bartholomae, C.C.; Ranzani, M.; Benedicenti, F.; Sergi, L.S.; Ambrosi, A.; Ponzoni, M.; et al. The genotoxic potential of retroviral vectors is strongly modulated by vector design and integration site selection in a mouse model of HSC gene therapy. J. Clin. Investig. 2009, 119, 964–975. [Google Scholar] [CrossRef] [PubMed]
- Modlich, U.; Navarro, S.; Zychlinski, D.; Maetzig, T.; Knoess, S.; Brugman, M.H.; Schambach, A.; Charrier, S.; Galy, A.; Thrasher, A.J.; et al. Insertional Transformation of Hematopoietic Cells by Self-inactivating Lentiviral and Gammaretroviral Vectors. Mol. Ther. 2009, 17, 1919–1928. [Google Scholar] [CrossRef] [PubMed]
- Aiuti, A.; Biasco, L.; Scaramuzza, S.; Ferrua, F.; Cicalese, M.P.; Baricordi, C.; Dionisio, F.; Calabria, A.; Giannelli, S.; Castiello, M.C.; et al. Lentiviral Hematopoietic Stem Cell Gene Therapy in Patients with Wiskott-Aldrich Syndrome. Science 2013, 341, 1233151. [Google Scholar] [CrossRef] [Green Version]
- Chu, J.I.; Henderson, L.A.; Armant, M.; Male, F.; Dansereau, C.H.; MacKinnon, B.; Burke, C.J.; Cavanaugh, M.E.; London, W.B.; Barlan, I.B.; et al. Gene Therapy Using a Self-Inactivating Lentiviral Vector Improves Clinical and Laboratory Manifestations of Wiskott-Aldrich Syndrome. Blood 2015, 126, 260. [Google Scholar] [CrossRef]
- Kohn, D.B.; Hershfield, M.S.; Puck, J.M.; Aiuti, A.; Blincoe, A.; Gaspar, H.B.; Notarangelo, L.D.; Grunebaum, E. Consensus approach for the management of severe combined immune deficiency caused by adenosine deaminase deficiency. J. Allergy Clin. Immunol. 2019, 143, 852–863. [Google Scholar] [CrossRef]
- De Ravin, S.S.; Wu, X.; Moir, S.; Kardava, L.; Anaya-O’Brien, S.; Kwatemaa, N.; Littel, P.; Theobald, N.; Choi, U.; Susan, M.; et al. Lentiviral hematopoietic stem cell gene therapy for X-linked severe combined immunodeficiency. Sci. Transl. Med. 2016, 8, 335ra57. [Google Scholar] [CrossRef] [Green Version]
- Kohn, D.B.; Booth, C.; Kang, E.M.; Pai, S.-Y.; Shaw, K.L.; Santilli, G.; Armant, M.; Buckland, K.F.; Choi, U.; De Ravin, S.S.; et al. Lentiviral gene therapy for X-linked chronic granulomatous disease. Nat. Med. 2020, 26, 200–206. [Google Scholar] [CrossRef]
- Cavazzana-Calvo, M.; Payen, E.; Negre, O.; Wang, G.; Hehir, K.; Fusil, F.; Down, J.; Denaro, M.; Brady, T.; Westerman, K.A.; et al. Transfusion independence and HMGA2 activation after gene therapy of human β-thalassaemia. Nat. Cell Biol. 2010, 467, 318–322. [Google Scholar] [CrossRef]
- Río, P.; Navarro, S.; Wang, W.; Sánchez-Domínguez, R.; Pujol, R.M.; Segovia, J.C.; Bogliolo, M.; Merino, E.; Wu, N.; Salgado, R.; et al. Successful engraftment of gene-corrected hematopoietic stem cells in non-conditioned patients with Fanconi anemia. Nat. Med. 2019, 25, 1396–1401. [Google Scholar] [CrossRef] [PubMed]
- Sessa, M.; Lorioli, L.; Fumagalli, F.; Acquati, S.; Redaelli, D.; Baldoli, C.; Canale, S.; Lopez, I.D.; Morena, F.; Calabria, A.; et al. Lentiviral haemopoietic stem-cell gene therapy in early-onset metachromatic leukodystrophy: An ad-hoc analysis of a non-randomised, open-label, phase 1/2 trial. Lancet 2016, 388, 476–487. [Google Scholar] [CrossRef]
- Biffi, A.; Montini, E.; Lorioli, L.; Cesani, M.; Fumagalli, F.; Plati, T.; Baldoli, C.; Martino, S.; Calabria, A.; Canale, S.; et al. Lentiviral Hematopoietic Stem Cell Gene Therapy Benefits Metachromatic Leukodystrophy. Science 2013, 341, 1233158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofling, A.A.; Devine, S.; Vogler, C.; Sands, M.S. Human CD34+ hematopoietic progenitor cell-directed lentiviral-mediated gene therapy in a xenotransplantation model of lysosomal storage disease. Mol. Ther. 2004, 9, 856–865. [Google Scholar] [CrossRef]
- Cartier, N.; Hacein-Bey-Abina, S.; Bartholomae, C.C.; Veres, G.; Schmidt, M.; Kutschera, I.; Vidaud, M.; Abel, U.; Dal-Cortivo, L.; Caccavelli, L.; et al. Hematopoietic Stem Cell Gene Therapy with a Lentiviral Vector in X-Linked Adrenoleukodystrophy. Science 2009, 326, 818–823. [Google Scholar] [CrossRef] [Green Version]
- Ribeil, J.-A.; Hacein-Bey-Abina, S.; Payen, E.; Magnani, A.; Semeraro, M.; Magrin, E.; Caccavelli, L.; Neven, B.; Bourget, P.; El Nemer, W.; et al. Gene Therapy in a Patient with Sickle Cell Disease. N. Engl. J. Med. 2017, 376, 848–855. [Google Scholar] [CrossRef]
- Coffin, J.M. Retroviridae and their replication. In Virology; Raven Press: New York, NY, USA, 1990; pp. 1347–1500. [Google Scholar]
- Sundquist, W.I.; Kräusslich, H.G. HIV-1 assembly, budding, and maturation. Cold Spring Harb. Perspect. Med. 2012, 2, a006924. [Google Scholar] [CrossRef]
- Pornillos, O.; Ganser-Pornillos, B.K.; Yeager, M. Atomic-level modelling of the HIV capsid. Nat. Cell Biol. 2011, 469, 424–427. [Google Scholar] [CrossRef]
- Temin, H.M. Homology between RNA from Rous Sarcoma Virus and DNA from Rous Sarcoma Virus-Infected Cells. Proc. Natl. Acad. Sci. USA 1964, 52, 323–329. [Google Scholar] [CrossRef] [Green Version]
- Hughes, S.H.; Shank, P.R.; Spector, D.H.; Kung, H.-J.; Bishop, J.; Varmus, H.E.; Vogt, P.K.; Breitman, M.L. Proviruses of avian sarcoma virus are terminally redundant, co-extensive with unintegrated linear DNA and integrated at many sites. Cell 1978, 15, 1397–1410. [Google Scholar] [CrossRef]
- Hughes, S.H.; Mutschler, A.; Bishop, J.M.; Varmus, H.E. A Rous sarcoma virus provirus is flanked by short direct repeats of a cellular DNA sequence present in only one copy prior to integration. Proc. Natl. Acad. Sci. USA 1981, 78, 4299–4303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majors, J.E.; Swanstrom, R.; DeLorbe, W.J.; Payne, G.S.; Hughes, S.H.; Ortíz, S.; Quintrell, N.; Bishop, J.M.; Varmus, H.E. DNA Intermediates in the Replication of Retroviruses Are Structurally (and Perhaps Functionally) Related to Transposable Elements. Cold Spring Harb. Symp. Quant. Biol. 1981, 45, 731–738. [Google Scholar] [CrossRef] [PubMed]
- Fassati, A.; Goff, S.P. Characterization of Intracellular Reverse Transcription Complexes of Human Immunodeficiency Virus Type 1. J. Virol. 2001, 75, 3626–3635. [Google Scholar] [CrossRef] [Green Version]
- Miller, M.D.; Farnet, C.M.; Bushman, F.D. Human immunodeficiency virus type 1 preintegration complexes: Studies of organization and composition. J. Virol. 1997, 71, 5382–5390. [Google Scholar] [CrossRef] [Green Version]
- Burdick, R.C.; Delviks-Frankenberry, K.; Chen, J.; Janaka, S.K.; Sastri, J.; Hu, W.-S.; Pathak, V.K. Dynamics and regulation of nuclear import and nuclear movements of HIV-1 complexes. PLoS Pathog. 2017, 13, e1006570. [Google Scholar] [CrossRef] [PubMed]
- Francis, A.C.; Melikyan, G.B. Single HIV-1 Imaging Reveals Progression of Infection through CA-Dependent Steps of Docking at the Nuclear Pore, Uncoating, and Nuclear Transport. Cell Host Microbe 2018, 23, 536–548.e6. [Google Scholar] [CrossRef] [Green Version]
- Dharan, A.; Bachmann, N.; Talley, S.; Zwikelmaier, V.; Campbell, E.M. Nuclear pore blockade reveals that HIV-1 completes reverse transcription and uncoating in the nucleus. Nat. Microbiol. 2020, 5, 1–8. [Google Scholar] [CrossRef]
- Jang, S.; Cook, N.J.; Pye, V.E.; Bedwell, G.J.; Dudek, A.M.; Singh, P.K.; Cherepanov, P.; Engelman, A.N. Differential role for phosphorylation in alternative polyadenylation function versus nuclear import of SR-like protein CPSF6. Nucleic Acids Res. 2019, 47, 4663–4683. [Google Scholar] [CrossRef] [Green Version]
- De Iaco, A.; Santoni, F.; Vannier, A.; Guipponi, M.; Antonarakis, S.E.; Luban, J. TNPO3 protects HIV-1 replication from CPSF6-mediated capsid stabilization in the host cell cytoplasm. Retrovirology 2013, 10, 20. [Google Scholar] [CrossRef] [Green Version]
