
Abstract
Infectious diseases, including the coronavirus disease 2019 (COVID-19) pandemic that has brought the world to a standstill, are emerging at an unprecedented rate with a substantial impact on public health and global economies. For many life-threatening global infectious diseases, such as human immunodeficiency virus (HIV) infection, malaria and influenza, effective vaccinations are still lacking. There are numerous roadblocks to developing new vaccines, including a limited understanding of immune correlates of protection to these global infections. To induce a reproducible, strong immune response against difficult pathogens, sophisticated nanovaccine technologies are under investigation. In contrast to conventional vaccines, nanovaccines provide improved access to lymph nodes, optimal packing and presentation of antigens, and induction of a persistent immune response. This Review provides a perspective on the global trends in emerging nanoscale vaccines for infectious diseases and describes the biological, experimental and logistical problems associated with their development, and how immunoengineering can be leveraged to overcome these challenges.
Similar content being viewed by others

Main
The outbreak of the 2009 influenza A virus subtype H1N1 pandemic caused an estimated global mortality of 200,000 within the first year1, and coronavirus disease 2019 (COVID-19) has rapidly claimed >900,000 deaths within about nine months at the time of writing this Review. Infectious diseases are unpredictable and can affect people of all ages; however, the fatality demographic may differ, as the 1918 Spanish flu claimed more lives of young adults. In contrast, COVID-19 has adversely impacted the elderly and immunocompromised individuals more than others2; however, infections among young adults are sharply rising, with 2.7% death among hospitalized patients in the United States between ages 18 and 34 (ref. 3). Unless there is a drug that is at least 95% effective to stop the outbreaks, normalcy in life relies on safe and effective vaccines. However, there are substantial challenges in developing effective vaccines, as described in Box 1, including failure to elicit optimally mutated antibodies4,5 and biases in the immune system through immunological imprinting to prior infections6. Antibody responses to severe acute respiratory syndrome coronavirus 1 (SARS-CoV-1) or Middle East respiratory syndrome coronavirus (MERS-CoV) waned after two to three years in individuals that survived lethal infections7, and post-mortem analysis of lymph node and spleen tissues in critically ill COVID-19 patients suggested a lack of lymphoid structures that lead to durable antibody responses8. These findings raise new challenges to the development of infectious disease vaccines that aim to induce a persistent immune response.
The live attenuated vaccines are complex and require a substantially long time for development, often involving tremendous revamping if the pathogen mutates. The seasonal influenza vaccine, for example, delivers inconsistent performance with as good as 60% effectiveness, and as low as 10% or 20% in mismatched years9. Therefore, the burden of disease shifts to the development of vaccines that promise broader protection than seasonal shots. To overcome the limitations of live attenuated vaccines, sophisticated vaccine technologies are being developed, including structurally engineered immunogens10,11, germline-targeting immunogens12,13,14, novel synthetic adjuvants15,16 and material-based vaccines of multiple length scales14,16,17,18. Engineered vaccines with natural or synthetic materials can induce broadly neutralizing antibodies and strong memory responses against infections. Among these, nanovaccines, which are the focus of this Review, provide distinct advantages of structural and size proximity to pathogens, tunable physiochemical and biophysical properties, protection of the vaccine antigen from degradation or rapid clearance, improved transport through lymphatics and into the immune follicles of lymph nodes, as well as co-delivery of immunomodulatory molecules to boost immune recognition.
Vaccine transport and spatial localization in lymph nodes
Defining where and in what form specialized immune cells, B and T lymphocytes, encounter vaccine antigens in their soluble or particulate form is fundamental to understanding how long-term, antigen-specific immune responses occur to nanovaccines. During the immune response to an infection, antigen-primed B cells clonally expand within B-cell follicles of lymph nodes and undergo secondary diversification of their immunoglobulin genes, followed by the selection of rare winner cells, called plasma cells and memory B cells19. Naive B cells in lymph nodes can encounter antigens in B-cell follicles either through direct binding of their immature B-cell receptors (BCR) or on the surfaces of resident antigen-presenting cells, including follicular dendritic cells (FDCs).
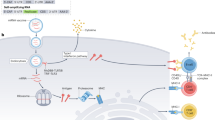
A key question is how do nanovaccines carrying complex antigens traffic inside the B-cell follicles to reach FDCs and whether this localization is necessary. After immunization, the nanovaccines are picked up in the flow of interstitial fluid and localize to various parts of a lymph node. Nanovaccines of the order of small antigens (<200 nm) enter the lymph nodes, and before the lymph fluid exits through the efferent lymphatics near the medullary sinus, the particulate antigens localize on the subcapsular sinus macrophages overlying B-cell follicles (Fig. 1a). However, targeting FDCs and inner structures within a B-cell follicle is not a common characteristic of nanovaccines, as particles of different sizes and material compositions tend to localize outside of the B-cell follicles. It is only recently that glycoengineered nanovaccines were shown to deposit within the B-cell follicles14. In contrast, small nanovaccines with a hydrodynamic radius of about 5 nm bypass subcapsular sinus macrophages and gain direct access to the B-cell follicles through a network of collagen-rich fibre conduits (Fig. 1a). The conduits are prevalent between B-cell follicles and in the T-cell zones and are wrapped by fibroblastic reticular cells20,21. Although the conduit openings are about 1 μm, collagen fibres have a spacing smaller than 10 nm, and therefore only allow passage of nanovaccines with a dynamic radius of less than about 5 nm (~70 kDa). In the case of larger nanoparticles (200–500 nm) that drain into the subcapsular sinus, the antigen may get proteolysed22 and transported through macrophages or through the conduits. In contrast, nanovaccines that are greater than 500 nm get internalized by the dendritic cells through Fc fragment of IgG receptor IIb (FcγRIIb) receptor, and the antigen is recycled to the dendritic cell surface to present to the B cells. Therefore, the nanoscale size range of the antigen vehicle is a critical design criterion and can determine the spatial location of antigen. The size range is not generalizable and depends on the dimensions and chemical properties of the nanovaccines23,24, opsonization of nanovaccines by complement and complement receptor14, and other factors25. In addition to macrophages, lymph node-resident stromal cells, such as lymphatic endothelial cells, can bind and endocytose antigens, including viruses, via mannose receptor and scavenger receptors26. However, the role of diverse lymph node stromal cells27 in nanovaccine capture, transport and presentation is yet to be thoroughly studied.

