
Abstract
Nutrient turnover in soils is strongly driven by soil properties, including clay mineral composition. One main nutrient is phosphorus (P), which is known to be easily immobilized in soil. Therefore, the specific surface characteristics of clay minerals might substantially influence P availability in soil and thus the microbial strategies for accessing P pools. We used a metagenomic approach to analyze the microbial potential to access P after 842 days of incubation in artificial soils with a clay mineral composition of either non-expandable illite (IL) or expandable montmorillonite (MT), which differ in their surface characteristics like soil surface area and surface charge. Our data indicate that microorganisms of the two soils developed different strategies to overcome P depletion, resulting in similar total P concentrations. Genes predicted to encode inorganic pyrophosphatase (ppa), exopolyphosphatase (ppx), and the pstSCAB transport system were higher in MT, suggesting effective P uptake and the use of internal poly-P stores. Genes predicted to encode enzymes involved in organic P turnover like alkaline phosphatases (phoA, phoD) and glycerophosphoryl diester phosphodiesterase were detected in both soils in comparable numbers. In addition, Po concentrations did not differ significantly. Most identified genes were assigned to microbial lineages generally abundant in agricultural fields, but some were assigned to lineages known to include oligotrophic specialists, such as Bacillaceae and Microchaetaceae.
Similar content being viewed by others


Introduction
In soils, minerals and microbiota are tightly associated and form highly reactive interfaces [1, 2], which represent activity hotspots. These hotspots can be considered highly structured, heterogeneous, and discontinuous [3]. The resulting soil properties strongly determine water and nutrient availability. The physicochemical and biological characteristics of reactive interfaces are strongly influenced by soil mineral types. For example, swelling clay minerals like montmorillonite are characterized by large specific surface areas, which result in water and nutrient retention. In contrast, non-swelling minerals like illite are characterized by relatively small reactive interfaces [4]. The importance of soil mineral composition as a driver for the formation of interfaces in soils has been proven in experiments with artificial soil mixtures where different combinations of clay minerals (illite and montmorillonite), iron and aluminum oxides (ferrihydrite and boehmite), and charcoal were investigated, while texture and incubation conditions were the same [5,6,7]. In a 2-year incubation experiment with repeated manure application, Vogel et al. [4] concluded that clay mineral identity was the main driver of microbial community composition. Despite the change in community composition, which was assessed by UPGMA cluster analysis of degenerating gradient gel electrophoresis profiles, the respiration rate, organic matter degradation rate, and quality of organic matter were comparable among the different artificial soil mixtures. Thus, the authors concluded that functionally redundant microbes were present in the different soil mixtures, at least those involved in organic matter degradation.
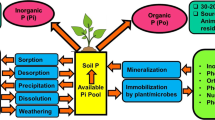
Phosphorus (P) is an important soil nutrient known to be easily immobilized in soil, and thus sensitive to clay mineral properties [8, 9]. The mineralization and solubilization of P are dependent on hydrolytic enzymes, such as phosphatases, phosphonatases, and C-P lyases [10]. In addition, microorganisms can increase orthophosphate ions in soil solutions or mobility of organic P (Po) via direct or indirect mechanisms [8]. Several studies have shown that soil bacteria belonging to different genera including Pseudomonas, Azotobacter, Burkholderia, Bacillus, Rhizobium, and Actinomyces have the ability to increase P availability in soil [11,12,13,14,15]. Clay minerals and their specific surface characteristics have recently been shown to be highly relevant for P binding in soils. At low P concentrations, the structural Al sites of clay minerals are most probably responsible for a comparatively high sorption capacity for P [5]. The specific surface characteristics might directly influence the availability of P in soil and consequently the solubilization and mineralization strategies adapted by microbes [16]. However, the role of clay minerals as drivers for microbiota, which catalyze P transformation processes, is still poorly understood.
In this study, we propose that soils with expandable clay minerals and larger soil surface area can both support a higher diversity of microbiota involved in P turnover and supply more interfaces for P immobilization, thus supporting more P recycling than non-expandable matrices. To test this hypothesis, we used two artificial soils that differ only in clay mineral type and represent a model for an early phase of soil development [4]. One soil contained the expandable clay mineral montmorillonite, the second the non-expandable illite. We used a metagenomics approach to identify microbial P turnover pathways and involved microorganisms.