- Schaller, T.; Ocwieja, K.E.; Rasaiyaah, J.; Price, A.J.; Brady, T.L.; Roth, S.L.; Hué, S.; Fletcher, A.J.; Lee, K.; KewalRamani, V.N.; et al. HIV-1 Capsid-Cyclophilin Interactions Determine Nuclear Import Pathway, Integration Targeting and Replication Efficiency. PLoS Pathog. 2011, 7, e1002439. [Google Scholar] [CrossRef]
- Lee, K.; Ambrose, Z.; Martin, T.D.; Oztop, I.; Mulky, A.; Julias, J.G.; Vandegraaff, N.; Baumann, J.G.; Wang, R.; Yuen, W.; et al. Flexible Use of Nuclear Import Pathways by HIV-1. Cell Host Microbe 2010, 7, 221–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewinski, M.K.; Yamashita, M.; Emerman, M.; Ciuffi, A.; Marshall, H.; Crawford, G.; Collins, F.; Shinn, P.; Leipzig, J.; Hannenhalli, S.; et al. Retroviral DNA Integration: Viral and Cellular Determinants of Target-Site Selection. PLoS Pathog. 2006, 2, e60. [Google Scholar] [CrossRef] [PubMed]
- Ciuffi, A. The benefits of integration. Clin. Microbiol. Infect. 2016, 22, 324–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paillart, J.-C.; Shehu-Xhilaga, M.; Marquet, R.; Mak, J. Dimerization of retroviral RNA genomes: An inseparable pair. Nat. Rev. Genet. 2004, 2, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Mak, J.; Kleiman, L. Primer tRNAs for reverse transcription. J. Virol. 1997, 71, 8087–8095. [Google Scholar] [CrossRef] [Green Version]
- Frankel, A.D.; Young, J.A.T. HIV-1: Fifteen Proteins and an RNA. Annu. Rev. Biochem. 1998, 67, 1–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Telesnitsky, A.; Goff, S. Reverse Transcriptase and the Generation of Retroviral DNA. In Retroviruses; Cold Spring Harbor Laboratory Press, Cold Spring Harbor: Long Island, NY, USA, 1997; ISBN 0879695714. [Google Scholar]
- Charneau, P.; Alizon, M.; Clavel, F. A second origin of DNA plus-strand synthesis is required for optimal human immunodeficiency virus replication. J. Virol. 1992, 66, 2814–2820. [Google Scholar] [CrossRef] [Green Version]
- Fischer, U.; Huber, J.; Boelens, W.C.; Mattajt, L.W.; Lührmann, R. The HIV-1 Rev Activation Domain is a nuclear export signal that accesses an export pathway used by specific cellular RNAs. Cell 1995, 82, 475–483. [Google Scholar] [CrossRef] [Green Version]
- Gilboa, E.; Mitra, S.W.; Goff, S.; Baltimore, D. A detailed model of reverse transcription and tests of crucial aspects. Cell 1979, 18, 93–100. [Google Scholar] [CrossRef]
- Zufferey, R.; Dull, T.; Mandel, R.J.; Bukovsky, A.; Quiroz, D.; Naldini, L.; Trono, D. Self-Inactivating Lentivirus Vector for Safe and Efficient In Vivo Gene Delivery. J. Virol. 1998, 72, 9873–9880. [Google Scholar] [CrossRef] [Green Version]
- Wyatt, R. The HIV-1 Envelope Glycoproteins: Fusogens, Antigens, and Immunogens. Science 1998, 280, 1884–1888. [Google Scholar] [CrossRef] [PubMed]
- Dalgleish, A.G.; Beverley, P.C.L.; Clapham, P.R.; Crawford, D.H.; Greaves, M.F.; A Weiss, R. The CD4 (T4) antigen is an essential component of the receptor for the AIDS retrovirus. Nat. Cell Biol. 1984, 312, 763–767. [Google Scholar] [CrossRef] [PubMed]
- Klatzmann, D.; Champagne, E.; Chamaret, S.; Gruest, J.; Guetard, D.; Hercend, T.; Gluckman, J.-C.; Montagnier, L. T-lymphocyte T4 molecule behaves as the receptor for human retrovirus LAV. Nat. Cell Biol. 1984, 312, 767–768. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Liu, R.; Ellmeier, W.; Choe, S.; Unutmaz, D.; Burkhart, M.; Marzio, P.D.; Marmon, S.; Sutton, R.E.; Hill, C.M.; et al. Identification of a major co-receptor for primary isolates of HIV-1. Nat. Cell Biol. 1996, 381, 661–666. [Google Scholar] [CrossRef] [PubMed]
- Doranz, B.J.; Rucker, J.; Yi, Y.; Smyth, R.J.; Samson, M.; Peiper, S.C.; Parmentier, M.; Collman, R.G.; Doms, R.W. A Dual-Tropic Primary HIV-1 Isolate That Uses Fusin and the β-Chemokine Receptors CKR-5, CKR-3, and CKR-2b as Fusion Cofactors. Cell 1996, 85, 1149–1158. [Google Scholar] [CrossRef] [Green Version]
- Alkhatib, G.; Combadiere, C.; Broder, C.C.; Feng, Y.; Kennedy, P.E.; Murphy, P.M.; Berger, E.A. CC CKR5: A RANTES, MIP-1, MIP-1 Receptor as a Fusion Cofactor for Macrophage-Tropic HIV-1. Science 1996, 272, 1955–1958. [Google Scholar] [CrossRef]
- Dragic, T.; Litwin, V.; Allaway, G.P.; Martin, S.R.; Huang, Y.; Nagashima, K.A.; Cayanan, C.S.; Maddon, P.J.; Koup, R.A.; Moore, J.P.; et al. HIV-1 entry into CD4+ cells is mediated by the chemokine receptor CC-CKR-5. Nat. Cell Biol. 1996, 381, 667–673. [Google Scholar] [CrossRef]
- Oberlin, E.; Amara, A.; Bachelerie, F.; Bessia, C.; Virelizier, J.-L.; Arenzana-Seisdedos, F.; Schwartz, O.; Heard, J.-M.; Clark-Lewis, I.; Legler, D.F.; et al. The CXC chemokine SDF-1 is the ligand for LESTR/fusin and prevents infection by T-cell-line-adapted HIV-1. Nat. Cell Biol. 1996, 382, 833–835. [Google Scholar] [CrossRef] [Green Version]
- Abrahamyan, L.G.; Markosyan, R.M.; Moore, J.P.; Cohen, F.S.; Melikyan, G.B. Human Immunodeficiency Virus Type 1 Env with an Intersubunit Disulfide Bond Engages Coreceptors but Requires Bond Reduction after Engagement to Induce Fusion. J. Virol. 2003, 77, 5829–5836. [Google Scholar] [CrossRef] [Green Version]
- Koshiba, T.; Chan, D.C. The Prefusogenic Intermediate of HIV-1 gp41 Contains Exposed C-peptide Regions. J. Biol. Chem. 2002, 278, 7573–7579. [Google Scholar] [CrossRef] [Green Version]
- Si, Z.; Madani, N.; Cox, J.M.; Chruma, J.J.; Klein, J.C.; Schön, A.; Phan, N.; Wang, L.; Biorn, A.C.; Cocklin, S.; et al. Small-molecule inhibitors of HIV-1 entry block receptor-induced conformational changes in the viral envelope glycoproteins. Proc. Natl. Acad. Sci. USA 2004, 101, 5036–5041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, D.C.; Fass, D.; Berger, J.M.; Kim, P.S. Core Structure of gp41 from the HIV Envelope Glycoprotein. Cell 1997, 89, 263–273. [Google Scholar] [CrossRef] [Green Version]
- Lu, M.; Blacklow, S.C.; Kim, P.S. A trimeric structural domain of the HIV-1 transmembrane glycoprotein. Nat. Struct. Mol. Biol. 1995, 2, 1075–1082. [Google Scholar] [CrossRef] [PubMed]
- Weissenhorn, W.; Dessen, A.; Harrison, S.C.; Skehel, J.J.; Wiley, D.C. Atomic structure of the ectodomain from HIV-1 gp41. Nat. Cell Biol. 1997, 387, 426–430. [Google Scholar] [CrossRef]
- Sengupta, P.; Seo, A.Y.; Pasolli, H.A.; Song, Y.E.; Johnson, M.C.; Lippincott-Schwartz, J. A lipid-based partitioning mechanism for selective incorporation of proteins into membranes of HIV particles. Nat. Cell Biol. 2019, 21, 452–461. [Google Scholar] [CrossRef]
- Nguyen, D.H.; Hildreth, J.E.K. Evidence for Budding of Human Immunodeficiency Virus Type 1 Selectively from Glycolipid-Enriched Membrane Lipid Rafts. J. Virol. 2000, 74, 3264–3272. [Google Scholar] [CrossRef] [Green Version]
- Varior-Krishnan, G.; Trescol-Biémont, M.C.; Naniche, D.; Rabourdin-Combe, C.; Gerlier, D. Glycosyl-phosphatidylinositol-anchored and transmembrane forms of CD46 display similar measles virus receptor properties: Virus binding, fusion, and replication; down-regulation by hemagglutinin; and virus uptake and endocytosis for antigen presentation by major histocompatibility complex class II molecules. J. Virol. 1994, 68, 7891–7899. [Google Scholar] [CrossRef] [Green Version]
- Komura, N.; Suzuki, K.G.N.; Ando, H.; Konishi, M.; Koikeda, M.; Imamura, A.; Chadda, R.; Fujiwara, T.K.; Tsuboi, H.; Sheng, R.; et al. Raft-based interactions of gangliosides with a GPI-anchored receptor. Nat. Chem. Biol. 2016, 12, 402–410. [Google Scholar] [CrossRef]
- Tsui-Pierchala, B.A.; Encinas, M.; Milbrandt, J.; Johnson, E.M. Lipid rafts in neuronal signaling and function. Trends Neurosci. 2002, 25, 412–417. [Google Scholar] [CrossRef]
