
Abstract
Photo-generated high-energy surface states can help to produce chlorine in aqueous environments. Here, aligned rutile (TiO2) nanocrystal arrays are grown onto fluorine-doped tin oxide (FTO) substrates and activated either by hydrothermal Sr/Ba surface doping and/or by vacuum-annealing. With vacuum-annealing, highly photoactive films are obtained with photocurrents of typically 8 mA cm−2 at 1.0 V vs. SCE in 1 M KCl (LED illumination with λ = 385 nm and approx. 100 mW cm−2). Photoelectrochemical chlorine production is demonstrated at proof-of-concept scale in 4 M NaCl and suggested to be linked mainly to the production of Ti(III) surface species by vacuum-annealing, as detected by post-catalysis XPS, rather than to Sr/Ba doping at the rutile surface. The vacuum-annealing treatment is proposed to beneficially affect (i) bulk semiconductor TiO2 nanocrystal properties and electron harvesting, (ii) surface TiO2 reactivity towards chloride adsorption and oxidation, and (iii) FTO substrate performance.

Photo-chlorine production is possible in aqueous chloride media under neutral conditions with rutile nanocrystal arrays under illumination from band gap–matched LEDs.
Similar content being viewed by others
Introduction
TiO2-based materials in doped or undoped forms are widely applied in photocatalysis, [1] but are generally found to be relatively inactive for photo-chlorine production [2]. This is likely to be linked, in part, to the insufficient adsorption of chloride anions onto the TiO2 surface. Studies in brine and metal cation–doped TiO2 materials have not been very successful [2]. Generally, titanium dioxide (TiO2) is a promising photoactive n-type semiconductor for use in solar energy conversion due to its high chemical stability, low cost, and non-toxicity [3]. TiO2 exhibits a relatively large bandgap, typically 3.0 to 3.2 eV [4], which allows effective energy absorption only in the blue/near-UV region. For TiO2-based materials, flat band potentials of approx. ‑ 0.40 V vs. SCE in 1 M acid [5] have been reported and observed to be sensitive to both pH and illumination [6].
The most widely suggested applications of TiO2 photocatalysis are in water decontamination and water splitting to hydrogen and oxygen [7, 8]. However, photocatalysts can also drive a diverse range of other mechanisms, including hydrogen peroxide formation [9], ammonia degradation [10], and photo-chlorine evolution [2, 11,12,13]. Commonly reported methods to improve TiO2 properties and performance are based on doping, usually by thermal annealing or by ion substitution/addition, and by tailoring the morphology and structure of the electrode to increase the surface area [14] and the extent of the more active crystal facets. Amorphous TiO2 has been reported for photoelectrochemistry [15]. Rare earth doping methods have also been developed to improve the photocatalytic activity of TiO2 materials [16]. A RuO2/TiO2/n-Si photoanode hybrid has been shown to provide high yields of photo-chlorine in brine [17]. Another alternative electrode reported for use in photo-chlorine evolution is based on n-type semiconductor WO3 films [18,19,20,21,22], with a relatively smaller band gap, better light absorption up to 500-nm wave ength, cost-effective manufacturing, and long-term stability. However, when employing effective band gap–matched LED light sources, TiO2-based materials become competitive and practical as there are no significant problems with spectral losses. TiO2 with a lower flat band potential, compared to that for WO3, is likely to operate at lower applied potentials. Furthermore, by employing a photoelectrochemical process as compared to dark electrolytic processes, the reactive surface species on the semiconductor surface (the “holes”) are produced locally with a light intensity–dependent rate and at relatively low applied potential. These conditions differ from those in dark electrocatalysis, for example on Ru/Ir-oxide type electrodes [23] and may aid chlorine production.
Chlorine is an essential chemical for modern society. For example, pure chlorine gas is used in a wide range of industrial manufacturing techniques, such as disinfection [24] and polymer production [25]. The dissociation of dissolved chlorine in aqueous solution forms hypochlorite (ClO−), which is a key component in disinfectants, bleaches, deodorisers, oxidising agents, and water treatment formulations. Due to the highly reactive nature of chlorine, it has virtually no natural abundance and improving production methods to satisfy the ever-increasing demand for this chemical is therefore of high interest. In particular, on-site production directly from brine with high flux is desirable. The production of chlorine from the ubiquitous sources of seawater and solar (or synthetic LED) radiation via photoelectrocatalysis could be beneficial in specific applications. There is also evidence in the literature on the applications of chlorine intermediates in the related photoelectrochemical pollutant degradation in chloride containing media [26,27,28]. The two biggest limitations preventing photoelectrocatalysis becoming a common industrial technique (e.g. in disinfection applications) are (i) finding a stable, but effective, anode material that does not need regular replacement, (ii) achieving an efficient and rapid rate of chlorine production, such that the demand can be fulfilled without additional running costs.
In this paper, the light-driven production of chlorine in neutral aqueous KCl or NaCl is investigated. The effect of pH on the production of chlorine has been extensively studied for the TiO2 thin film photocatalyst [29]. A basic pH (pH > 7) showed a negligible chlorine production, while increasing acidity (pH < 6) resulted in increasing chlorine production. This might suggest some conversion of chlorine to hypochlorite, but also more importantly, that chloride adsorption is favoured only in more acidic environments with less hydroxide to compete with. As the pH is increased further into a basic regime, hypochlorite may also undergo further disproportionation reactions into chlorate [30].
Catalytic reactions occur on active sites on the surface of the catalyst materials, indicating that a large surface area is vital for a highly efficient catalytic process. An important step in designing the structure of a catalyst is therefore identifying the size and shape requirement of the material to maximise its efficiency. TiO2 has been shown to have a photoelectrochemical activity that is dependent on its shape and crystal orientation [31, 32]. Quasi-1-dimensional TiO2 nanorods and nanotubes are promising nanostructures not only due, in part, to their exceptionally high surface area but also importantly because of heightened charge transport properties. The increased charge transport results in faster charge carrier diffusion (e− over longer distances and h+ over shorter distances), thereby extending the charge carrier lifetime and energy harvesting efficiency [33]. When applied to an oxidation catalysis reaction, such as photo-chlorine evolution, the reduced required diffusion distance for the hole to reach the surface in nanorods, compared to alternative structures such as crystalline thin films, is likely to result in an increased rate of catalysis and higher photocurrents. More holes will reach the surface and react with weakly adsorbed chloride/hydroxide anions before any recombination can occur. The more mobile electrons can be harvested along the longer axis of the nanorods.