Materials and Methods
Preparation and Maturation of Artificial Soils
Two artificial soil mixtures containing montmorillonite (MT) (Ceratosil WG, Süd-Chemie AG, Moosburg, Germany) or illite (IL) (Inter-ILI Mérnöki Iroda, Hungary) were prepared and incubated. To achieve similar soil texture, each mixture was based on 40-42% sand (Quartz Sand Haltern, H33), 52-54% silt (Millisil W11H, Quarzwerke GmbH, Frechen, Germany), and clay (< 6.3 μm). Montmorillonite (6.3%) and illite (8.0%) were used as clay minerals. Detailed information on the experimental design and model material was previously described [4, 17]. In brief, for each artificial soil mixture, 500 g mixtures were prepared and treated as true replicates throughout. Each mixture was inoculated with a water extractable microbial fraction obtained from the Ap horizon of a Luvisol (Scheyern, Germany) and with a dry and sterilized manure (4.5 wt%, organic carbon (OC) content of 338.5 ± 6.9 mg g−1, N content of 30.7 ± 1.6 mg g−1, C/N ratio 11.0 ± 0.4) and sieved to < 2 mm as a nutrient source. The artificial soil mixtures were incubated in the dark at 20 °C for 842 days. The water content was kept at 60% of the maximum water holding capacity, which was adjusted at weekly intervals using 0.01 M CaCl2.. To avoid nutrient depletion, fresh sterile manure was reapplied on day 562 (4.5 wt%, OC content of 165.4 ± 2.7 mg g−1, N content of 11.5 ± 0.2 mg g−1, and a C/N ratio of 14.4 ± 0.2). Samples were obtained from three independent replicates at the end of the incubation period after 842 days and stored at − 20 °C immediately after sampling.
Analyses of bulk properties and macro-aggregation were performed at the same time point (842 days) and have been recently published [4]. Briefly, macro-aggregates (> 2 mm) differed significantly between the two treatments with higher amounts found in MT (65%) compared to IL (48%). The pH of MT was slightly lower than measured in IL (7.2 ± 0.2 and 7.6 ± 0.3, respectively). The MT had slightly higher OC and N contents than IL soils, but no significant differences were found.
Measurement of P Concentrations
The bioavailable phosphorus (PNaHCO3) was extracted from 4 g dry soil with 40 ml 0.5 M NaHCO3. The stable P pools were determined by the ignition-acid extraction method [18]. Briefly, 0.5 g of dry soil was extracted with 25 ml 0.5 M H2SO4 before (only stable inorganic P, Pi) and after ignition at 550 °C (total P, Ptotal). In both H2SO4 extracts, the molybdate-reactive P was analyzed photometrically (UV-VIS spectral photometer Specord®200, Analytik Jena AG, Jena, Germany) using the molybdenum-blue method. The organic P (Po) has been estimated by calculating the difference between Ptotal and Pi. Each method was carried out in three technical replicates.
Total Community DNA Extraction, Library Preparation, and Sequencing
Total community DNA was extracted from 0.5 g of each replicate using the NucleoSpin Soil Kit (Macherey-Nagel, Germany) according to the manufacturer’s instructions. To optimize the DNA yield, the proposed protocol with lysis buffer SL2 and enhancer SX was used as described in the user manual. Bead beating was performed twice (5.5 ms−1, 30 s) using a Precellys 24 (Bertin Technologies, France). The DNA quality was checked photometrically (Nanodrop ND-1000, Thermo Fisher Scientific, USA) and quantified with the Quant-iT PicoGreen dsDNA Assay Kit (Thermo Fisher Scientific, USA). DNA extracts were stored at − 20 °C.