- Miyagawa-Yamaguchi, A.; Kotani, N.; Honke, K. Expressed Glycosylphosphatidylinositol-Anchored Horseradish Peroxidase Identifies Co-Clustering Molecules in Individual Lipid Raft Domains. PLoS ONE 2014, 9, e93054. [Google Scholar] [CrossRef]
- Marschang, P.; Sodroski, J.; Würzner, R.; Dierich, M.P. Decay-accelerating factor (CD55) protects human immunodeficiency virus type 1 from inactivation by human complement. Eur. J. Immunol. 1995, 25, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Bohanakashtan, O. Cell signals transduced by complement. Mol. Immunol. 2004, 41, 583–597. [Google Scholar] [CrossRef] [PubMed]
- Saifuddin, M.; Holguin, M.H.; Parker, C.J.; Hedayati, T.; Atkinson, J.P.; Spear, G.T. Human immunodeficiency virus type 1 incorporates both glycosyl phosphatidylinositol-anchored CD55 and CD59 and integral membrane CD46 at levels that protect from complement-mediated destruction. J. Gen. Virol. 1997, 78, 1907–1911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amet, T.; Lan, J.; Shepherd, N.; Yang, K.; Byrd, D.; Xing, Y.; Yu, Q. Glycosylphosphatidylinositol Anchor Deficiency Attenuates the Production of Infectious HIV-1 and Renders Virions Sensitive to Complement Attack. AIDS Res. Hum. Retrovir. 2016, 32, 1100–1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wyma, D.J.; Kotov, A.; Aiken, C. Evidence for a Stable Interaction of gp41 with Pr55Gag in Immature Human Immunodeficiency Virus Type 1 Particles. J. Virol. 2000, 74, 9381–9387. [Google Scholar] [CrossRef] [Green Version]
- Zacharias, D.A.; Violin, J.D.; Newton, A.C.; Tsien, R.Y. Partitioning of Lipid-Modified Monomeric GFPs into Membrane Microdomains of Live Cells. Science 2002, 296, 913–916. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Schey, K.L. Proteomic Analysis of Lipid Raft-Like Detergent-Resistant Membranes of Lens Fiber Cells. Investig. Opthalmol. Vis. Sci. 2015, 56, 8349–8360. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, M.F.; Schlesinger, M.J. Fatty acid binding to vesicular stomatitis virus glycoprotein: A new type of post-translational modification of the viral glycoprotein. Cell 1979, 17, 813–819. [Google Scholar] [CrossRef]
- Schmidt, M.F. Acylation of virol. spike glycoproteins: A feature of enveloped RNA viruses. Virology 1982, 116, 327–338. [Google Scholar] [CrossRef]
- Schmidt, M.F.; Bracha, M.; Schlesinger, M.J. Evidence for covalent attachment of fatty acids to Sindbis virus glycoproteins. Proc. Natl. Acad. Sci. USA 1979, 76, 1687–1691. [Google Scholar] [CrossRef] [Green Version]
- Finkelshtein, D.; Werman, A.; Novick, D.; Barak, S.; Rubinstein, M. LDL receptor and its family members serve as the cellular receptors for vesicular stomatitis virus. Proc. Natl. Acad. Sci. USA 2013, 110, 7306–7311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burns, J.C.; Friedmann, T.; Driever, W.; Burrascano, M.; Yee, J.K. Vesicular stomatitis virus G glycoprotein pseudotyped retroviral vectors: Concentration to very high titer and efficient gene transfer into mammalian and nonmammalian cells. Proc. Natl. Acad. Sci. USA 1993, 90, 8033–8037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lentz, T.L.; Burrage, T.G.; Smith, A.L.; Crick, J.; Tignor, G.H. Is the acetylcholine receptor a rabies virus receptor? Science 1982, 215, 182–184. [Google Scholar] [CrossRef] [PubMed]
- Thoulouze, M.-I.; Lafage, M.; Schachner, M.; Hartmann, U.; Cremer, H.; Lafon, M. The Neural Cell Adhesion Molecule Is a Receptor for Rabies Virus. J. Virol. 1998, 72, 7181–7190. [Google Scholar] [CrossRef] [Green Version]
- Tuffereau, C.; Bénéjean, J.; Blondel, D.; Kieffer, B.; Flamand, A. Low-affinity nerve-growth factor receptor (P75NTR) can serve as a receptor for rabies virus. EMBO J. 1998, 17, 7250–7259. [Google Scholar] [CrossRef]
- Wang, J.; Wang, Z.; Liu, R.; Shuai, L.; Wang, X.; Luo, J.; Wang, C.; Chen, W.; Wang, X.; Ge, J.; et al. Metabotropic glutamate receptor subtype 2 is a cellular receptor for rabies virus. PLoS Pathog. 2018, 14, e1007189. [Google Scholar] [CrossRef]
- Bartz, R.; Firsching, R.; Ter Meulen, V.; Schneider-Schaulies, J.; Rima, B. Differential receptor usage by measles virus strains. J. Gen. Virol. 1998, 79, 1015–1025. [Google Scholar] [CrossRef] [Green Version]
- Hsu, E.C.; Sarangi, F.; Iorio, C.; Sidhu, M.S.; Udem, S.A.; Dillehay, D.L.; Xu, W.; Rota, P.A.; Bellini, W.J.; Richardson, C.D. A Single Amino Acid Change in the Hemagglutinin Protein of Measles Virus Determines Its Ability to Bind CD46 and Reveals Another Receptor on Marmoset B Cells. J. Virol. 1998, 72, 2905–2916. [Google Scholar] [CrossRef] [Green Version]
- Buckland, R.; Wild, T.F. Is CD46 the cellular receptor for measles virus? Virus Res. 1997, 48, 1–9. [Google Scholar] [CrossRef]
- Hsu, E.C.; Iorioab, C.; Sarangiab, F.; AyeKhineab, A.; Richardson, C.D. CDw150(SLAM) Is a Receptor for a Lymphotropic Strain of Measles Virus and May Account for the Immunosuppressive Properties of This Virus. Virology 2001, 279, 9–21. [Google Scholar] [CrossRef] [Green Version]
- Murabayashi, N.; Kurita-Taniguchi, M.; Ayata, M.; Matsumoto, M.; Ogura, H.; Seya, T. Susceptibility of human dendritic cells (DCs) to measles virus (MV) depends on their activation stages in conjunction with the level of CDw150: Role of Toll stimulators in DC maturation and MV amplification. Microbes Infect. 2002, 4, 785–794. [Google Scholar] [CrossRef]
- Tatsuo, H.; Ono, N.; Tanaka, K.; Yanagi, Y. SLAM (CDw150) is a cellular receptor for measles virus. Nat. Cell Biol. 2000, 406, 893–897. [Google Scholar] [CrossRef] [PubMed]
- Verhoeyen, E.; Cosset, F.-L. Surface-engineering of lentiviral vectors. J. Gene Med. 2004, 6, S83–S94. [Google Scholar] [CrossRef] [PubMed]
- Lévy, C.; Amirache, F.; Girard-Gagnepain, A.; Frecha, C.; Roman-Rodríguez, F.J.; Bernadin, O.; Costa, C.; Nègre, D.; Gutierrez-Guerrero, A.; Vranckx, L.S.; et al. Measles virus envelope pseudotyped lentiviral vectors transduce quiescent human HSCs at an efficiency without precedent. Blood Adv. 2017, 1, 2088–2104. [Google Scholar] [CrossRef] [PubMed]
- Witting, S.R.; Vallanda, P.; Gamble, A.L. Characterization of a third generation lentiviral vector pseudotyped with Nipah virus envelope proteins for endothelial cell transduction. Gene Ther. 2013, 20, 997–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palomares, K.; Vigant, F.; Van Handel, B.; Pernet, O.; Chikere, K.; Hong, P.; Sherman, S.P.; Patterson, M.; An, D.S.; Lowry, W.E.; et al. Nipah Virus Envelope-Pseudotyped Lentiviruses Efficiently Target ephrinB2-Positive Stem Cell Populations In Vitro and Bypass the Liver Sink When Administered In Vivo. J. Virol. 2012, 87, 2094–2108. [Google Scholar] [CrossRef] [Green Version]
- Wintachai, P.; Wikan, N.; Kuadkitkan, A.; Jaimipuk, T.; Ubol, S.; Pulmanausahakul, R.; Auewarakul, P.; Kasinrerk, W.; Weng, W.-Y.; Panyasrivanit, M.; et al. Identification of prohibitin as a Chikungunya virus receptor protein. J. Med. Virol. 2012, 84, 1757–1770. [Google Scholar] [CrossRef]
- Zhang, R.; Kim, A.S.; Fox, J.M.; Nair, S.; Basore, K.; Klimstra, W.B.; Rimkunas, R.; Fong, R.H.; Lin, H.; Poddar, S.; et al. Mxra8 is a receptor for multiple arthritogenic alphaviruses. Nat. Cell Biol. 2018, 557, 570–574. [Google Scholar] [CrossRef]
- Matusali, G.; Colavita, F.; Bordi, L.; Lalle, E.; Ippolito, G.; Capobianchi, M.R.; Castilletti, C. Tropism of the Chikungunya Virus. Viruses 2019, 11, 175. [Google Scholar] [CrossRef] [Green Version]