a, Nanovaccines are carried through the lymphatics into the subcapsular sinuses located between the collagenous capsule and the immune cell-rich cortex region of the lymph node. The nanovaccines can localize on subcapsular macrophages overlying B-cell follicles. Nanovaccines are transported to FDCs in B-cell follicles by the relay of complexes from subcapsular sinus macrophages to migrating B cells, which in turn transfer antigen to FDCs, in a complement- and complement receptor-dependent manner. After encountering vaccine antigens, primed B cells decrease their migration velocity and relocate to the border of B-cell follicles (a C–X–C chemokine receptor type 5 (CXCR5) and CC-chemokine receptor 7 (CCR7)-dependent migration), where they encounter a specific subclass of CD4+ T cells, the TFH cells, eventually leading to germinal centre reactions. Nanovaccine size may regulate transport mechanisms in the lymph nodes, with ~5-nm nanoparticles entering the B-cell follicle through collagen conduits and >500 nm particles transported by dendritic cells. b, Naïve B cells differentiate into germinal centre B cells (1) or short-lived plasma cells (2) as two possible outcomes of antigen and T-cell encounter. If successfully induced by nanovaccines, the germinal centre could lead to a high-affinity antibody response through a complex iterative process of somatic hypermutation, affinity maturation and selection. c, Multiple distinct B-cell clones seed each germinal centre in a vaccinated individual, and these specialized cells lose clonal diversity at widely disparate rates. Multiple clones can evolve in parallel within the same germinal centre, making it a highly heterogeneous structure, and a fraction of germinal centres become heavily dominated by the substantial expansion of the descendants of a single somatic hypermutation variant arising at or after the onset of germinal centre selection over cells of the same and of different clones. NP, nanoparticles; SCS MΦ, subcapsular sinus macrophage; CSR, class switch recombination.
Germinal centres and B-cell stimulation by nanovaccines
The ultimate goal of an antiviral nanovaccine is to elicit a durable, antigen-specific, high-affinity antibody response, which depends on the response of the germinal centre B cells in the B-cell follicles of lymph nodes. After encountering vaccine antigens, primed B cells relocate to the border of B-cell follicles to interact with the follicular helper T (TFH) cells (Fig. 1a). Depending on the nature of the resulting interactions between B cells and TFH cells, which include CD40L binding to CD40 on B cells, naive B cells could differentiate into specialized germinal centre B cells or short-lived plasma cells (Fig. 1b). The germinal centre is a subanatomical compartment within B-cell follicles that is dynamically formed whenever an antigen is present and B cells start dividing. Through an epigenetically and transcriptionally controlled process28, the germinal centre grows with proliferating B cells and polarizes into the dark and light zone within seven to ten days from immunization (Fig. 1b). In the dark zone, the rapidly proliferating B cells undergo somatic hypermutation and diversify the antibody repertoire to select for better, fitter antigen-reactive B-cell clones (Fig. 1b). After migration to the light zone, the B cells ‘test’ their BCRs against the antigens/immune complexes presented by the FDCs. At this point, most of the primed B cells undergo apoptosis; however, some clones receive enough activation—by a combination of the FDCs and TFH cells—to migrate to the dark zone where they proliferate and mutate further. B cells experience several rounds of BCR testing, which determines their fate: proliferation or apoptosis. Surviving B cells undergo cycles of somatic hypermutations of the antigen-binding variable regions of their immunoglobulin genes29, which produce class-switched high-affinity immunoglobulin-α (IgA)- and immunoglobulin-γ (IgG)-type antibodies. Eventually, activated B cells exit germinal centres to become long-lived plasma cells and memory B cells (Fig. 1a). The long-lived antibody-secreting plasma B cells relocate to bone marrow and protect against re-infection for months and years, sometimes for the entire life. The memory B cells, however, do not secrete antibodies and instead become plasmablast in case of a re-infection.
The germinal centre is reminiscent of Darwinian selection19. The use of engineered confetti mice has shed light on clonal competition among diverse B cells during germinal centre responses30 that can have direct implications for the development of nanovaccines and adjuvants where antibodies with non-immunodominant specificities need to be elicited (for example, human immunodeficiency virus (HIV)-1 and influenza) (Fig. 1c). Tas et al. showed that multiple B-cell clones seed individual germinal centres and lose diversity at disparate rates, suggesting the possibility that the germinal centre competition may restrict the emergence of non-competitive clones and promote somatic diversification that must be elicited for the generation of broadly neutralizing antibodies30. Understanding how nanovaccines elicit clonal bursts have implications for the design of better-engineered vaccines against highly variable viruses, where the success of immunization depends on whether broadly protective antibodies targeting conserved, non-immunodominant epitopes are elicited.
Overcoming transport barriers to B-cell follicles
Targeting vaccines to specific immune or stromal cells in the lymph nodes or, more specifically, the B-cell follicles provides the possibility to programme the humoral immune response. However, the structure of a lymph node, zonal localization of lymph node-resident cells and their intra-lymph node migration makes it challenging to access the target cell population31. When compared with vaccine antigens, polymeric materials can be more easily tuned into specific sizes or shapes, or modified by functional groups that target unique receptors on immune cells without compromising the functionality of immunogenic epitopes. The human immune system has a wide range of pattern recognition receptors—C-type lectin receptors, such as dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin (DC-SIGN), mannose receptors and scavenger receptors, which recognize polysaccharide motifs on microbes. As such, glycoengineering of nanoparticles has emerged as a powerful vaccine design tool. Wilson et al.16 recently reported a synthetic polymeric glyco-adjuvant p(Man–TLR7) that targets dendritic cells via mannose-binding receptors and activates them via Toll-like receptor 7 (TLR7) stimulation. When conjugated to antigens, p(Man–TLR7) elicited robust humoral and cellular immunity against malaria16. Synthetic glycosylation is fully characterizable and stable, and does not induce an unnecessary immune response to an antibody-based targeting moiety. The approach also contrasts with covalently linked glycosylation approaches that can hinder the intracellular processing of antigens for major histocompatibility complex presentation.