The synthesis of rutile TiO2 nanorods or nanocrystal arrays by hydrothermal methods is a relatively convenient technique that is currently employed to produce nanorods for a variety of applications [34, 35]. Rutile nanocrystals can be produced with defined shape and size. Using hydrothermal methods, the duration and temperature of synthesis can be used to tailor the size of the nanorods and the photoelectrochemical activity. Many methods other than hydrothermal synthesis have been tested; however, the hydrothermally synthesised nanocrystals generally provide a greater photoelectrochemical activity [32]. Anatase, a different TiO2 phase, has also been reported to be synthesised in a range of geometries (but without attachment to a substrate), including nanorods, nanocubes, and nanosheets, by tailoring the specific concentrations of surface complexing ligands [36]. Here, rutile nanorods are employed grown directly onto a fluorine-doped tin oxide (FTO) substrate.
Improved TiO2 nanocrystal array photocatalytic performance has been reported through the use of a thermal annealing treatment after synthesis, used for many functions including the removal of surface hydroxyl groups [37] and creating oxygen vacancies [38]. Annealing in a hydrogen environment was tested by Wang et al. [39] and found to increase the density of oxygen vacancies, as well as increasing the generated photocurrent densities compared to pristine TiO2 nanocrystal arrays. The impact of oxygen vacancies on the photoelectrochemical activity of TiO2 was discussed by Huang and coworkers [40]. Four different annealing environments were used to treat pristine TiO2 nanocrystal arrays to produce structurally and morphologically identical nanorods, but with different TiO2 oxygen contents. Vacuum-annealing offers a convenient methodology more generally to post-synthetically “activate” semiconductor materials. TiO2 has been shown previously to become more photoactive [41]. However, SnO2-based substrates have also been shown to provide better performance [42, 43] due to their increased conductivity due to oxygen vacancy doping. In this report, we compare Sr/Ba surface doping and vacuum-annealing for the activation of rutile nanocrystal arrays on fluorine-doped tin oxide (FTO) for applications in photoelectrochemistry. We demonstrate vacuum-annealing of rutile nanocrystal arrays as a convenient method to “activate” the rutile surface for photo-chlorine production and to enhance charge carrier transport and collection.
Experimental
Chemical Reagents
KCl (99–100.5%), NaCl (99.5%), BaCl2 dihydrate (> 99%), SrCl2 hexahydrate (99%), HCl (37%), and titanium(IV) butoxide (97%) were obtained in analytical or the purist available grade from Sigma-Aldrich and used without further purification. Ultra-pure water (demineralised and filtered, resistivity 18.2 MΩ cm at 22 °C) was taken from an ELGA Purelab Classic system.
Instrumentation
Voltammetry
A blue LED light source (Thorlabs ML385, λ = 385 nm, approx. 100 (± 20) mW cm−2 depending on the positioning of the electrode and light source) was employed in pulse mode (usually 5-s on, 5-s off) to demonstrate photocurrents and dark background currents during cyclic voltammetry scans with a 10 mV s−1 scan rate, measured employing an AUTOLAB Type III potentiostat (Metrohm, UK). A three-electrode electrochemical cell was employed (made from Pyrex glass transparent > 350 nm with the LED positioned approx. 4 cm from the working electrode) containing the nanocrystal array photocatalyst working electrode (WE), a platinum wire counter electrode (CE), and a saturated calomel electrode (SCE) as the reference electrode (Fig. 1E). The electrolyte was either a potassium chloride (KCl) or sodium chloride (NaCl, used only for higher concentration experiments) solution of varying concentrations (ranging from 10 mM to 4 M).

Photographic appearance of (A) FTO-coated glass substrate, (B) pristine TiO2 nanocrystal array deposit, (C) Sr/Ba-TiO2 as-deposited nanocrystal array, and (D) vacuum-annealed Sr/Ba-doped TiO2 nanocrystal array. The scale is indicated by the horizontal lines, which are spaced by 7.1 mm. (E) Schematic of the photoelectrochemical cell with working, counter, and reference electrode
Characterisation
The morphology of the nanocrystals was measured using a field-emission scanning electron microscope (FESEM, JEOL JSM-6301F) and a transmission electron microscope (TEM, JEOL JEM-2100Plus). The TEM was used in conjunction with an Oxford instruments X-MaxN TSR silicon drift detector EDX. Raman spectroscopy was carried out on a Renishaw inVia system, using both visible and UV wavelength lasers, in order to probe molecular composition and structure. Finally, X-ray photoelectron spectroscopy (XPS, Thermo Fisher K-Alpha+) was used to probe the surface elemental compositions, distributions, and oxidation states.
Photoelectrode Preparation
A titanium dioxide (TiO2) nanocrystal array was first grown on a fluorine-doped tin oxide (FTO, AGC type U TCO glass, Japan) coated glass substrate by a hydrothermal synthesis reaction outlined by Zhang and coworkers [34]. Briefly, an FTO-coated glass substrate was ultrasonically cleaned for 30 min in a 1:1 solution of ethanol and acetone and placed inside a 100-mL Teflon lined stainless steel autoclave with the conductive FTO-coated side facing upwards. A solution containing 0.7 g of titanium(IV) butoxide in a 50-mL mixture of 1:1 concentrated hydrochloric acid (37%), and de-ionized water was stirred for 30 min and pipetted onto the FTO-coated substrate. The autoclave was sealed and heated in an oven at 180 °C for 3 h. The uniform nanocrystal array growth occurs due to lattice mismatches between the rutile TiO2 and the tetragonal FTO structures [34].