- Salvador, B.; Zhou, Y.; Michault, A.; Muench, M.O.; Simmons, G. Characterization of Chikungunya pseudotyped viruses: Identification of refractory cell lines and demonstration of cellular tropism differences mediated by mutations in E1 glycoprotein. Virology 2009, 393, 33–41. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.S.; Kuhn, R.J.; Strauss, E.G.; Ou, S.; Strauss, J.H. High-affinity laminin receptor is a receptor for Sindbis virus in mammalian cells. J. Virol. 1992, 66, 4992–5001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rose, P.P.; Hanna, S.L.; Spiridigliozzi, A.; Wannissorn, N.; Beiting, D.P.; Ross, S.R.; Hardy, R.W.; Bambina, S.A.; Heise, M.T.; Cherry, S. Natural Resistance-Associated Macrophage Protein Is a Cellular Receptor for Sindbis Virus in Both Insect and Mammalian Hosts. Cell Host Microbe 2011, 10, 97–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elegheert, J.; Behiels, E.; Bishop, B.; Scott, S.; Woolley, R.E.; Griffiths, S.C.; Byrne, E.F.; Chang, V.T.; Stuart, D.I.; Jones, E.Y.; et al. Lentiviral transduction of mammalian cells for fast, scalable and high-level production of soluble and membrane proteins. Nat. Protoc. 2018, 13, 2991–3017. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, K.; Mirnikjoo, B.; Schroit, A.J. Regulated Externalization of Phosphatidylserine at the Cell Surface. J. Biol. Chem. 2007, 282, 18357–18364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bevers, E.M.; Williamson, P.L. Getting to the Outer Leaflet: Physiology of Phosphatidylserine Exposure at the Plasma Membrane. Physiol. Rev. 2016, 96, 605–645. [Google Scholar] [CrossRef]
- Coil, D.A.; Miller, A.D. Phosphatidylserine Is Not the Cell Surface Receptor for Vesicular Stomatitis Virus. J. Virol. 2004, 78, 10920–10926. [Google Scholar] [CrossRef] [Green Version]
- Brown, M.S.; Goldstein, J.L. A receptor-mediated pathway for cholesterol homeostasis. Science 1986, 232, 34–47. [Google Scholar] [CrossRef] [Green Version]
- Go, G.-W.; Mani, A. Low-density lipoprotein receptor (LDLR) family orchestrates cholesterol homeostasis. Yale J. Biol. Med. 2012, 85, 19–28. [Google Scholar]
- Sun, X.; Roth, S.L.; Bialecki, M.A.; Whittaker, G.R. Internalization and fusion mechanism of vesicular stomatitis virus and related rhabdoviruses. Futur. Virol. 2010, 5, 85–96. [Google Scholar] [CrossRef] [Green Version]
- Clague, M.J.; Schoch, C.; Zech, L.; Blumenthal, R. Gating kinetics of pH-activated membrane fusion of vesicular stomatitis virus with cells: Stopped-flow measurements by dequenching of octadecylrhodamine fluorescence. Biochemistry 1990, 29, 1303–1308. [Google Scholar] [CrossRef]
- Paternostre, M.-T.; Lowy, R.J.; Blumenthal, R. pH-dependent fusion of reconstituted vesicular stomatitis virus envelopes with vero cells Measurement by dequenching of fluorescence. FEBS Lett. 1989, 243, 251–258. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Yau, V.K.; Briggs, B.J.; Whittaker, G.R. Role of clathrin-mediated endocytosis during vesicular stomatitis virus entry into host cells. Virology 2005, 338, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Albertini, A.A.; Baquero, E.; Ferlin, A.; Gaudin, Y. Molecular and Cellular Aspects of Rhabdovirus Entry. Viruses 2012, 4, 117–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, I.S.; Jenni, S.; Stanifer, M.L.; Roth, E.; Whelan, S.P.J.; Van Oijen, A.M.; Harrison, S.C. Mechanism of membrane fusion induced by vesicular stomatitis virus G protein. Proc. Natl. Acad. Sci. USA 2017, 114, E28–E36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantore, A.; Nair, N.; Della Valle, P.; Di Matteo, M.; Màtrai, J.; Sanvito, F.; Brombin, C.; Di Serio, C.; D’Angelo, A.; Chuah, M.K.L.; et al. Hyperfunctional coagulation factor IX improves the efficacy of gene therapy in hemophilic mice. Blood 2012, 120, 4517–4520. [Google Scholar] [CrossRef] [Green Version]
- Cantore, A.A.; Ranzani, M.M.; Bartholomae, C.C.; Volpin, M.M.; Valle, P.D.P.; Sanvito, F.F.; Sergi, L.S.L.; Gallina, P.P.; Benedicenti, F.F.; Bellinger, D.D.; et al. Liver-directed lentiviral gene therapy in a dog model of hemophilia B. Sci. Transl. Med. 2015, 7, 277ra28. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, H.; Kimura, T.; Haga, K.; Kasahara, N.; Anton, P.A.; McGowan, I. Effective In Vivo and Ex Vivo gene transfer to intestinal mucosa by VSV-G-pseudotyped lentiviral vectors. BMC Gastroenterol. 2010, 10, 44. [Google Scholar] [CrossRef] [Green Version]
- Kawabata, K.; Migita, M.; Mochizuki, H.; Miyake, K.; Igarashi, T.; Fukunaga, Y.; Shimada, T. Ex vivo cell-mediated gene therapy for metachromatic leukodystrophy using neurospheres. Brain Res. 2006, 1094, 13–23. [Google Scholar] [CrossRef]
- Suzuki, A.; Obi, K.; Urabe, T.; Hayakawa, H.; Yamada, M.; Kaneko, S.; Onodera, M.; Mizuno, Y.; Mochizuki, H. Feasibility of Ex Vivo gene therapy for neurological disorders using the new retroviral vector GCDNsap packaged in the vesicular stomatitis virus G protein. J. Neurochem. 2002, 82, 953–960. [Google Scholar] [CrossRef] [Green Version]
- Mazarakis, N.D.; Azzouz, M.; Rohll, J.B.; Ellard, F.M.; Wilkes, F.J.; Olsen, A.L.; Carter, E.E.; Barber, R.D.; Baban, D.F.; Kingsman, S.M.; et al. Rabies virus glycoprotein pseudotyping of lentiviral vectors enables retrograde axonal transport and access to the nervous system after peripheral delivery. Hum. Mol. Genet. 2001, 10, 2109–2121. [Google Scholar] [CrossRef]
- Piccinotti, S.; Whelan, S.P. Rabies Internalizes into Primary Peripheral Neurons via Clathrin Coated Pits and Requires Fusion at the Cell Body. PLoS Pathog. 2016, 12, e1005753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burrage, T.G.; Tignor, G.H.; Smith, A.L. Rabies virus binding at neuromuscular junctions. Virus Res. 1985, 2, 273–289. [Google Scholar] [CrossRef]
- Tuffereau, C.; Schmidt, K.; Langevin, C.; Lafay, F.; DeChant, G.; Koltzenburg, M. The Rabies Virus Glycoprotein Receptor p75NTR Is Not Essential for Rabies Virus Infection. J. Virol. 2007, 81, 13622–13630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kyrkanides, S.; Miller, J.H.; Brouxhon, S.M.; Olschowka, J.A.; Federoff, H.J. β-hexosaminidase lentiviral vectors: Transfer into the CNS via systemic administration. Mol. Brain Res. 2005, 133, 286–298. [Google Scholar] [CrossRef]
- Chang, A.; Dutch, R.E. Paramyxovirus Fusion and Entry: Multiple Paths to a Common End. Viruses 2012, 4, 613–636. [Google Scholar] [CrossRef]
- Plattet, P.; Alves, L.; Herren, M.; Aguilar, H.C. Measles Virus Fusion Protein: Structure, Function and Inhibition. Viruses 2016, 8, 112. [Google Scholar] [CrossRef] [Green Version]
- Yanagi, Y.; Takeda, M.; Ohno, S. Measles virus: Cellular receptors, tropism and pathogenesis. J. Gen. Virol. 2006, 87, 2767–2779. [Google Scholar] [CrossRef]
- Dörig, R.E.; Marcil, A.; Chopra, A.; Richardson, C.D. The human CD46 molecule is a receptor for measles virus (Edmonston strain). Cell 1993, 75, 295–305. [Google Scholar] [CrossRef]
- Naniche, D.; Varior-Krishnan, G.; Cervoni, F.; Wild, T.F.; Rossi, B.; Rabourdin-Combe, C.; Gerlier, D. Human membrane cofactor protein (CD46) acts as a cellular receptor for measles virus. J. Virol. 1993, 67, 6025–6032. [Google Scholar] [CrossRef] [Green Version]
- Liszewski, M.K.; Post, T.W.; Atkinson, J.P. Membrane cofactor protein (MCP or CD46): Newest member of the regulators of complement activation gene cluster. Annu. Rev. Immunol. 1991, 9, 431–455. [Google Scholar] [CrossRef]
- Noyce, R.S.; Bondre, D.G.; Ha, M.N.; Lin, L.-T.; Sisson, G.; Tsao, M.-S.; Richardson, C.D. Tumor Cell Marker PVRL4 (Nectin 4) Is an Epithelial Cell Receptor for Measles Virus. PLoS Pathog. 2011, 7, e1002240. [Google Scholar] [CrossRef]