However, most nanovaccines do not easily enter B-cell follicles in the lymph nodes. To overcome this challenge, Tokatlian et al. glycoengineered multivalent protein nanoparticles with HIV envelope antigens and compared them against soluble monomers14. These germline-targeting immunogens, gp120 (eOD-GT8) and gp140 envelope trimer (MD39), offer distinct advantages, including improved thermal stability12,13,32,33. The study elucidated how the immune system generates a response to multimeric nanoparticles, a phenomenon that is poorly understood, while simultaneously demonstrating that nanoparticle glycosylation is key to enhanced humoral immunity. Glycosylation spurs binding to mannose-binding lectin complement fixation and antigen trafficking to FDCs (Fig. 2a). The eOD-60-mers are predominantly decorated with mannose glycans, which, even in low amounts, are sufficient to transport nanovaccines to B-cell follicles and FDCs, via a mannose binding lectin-mediated process. Simple ferritin nanoparticles14, even without antigen, when decorated with a synthetic trimannose moiety at as low as about 96 mannose per nanoparticle, can markedly improve delivery to B-cell follicles, induction of germinal centre response, B cell–TFH cell interactions, as well as the generation of neutralizing antibodies when compared with those lacking glycosylation (Fig. 2a). With respect to size, it appears that approximately 40 nm nanoparticles are optimal for vaccine design, as they deposit within the B-cell follicles and generate a strong immune response. However, a more rigorous analysis is needed to understand whether size alone can regulate entry into the follicles34 or whether glycosylation of larger particles (100–500 nm) and polymeric particles can also facilitate transport to B-cell follicles. Interestingly, persistent long-term germinal centres have been reported in mouse models of influenza and yellow fever immunized with 300 nm poly(lactic-co-glycolic acid) (PLGA) nanovaccines without glycoengineering35. Immunization with PLGA nanovaccine that mimicked H1N1 influenza A in their ability to stimulate the first-responder immune cells in the body, induced persistent germinal centre B-cell response in draining lymph nodes and strong antibody responses in a rhesus macaque model of the 2009 pandemic H1N1 influenza A along with long-lived memory responses35.

a, Glycoengineered nanovaccines, when immunized, enhance antigen trafficking to FDCs in B-cell follicles. The glycoengineered (Glycosylated) nanovaccines elicit a higher number of antigen-specific B cells, and increased germinal centre B cell–TFH cell interactions and neutralizing antibodies than non-glycoengineered (aGlycosylated) nanovaccines. b, The gut microbiome, either through TLRs or other means, such as metabolites, secondary bile acids and inflammasome regulation, enhances antigen-specific germinal centre and antibody response to influenza vaccines in healthy individuals. Disruption of the gut microbiome through antibiotics or lack of sensing of TLR5, leads to poor vaccine outcomes. Rationally designed nanovaccines using immunomodulatory nanomaterials or co-delivery of TLR agonists may enhance the immune response under altered gut microbiome conditions, leading to a universal response. Immunization image in b reproduced with permission from ref. 64, Springer Nature Ltd.
In contrast to the size, the shape of nanoparticles does not appear to influence particle localization to B-cell follicles. Similar-sized spherical-shaped eOD-60-mer and flower-shaped MD39-8-mer nanoparticulate gp140 vaccines, engineered by fusing the antigen to archaeal ferritin in a multimeric form, elicit markedly higher antibody titres and germinal centre response than soluble vaccines and localize to a similar extent in B-cell follicles. However, nanodisks are preferentially uptaken over nanorods by other immune cells36, and therefore more rigorous shape analysis is needed to distinguish possible shape effects on particle localization to B-cell follicles. It would be intriguing to see whether, in addition to size, shape and glycosylation, optimal spacing and high density of epitopes, mimicking the distribution of spike proteins on viruses (spacing 5–10 nm, density 15–20 immunogens), can facilitate nanovaccine access to B-cell follicles and enhance the germinal centre response. Nanomaterial chemistry offers a unique opportunity for antigen multimerization and precise dosing on a single particle, which can enable activation of low-to-high affinity B cells and investigation of how immunogens interact with the BCR to induce strong immunogenic signals in B cells. For most conventional antigens, binding to the BCR is necessary but not sufficient to drive the full activation of B cells, including proliferation and differentiation into antibody-producing plasma cells. A temporally distinct second signal is required that could be provided by TFH cells or via pattern-recognition receptors expressed by B cells, such as TLR937. Nanovaccines could simultaneously induce this twofold signalling through BCR engagement and co-delivery of TLR agonists. Nevertheless, optimizing vaccines would require help from TFH cell, which could be partially triggered by exposing the CD4 T cells to good epitopes and stimulatory signals.