Vacuum-annealing and/or metal cation (strontium and barium) doping methods were then applied in order to modify the surface activity to achieve improved photoelectrochemical properties. Briefly, a 50-mL mixture of 0.02 M barium chloride and 0.02 M strontium chloride was prepared by adding concentrated HCl (37%) dropwise (approx. 0.50–0.75 mL total) to a 0.02 M barium hydroxide and 0.02 M strontium hydroxide solution until they were fully dissolved. The solution was then left to stir for approximately 12 h and sealed inside an autoclave with a TiO2 nanocrystal array sample for 10 h at 200 °C. Finally, the resulting strontium-/barium-doped TiO2 (Sr/Ba-TiO2) was washed with 0.2 M HCl, demineralised water, and ethanol. Both the TiO2 nanocrystal array and the as-deposited Sr/Ba-TiO2 nanocrystal array were annealed in a vacuum environment (Speedivac Edwards High Vacuum Pump ED50, Crawley, England) at 550 °C (the maximum temperature tolerated by these FTO substrates) for 4 h to create oxygen vacancies in the structure, resulting in vacuum-annealed TiO2 (VA-TiO2) and vacuum-annealed Sr/Ba-TiO2 (VA-Sr/Ba-TiO2) respectively. Initial appearances of the samples, as seen in Fig. 1, show a transition from transparent to opaque upon growth of the nanorods and some darkening after vacuum-annealing. All electrodes were coated in silver paint at one end to form a conductive contact with the FTO layer beneath the nanorods. Copper tape was applied over the silver paint, followed by an insulating silicon sealant and Kapton tape in order to prevent electrolyte from contacting the conductive copper or silver. An active solution exposed area of 1 cm2 was ensured for all electrodes.
Bipotentiostatic Chlorine Detection
Detection of chlorine was carried out using the same cell and the same working electrode (WE1), reference electrode (SCE), and counter electrode (CE) as described above, but with an additional 125-μm-diameter Teflon-coated platinum wire (Advent Research Materials, UK) as a second working electrode (WE2). An Ivium Technologies Compactstat Electrochemical Interface was used as the bipotentiostat. The electrolyte consisted of a 4 M NaCl aqueous solution. One end of the Teflon-coated wire was placed in a Bunsen burner to remove the Teflon coating, followed by the application of a copper tape contact onto the revealed platinum wire. The other end was cut perpendicularly with a sharp knife to reveal approx. a 125-μm-diameter cross-section of the platinum wire, which was placed very near (in approx. 250 μm distance) to the surface of the first working electrode immersed in the electrolyte. The potential of the second working electrode was set to potentials from 0.5 V vs. SCE to 0.80 V vs. SCE and to 0.85 V vs. SCE. In these experiments, the LED light source was pulsed for 10 s on 10 s off, with a potential scan rate of 5 mV s−1.
Results and Discussion
Formation and Characterisation of Surface-Doped TiO2
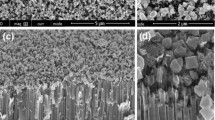
TiO2 nanocrystal arrays were grown following a procedure developed by Zhang et al. [34], see experimental. Figure 2a shows typical cross-sectional electron microscopy images of the aligned nanocrystal arrays, which are typically 1 μm in length and 40–80 nm in diameter, with an aspect ratio of 20. The top view (Fig. 2b) demonstrates the uniform distribution of the nanocrystals, with sufficient space for electrolyte to permeate. The Sr/Ba treatment under hydrothermal conditions leads to no significant change in appearance (see Fig. 2c) apart from some probably carbonate-containing debris. A wash with aqueous HCl and vacuum-annealing then produces the material shown in Fig. 2d.

Scanning electron microscopy (SEM) cross-sectional image for as-grown hydrothermal TiO2 nanocrystals (a); top view for as-grown hydrothermal TiO2 nanocrystals (b); Sr/Ba-doped TiO2 nanocrystals (c); and activated and vacuum-annealed TiO2 nanocrystals (d)
TEM analysis of TiO2 nanocrystals, shown in Figure SI1, after ultrasonic lift-off from the FTO substrate reveals a high degree of crystallinity for all four materials: (i) as-grown TiO2, (ii) Sr/Ba-treated TiO2, (iii) Sr/Ba-treated and vacuum-annealed TiO2, and (iv) only vacuum-annealed TiO2. There is no significant difference based on TEM data between the different materials. Figure 3 shows TEM images and elemental mapping for Sr/Ba-treated and vacuum-annealed TiO2. The elemental content for Ti and O is high, but for Sr and Ba, the mapping suggests elemental levels that are just at, or possibly below, the detection limit. In part, this could be linked to the washing step with 0.2 M HCl, but fundamentally there is very little Sr/Ba to affect the reactivity (see also XPS data presented below).

a–e TEM image and elemental mapping for Sr/Ba-treated and vacuum-annealed TiO2 nanocrystals
When investigating the electron diffraction data in Fig. 4a, there is evidence that rutile crystal structures are obtained for all materials with diffraction spots matching stronger diffraction lines from the corresponding JCPDS data sets (observed: 2.94 nm−1, 3.99 nm−1, 4.37 nm−1, 5.87 nm−1, 7.30 nm−1; expected for rutile: 3.080 nm−1, 4.021 nm−1, 4.353 nm−1, 4.570 nm−1, 5.928 nm−1, 7.354 nm−1 [JCPDS 21–1276]: expected for anatase: 2.841 nm−1, 4.113 nm−1, 4.205 nm−1, 4.288 nm−1, 5.285 nm−1, 5.883 nm−1, 6.001 nm−1, 6.753 nm−1 [JCPDS 21–1272]). This is confirmed from the Raman data analysis, where the results in Fig. 4b–e confirm that the TiO2 nanocrystal array was rutile, as opposed to other TiO2 phases [44]. The characteristic Raman peak positions and reciprocal lattice spacings are obtained. Neither the visible nor UV wavelength spectra showed any peaks other than rutile TiO2.