- Mühlebach, M.D.; Mateo, M.; Sinn, P.L.; Prüfer, S.; Uhlig, K.M.; Leonard, V.H.J.; Navaratnarajah, C.K.; Frenzke, M.; Wong, X.X.; Sawatsky, B.; et al. Adherens junction protein nectin-4 is the epithelial receptor for measles virus. Nat. Cell Biol. 2011, 480, 530–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeRycke, M.S.; Pambuccian, S.E.; Gilks, C.B.; Kalloger, S.E.; Ghidouche, A.; Lopez, M.; Bliss, R.L.; Geller, M.A.; Argenta, P.A.; Harrington, K.M.; et al. Nectin 4 Overexpression in Ovarian Cancer Tissues and Serum. Am. J. Clin. Pathol. 2010, 134, 835–845. [Google Scholar] [CrossRef] [PubMed]
- Takano, A.; Ishikawa, N.; Nishino, R.; Masuda, K.; Yasui, W.; Inai, K.; Nishimura, H.; Ito, H.; Nakayama, H.; Miyagi, Y.; et al. Identification of Nectin-4 Oncoprotein as a Diagnostic and Therapeutic Target for Lung Cancer. Cancer Res. 2009, 69, 6694–6703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vongpunsawad, S.; Oezgun, N.; Braun, W.; Cattaneo, R. Selectively Receptor-Blind Measles Viruses: Identification of Residues Necessary for SLAM or CD46-Induced Fusion and Their Localization on a New Hemagglutinin Structural Model. J. Virol. 2004, 78, 302–313. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, T.; Peng, K.-W.; Harvey, M.; Greiner, S.; Lorimer, I.A.J.; James, C.D.; Russell, S.J. Rescue and propagation of fully retargeted oncolytic measles viruses. Nat. Biotechnol. 2005, 23, 209–214. [Google Scholar] [CrossRef]
- Münch, R.C.; Mühlebach, M.D.; Schaser, T.; Kneissl, S.; Jost, C.; Plückthun, A.; Cichutek, K.; Buchholz, C.J. DARPins: An Efficient Targeting Domain for Lentiviral Vectors. Mol. Ther. 2011, 19, 686–693. [Google Scholar] [CrossRef]
- De Swart, R.L.; Yüksel, S.; Osterhaus, A.D.M.E. Relative Contributions of Measles Virus Hemagglutinin- and Fusion Protein-Specific Serum Antibodies to Virus Neutralization. J. Virol. 2005, 79, 11547–11551. [Google Scholar] [CrossRef] [Green Version]
- Kneissl, S.; Abel, T.; Rasbach, A.; Brynza, J.; Schneider-Schaulies, J.; Buchholz, C.J. Measles Virus Glycoprotein-Based Lentiviral Targeting Vectors That Avoid Neutralizing Antibodies. PLoS ONE 2012, 7, e46667. [Google Scholar] [CrossRef] [Green Version]
- Khetawat, D.; Broder, C.C. A Functional Henipavirus Envelope Glycoprotein Pseudotyped Lentivirus Assay System. Virol. J. 2010, 7, 312. [Google Scholar] [CrossRef] [Green Version]
- Bruhn, J.F.; Barnett, K.C.; Bibby, J.; Thomas, J.M.H.; Keegan, R.M.; Rigden, D.J.; Bornholdt, Z.A.; Saphire, E.O. Crystal Structure of the Nipah Virus Phosphoprotein Tetramerization Domain. J. Virol. 2013, 88, 758–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bender, R.R.; Muth, A.; Schneider, I.C.; Friedel, T.; Hartmann, J.; Plückthun, A.; Maisner, A.; Buchholz, C.J. Receptor-Targeted Nipah Virus Glycoproteins Improve Cell-Type Selective Gene Delivery and Reveal a Preference for Membrane-Proximal Cell Attachment. PLoS Pathog. 2016, 12, e1005641. [Google Scholar] [CrossRef] [PubMed]
- Enkirch, T.; Kneissl, S.; Hoyler, B.; Ungerechts, G.; Stremmel, W.; Buchholz, C.J.; Springfeld, C. Targeted lentiviral vectors pseudotyped with the Tupaia paramyxovirus glycoproteins. Gene Ther. 2012, 20, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Funke, S.; Maisner, A.; Mühlebach, M.D.; Koehl, U.; Grez, M.; Cattaneo, R.; Cichutek, K.; Buchholz, C.J. Targeted Cell Entry of Lentiviral Vectors. Mol. Ther. 2008, 16, 1427–1436. [Google Scholar] [CrossRef] [PubMed]
- Bonaparte, M.I.; Dimitrov, D.S.; Bossart, K.N.; Crameri, G.; Mungall, B.A.; Bishop, K.A.; Choudhry, V.; Wang, L.-F.; Eaton, B.T.; Broder, C.C. From the Cover: Ephrin-B2 ligand is a functional receptor for Hendra virus and Nipah virus. Proc. Natl. Acad. Sci. USA 2005, 102, 10652–10657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Negrete, O.A.; Wolf, M.C.; Aguilar, H.C.; Enterlein, S.; Wang, W.; Mühlberger, E.; Su, S.V.; Bertolotti-Ciarlet, A.; Flick, R.; Lee, B. Two Key Residues in EphrinB3 Are Critical for Its Use as an Alternative Receptor for Nipah Virus. PLoS Pathog. 2006, 2, 0078–0086. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.; Garcia-Cardena, G.; Hayashid, S.-I.; Geretya, S.; Asaharad, T.; Stavrakisb, G.; Isnerd, J.; Folkman, J.; Gimbrone, M.A.; Anderson, D.J. Expression of EphrinB2 Identifies a Stable Genetic Difference Between Arterial and Venous Vascular Smooth Muscle as Well as Endothelial Cells, and Marks Subsets of Microvessels at Sites of Adult Neovascularization. Dev. Biol. 2001, 230, 139–150. [Google Scholar] [CrossRef] [Green Version]
- Foo, S.S.; Turner, C.J.; Adams, S.; Compagni, A.; Aubyn, D.; Kogata, N.; Lindblom, P.; Shani, M.; Zicha, D.; Adams, R.H. Ephrin-B2 Controls Cell Motility and Adhesion during Blood-Vessel-Wall Assembly. Cell 2006, 124, 161–173. [Google Scholar] [CrossRef] [Green Version]
- Martowicz, A.; Seeber, A.; Untergasser, G. The role of EpCAM in physiology and pathology of the epithelium. Histol. Histopathol. 2015, 31, 349–355. [Google Scholar]
- Wengler, G.; Koschinski, A.; Wengler, G.; Dreyer, F. Entry of alphaviruses at the plasma membrane converts the viral surface proteins into an ion-permeable pore that can be detected by electrophysiological analyses of whole-cell membrane currents. J. Gen. Virol. 2003, 84, 173–181. [Google Scholar] [CrossRef]
- Lee, R.C.H.; Hapuarachchi, H.C.; Chen, K.C.; Hussain, K.M.; Chen, H.; Low, S.L.; Ng, L.C.; Lin, R.; Ng, M.M.-L.; Chu, J.J.H. Mosquito Cellular Factors and Functions in Mediating the Infectious entry of Chikungunya Virus. PLoS Negl. Trop. Dis. 2013, 7, e2050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Duijl-Richter, M.K.S.; Hoornweg, T.E.; Rodenhuis-Zybert, I.A.; Smit, J.M. Early Events in Chikungunya Virus Infection—From Virus CellBinding to Membrane Fusion. Viruses 2015, 7, 3647–3674. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.J.; Cheng, R.H.; Olson, N.H.; Peterson, P.; Chase, E.; Kuhn, R.J.; Baker, T.S. Putative receptor binding sites on alphaviruses as visualized by cryoelectron microscopy. Proc. Natl. Acad. Sci. USA 1995, 92, 10648–10652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uchime, O.; Fields, W.; Kielian, M. The Role of E3 in pH Protection during Alphavirus Assembly and Exit. J. Virol. 2013, 87, 10255–10262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carleton, M.; Lee, H.; Mulvey, M.; Brown, D.T. Role of glycoprotein PE2 in formation and maturation of the Sindbis virus spike. J. Virol. 1997, 71, 1558–1566. [Google Scholar] [CrossRef] [Green Version]
- Snyder, A.J.; Mukhopadhyay, S. The Alphavirus E3 Glycoprotein Functions in a Clade-Specific Manner. J. Virol. 2012, 86, 13609–13620. [Google Scholar] [CrossRef] [Green Version]
- Sahoo, B.; Chowdary, T.K. Conformational changes in Chikungunya virus E2 protein upon heparan sulfate receptor binding explain mechanism of E2–E1 dissociation during viral entry. Biosci. Rep. 2019, 39, 39. [Google Scholar] [CrossRef] [Green Version]
- Hua, R.L.C.; Hussain, K.M.; Chu, J.J.H.; Lee, C.H.R. Macropinocytosis dependent entry of Chikungunya virus into human muscle cells. PLoS Negl. Trop. Dis. 2019, 13, e0007610. [Google Scholar] [CrossRef]
- Cerny, T.; Schwarz, M.; Lemant, J.; Gérardin, P.; Keller, E. The Range of Neurological Complications in Chikungunya Fever. Neurocrit. Care 2017, 27, 447–457. [Google Scholar] [CrossRef]
- Thiberville, S.-D.; Moyen, N.; Dupuis-Maguiraga, L.; Nougairede, A.; Gould, E.A.; Roques, P.; De Lamballerie, X. Chikungunya fever: Epidemiology, clinical syndrome, pathogenesis and therapy. Antivir. Res. 2013, 99, 345–370. [Google Scholar] [CrossRef]