From each sample, 2 μg of DNA was fragmented using an ultrasonicator (peak instrument power 175 W, duty factor 10%, 200 cycles per burst, treatment time 100 s; E220 Covaris, MA, USA). Fragmented DNA was purified with Agencourt AMPure XP beads (Beckman Coulter, CA, USA) and fragment size was assessed using a Bioanalyzer 2100 (Agilent Technologies, Germany) and Agilent DNA 7500 Kit (Agilent Technologies, Germany). Six metagenomic libraries were prepared from 100 ng DNA per library using the NEBNext Ultra DNA Library Prep Kit (New England BioLabs, MA, USA) according to the manufacturer’s instructions. For adaptor ligation, a 10-fold dilution of NEBNext Adaptor for Illumina with a final concentration of 1.5 μM was used. In the final enrichment, PCR libraries were indexed utilizing NEBNext Multiplex Oligos for Illumina (Dual Index Primers Set 1; New England BioLabs, MA, USA). The PCR reaction contained 2.5 μl of each indexed primer, 25 μl of NEBNext High-Fidelity 2x PCR master mix, and 20 μl of adapter ligated DNA. PCR cycles included an initial denaturation step at 98 °C for 30 s, 6 cycles of denaturation at 98 °C for 10 s, annealing at 65 °C for 30 s, and extension at 72 °C for 30 s followed by a final extension step at 72 °C for 5 min. Metagenomic libraries were electrophoretically size-selected using a Pippin Prep (Sage Science, MA, USA; setting “range mode” to select for 300-580 bp). The final libraries were checked for size using a Bioanalyzer 2100 (Agilent Technologies, Germany) and the Agilent High Sensitivity DNA Kit (Agilent Technologies, Germany) followed by quantification with Quant-iT PicoGreen dsDNA Assay Kit (Thermo Fisher Scientific, USA). Libraries were diluted to 2 nM each. The sample pool was denatured with 0.2 N NaOH, diluted to 10 pM and spiked with 2.5% (v/v) PhiX. Paired-end sequencing was performed on an Illumina MiSeq platform (Illumina, USA), using the MiSeq® 2x 300-cycle V3 kit, following the standard Illumina sequencing protocol. The obtained sequences were deposited to NCBI SRA (Sequence Read Archive) under accession number PRJNA 556907.
Bioinformatics and Statistical Analysis
Any remaining adaptor sequences or reads shorter than 50 bp and reads with Phred score < 15 were removed from the dataset using AdapterRemoval v2 [19]. Since most improvement in accuracy is achieved by trimming at the level Phred score > 5, a less aggressive trimming approach was chosen to avoid unnecessary read loss [20]. Further processing included removal of PhiX sequences with DeconSeq version 0.4.3 [21]. For taxonomic annotation, high quality fasta sequences were aligned against the NCBI non-redundant (nr) protein sequence data base using DIAMOND [22] and the KEGG database [23] with default settings. Blastn results were mapped and taxonomically and functionally annotated using MEGAN5 (version 5.6.5) [24] with the following parameters: min score = 50, top percent = 10, min support = 1, and min complexity = 0. Sequences of enzymes with corresponding KEGG orthology (KO) numbers involved in phosphorus turnover were extracted from the functionally annotated dataset obtained from the KEGG database and taxonomically annotated by using DIAMOND against NCBI non-redundant (nr) protein sequence database. The 42 enzymes investigated are listed with their corresponding KO numbers in Supplementary Table S1.
Statistical analyses were computed using R [25]. Packages “stats,” “graphics,” “grDevices,” [25] “vegan,” [26] “GUniFrac,” [27] “gridExtra,” [28] “cluster,” [29] “calibrate,” [30] “RColorBrewer,” [31] “gplots,” [32] and “vcd” [33] were used. The metagenomic datasets were subsampled to the lowest number of reads per sample by using the rarify function from the “GUniFrac” package. Significant differences for the taxonomic and functional annotation were determined by unpaired t test, where p values were adjusted by Bonferroni test.
Results
P Concentrations of the Two Artificial Soils
The results of the Ptotal, Po, Pi, and PNaHCO3 measurements are summarized in Table 1. Ptotal did not significantly differ between the two soils. Pi was significantly higher in MT (304 mg kg−1) than IL (264 mg kg−1), while Po concentrations did not significantly differ between the two soil mixtures, and were only slightly higher in IL (120 mg kg−1). PNaHCO3 concentrations were significantly lower in IL (134 mg kg−1) than MT (153 mg kg−1).