a TEM electron diffraction pattern with rutile (TiO2) indexing. b Raman (λexcitation = 532 nm) for as-grown TiO2. c Raman (λexcitation = 532 nm) for Sr/Ba-treated and vacuum-annealed TiO2. d Raman (λexcitation = 325 nm) for as-grown TiO2. e Raman (λexcitation = 325 nm) for Sr/Ba-treated and vacuum-annealed TiO2
Photoelectrochemical Characterisation of Surface-Doped TiO2
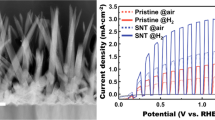
Initial photoelectrochemical experiments were performed with a 1 cm2 TiO2-coated FTO electrode immersed into an aqueous 0.1 M KCl solution. Light pulses were generated from an LED (λ = 385 nm) in the blue region of the visible spectrum. The typical rutile band gap is 3.0 eV [45], corresponding to 412 nm and therefore in the region of excitation with 385-nm light. When scanning the applied potential from − 0.25 V vs. SCE to 1.25 V vs. SCE, clear anodic photocurrent pulses are observed (Fig. 5a). The onset of photocurrent response should occur close to the flatband potential, which is close to − 0.1 V vs. SCE in neutral solution and in the dark [46]. For as-grown TiO2, the photocurrents reach typically 0.5 mA cm−2 at 1 V vs. SCE, but for the Sr/Ba-treated and vacuum-annealed material, an order of magnitude higher photocurrents are detected, as seen in trace (i) of Fig. 5a.

a Photoelectrochemical voltammograms (scan rate 10 mVs−1, light on 5 s and light off 5 s, LED with λ = 385 nm, approx. 100 mW cm−2) for a 1 cm2 TiO2 nanocrystal electrode (i) Sr/Ba-treated and vacuum-annealed and (ii) as-grown TiO2 immersed in aqueous 0.1 M KCl. b Photoelectrochemical voltammograms (as above) for a 1 cm2 TiO2 nanocrystal electrode (Sr/Ba-treated and vacuum-annealed) immersed in aqueous (i) 1 M and (ii) 0.01 M KCl. c Plot of photocurrent data at 1 V vs. SCE (for a Sr/Ba-treated and vacuum-annealed and for only vacuum-annealed TiO2 electrodes; device to device error estimated as ± 10%) as a function of chloride concentration
The effect of the chloride concentration on photocurrent responses is demonstrated in Fig. 5b. Photocurrent data obtained for as-grown TiO2 nanocrystals are only weakly affected by the chloride concentration, but for Sr/Ba-treated and vacuum-annealed electrodes, a strong increase occurs, reaching up to 10 mA cm−2 at high salinity. Interestingly, the same effect occurs for TiO2 nanocrystals that have only been treated by vacuum-annealing. Therefore, these effects are not necessarily due to Sr/Ba doping. The plot in Fig. 5c summarises the photocurrent data at 1.0 V vs. SCE. Device-to-device errors are estimated as ± 10%, but trends in the data are clear. An approximate light intensity of 100 mW cm−2 assumed at the location of the working electrode suggests a flux of 1.9 × 1017 photons s−1, or correspondingly 31-mA photocurrent. The opaque appearance of electrodes in Fig. 1 suggests effective photon absorption and low transparency. Therefore, at 1.0 V vs. SCE, approximately a third of the photons are converted to a photocurrent. Note, that the nature of the products from the photocurrents in Fig. 5 is not experimentally verified. The increase in photocurrent with chloride concentration could be an indication of chlorine production, but without direct evidence for this, it has to be assumed that oxygen evolution (which is kinetically more difficult but within the accessible pH range thermodynamically preferred over chlorine evolution [47]) may also occur.
Next, the annealing method is investigated to reveal more insights on the origin of the exceptionally high photocurrents. The Sr/Ba-treated electrodes were either annealed in ambient air or under vacuum conditions, in both cases at 550 °C for 4 h. Figure 6a shows the significant effect of vacuum-annealing on photoelectrochemical responses. Vacuum conditions have been shown previously to result in the formation of oxygen vacancies and thereby in increased electron mobility and photo-reactivity in TiO2 [41], as well as in other oxides. The data in Fig. 6b have been obtained for as-grown TiO2 nanocrystals after vacuum-annealing and for Sr/Ba-treated and vacuum-annealed TiO2 nanocrystals. Clearly, the presence of Sr or Ba is not essential, and the vacuum-annealing step is the most important part of the process to result in highly photoactive TiO2 film electrodes. An important question is therefore how does the vacuum-annealing step enhance the photoelectrochemical activity?

a Photoelectrochemical voltammograms (scan rate 10 mV s−1, light on 5 s and light off 5 s, LED with λ = 385 nm, approx. 100 mW cm−2) for a 1 cm2 TiO2 nanocrystal electrode (Sr/Ba-treated and (i) vacuum-annealed or (ii) air annealed) immersed in aqueous 1 M KCl. b Photoelectrochemical voltammograms (as above) for a 1 cm2 TiO2 nanocrystal electrode (Sr/Ba-treated and vacuum-annealed (i) or only vacuum-annealed without Sr/Ba treatment (ii)) immersed in aqueous 1 M KCl
Figure 7 provides a summary of the suggested hypothetical mechanism by which vacuum-annealing can increase the collection of charge carriers and the resulting photocurrents. Due to additional oxygen vacancies in the rutile structure, electrons can be transported over longer distances and the applied bias at the working electrode can be employed to “harvest” these electrons from the entire rutile nanocrystals. A point of inflection occurs at approx. 0.9 V vs. SCE, which suggests a gradual approach towards a plateau outside of the measurement range (> 1.25 V vs. SCE). The plateauing could suggest that, at high applied potentials, electron harvesting is extended over the complete length of the nanocrystal. This may lead to photo-chlorine production over the complete nanorod surface, but direct experimental evidence for chlorine production is necessary.

Photoelectrochemical voltammogram (scan rate 10 mV s−1, light on 5 s and light off 5 s, LED with λ = 385 nm, approx. 100 mW cm−2) for a 1 cm2 TiO2 nanocrystal electrode (Sr/Ba-treated and vacuum-annealed) immersed in aqueous 1 M KCl with schematics to illustrate the field-driven electron harvesting in TiO2 nanocrystals
Chlorine Evolution on Surface-Doped TiO2
There have been reports in the literature on the possibility of chlorine generation from brine using titania photocatalyst materials [2]. However, success to date has been limited and it was observed that even doping with a range of cations [2] was insufficient to achieve a reproducible photo-response for practical applications. TiO2 nanotubes on titanium substrates have been reported to give rise to photo-chlorine evolution in the presence of UV light [48]. Here, a platinum detector electrode is employed (as a secondary working electrode) to directly monitor chlorine production, at least in a qualitative manner.