- Martinez-Pulgarin, D.F.; Chowdhury, F.R.; Villamil-Gómez, W.E.; Rodríguez-Morales, A.J.; Blohm, G.M.; Paniz-Mondolfi, A.E. Ophthalmologic aspects of chikungunya infection. Travel Med. Infect. Dis. 2016, 14, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Eleftheriadou, I.; Dieringer, M.; Poh, X.Y.; Sanchez-Garrido, J.; Gao, Y.; Sgourou, A.; Simmons, L.E.; Mazarakis, N.D. Selective transduction of astrocytic and neuronal CNS subpopulations by lentiviral vectors pseudotyped with Chikungunya virus envelope. Biomaterials 2017, 123, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Bavelloni, A.; Piazzi, M.; Raffini, M.; Faenza, I.; Blalock, W. Prohibitin 2: At a communications crossroads. IUBMB Life 2015, 67, 239–254. [Google Scholar] [CrossRef] [PubMed]
- Bentayeb, H.; Aitamer, M.; Petit, B.; Dubanet, L.; Elderwish, S.; Désaubry, L.; De Gramont, A.; Raymond, E.; Olivrie, A.; Abraham, J.; et al. Prohibitin (PHB) expression is associated with aggressiveness in DLBCL and flavagline-mediated inhibition of cytoplasmic PHB functions induces anti-tumor effects. J. Exp. Clin. Cancer Res. 2019, 38, 450. [Google Scholar] [CrossRef] [PubMed]
- Powers, A.M.; Brault, A.C.; Shirako, Y.; Strauss, E.G.; Kang, W.; Strauss, J.H.; Weaver, S.C. Evolutionary Relationships and Systematics of the Alphaviruses. J. Virol. 2001, 75, 10118–10131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khalfaoui, T.; Groulx, J.-F.; Sabra, G.; Guezguez, A.; Basora, N.; Vermette, P.; Beaulieu, J.-F. Laminin Receptor 37/67LR Regulates Adhesion and Proliferation of Normal Human Intestinal Epithelial Cells. PLoS ONE 2013, 8, e74337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montuori, N.; Pesapane, A.; Giudice, V.; Serio, B.; Rossi, F.W.; De Paulis, A.; Selleri, C. 67 kDa laminin receptor (67LR) in normal and neoplastic hematopoietic cells: Is its targeting a feasible approach? Transl. Med. 2016, 15, 8–14. [Google Scholar]
- Berno, V. The 67 kDa laminin receptor increases tumor aggressiveness by remodeling laminin-1. Endocr. Relat. Cancer 2005, 12, 393–406. [Google Scholar] [CrossRef]
- Gruenheid, S.; Canonne-Hergaux, F.; Gauthier, S.; Hackam, D.J.; Grinstein, S.; Gros, P. The Iron Transport Protein NRAMP2 Is an Integral Membrane Glycoprotein That Colocalizes with Transferrin in Recycling Endosomes. J. Exp. Med. 1999, 189, 831–841. [Google Scholar] [CrossRef]
- Gruenheid, S.; Cellier, M.; Vidal, S.; Gros, P. Identification and characterization of a second mouse Nramp gene. Genomics 1995, 25, 514–525. [Google Scholar] [CrossRef]
- Gunshin, H.; MacKenzie, B.; Berger, U.V.; Gunshin, Y.; Romero, M.F.; Boron, W.F.; Nussberger, S.; Gollan, J.L.; Hediger, M.A. Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nat. Cell Biol. 1997, 388, 482–488. [Google Scholar] [CrossRef] [PubMed]
- Paredes, A.M.; Ferreira, D.; Horton, M.; Saad, A.; Tsuruta, H.; Johnston, R.; Klimstra, W.; Ryman, K.; Hernandez, R.; Chiu, W.; et al. Conformational changes in Sindbis virions resulting from exposure to low pH and interactions with cells suggest that cell penetration may occur at the cell surface in the absence of membrane fusion. Virolology 2004, 324, 373–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vancini, R.; Wang, G.; Ferreira, D.; Hernandez, R.; Brown, D.T. Alphavirus Genome Delivery Occurs Directly at the Plasma Membrane in a Time- and Temperature-Dependent Process. J. Virol. 2013, 87, 4352–4359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohno, K.; Sawai, K.; Lijima, Y.; Levin, B.; Meruelo, D. Cell-specific targeting of Sindbis virus vectors displaying IgG-binding domains of protein A. Nat. Biotechnol. 1997, 15, 763–767. [Google Scholar] [CrossRef] [PubMed]
- Morizono, K.; Bristol, G.; Xie, Y.-M.; Kung, S.K.-P.; Chen, I.S.Y. Antibody-Directed Targeting of Retroviral Vectors via Cell Surface Antigens. J. Virol. 2001, 75, 8016–8020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morizono, K.; Xie, Y.; Ringpis, G.-E.; Johnson, M.; Nassanian, H.; Lee, B.; Wu, L.; Chen, I.S.Y. Lentiviral vector retargeting to P-glycoprotein on metastatic melanoma through intravenous injection. Nat. Med. 2005, 11, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Morizono, K.; Ku, A.; Xie, Y.; Harui, A.; Kung, S.K.P.; Roth, M.D.; Lee, B.; Chen, I.S.Y. Redirecting Lentiviral Vectors Pseudotyped with Sindbis Virus-Derived Envelope Proteins to DC-SIGN by Modification of N-Linked Glycans of Envelope Proteins. J. Virol. 2010, 84, 6923–6934. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Organization of the human immunodeficiency virus type 1 (HIV-1) genomic RNA. U3, unique sequence 3’; R, repeated sequence; U5, unique sequence 5’; Ψ, indicates the packaging and dimerization sequences; RRE, Rev responsive element. The PPT sequences as well as the primer binding sequence (PBS) region are not shown.
Figure 1.
Organization of the human immunodeficiency virus type 1 (HIV-1) genomic RNA. U3, unique sequence 3’; R, repeated sequence; U5, unique sequence 5’; Ψ, indicates the packaging and dimerization sequences; RRE, Rev responsive element. The PPT sequences as well as the primer binding sequence (PBS) region are not shown.

Figure 2.
Schematic representation of the structure of the genomic forms of the viral DNA during natural infection (panel (A)) or during transfection and transduction by a self-inactivating (SIN) lentiviral-derived (LV) vector (panel (B)).
Figure 2.
Schematic representation of the structure of the genomic forms of the viral DNA during natural infection (panel (A)) or during transfection and transduction by a self-inactivating (SIN) lentiviral-derived (LV) vector (panel (B)).

Figure 3.
The various generations of lentiviral vectors. Top panel. Plasmids employed for constructing first generation lentiviral vectors. Three plasmids are employed. (1) A packaging (or transcomplementation) plasmid encodes the Gag, Pol, Vif, Tat, Rev, Nef, Vpr proteins under the control of the Cytomegalovirus (CMV) promoter. (2) The envelope protein(s) (the vescicular stomatitis virus (VSV) G protein in the example given) is encoded by an expression plasmid under the control of the CMV promoter. (3) Finally, the plasmid leading to the synthesis of the genomic RNA (“genomic plasmid”) contains the sequences required in cis for the packaging and reverse transcription of the RNA. It also contains the sequence of the transgene under the control of an internal promoter (EF1α in the example). The expression of the genomic RNA is driven by the 5’ terminal repeated regions (LTR). The sequence U3 in the 3’ LTR is partially deleted, inactivating the promoter present in U3 generating self-inactivating (SIN) vectors. Middle panel: 2nd generation vectors are built by triple transfection of the producer cells. In this generation of LV vectors, the packaging plasmid only encodes Gag, Pol, Tat and Rev, increasing the level of biosafety (the auxiliary proteins Vif, Nef and Vpr are absent). Bottom panel, third generation of vectors. The packaging plasmid is split in two plasmids, one encoding (under the control of the CMV promoter) the Gag and Pol sequences and carrying the Rev responsive element (RRE), the second encoding the Rev protein (also under the control of the CMV, in the example given). In the genomic plasmid the 5’ LTR sequence is replaced by the sequence of a chimeric LTR where the U3 sequence is replaced by that of a heterologous promoter (the Rous Sarcoma Virus-RSV-promoter in the example given). Finally, "next generation" vectors have also been elaborated, but since they differ considerably from one another, no "synthetic" drawing summarizing them is provided. The main improvement consists of the splitting of the coding sequences in a larger number of plasmids, for increasing biosafety.
Figure 3.