Phylogenetic Annotation of Artificial Soil Metagenomes
To identify microbial key players involved in P turnover, a metagenomic approach was used. All metagenomic datasets were randomly subsampled to the lowest number of reads per sample (n = 3,074,662). The coverage of metagenomes based on the read redundancy values as calculated by the Nonpareil algorithm ranged between 0.657 and 0.773 (data not shown). Of all reads, 55.84% could be annotated using the NCBI non-redundant protein sequence database. Later relative abundances were calculated based on assigned reads. Of those, 93.72% were bacterial reads, 4.21% eukaryotic, and 2.07% archaeal. Overall, the obtained reads were assigned to 477 prokaryotic families. Only 7 of those predicted families showed a relative abundance > 1.0% in both artificial soils, namely Microchaetaceae (4.50%), Bacillaceae (3.51%), Cytophagaceae (2.95%), Flavobacteriaceae (2.57%), Flammeovirgaceae (2.48%), Ignavibacteriaceae (1.96%), and Chitinophagaceae (1.81%) (Fig. 1a). Besides those 7 families, an additional 339 families were shared between both artificial soils, but in lower abundance. To detect significant differences between the prokaryotic communities in MT and IL, the abundance of all families was compared. To be more stringent, only taxa with a relative abundance of at least 0.005% in one of the samples were included. The relative number of significantly different reads at the family level is shown in the Supplementary Table S2. In general, 54 bacterial families differed significantly in the number of assigned reads. Among the 20 most abundant families, Ignavibacteriaeceae, Bacillaceae, and Paenibacillaceae were significantly higher in MT (Bonferroni adjusted unpaired t test, p = 0.010, p = 0.045, and p = 0.003, respectively) while no bacterial family was significantly more abundant in IL.

Relative abundance of overall prokaryotic families (a) and families potentially involved in P turnover (b) in the metagenomes of two artificial soils. Sequences were aligned against the NCBI-nr database and annotated with MEGAN5. Relative abundances were calculated based on the number of all assigned reads. Twenty most abundant taxa are shown. Asterisks indicate significant differences (p < 0.05, n = 3)
Functional Annotation of Artificial Soil Metagenomes
The functional annotation of reads was performed by aligning the subsampled dataset against the KEGG database [23] and visualizing the results using MEGAN5 [24]. In total, 92,430 reads were assigned to genes predicted to code for enzymes involved in P turnover. Rarefaction analysis of KO numbers associated with P turnover revealed comparable functional richness for both metagenomes of MT and IL (Supplementary Figure S1).
Of the 42 investigated genes, 29 could be detected in both soils (Fig. 2, Supplementary Table S1). The majority of reads could be assigned to the genes involved in phosphorus transport and uptake (33,720), followed by mineralization (26,461), regulatory systems (18,774), and solubilization (13,525). Among those predicted genes, three were significantly more abundant in MT: phnX, phnK, and pstC. In addition, other predicted genes were more abundant in MT, but not on a significant level of 5%. Those genes included further subunits of the phosphate-specific transport system (pstSCAB), inorganic pyrophosphatase (ppa), and exopolyphosphatase (ppx, p = 0.059). Genes coding for the glycerol-3-phosphatase transport system (ugpBAEC), alkaline phosphatases (phoA, phoD), glycerophosphoryl diester phosphodiesterase (ugpQ), and quinoprotein glucose dehydrogenase (gcd) were more abundant in IL, although this was not significant. Genes associated with P regulation (phoR, phoB) showed nearly identical relative abundances in both soils.

Relative abundance of prokaryotic genes predicted to be involved in phosphorus turnover in both artificial soils. Obtained sequences were aligned against KEGG database and annotated with MEGAN5. Relative abundances were calculated based on the number of all assigned KEGG reads. Asterisks indicate significant differences (p < 0.05, n = 3) in the number of annotated reads between the investigated soils
The Microbiota Driving the Phosphorus Turnover in MT and IL Soils
To predict key drivers of phosphorus turnover, the 29 genes detected in both soils were extracted from the KEGG annotated dataset, and taxonomically annotated using DIAMOND and NCBI non-redundant (nr) protein sequence database. In general, families predicted to be involved in most P turnover processes in both soils mostly belonged to the highly abundant families, such as Nitrososphaeraceae, Cytophagaceae, Bacillaceae, Chitinophagaceae, and Flammeovirgaceae (Fig. 1b, Fig. 3, Supplementary Figure S2). More specifically, it was predicted that P solubilization in MT was driven by Ignavibacteriaceae, Cytophagaceae, Chitinophagaceae, candidate division WWE3, and Nitrososphaeraceae. The mineralization of P was predicted to be dominated by Bacillaceae and Microchaetaceae and uptake by Anaerolinaceae, Bacillaceae, and Meliobacteriaceae in MT. Similar families were potentially involved in P turnover in IL.