Figure 8 shows a cyclic voltammogram obtained at a 125-μm-diameter Pt electrode (Teflon-coated) immersed in aqueous 4 M NaCl. Under these conditions, scanning the potential from 0.5 V vs. SCE initially leads to oxygen reduction (see [49]). Following this, a broad reduction in current response is observed at potentials negative of − 0.5 V vs, SCE, which is associated with hydrogen production. This process is accompanied with hydroxide formation in the unbuffered salt solution. Upon reversal of the scan direction, hydrogen oxidation in the presence of hydroxide is observed and is followed by a swing in pH and a secondary hydrogen oxidation response in the presence of locally produced acid at − 0.25 V vs. SCE. The formation of chlorine is observed as a chemically reversible signal at 1.2 V vs. SCE. However, the chlorine reduction response is weak, mainly due to (i) a high degree of spherical diffusion that diminishes product peaks and (ii) chlorine under these conditions reacting to give rise to secondary products, such as hypochlorite. Importantly, a reduction signal detected at the platinum electrode with a potential positive of 0.5 V vs. SCE has to be linked to the formation of chlorine (or hypochlorite). Therefore, the 125-μm-diameter platinum electrode is employed as a detector electrode (secondary working electrode) for chlorine in the photoelectrochemical experiments.

Cyclic voltammogram (scan rate 0.1 Vs−1) using a 125-μm-diameter Pt wire electrode (Teflon-coated; used as WE2 in Fig. 10) as the working electrode. Positions of hydrogen, oxygen, and chlorine redox reactions are shown. Aqueous 4 M NaCl was used as the electrolyte
With the ability to distinguish oxygen photo-products from photo-chlorine production, a generator-collector type experiment (see Fig. 9) can be designed to demonstrate chlorine production. Figure 9d illustrates the experimental design with a 125-μm-diameter Teflon-coated Pt wire electrode placed close to the photoanode (approx. 250 μm in distance). Each light pulse is supposed to lead to a pulse of chlorine, which is then observed after a slight delay at the second working electrode located in close proximity to the TiO2 electrode.

a Photoelectrochemical voltammograms (scan rate 5 mV s−1, light on 10 s and light off 10 s, LED with λ = 385 nm, approx. 100 mW cm−2) for a 1 cm2 TiO2 nanocrystal electrode (Sr/Ba-treated and vacuum-annealed, WE1) immersed in aqueous 4 M NaCl. A platinum wire (125 μm diameter, Teflon-coated, WE2) served as a collector electrode in approx. 250-μm distance from WE1 (potential WE2 = 0.5 V vs. SCE). b As above with WE2 = 0.8 V vs. SCE. c As above with WE2 = 0.85 V vs. SCE. d Schematic to explain the generator—collector photoelectrochemical experiment for chlorine detection
Data in Fig. 9a show photocurrents for a Sr/Ba-treated and vacuum-annealed TiO2 nanocrystal electrode immersed in aqueous 4 M NaCl. The second working electrode biased at 0.5 V vs. SCE clearly shows a reduction current with the same peak pattern that is linked to the pulsed light excitation. Due to the lack of calibration of the current at the secondary electrode and the uncertain experimental geometry, the data need to be considered qualitative. The current reaches 1.5 μA and, for smaller diameter electrodes, this could be used to estimate the local concentration as cchlorine = Ilim/4nFDr = 15 mol m−3 [50], which seems reasonable, although this has to be considered an approximate value under these conditions. A limiting current of 6 mA for a 1 cm2 area for a natural convection based localised Nernst diffusion layer [51] (with diffusion layer thickness δ = 0.5 mm) should produce an average concentration of cchlorine = Ilim,photo δ/nFDA = 80 mol m−3 at the photoelectrode surface or approx. 40 mol m−3 at the location of the platinum sensor electrode. Given the approximations employed, this seems to be similar to the value estimated from the detector current data and indicative of a high degree of chlorine and/or hypochlorite formation. When setting the potential bias for the platinum detector electrode to 0.80 V vs. SCE or 0.85 V vs. SCE, there is still a reduction in current detected, although less significant than that observed at 0.5 V vs. SCE (see Fig. 9b and c, respectively). TiO2 nanocrystal array electrodes produced with Sr/Ba treatment and vacuum-annealing clearly are just as effective as electrodes that are simply vacuum-annealed. It is therefore of interest to explore the XPS surface analysis for electrode after performing photoelectrochemical chlorine evolution experiments, which are reported next.
The surface elemental analysis with XPS, which is applied after using the electrodes in photo-chlorine generation, can help revealing the surface chemistry for active photoelectrocatalysts with respect to observed photoelectrochemistry. Figure 10 shows survey data, Ti2p data, and O1s data for three types of samples: (a–c) as-grown TiO2 after use in photoelectrochemistry, (d–f) vacuum-annealed TiO2 after use in photoelectrochemistry, and (g–i) Sr/Ba-treated and vacuum-annealed TiO2 after use in photoelectrochemistry. Samples that should contain Sr/Ba only show a very small signal for trace Sr (not shown) indicating that Sr/Ba treatment has little or no effect on the surface composition/reactivity during photoelectrochemistry. However, vacuum-annealing appears to have a strong effect on the presence of Ti3+ and Ti2O3, including the presence of non-lattice oxygen. Data are summarised in Table 1.

a–c XPS survey data, Ti2p data, and O1s data for as-grown TiO2 after use in photoelectrochemistry. d–f XPS survey data, Ti2p data, and O1s data for vacuum-annealed TiO2 after use in photoelectrochemistry. g–i XPS survey data, Ti2p data, and O1s data for Sr/Ba-treated and vacuum-annealed TiO2 after use in photoelectrochemistry.