The microbiome effect on nanovaccines
The considerable variation in human microbiota and metabolism imposes a substantial challenge to the development and translation of vaccines38. How nanovaccines perform in the altered gut microbiome or modulate the gut microbiome is poorly understood. Recent preclinical and clinical studies have suggested that depletion of gut bacteria by broad-spectrum antibiotics can weaken the immune system capability to respond to vaccines, including some nanovaccines38,39,40,41. Pulendran and colleagues first reported that engineered mice that fail to recognize flagella on gut bacteria result in a poor germinal centre and antibody response against human influenza and polio vaccinations40. These studies elucidated that a TLR5-mediated sensing of flagellin promoted plasma cell differentiation directly and by stimulating lymph node macrophages to produce plasma cell growth factors. TLR5 has a vital role in the inflammation response to flagellated pathogens that breach the epithelial barrier. Notably, the decimation of the gut microbiome in mice using antibiotics also led to inadequate vaccine outcomes40. A follow-up clinical trial investigated the role of perturbing the gut microbiome using broad-spectrum antibiotics on the efficiency of the H1N1 influenza vaccine39. Importantly, these studies showed that lack of prior vaccination or exposure to flu strains is critical to the impairment of H1N1-specific neutralization and IgG1 and IgA binding responses when antibiotics alter the gut microbiome. These studies provide unprecedented insights into the gut microbiome role in the activity of conventional vaccines. They suggest that antibiotic treatment can enhance inflammatory signatures, increase dendritic cell activation and induce divergent metabolic trajectories, such as reduced levels of serum secondary bile acids (Fig. 2b). In particular, transcriptional signatures have revealed that alteration to the gut microbiome through antibiotic treatment can enhance innate immune responses and gene expression programmes associated with the transcription factors activating protein 1 (AP-1, comprising FOS and JUN) and nuclear receptor 4A1 (NR4A1), which are central to inflammatory responses39. Antibiotic administration can alter the blood metabolome of patients receiving the inactivated seasonal influenza vaccine, including changes in bile acids, such as lithocholic acid, and are possibly associated with increased inflammation and regulation of vaccine responses.
We showed that alteration in gut microbiome sensing through TLR5 and the resulting metabolic syndrome in TLR5−/− mice diminishes the germinal centre immune response induced by PLGA nanovaccines41 (Fig. 2b). The nanovaccines, unexpectedly, changed gut microbiome diversity, potentially creating a feedback loop in the immune response. By chronically treating mice with antibiotics, we showed that disrupting the gut microbiome leads to poor vaccine response, likely attributable to increased interleukin-6 levels in mice. More importantly, the low immune response can be rescued by an immunoengineered pyridine-poly(hydroxyethyl methacrylate) (Pyr-pHEMA) nanogel vaccine, which functions through the TLR2 stimulation and induced a more robust germinal centre response than alum-supplemented PLGA nanovaccines41. This is the first study to highlight the advantage of using a material-based nanovaccine in gut-associated metabolic syndrome, where the material itself offers immunomodulatory properties in overcoming the immunological restrictions imposed by gut-mediated inflammatory disease conditions and generating a more robust response than alum-adjuvanted vaccines.
Nanovaccines to elicit broadly neutralizing antibodies
HIV and influenza viruses can evade effective neutralizing antibody responses, and only a proportion of infected individuals generate broad and potent neutralizing antibody responses. Therefore, vaccine research has steered its focus towards eliciting broadly neutralizing antibodies (bnAbs) to recognize and neutralize the majority of pathogen’s quasispecies.
Harnessing the advantages of nanovaccine delivery, bioavailability and multimeric antigen presentation of rationally designed B-cell lineage immunogens is revolutionizing the field of HIV. The eOD-GT8 60-mer nanoparticle vaccines elicit VRC01-class bnAbs, which have garnered particular attention for epitope-directed HIV vaccine design as they can neutralize up to 98% of HIV strains, and have entered phase 1 of the first-in-human clinical trial32,33,42,43,44. Nevertheless, most of the other HIV nanovaccines in development need to overcome the challenges of generating bnAbs that first prime B cells encoding bnAb precursors (with a low affinity for the virus), followed by immunogens that guide antibody affinity maturation. The bnAbs can puncture the glycan-shield defence of the HIV envelope (Env) trimer in five regions, each of which may be involved in Env function (see detailed structure45). HIV poses unique challenges attributed to the antigenic diversity, glycosylation and immune evasion of its Env, together with poor bnAbs generation. In addition, HIV Env consists of ~50% glycans by mass and the bnAb–glycan binding is weak; therefore, nanovaccines must be designed to elicit bnAbs that strongly bind with glycans to access the native trimer and neutralize viral infection. Indeed, very few patients infected with HIV produce high levels of bnAbs46,47. Understanding why such antibodies are not produced in patients or vaccinated individuals will enable better nanovaccine design.
Detailed analysis of HIV-specific antibodies from infected individuals alludes to bnAbs having all or some of the following infrequent qualities: long antibody combining heavy-chain third complementarity-determining regions, high levels of somatic mutations and autoreactivity with host non-HIV antigens48. As a result, the complex antibody traits predispose the antibody-producing B cells to immune tolerance-mediated elimination or suppress the B-cell activation by making them anergic. The bnAbs are unusually somatically hypermutated for affinity maturation to form improbable mutations (Fig. 3a) and require long periods of germinal centre reaction5,48. Therefore, key criteria to be considered in nanovaccine design would be (1) prolonged exposure and retention in B-cell follicle for a persistent germinal centre reaction, and (2) overcoming somatic mutation roadblocks by specifically engaging bnAb precursors and selecting for improbable mutations critical for successful vaccine induction of potent bnAb B-cell lineages (Fig. 3a). The first criterion can be achieved through controlled release systems and the second by vaccination with nanoparticles carrying immunogens that bind with moderate to high affinity to bnAb B-cell precursors, and with higher affinity to B-cell precursors that have acquired improbable mutations4,48.

a, bnAbs require complex somatic hypermutations to acquire improbable mutations for neutralizing activity. Conventional vaccines face this somatic hypermutation roadblock and do not typically elicit bnAbs like an infectious response. Immunoengineered nanovaccines can elicit antibodies that exhibit neutralization activity similar to that of intermediate-stage bnAbs. By rationally combining nanoparticles with structurally engineered immunogens followed by serial immunizations, improbable mutations, akin to infection, may be achievable. b, ADE may impose a challenge in nanovaccine design and elicited immune response. Nanovaccines may induce high-quality, mutated, neutralizing antibodies that would bind to targeted proteins on pathogen surface and inhibit host–pathogen interaction. Nanovaccines may reduce production of non-neutralizing antibodies, which can use their Fab domain to attach to pathogens, without neutralizing them he, and their Fc domain to bind to the corresponding receptors on innate immune cells, actually facilitating infection and/or induction of a cytokine storm through the stimulation of TLRs.