Relative abundance of families involved in P turnover in the montmorillonite (MT) and illite (IL) artificial soils. Obtained sequences were assigned on the functional level by aligning against the KEGG database and on taxonomic level against the NCBI-nr database. Relative abundances were calculated based on the number of reads assigned to P turnover. Twenty most abundant families are depicted. Shown are mean values of all genes involved in solubilization, mineralization, organic uptake, inorganic uptake, and regulation. Families marked bold represent families that differ significantly between artificial soils (p < 0.05, n = 3)
Interestingly, most of the families predicted to be involved in P turnover were shared between the two soils (Fig. 4). Among the 50 most abundant families, 17 were similar in IL and MT for solubilization, 41 for mineralization, and 43 for the uptake of P. The relative abundance of unique families was generally very low. In IL, unique families potentially involved in P solubilization were assigned to Prolixybacteraceae and Halobacteriaceae, and for P mineralization to Ignavibacteriaceae and Sphaerobacteraceae. Unique families in MT predicted to be involved in P solubilization (Streptomycetaceae, Saprospiraceae, Deinococcaceae, Thermaceae, Paenibacillaceae, and Pseudomonadaceae) and P mineralization (Chlamydiaceae, Herpetosiphonaceae, Thermaceae, Syntrophaceae, and Thermotogaceae) were slightly more diverse when compared to IL. Regarding P uptake, only one family per soil was predicted to be unique, namely Leptospiraceae in MT and Cryomorphaceae in IL. Although both soils had many families in common, the relative abundance of 13 bacterial families that were predicted to be involved in P turnover was significantly different, with only Meliobacteriaceae (p = 0.002) and Paenibacillaceae (p = 0.042) belonging to the 20 most abundant families (Supplementary Table S3). Only Bacillaceae potentially covered all processes in both soils, while the other families were predicted to drive specific transformation steps in P turnover (Fig. 3).

Distribution of shared and unique families involved in P turnover in the montmorillonite (MT) and illite (IL) artificial soils. Obtained sequences were functionally assigned using the KEGG database and taxonomically using the NCBI-nr database. Shown are mean values of the relative abundance of the fifty most abundant families involved in P solubilization, mineralization, and uptake. Relative abundances were calculated based on the number of all assigned reads
Discussion
Microbial Diversity and Community Establishment Are Affected by Soil Mineral Composition
The effects of mineral composition on the establishment of microbial communities in the initial phase of soil development have been described in a number of publications focusing on several short-term [34, 35] and long-term [4] experiments. For example, Ding et al. [35] showed for the same artificial soil mixtures we used that genera belonging to the phyla Proteobacteria, Firmicutes, Actinobacteria, and Bacteroidetes were significantly higher in IL compared to MT after 90 days of incubation. We demonstrated in our study based on metagenomic sequences that the relative abundances of 35 families were significantly higher in MT, while only 19 were significantly higher in IL. This is in line with our assumption that MT containing soil mixtures promote a more diverse community. Vogel et al. [4] showed that aggregation in matured artificial soils differed depending on clay minerals, while total nutrient concentrations were similar. Ptotal concentrations in our experiment also confirmed their findings (Table 1). After 842 days, the content of macro-aggregates was higher in MT than IL, suggesting a faster aggregate turnover in artificial soils with IL. Moreover, differences in the microbiome composition might be explained by the protective influence of mineral surfaces on organic matter and consequently lower mineralization rates inducing the slower release of nutrients in MT, shown by the higher OC content as well as significantly higher Po and bioavailable PNaHCO3 concentrations in MT. We assume that this might have influenced members of Bacillaceae, Ignavibacteriaceae, and Chlorobiaceae that have the ability to utilize recalcitrant compounds from organic matter [36, 37]. In contrast, a weaker immobilization potential in IL could promote a faster mineralization of OC by members of Bacteroidetes like Cytophagaceae, Flammeovirgaceae, and Pseudomonadaceae, which are frequently found in copiotrophic environments [38] and may have benefited from the addition of manure, which introduced a huge amount of nutrients (Table 1). Moreover, Vogel et al. [4] also observed the increase of Pseudomonas species after the second manure addition.