Peak fitting for the titanium oxide surface environments for the Ti2p signal (Fig. 10b, e, h) shows strong Ti4+ peaks, identified as the TiO2 titanium species by its characteristic binding energies [52]. Two distinct peaks are seen (2p1/2 and 2p3/2) due to spin-orbit coupling. In pristine TiO2 (b), the peaks for Ti3+ are negligibly small; however, in the vacuum-annealed samples (e and h), there are substantial intensities for Ti3+. Vacuum-annealing is known to give rise to oxygen vacancies, which act as 2-electron donors and reduce Ti4+ to Ti3+. The maximum penetration depth in XPS is around 5 nm; therefore, the species identified must be present in the outermost surface layers. Usually, oxygen vacancies associated with Ti3+ move into the bulk to become more stable. However, in this case, it can be seen that Ti3+ sites are to some degree retained on the surface even after use in photoelectrochemistry (all samples characterised by XPS had been previously employed in photo-chlorine production), implying that use in a photoelectrocatalytic reaction does not cause the Ti3+ to be lost from the surface. The retention of the Ti3+ at the surface will likely have a significant impact on the surface properties and behaviour of the electrode.
Oxygen surface environments also show a dramatic change upon vacuum-annealing, see Fig. 10c, f, and i. The non-lattice oxygen peak, which is likely to be an amorphous or surface oxide species with a higher binding energy (lower electron density) than the O2− (from TiO2) peak, becomes more intense after vacuum-annealing. Photoelectrochemical reactions of these electrodes include an applied bias potential to an electrode immersed in a salt solution; this could lead to surface contamination, for example with some silicate which could contribute to the non-lattice oxygen signal. The Ti2O3 peak corresponds to the interactions between the Ti3+ cations and nearby oxygen. The non-lattice oxygen is likely to be linked (at least in part) to silicate or stannate surface species [53]. Si2p (102.5 eV) and Si1s (approx. 152 eV) signals are seen after annealing and this is associated with a large increase in O1s at the location of silicate oxygen peaks [54].
The photo-chlorine production appears to take place in a high concentration NaCl solution. In this exploratory study, photocurrents are enhanced by vacuum-annealing of rutile nanocrystal arrays and this effect can be linked hypothetically to (i) Ti3+ sites at the rutile surface allowing chloride anions to bind and react with photo-generated holes, and (ii) the improved transport of conduction band electrons in the presence of oxygen vacancies allowing better electron harvesting. In addition, (iii) the interface of TiO2 to SnO2 could be affected by annealing to provide improved electron harvesting due to band bending [55] as well as improving FTO conduction. Importantly, more work will be required to better investigate the long-term persistence of Ti3+ surface states under more harsh conditions and the mechanism of the rutile nanorod photo-chlorine production in terms of effective use of light energy in the chlorine production.
Conclusions
It has been shown that vacuum-annealing can be used to significantly enhance the efficiency of photo-chlorine production with rutile nanocrystal arrays on FTO substrates. This has been attributed to a combination of effects based on oxygen vacancy states forming and enhancing rutile majority carrier transport, formation of Ti3+ surface sites at the rutile | aqueous interface to allow effective chloride binding and transformation, improved conduction across the whole length of the rutile nanocrystal with sufficiently high applied positive potential bias, and improvements due to the underlying FTO substrate becoming more conductive. The contribution from each of these factors and the rate of chlorine production are currently not fully quantified. However, the results seem to suggest that vacuum-annealing produces the over-riding effect on the behaviour of these photocatalysts. XPS data suggest that the Ti3+ surface sites are persistent at least over a period of hours under photoelectrochemistry conditions, but for longer periods of use further study will be necessary to establish persistence and performance. Further improvements by optimisation are possible and more work will be required to develop the photo-chlorine production. Importantly, in the future, an up-scaled version of the reactor will be necessary for more quantitative evaluation of chlorine and hypochlorite production under more realistic conditions and for assessing the behaviour of the electrode during long-term operation conditions required for industrial purposes.
References
O. Carp, C.L. Huisman, A. Reller, Photoinduced reactivity of titanium dioxide. Prog. Solid State Chem. 32(1-2), 33–177 (2004)
M.V.B. Zanoni, J.J. Sene, H. Selcuk, M.A. Anderson, Environ. Sci. Technol. 38, 3203 (2004)
S.H. Shen, J. Chen, M. Wang, X. Sheng, X.Y. Chen, X.J. Feng, S.S. Mao, Titanium dioxide nanostructures for photoelectrochemical applications. Prog. Mater. Sci. 98, 299–385 (2018)
X.L. Kang, S.H. Liu, Z.D. Dai, Y.P. He, X.Z. Song, Z.Q. Tan, Catalysis 9, 191 (2019)