Nanoparticles offer a unique structure-based design opportunity to package antigens, which is less feasible with standalone vaccines. For instance, the structure of ferritin, a ubiquitous iron storage protein that self-assembles into nanoparticles, allows for the insertion of influenza virus haemagglutinin (HA) in its physiologically relevant trimeric viral spike form49. In a landmark study49, the HA–ferritin nanovaccine (~37 nm diameter) was shown to elicit approximately 34 times higher neutralization titres in immunized animals than the commercially available flu vaccine, a trivalent inactivated influenza vaccine (TIV), prompting the start of a clinical trial (ClinicalTrials.gov identifier: NCT03186781). The HA–ferritin nanovaccine also resulted in reduced side effects, such as weight loss, and can induce, with a single shot, an immune response comparable to the broadly neutralizing antibody immune responses generated by multiple immunizations of TIV49.
Overcoming disease enhancement, virus mutations and biomanufacturing challenges
In the quest for bnAbs, a suboptimal quality antibody response against a nanovaccine is probable and can promote pathology through antibody-dependent enhancement (ADE) of disease (Fig. 3b), whereby antibodies that bind viral spike protein can facilitate uptake by macrophages and B cells via their Fcγ receptors. Specifically, non-neutralizing antibodies have the potential to mediate enhancement of respiratory disease in influenza vaccination, with non-neutralized virus–antibody complexes finding alternative receptors and entry routes into the cell via the Fc-receptor pathway. One study50 described ADE with two different functional monoclonal antibodies that increased influenza virus fusion kinetics and led to enhanced lung pathology and respiratory disease in a dose-dependent manner in mice following the H3N2 virus challenge (haemagglutinin (H) protein; viral protein, neuraminidase (N)). The ADE-mediated pathology is not limited to influenza; SARS-CoV studies have reported that the immunization of mice and non-human primates with inactivated whole SARS-CoV, virus-like-particle vaccine or various forms of S protein could induce ADE51,52,53. ADE of viral pathology can cause an inflammatory response through stimulation of RNA-sensing TLR pathways in the infected cell, leading to a cytokine storm54. Wang et al.55 systemically tested immunodominant B-cell peptide epitopes of SARS-CoV-1 spike protein in non-human primates and found that the spike glycoprotein peptides S471–503, S604–625 and S1164–1191 elicited antibodies that efficiently prevented infection in rhesus macaques, whereas peptide S597–603 induced ADE-like behaviour. Therefore, while it is true that the quality of antibody production is one aspect that needs careful optimization, the antigen itself may also need to be edited to steer clear of epitopes that, upon binding of the antibody, may enhance viral infectivity. Whether SARS-CoV-2 or vaccines currently in clinical trials can cause ADE remains unclear. Nanoparticles can play an essential role in overcoming ADE as they can potentially be engineered to shield the effect of ADE-promoting epitopes or maximize safety through controlled delivery (Fig. 3b). Nanoparticles become important if epitopes that show low ADE potential are also poorly immunogenic; then, the immunogenicity of these antigens, and the resulting immune response, can be enhanced through simultaneous packaging of adjuvants in nanovaccines. As a cautionary note, whether or not nanoparticle configuration, materials, and formulations can themselves add to ADE remains to be carefully investigated.
Most of the immune response in influenza is thought to be targeted at the virus-exposed head, which contains features that elicit a strong antibody response, instead of the slender stalk. However, an influenza virus that is highly diverse, such as influenza A, can change within two years, therefore leaving flu vaccine recipients largely unprotected56,57. To overcome these issues, multiple vaccine and nanovaccine programmes are targeting ‘universal influenza vaccines’ that can induce broad cross-protection against divergent viruses. Nanoparticles such as ferritin, with their unique structural features, are useful vaccine platforms because they can display multiple copies of influenza HA spikes on their surface, mimicking the natural organization of HA on the influenza virus49. The universal flu nanovaccine approaches have leveraged on these nanoparticle properties to vaccinate against conserved domains, such as the slender stalk or the immunogenic subdominant stem region of HA. This region is highly conserved and recognized by antibodies capable of binding multiple HA subtypes. Yassine et al.58 developed HA-stem nanoparticles that generated heterosubtypic influenza protection. Using an iterative structure-based design, the team developed H1-based HA-stabilized stem (HA-SS) glycoprotein immunogens that lack the immunodominant head domain. With the stem immunogens, a C-terminal fusion to the ferritin nanoparticle created self-assembling HA-SS nanoparticles (HA-SS NPs) and reduced the splaying of the membrane-proximal regions of the stem. The HA-SS NP vaccine elicited broadly cross-reactive antibodies in mice and ferret models with complete to partial protection against the lethal heterosubtypic H5N1 influenza virus challenge58. The HA-SS NP vaccine design is now undergoing a phase I clinical trial and could, in principle, confer protection against a broad range of pandemic influenza virus subtypes9.
A nanoparticle vaccine, which offers added benefits of excluding any vector components and tunability towards antigens, has also been developed against two other conserved domains of influenza—the matrix protein 2 ectodomain (M2e) and the neuraminidase (NA) membrane glycoprotein containing four identical polypeptides. The M2e immunogen, however, has low immunogenicity due to the small size and low abundance in virions compared with HA and NA. To overcome this, a layered protein nanoparticle system comprising structure-stabilized HA stalk domains and a vaccine construct M2e has been engineered, and shown to induce high immunogenicity and protection against homosubtypic and heterosubtypic influenza A virus challenges59. These nanoparticles were composed exclusively of the antigens without any vector components. The nanovaccine-induced M2e antibodies showed strong cross-reactivity to a diverse set of influenza strains, followed by complete protection. Using crosslinkers that link a disulfide bond between primary amines in a protein, one could regulate the prolonged release of antigen in physiological redox conditions, potentially promoting B-cell responses59. The binding of soluble HA antigen to the desolvated double-layered nanoparticles prevents the risk of solution instability shown by virus-like particles as well as off-target immune responses against self-assembly motifs, such as the ferritin nanoparticles49,58,59.