The various generations of lentiviral vectors. Top panel. Plasmids employed for constructing first generation lentiviral vectors. Three plasmids are employed. (1) A packaging (or transcomplementation) plasmid encodes the Gag, Pol, Vif, Tat, Rev, Nef, Vpr proteins under the control of the Cytomegalovirus (CMV) promoter. (2) The envelope protein(s) (the vescicular stomatitis virus (VSV) G protein in the example given) is encoded by an expression plasmid under the control of the CMV promoter. (3) Finally, the plasmid leading to the synthesis of the genomic RNA (“genomic plasmid”) contains the sequences required in cis for the packaging and reverse transcription of the RNA. It also contains the sequence of the transgene under the control of an internal promoter (EF1α in the example). The expression of the genomic RNA is driven by the 5’ terminal repeated regions (LTR). The sequence U3 in the 3’ LTR is partially deleted, inactivating the promoter present in U3 generating self-inactivating (SIN) vectors. Middle panel: 2nd generation vectors are built by triple transfection of the producer cells. In this generation of LV vectors, the packaging plasmid only encodes Gag, Pol, Tat and Rev, increasing the level of biosafety (the auxiliary proteins Vif, Nef and Vpr are absent). Bottom panel, third generation of vectors. The packaging plasmid is split in two plasmids, one encoding (under the control of the CMV promoter) the Gag and Pol sequences and carrying the Rev responsive element (RRE), the second encoding the Rev protein (also under the control of the CMV, in the example given). In the genomic plasmid the 5’ LTR sequence is replaced by the sequence of a chimeric LTR where the U3 sequence is replaced by that of a heterologous promoter (the Rous Sarcoma Virus-RSV-promoter in the example given). Finally, "next generation" vectors have also been elaborated, but since they differ considerably from one another, no "synthetic" drawing summarizing them is provided. The main improvement consists of the splitting of the coding sequences in a larger number of plasmids, for increasing biosafety.

Figure 4.
Outline of the mechanisms of viral entry by the main envelope proteins that can be used to pseudotype LV vectors. ECM: Extracellular Medium; MVB: Multivesicular Body.
Figure 4.