T. Berger, D. Monllor-Satoca, M. Jankulovska, T. Lana-Villarreal, R. Gomez, The electrochemistry of nanostructured titanium dioxide electrodes. ChemPhysChem 13(12), 2824–2875 (2012)
T. Yoko, A. Yuasa, K. Kamiya, S. Sakka, Sol‐gel‐derived TiO2 film semiconductor electrode for photocleavage of water: preparation and effects of postheating treatment on the photoelectrochemical behavior. J. Electrochem. Soc. 138(8), 2279–2285 (1991)
X. Zhang, H. Cui, M. Humayun, Y. Qu, N. Fan, X. Sun, L. Jing, Sci. Report. 6, 21430 (2016)
K. Maeda, Photocatalytic water splitting using semiconductor particles: history and recent developments. J Photochem Photobiol C: Photochem Rev 12(4), 237–268 (2011)
T. Baran, S. Wojtyła, A. Vertova, A. Minguzzi, S. Rondinini, Photoelectrochemical and photocatalytic systems based on titanates for hydrogen peroxide formation. J. Electroanal. Chem. 808, 395–402 (2018)
S. Xiao, D. Wan, K. Zhang, H. Qu, J. Peng, Enhanced photoelectrocatalytic degradation of ammonia by in situ photoelectrogenerated active chlorine on TiO 2 nanotube electrodes. J. Environ. Sci. 50, 103–108 (2016)
D. Rosestolato, J. Fregoni, S. Ferro, A. De Battisti, Influence of the nature of the electrode material and process variables on the kinetics of the chlorine evolution reaction. The case of IrO2-based electrocatalysts. Electrochim. Acta 139, 180–189 (2014)
I.A. Moreno-Hernandez, B.S. Brunschwig, N.S. Lewis, Crystalline nickel, cobalt, and manganese antimonates as electrocatalysts for the chlorine evolution reaction. Energy Environ. Sci. 12(4), 1241–1248 (2019)
J. Prez-Ramrez, C. Mondelli, T. Schmidt, O.F.K. Schluter, A. Wolf, L. Mleczko, T. Dreier, Energy Environ. Sci. 4, 4786 (2011)
C. Boyle, N. Skillen, H.Q.N. Gunaratne, P.K. Sharma, J.A. Byrne, P.K.J. Robertson, The use of titanium dioxide nanotubes as photoanodes for chloride oxidation. Mater. Sci. Semicond. Process. 109, 104930 (2020)
S.D. Sun, P. Song, J. Cui, S.H. Liang, Amorphous TiO2nanostructures: synthesis, fundamental properties and photocatalytic applications. Catal. Sci. Technol. 9(16), 4198–4215 (2019)
N.U. Saqib, R. Adnan, I. Shah, Environ. Sci. Pollut. Res. 23, 15941 (2016)
Z.L. Liu, Z.L. Yin, Z.M. Wan, L. Xiao, H.L. Ma, J.T. Lu, L. Zhuang, Efficient solar-to-chemical conversion with chlorine photoanode. Electrochem. Commun. 67, 69–72 (2016)
S. Ahmed, I.A.I. Hassan, H. Roy, F. Marken, Photoelectrochemical transients for chlorine/hypochlorite formation at “roll-on” nano-WO3film electrodes. J. Phys. Chem. C 117(14), 7005–7012 (2013)
M. Jadwiszczak, K. Jakubow-Piotrowska, P. Kedzierzawski, K. Bienkowski, J. Augustynski, Highly efficient sunlight‐driven seawater splitting in a photoelectrochemical cell with chlorine evolved at nanostructured WO3photoanode and hydrogen stored as hydride within metallic cathode. Adv. Energy Mater. 10(3), 1903213 (2020)
Y. Shi, Y. Li, X. Wei, J. Feng, H. Li, W. Zhou, Facile preparation of porous WO3 film for photoelectrochemical splitting of natural seawater. J. Electron. Mater. 46(12), 6878–6883 (2017)
M.S. Koo, X.F. Chen, K. Cho, T.C. An, W. Choi, Environ. Sci. Technol. 53(16), 9926–9936 (2019)
R. Solarska, B.D. Alexander, A. Braun, R. Jurczakowski, G. Fortunato, M. Stiefel, T. Graule, J. Augustynski, Tailoring the morphology of WO3 films with substitutional cation doping: effect on the photoelectrochemical properties. Electrochim. Acta 55(26), 7780–7787 (2010)
D. Pletcher, F. C. Walsh, Industrial electrochemistry, (Springer, Dordrecht, 1993)
L.E. Fraga, M.A. Anderson, M.L.P.M.A. Beatriz, F.M.M. Paschoal, L.P. Romao, M.V.B. Zanoni, Electrochim. Acta 54(2069) (2009)
D. Wintrich, D. Öhl, S. Barwe, A. Ganassin, S. Möller, T.S. Tarnev, A. Botz, A. Ruff, J. Clausmeyer, J. Masa, W. Schuhmann, Enhancing the selectivity between oxygen and chlorine towards chlorine during the anodic chlorine evolution reaction on a dimensionally stable anode. ChemElectroChem 6(12), 3108–3112 (2019)
R. Leite de Oliveira, M.A. Anderson, G. de Aragão Umbuzeiro, G.J. Zocolo, M.V.B. Zanoni, Assessment of by-products of chlorination and photoelectrocatalytic chlorination of an azo dye. J. Hazard. Mater. 205-206, 1–9 (2012)
I. Olvera-Rodríguez, R. Hernández, A. Medel, C. Guzmán, L. Escobar-Alarcón, E. Brillas, I. Sirés, K. Esquivel, Sep. Purif. Technol. 224, 189 (2019)
K. Changanaquia, H. Alarcón, E. Brillas, I. Sirésa, Blue LED light-driven photoelectrocatalytic removal of naproxen from water: kinetics and primary by-products. J. Electroanal. Chem. 867, 114192 (2020)
T.X. Wang, D.W. Margerum, Kinetics of reversible chlorine hydrolysis: temperature dependence and general-acid/base-assisted mechanisms. Inorg. Chem. 33(6), 1050–1055 (1994)
H. Selcuk, M.A. Anderson, Effect of pH, charge separation and oxygen concentration in photoelectrocatalytic systems: active chlorine production and chlorate formation. Desalination 176(1-3), 219–227 (2005)
T.R. Gordon, M. Cargnello, T. Paik, F. Mangolini, R.T. Weber, P. Fornasiero, C.B. Murray, Nonaqueous synthesis of TiO2 nanocrystals using TiF4 to engineer morphology, oxygen vacancy concentration, and photocatalytic activity. J. Amer. Chem. Soc. 134(15), 6751–6761 (2012)