Reassessing how we target the induction of B-cell immunity and improve associated T-cell response is the future of nanovaccines. The approaches discussed in this Review can also be applied to other infectious diseases, such as malaria16, Lyme disease60 and potentially COVID-19. Several nanovaccine candidates are in development and in pre-clinical phases of testing against COVID-19. Novavax, Inc., a late-stage biotechnology company, had developed a proprietary virus-like particle vaccine for use against MERS-CoV comprising a MERS-CoV that contained a minimum of one trimer of an S protein and their proprietary Matrix-M adjuvant. They used the same technology to produce a vaccine candidate against SARS-CoV-2, NVX-CoV2373, and, at the end of May 2020, announced the enrolment of the first participants in a phase I/II clinical trial. An alternative to protein vaccines, messenger RNA nanovaccine platforms, such as a lipid nanoparticle encapsulated RNA vaccine (reviewed elsewhere61), have been proposed for several diseases. For example, the 2019-nCoV vaccine (mRNA-1273) developed by Moderna, Inc. is currently in phase III trial for COVID-19. Moderna, Inc. has also reported that lipid nanoparticle-based mRNA vaccines against H10N8 and H7N9 demonstrate favourable safety and reactogenicity profiles in healthy adults in phase I randomized clinical trials62 and has tested its mRNA-based platform against chikungunya in mice and macaques63.
The challenges in nanovaccines do not end with the induction of humoral immunity. Once validated for pre-clinical studies in larger animal models, clinical translation of nanovaccines will require a complex safety testing, clinical trial, and setting up bioprocess and analytical pipeline for supporting engineered vaccine development. The safety testing process requires compliance with good laboratory practice and cannot be skipped because there are insufficient data available for nanovaccine production processes. Nanovaccines will need to be then produced in facilities that comply with current good manufacturing practice to ensure continuous quality and safety. It is unlikely that these processes could be incorporated fully within existing viral vaccine pipelines, and therefore they need to be completely developed. Nevertheless, polymeric nanovaccines offer unique advantages: they can be characterized biochemically; could be less reactogenic than live or inactivated virus vaccines; and can be purified to a high degree owing to the relatively large size of nanoparticles compared with the other components of the culture medium. However, unlike the manufacturing of live, attenuated organisms, which readily accumulate to high concentrations in virus growth conditions, including eggs, nanovaccines would require the production of distinct components separately (antigen, delivery system and adjuvant), and then their assembly in the final product. The antigens could be produced in a similar manner as other subunit vaccines, but little industrial procedure exists for manufacturing immune potentiator and delivery system for nanovaccines.
Finally, the nanovaccine field has many outstanding biological questions to answer. These include—but are not limited to—whether nanovaccines can induce T-cell subsets that enhance antigen-specific bnAb development, whether they have an impact on B-cell somatic hypermutation and TFH cell functions, and whether they can break through clonality bottlenecks that restrict the engagement of the large diversity of B-cell repertoire and memory responses. Emerging ex vivo cellular and acellular technologies (for example, organoids28,64,65,66 and acellular antibody discovery pipeline67) that could be integrated with nanovaccine research in both in vivo and ex vivo settings can reduce the nanovaccine discovery timeline, impact on the dynamics of germinal centres and memory B-cell re-activation, and elucidate mechanisms to overcome poor immunity in the elderly population68.
References
Dawood, F. S. et al. Estimated global mortality associated with the first 12 months of 2009 pandemic influenza A H1N1 virus circulation: a modelling study. Lancet Infect. Dis. 12, 687–695 (2012).
Amanat, F. & Krammer, F. SARS-CoV-2 vaccines: status report. Immunity 52, 583–589 (2020).
Cunningham, J. W. et al. Clinical outcomes in young US adults hospitalized with COVID-19. JAMA Intern. Med. https://doi.org/10.1001/jamainternmed.2020.5313 (2020).
Saunders, K. O. et al. Targeted selection of HIV-specific antibody mutations by engineering B cell maturation. Science 366, eaay7199 (2019).
Wiehe, K. et al. Functional relevance of improbable antibody mutations for HIV broadly neutralizing antibody development. Cell Host Microbe 23, 759–765.e6 (2018).
Gostic, K. M., Ambrose, M., Worobey, M. & Lloyd-Smith, J. O. Potent protection against H5N1 and H7N9 influenza via childhood hemagglutinin imprinting. Science 354, 722–726 (2016).
Liu, W. et al. Two-year prospective study of the humoral immune response of patients with severe acute respiratory syndrome. J. Infect. Dis. 193, 792–795 (2006).
Kaneko, N. et al. Loss of Bcl-6-expressing T follicular helper cells and germinal centers in COVID-19. Cell 183, 143–157 (2020). The study shows that SARS-CoV-2 infection blunts long-lived antibody responses in critically ill patients.
Eisenstein, M. Towards a universal flu vaccine. Nature 573, S50–S52 (2019).
Correia, B. E. et al. Proof of principle for epitope-focused vaccine design. Nature 507, 201–206 (2014).
Xu, K. et al. Epitope-based vaccine design yields fusion peptide-directed antibodies that neutralize diverse strains of HIV-1. Nat. Med. 24, 857–867 (2018).
Jardine, J. G. et al. HIV-1 broadly neutralizing antibody precursor B cells revealed by germline-targeting immunogen. Science 351, 1458–1463 (2016). Seminal work demonstrating that engineered immunogen could engage B cells from HIV-uninfected individuals, generating a possibility that immunogen vaccine may elicit bnAbs in people.
Steichen, J. M. et al. HIV vaccine design to target germline precursors of glycan-dependent broadly neutralizing antibodies. Immunity 45, 483–496 (2016).
Tokatlian, T. et al. Innate immune recognition of glycans targets HIV nanoparticle immunogens to germinal centers. Science 363, 649–654 (2019). The study shows that glycosylation is critical for enhanced localization to B-cell follicles and inducing humoral immunity.
Kasturi, S. P. et al. 3M-052, a synthetic TLR-7/8 agonist, induces durable HIV-1 envelope-specific plasma cells and humoral immunity in nonhuman primates. Sci. Immunol. 5, eabb1025 (2020).