Outline of the mechanisms of viral entry by the main envelope proteins that can be used to pseudotype LV vectors. ECM: Extracellular Medium; MVB: Multivesicular Body.


{kind=link}
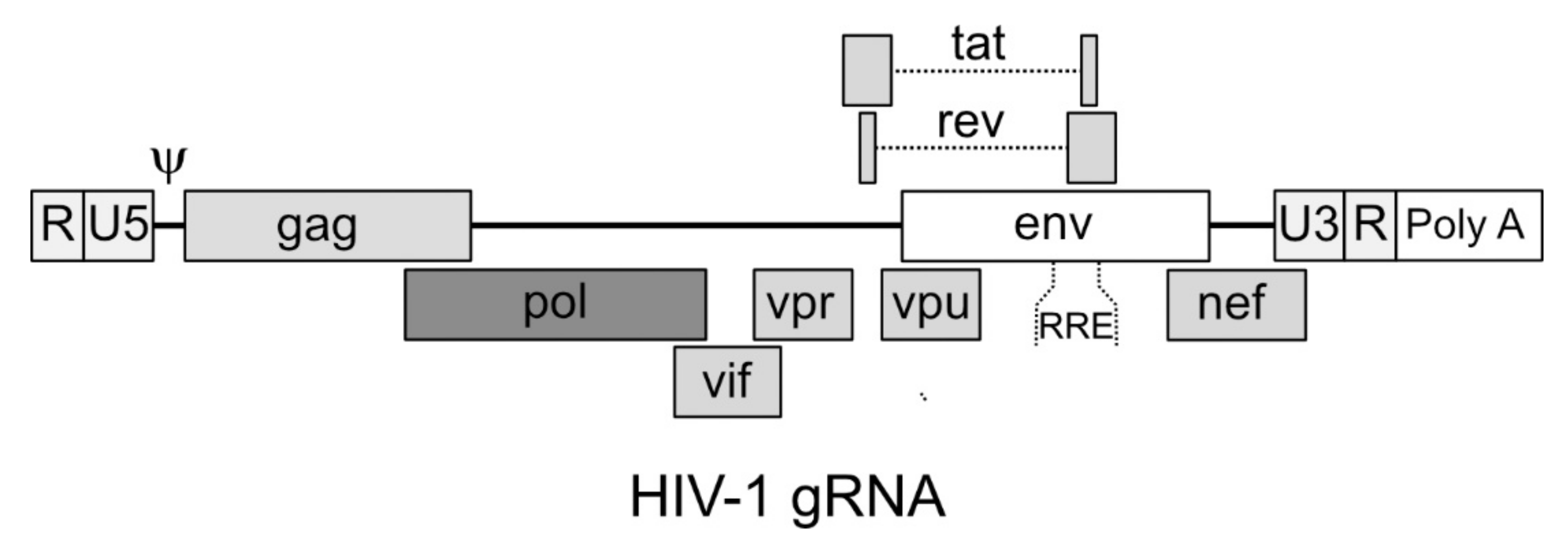{kind=link}
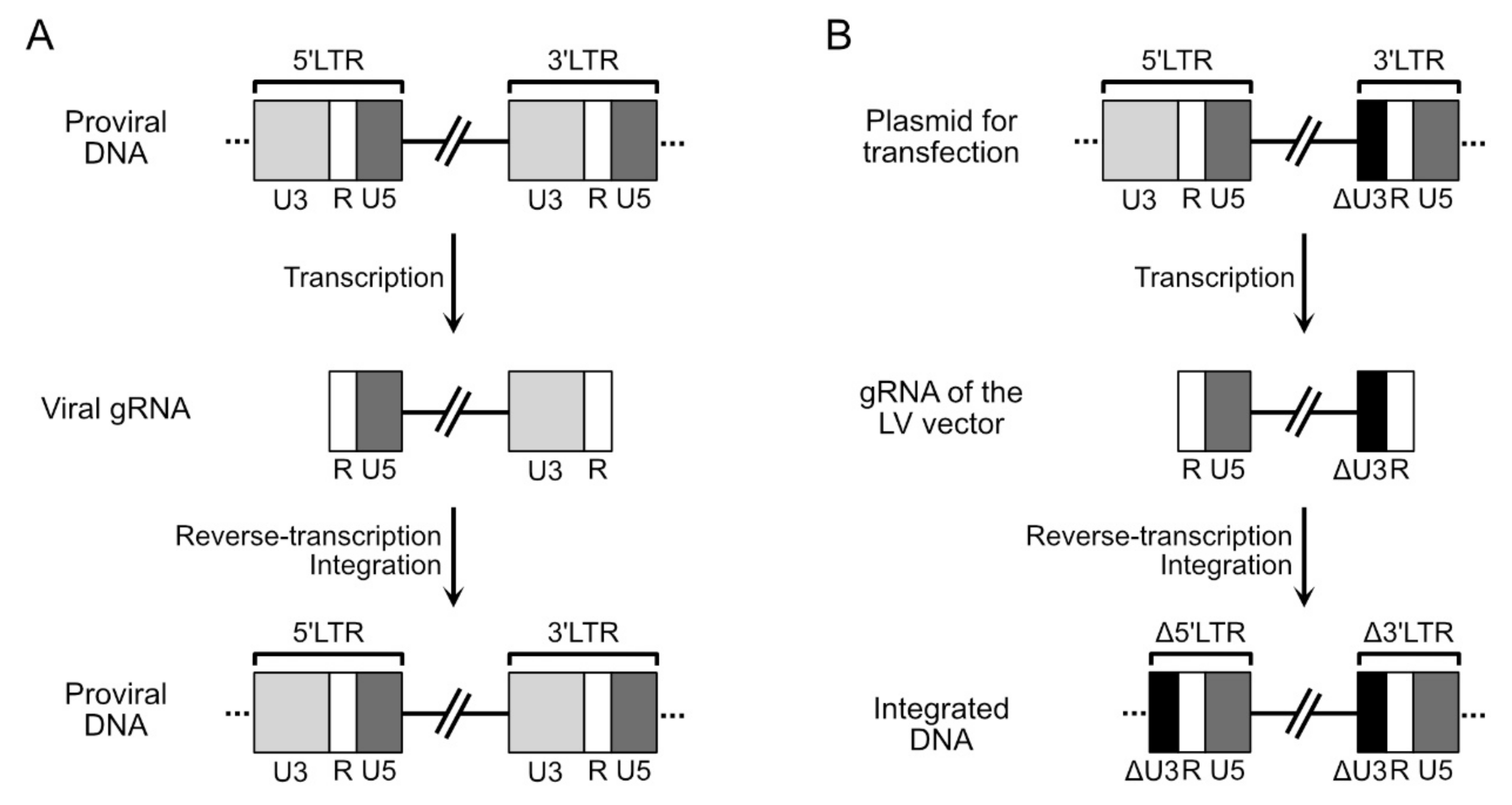{kind=link}
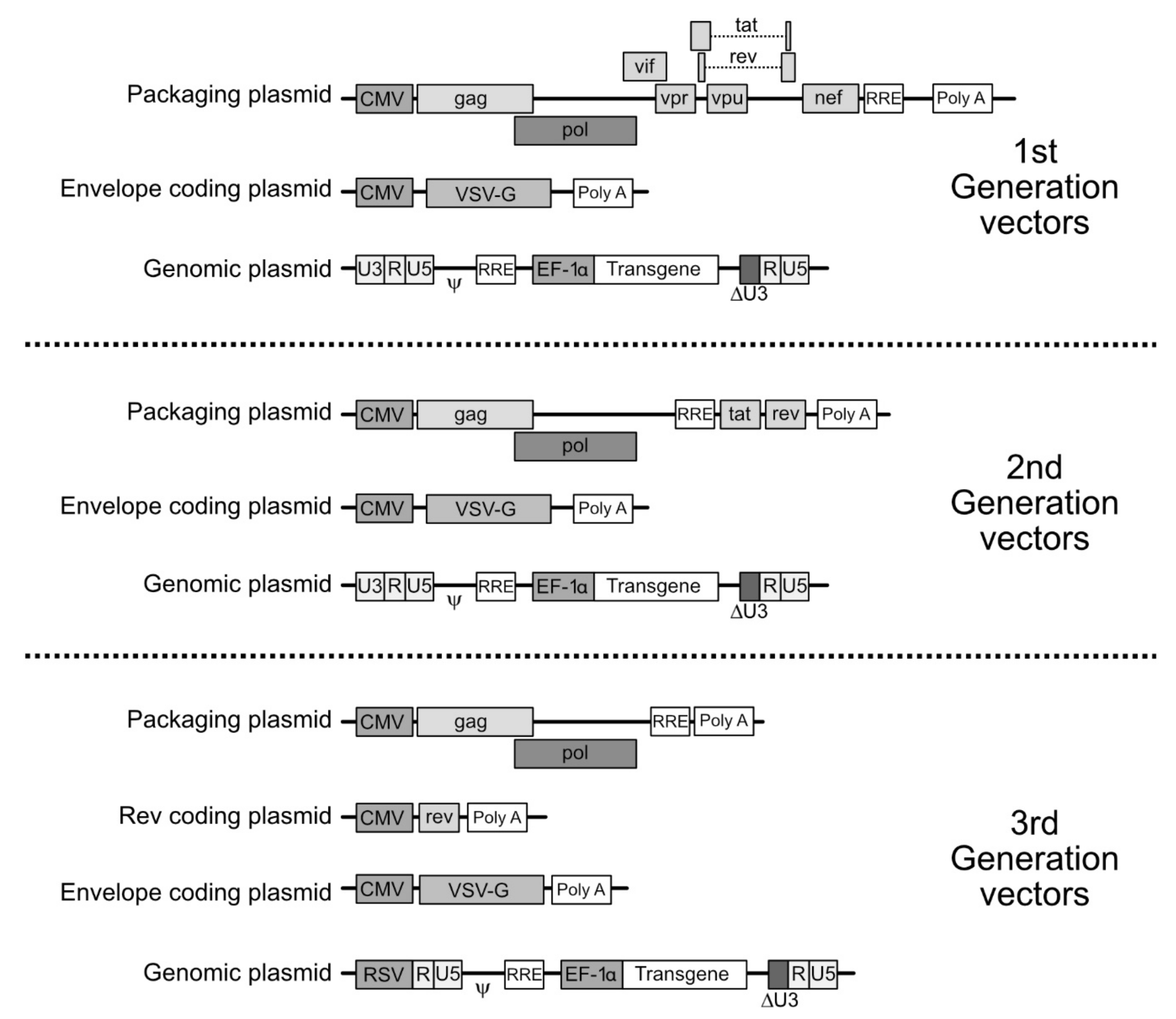{kind=link}
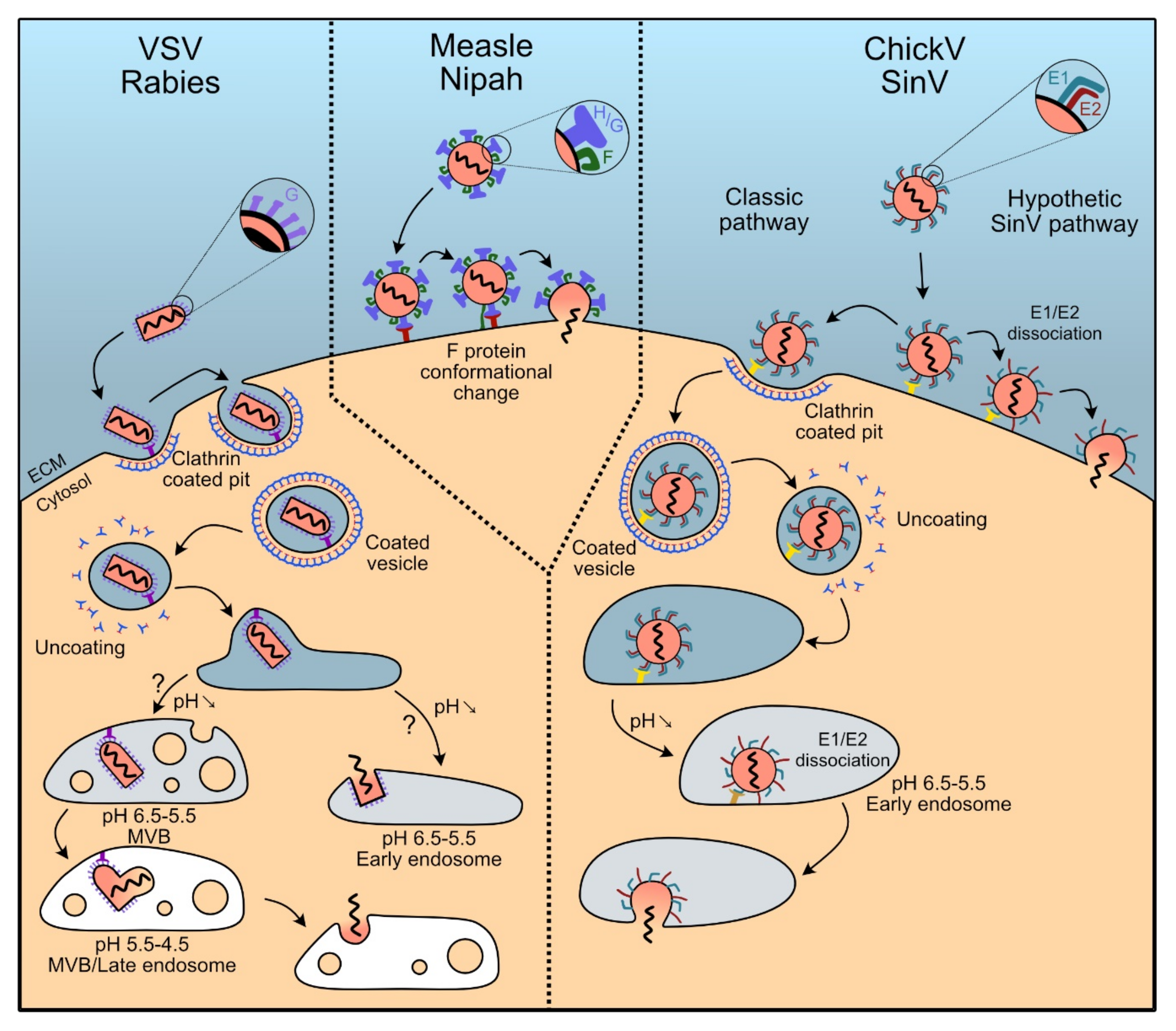{kind=link}
Table 1.
Overview table summarizing the main characteristics of the pseudotypes discussed in this review.
Table 1.
Overview table summarizing the main characteristics of the pseudotypes discussed in this review.
| Original Virus | Pseudotype | Main Characteristics of the Pseudotyped LV | Natural Cell Tropism | Receptor | Transduction Efficiency | References |
|---|---|---|---|---|---|---|
| Vesicular Stomatitis virus | VSV-G | Quasi-universal tropism, high efficiency | Broad | LDL-R | High | [95,96] |
| Rabies virus | RabV-G | Natural ability to efficiently targets neurons | Neurons | nAChR, CD56, p75NTR, mGluR2 | Up to 50% | [97,98,99,100] |
| Measle virus | H/F | High efficiency, tolerant to peptide insertion, can be neutralized by vaccines | B cells, T cells, Epithelial cells, Dendritic cells, HSPC | CD46, SLAM, nectin-4 | Up to 50–70% | [101,102,103,104,105,106,107,108] |
| Nipah virus | G/F | Low prevalence: low neutralization hazard | Pericytes, tumor endothelium | EphrinB2, EphrinB4 | 20–40% | [109,110] |
| Chickungunya virus | E1/E2 | Versatile basis for engineering/reprograming | Broad | PHB1, Mxra8, integrins, Heparan sulfates | Low on non-adherent cells, high on adherent cells (related to VSV-G) | [111,112,113,114] |
| Sindbis virus | E1/E2 | Versatile basis for engineering/reprograming, Low immunogenicity | Broad | 67LR, NRAMP2 | Variable | [115,116] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Duvergé, A.; Negroni, M. Pseudotyping Lentiviral Vectors: When the Clothes Make the Virus. Viruses 2020, 12, 1311. https://doi.org/10.3390/v12111311
AMA Style
Duvergé A, Negroni M. Pseudotyping Lentiviral Vectors: When the Clothes Make the Virus. Viruses. 2020; 12(11):1311. https://doi.org/10.3390/v12111311
Chicago/Turabian StyleDuvergé, Alexis, and Matteo Negroni. 2020. "Pseudotyping Lentiviral Vectors: When the Clothes Make the Virus" Viruses 12, no. 11: 1311. https://doi.org/10.3390/v12111311
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.