H. Kmentova, S. Kment, L. Wang, S. Pausova, T. Vaclavu, R. Kuzel, H. Han, Z. Hubicka, M. Zlamal, J. Olejnicek, M. Cada, J. Krysa, R. Zboril, Photoelectrochemical and structural properties of TiO 2 nanotubes and nanorods grown on FTO substrate: comparative study between electrochemical anodization and hydrothermal method used for the nanostructures fabrication. Catal. Today 287, 130–136 (2017)
A. Prathan, C. Bhoomanee, P. Ruankham, S. Choopun, A. Gardchareon, S. Phadungdhitidhada, D. Wongratanaphisan, Mater. Today Proceed. 17, 1514 (2019)
G. Zhang, X. Zhang, H. Huang, J. Wang, Q. Li, L.Q. Chen, Q. Wang, Toward wearable cooling devices: highly flexible electrocaloric Ba0.67Sr0.33TiO3 nanowire arrays. Adv. Mater. 28(24), 4811–4816 (2016)
W. Qi, J. Du, Y. Peng, W. Wu, Z. Zhang, X. Li, K. Li, K. Zhang, C. Gong, M. Luo, H. Peng, Hydrothermal synthesis of TiO2 nanorods arrays on ITO. Mater. Chem. Phys. 207, 435–441 (2018)
M. Synowiec, A. Micek-Ilnicka, K. Szczepanowicz, A. Różycka, A. Trenczek-Zajac, K. Zakrzewska, M. Radecka, Functionalized structures based on shape-controlled TiO2. Appl. Surf. Sci. 473, 603–613 (2019)
S. Jiang, Y. Li, X. Zhang, Y. Li, Enhancing the photoelectrochemical water splitting activity of rutile nanorods by removal of surface hydroxyl groups. Catal. Today 259, 360–367 (2016)
X. Pan, M.Q. Yang, X. Fu, N. Zhang, Y.J. Xu, Defective TiO2 with oxygen vacancies: synthesis, properties and photocatalytic applications. Nanoscale 5(9), 3601–3614 (2013)
G. Wang, H. Wang, Y. Ling, Y. Tang, X. Yang, R.C. Fitzmorris, C. Wang, J.Z. Zhang, Y. Li, Hydrogen-treated TiO2nanowire arrays for photoelectrochemical water splitting. Nano Lett. 11(7), 3026–3033 (2011)
H. Huang, X. Hou, J. Xiao, L. Zhao, Q. Huang, H. Chen, Y. Li, Effect of annealing atmosphere on the performance of TiO2 nanorod arrays in photoelectrochemical water splitting. Catal. Today 330, 189–194 (2019)
M.A. de Araujo, M.F. Gromboni, F. Marken, S.C. Parker, L.M. Peter, J. Turner, H.C. Aspinall, K. Black, L.H. Mascaro, Contrasting transient photocurrent characteristics for thin films of vacuum-doped “grey” TiO2 and “grey” Nb2O5. Appl. Catal. B Environ. 237, 339–352 (2018)
L.L. Kazmerski, D.M. Racine, The effects of annealing on the conduction mechanisms in vacuum-deposited SnO2 thin films. Thin Solid Films 30(2), L19–L22 (1975)
A.N. Bondarchuk, L.M. Peter, G.P. Kissling, E. Madrid, J.A. Aguilar-Martinez, Z. Rymansaib, P. Iravani, M. Gromboni, L.H. Mascaro, A. Walsh, F. Marken, Vacuum-annealing induces sub-surface redox-states in surfactant-structured α-Fe2O3 photoanodes prepared by ink-jet printing. Appl. Catal. B Environ. 211, 289–295 (2017)
D.A.H. Hanaor, C.C. Sorrell, Review of the anatase to rutile phase transformation. J. Mater. Sci. 46(4), 855–874 (2011)
M. Pelaez, N.T. Nolan, S.C. Pillai, M.K. Seery, P. Falaras, A.G. Kontos, P.S.M. Dunlop, J.W.J. Hamilton, J.A. Byrne, K. O'Shea, M.H. Entezari, D.D. Dionysiou, A review on the visible light active titanium dioxide photocatalysts for environmental applications. Appl. Catal. B Environ. 125, 331–349 (2012)
X. Cheng, D.S. Kong, Z. Wang, Y.Y. Feng, W.J. Li, Inhibiting effect of carbonate on the photoinduced flatband potential shifts during water photooxidation at TiO2/solution interface. J. Solid State Electrochem. 21(5), 1467–1475 (2017)
M. Pourbaix, Atlas of electrochemical equilibria in aqueous solutions (National Association of Corrosion Engineers, New York, 1974)
S.H. Xiao, J.H. Qu, H.J. Liu, X. Zhao, D.J. Wan, Fabrication of TiO2/Ti nanotube electrode and the photoelectrochemical behaviors in NaCl solutions. J. Solid State Electrochem. 13(12), 1959–1964 (2009)
J. Weber, A.J. Wain, F. Marken, Microwire chronoamperometric determination of concentration, diffusivity, and salinity for simultaneous oxygen and proton reduction. Electroanalysis 27(8), 1829–1835 (2015)
F. Scholz, Electroanalytical methods, 2nd edn. (Springer, Berlin, 2010), p. 76
P.H. Rieger, Electrochemistry (Chapman and Hall, New York, 1994), p. 208
N.D. Diby, Y.Q. Duan, P.A. Grah, F.S. Cai, Z.H. Yuan, Enhanced photoelectrochemical performance for hydrogen generation via introducing Ti3+ and oxygen vacancies into TiO2 nanorod arrays. J. Mater. Sci. Mater. Electron. 29(23), 20236–20246 (2018)
A.C. Bronneberg, C. Höhn, R. van de Krol, Probing the interfacial chemistry of ultrathin ALD-grown TiO2films: an in-line XPS study. J. Phys. Chem. C 121(10), 5531–5538 (2017)
J.G. Yu, H.G. Yu, B. Cheng, X.J. Zhao, J.C. Yu, W.K. Ho, The effect of calcination temperature on the surface microstructure and photocatalytic activity of TiO2 thin films prepared by liquid phase deposition. J. Phys. Chem. B 107(50), 13871–13879 (2003)
B. Levy, W. Liu, S.E. Gilbert, Directed photocurrents in nanostructured TiO2/SnO2 heterojunction diodes. J. Phys. Chem. B 101(10), 1810–1816 (1997)
Acknowledgements
Y.Z. (201809350006) and Z.G. (201809350005) thank the China Scholarship Council for PhD scholarships.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Thom R. Harris-Lee and Yan Zhang are joint first authors.
Electronic Supplementary Material
ESM 1
(DOCX 330 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Harris-Lee, T.R., Zhang, Y., Bowen, C.R. et al. Photo-Chlorine Production with Hydrothermally Grown and Vacuum-Annealed Nanocrystalline Rutile. Electrocatalysis 12, 65–77 (2021). https://doi.org/10.1007/s12678-020-00630-x
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12678-020-00630-x