Wilson, D. S. et al. Antigens reversibly conjugated to a polymeric glyco-adjuvant induce protective humoral and cellular immunity. Nat. Mater. 18, 175–185 (2019). The study highlighted a synthetic polymeric glyco-adjuvant vaccine to elicit cellular and humoral immune responses against malaria.
Purwada, A., Roy, K. & Singh, A. Engineering vaccines and niches for immune modulation. Acta Biomater. 10, 1728–1740 (2014).
Singh, A. & Peppas, N. A. Hydrogels and scaffolds for immunomodulation. Adv. Mater. 26, 6530–6541 (2014).
Mesin, L., Ersching, J. & Victora, G. D. Germinal center B cell dynamics. Immunity 45, 471–482 (2016).
Roozendaal, R. et al. Conduits mediate transport of low-molecular-weight antigen to lymph node follicles. Immunity 30, 264–276 (2009).
Sixt, M. et al. The conduit system transports soluble antigens from the afferent lymph to resident dendritic cells in the T cell area of the lymph node. Immunity 22, 19–29 (2005).
Cyster, J. G. B cell follicles and antigen encounters of the third kind. Nat. Immunol. 11, 989–996 (2010).
Reddy, S. T. et al. Exploiting lymphatic transport and complement activation in nanoparticle vaccines. Nat. Biotechnol. 25, 1159–1164 (2007).
Reddy, S. T., Rehor, A., Schmoekel, H. G., Hubbell, J. A. & Swartz, M. A. In vivo targeting of dendritic cells in lymph nodes with poly(propylene sulfide) nanoparticles. J. Control. Release 112, 26–34 (2006).
Swartz, M. A., Hubbell, J. A. & Reddy, S. T. Lymphatic drainage function and its immunological implications: from dendritic cell homing to vaccine design. Semin. Immunol. 20, 147–156 (2008).
Prevo, R., Banerji, S., Ni, J. & Jackson, D. G. Rapid plasma membrane-endosomal trafficking of the lymph node sinus and high endothelial venule scavenger receptor/homing receptor stabilin-1 (FEEL-1/CLEVER-1). J. Biol. Chem. 279, 52580–52592 (2004).
Rodda, L. B. et al. Single-cell RNA sequencing of lymph node stromal cells reveals niche-associated heterogeneity. Immunity 48, 1014–1028 e1016 (2018).
Beguelin, W. et al. EZH2 enables germinal centre formation through epigenetic silencing of CDKN1A and an Rb-E2F1 feedback loop. Nat. Commun. 8, 877 (2017). The study reported a three-dimensional B-cell follicular organoid system that mimics the germinal centre reaction, to define a positive feedback loop in which B-cell epigenetics controls cell proliferation and humoral immunity.
Gitlin, A. D., Shulman, Z. & Nussenzweig, M. C. Clonal selection in the germinal centre by regulated proliferation and hypermutation. Nature 509, 637–640 (2014).
Tas, J. M. et al. Visualizing antibody affinity maturation in germinal centers. Science 351, 1048–1054 (2016). The study used brainbow-confetti mice and sequencing to demonstrate B-cell clonal competition with one another.
Schudel, A. et al. Programmable multistage drug delivery to lymph nodes. Nat. Nanotechnol. 15, 491–499 (2020).
Jardine, J. et al. Rational HIV immunogen design to target specific germline B cell receptors. Science 340, 711–716 (2013).
Jardine, J. G. et al. HIV-1 vaccines. Priming a broadly neutralizing antibody response to HIV-1 using a germline-targeting immunogen. Science 349, 156–161 (2015).
Zhang, Y. N. et al. Nanoparticle size influences antigen retention and presentation in lymph node follicles for humoral immunity. Nano Lett. 19, 7226–7235 (2019).
Kasturi, S. P. et al. Programming the magnitude and persistence of antibody responses with innate immunity. Nature 470, 543–547 (2011). A seminal study that showed nanovaccine containing immunomodulatory adjuvants can boost the magnitude and persistence of germinal centre and antibody responses in non-human primates.
Agarwal, R. et al. Mammalian cells preferentially internalize hydrogel nanodiscs over nanorods and use shape-specific uptake mechanisms. Proc. Natl Acad. Sci. USA 110, 17247–17252 (2013).
Akkaya, M. et al. Second signals rescue B cells from activation-induced mitochondrial dysfunction and death. Nat. Immunol. 19, 871–884 (2018).
Pulendran, B. Immunology taught by vaccines. Science 366, 1074–1075 (2019).
Hagan, T. et al. Antibiotics-driven gut microbiome perturbation alters immunity to vaccines in humans. Cell 178, 1313–1328 e1313 (2019). The study shows that the loss of microbiome impairs seasonal influenza vaccination antibody response in human subjects with low pre-existing immunity.
Oh, J. Z. et al. TLR5-mediated sensing of gut microbiota is necessary for antibody responses to seasonal influenza vaccination. Immunity 41, 478–492 (2014).
Mosquera, M. J. et al. Immunomodulatory nanogels overcome restricted immunity in a murine model of gut microbiome-mediated metabolic syndrome. Sci. Adv. 5, eaav9788 (2019). The study highlights immunomodulation under gut-mediated metabolic syndrome conditions using advanced nanomaterials.
Dosenovic, P. et al. Immunization for HIV-1 broadly neutralizing antibodies in human Ig knockin mice. Cell 161, 1505–1515 (2015).
Tian, M. et al. Induction of HIV neutralizing antibody lineages in mice with diverse precursor repertoires. Cell 166, 1471–1484.e18 (2016).
Abbott, R. K. et al. Precursor frequency and affinity determine B cell competitive fitness in germinal centers, tested with germline-targeting HIV vaccine immunogens. Immunity 48, 133–146.e6 (2018).
Sok, D. & Burton, D. R. Recent progress in broadly neutralizing antibodies to HIV. Nat. Immunol. 19, 1179–1188 (2018).
Kwong, P. D., Mascola, J. R. & Nabel, G. J. Broadly neutralizing antibodies and the search for an HIV-1 vaccine: the end of the beginning. Nat. Rev. Immunol. 13, 693–701 (2013).
Moody, M. A. et al. Immune perturbations in HIV-1-infected individuals who make broadly neutralizing antibodies. Sci. Immunol. 1, aag0851 (2016).
Haynes, B. F., Kelsoe, G., Harrison, S. C. & Kepler, T. B. B-cell-lineage immunogen design in vaccine development with HIV-1 as a case study. Nat. Biotechnol. 30, 423–433 (2012).
Kanekiyo, M. et al. Self-assembling influenza nanoparticle vaccines elicit broadly neutralizing H1N1 antibodies. Nature 499, 102–106 (2013).
Winarski, K. L. et al. Antibody-dependent enhancement of influenza disease promoted by increase in hemagglutinin stem flexibility and virus fusion kinetics. Proc. Natl Acad. Sci. USA 116, 15194–15199 (2019).
Bolles, M. et al. A double-inactivated severe acute respiratory syndrome coronavirus vaccine provides incomplete protection in mice and induces increased eosinophilic proinflammatory pulmonary response upon challenge. J. Virol. 85, 12201–12215 (2011).
Yang, Z. Y. et al. Evasion of antibody neutralization in emerging severe acute respiratory syndrome coronaviruses. Proc. Natl Acad. Sci. USA 102, 797–801 (2005).
Tseng, C. T. et al. Immunization with SARS coronavirus vaccines leads to pulmonary immunopathology on challenge with the SARS virus. PLoS ONE 7, e35421 (2012).
Iwasaki, A. & Yang, Y. The potential danger of suboptimal antibody responses in COVID-19. Nat. Rev. Immunol. 20, 339–341 (2020).
Wang, Q. et al. Immunodominant SARS coronavirus epitopes in humans elicited both enhancing and neutralizing effects on infection in non-human primates. ACS Infect. Dis. 2, 361–376 (2016).
Chambers, B. S., Parkhouse, K., Ross, T. M., Alby, K. & Hensley, S. E. Identification of hemagglutinin residues responsible for H3N2 antigenic drift during the 2014–2015 influenza season. Cell Rep. 12, 1–6 (2015).
Flannery, B. et al. Early estimates of seasonal influenza vaccine effectiveness — United States, January 2015. MMWR Morb. Mortal. Wkly Rep. 64, 10–15 (2015).
Yassine, H. M. et al. Hemagglutinin-stem nanoparticles generate heterosubtypic influenza protection. Nat. Med. 21, 1065–1070 (2015).
Deng, L. et al. Double-layered protein nanoparticles induce broad protection against divergent influenza A viruses. Nat. Commun. 9, 359 (2018).
Kamp, H. D. et al. Design of a broadly reactive Lyme disease vaccine. npj Vaccines 5, 33 (2020).
Pardi, N., Hogan, M. J., Porter, F. W. & Weissman, D. mRNA vaccines — a new era in vaccinology. Nat. Rev. Drug Discov. 17, 261–279 (2018).
Feldman, R. A. et al. mRNA vaccines against H10N8 and H7N9 influenza viruses of pandemic potential are immunogenic and well tolerated in healthy adults in phase 1 randomized clinical trials. Vaccine 37, 3326–3334 (2019).
Kose, N. et al. A lipid-encapsulated mRNA encoding a potently neutralizing human monoclonal antibody protects against chikungunya infection. Sci. Immunol. 4, eaaw6647 (2019).
Kim, S., Shah, S. B., Graney, P. & Singh, A. Multiscale engineering of immune cells and lymphoid organs. Nat. Rev. Mater. 4, 355–378 (2019). Review highlighting material strategies to recreate primary, secondary and tertiary immune organs in vivo and in vitro.
Purwada, A. et al. Ex vivo synthetic immune tissues with T cell signals for differentiating antigen-specific, high affinity germinal center B cells. Biomaterials 198, 27–36 (2019).
Purwada, A. & Singh, A. Immuno-engineered organoids for regulating the kinetics of B-cell development and antibody production. Nat. Protoc. 12, 168–182 (2017).
Jaroentomeechai, T. et al. Single-pot glycoprotein biosynthesis using a cell-free transcription-translation system enriched with glycosylation machinery. Nat. Commun. 9, 2686 (2018).
Graney, P. et al. Organoid polymer functionality and mode of Klebsiella neumoniae membrane antigen presentation regulates ex vivo germinal center epigenetics in young and aged B cells. Adv. Funct. Mater. https://doi.org/10.1002/adfm.202001232 (2020).
Havenar-Daughton, C. et al. Direct probing of germinal center responses reveals immunological features and bottlenecks for neutralizing antibody responses to HIV Env trimer. Cell Rep. 17, 2195–2209 (2016).
Acknowledgements
I acknowledge financial support from the National Institute of Allergy and Infectious Diseases of the US National Institutes of Health (7R01AI132738-04 awarded to A.S.) for the research covered.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
A.S. holds stocks or shares in Moderna, Inc.
Additional information
Peer review information Nature Nanotechnology thanks James Moon and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Singh, A. Eliciting B cell immunity against infectious diseases using nanovaccines. Nat. Nanotechnol. 16, 16–24 (2021). https://doi.org/10.1038/s41565-020-00790-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41565-020-00790-3
This article is cited by
-
Nanotechnology’s frontier in combatting infectious and inflammatory diseases: prevention and treatment
Signal Transduction and Targeted Therapy (2024)
-
Enhancing antibody responses by multivalent antigen display on thymus-independent DNA origami scaffolds
Nature Communications (2024)
-
Nanovaccines to combat drug resistance: the next-generation immunisation
Future Journal of Pharmaceutical Sciences (2023)
-
Facile synthesis of multi-faceted, biomimetic and cross-protective nanoparticle-based vaccines for drug-resistant Shigella: a flexible platform technology
Journal of Nanobiotechnology (2023)
-
Immunoregulatory nanomedicine for respiratory infections
Nature Reviews Bioengineering (2023)