Microbial P Turnover in Montmorillonite and Illite Containing Artificial Soils
Although the last nutrient addition was more than 300 days ago, both soils showed surprisingly high concentrations of available inorganic P (Table 1), comparable for example to forest soils [39]. This might be linked to the low microbial respiration rates at the time of sampling [4] indicating low microbial activity and highly efficient strategies for the remobilization of P from the microbial biomass, as the community composition significantly changed in the 300 days after the last manure addition [4]. Furthermore, the preliminary limitation of other nutrients like nitrogen and carbon might have caused high remaining P concentrations, which is underlined by very low C:P and N:P ratios of less than 3.8 and 34.6, respectively (calculation based on [4] and Table 1). These values are much lower than those observed from Cleveland and Liptzin [40], who postulated a well constrained C:P ratio of 186:1 and N:P ratio of 13:1. This is further in line with findings of Heuck et al. [41], who demonstrated that under C limitation, organic P resources are mineralized with the aim to take up the released C first, which explains higher C:P ratios in our experiment. Moreover, the use of internally stored P might be an alternative, for example in acidocalcisomes. In both soils, genes predicted to encode the phosphate-specific transport system (pst) were highly abundant, while no reads were assigned to the phosphate inorganic transporter (pitA). This suggests that P is a highly competitive nutrient in those soils, as this transporter is typically expressed under P starvation [42]. It is not surprising that bacterial families predicted to be involved in P cycling belonged to the most abundant families, as P mobilization and uptake are of high importance in nutrient-limited soils. Members of Bacillaceae were among the most abundant bacteria in both soils and have the potential to perform all processes of P turnover. These results nicely confirm previous data from cultured members of Bacillaceae. For example, Bacillus spp. include well-known phosphorus-solubilizing bacteria (PSB). They harbor a variety of P-solubilizing mechanisms (e.g., changing pH, excretion of organic acids, siderophores, and phosphatase) [15]. Another advantage is the ability of Bacillaceae to sporulate under unfavorable conditions, thus helping them to survive and outcompete other microbiota in MT and IL soils [36].
Since P is one of the most important macronutrients, essential in energy metabolism and other cellular processes, enzymes related to the use of external P sources are harbored by a wide range of microorganisms [43], which we also show in this study (Fig. 1b). Genes predicted to be involved in external P turnover were assigned to microbiota belonging to Bacteroidetes, Firmicutes, Proteobacteria, and Planctomycetes in both soils, which is comparable to findings of Grafe et al. [44] in mature agricultural soils. This is not surprising, as the artificial soils had been inoculated with a microbial community extracted from an agricultural soil at the beginning of the maturation phase. In contrast, Bergkemper et al. [45] found that most reads assigned to P turnover in metagenomes from P-depleted forest soils belonged to the Acidobacteria (Acidobacteriales, Solibacterales), Actinobacteria (Actinomycetales), and Proteobacteria (Rhodospirillales, Burkholderiales). This difference could be due to the complexity of forest soil and the resulting differences in physicochemical properties, as well as to the different mineral composition, which selects for different families. In the MT, the most abundant predicted genes associated with P mineralization were ppa and ppx; the respective enzymes are predominantly involved in the utilization of inorganic P. The ppa gene codes for an inorganic pyrophosphatase, an enzyme that hydrolyzes polyphosphate compounds (poly-P), and ppx for an exopolyphosphatase which in turn releases inorganic P from poly-P chains [46] in prokaryotes under the condition of P starvation [47]. Thus, a first mechanism in overcoming P starvation might be the use of internal P storage pools like acidocalcisomes, which might have been developed after the addition of manure. Acidocalcisomes are well known and widespread both in prokaryotes and eukaryotes [48]. Moreover, MT has a higher storage capacity than IL due to its bigger soil surface area. Thus, inorganic P from external sources like manure might persist longer. Interestingly, in MT, the reads predicted to code for the ppx gene (exopolyphosphatase) were exclusively assigned to members of Cyanobacteria. Recent studies have shown that the expression of the ppx gene allows Cyanobacteria to adapt to the environmental fluctuation of P and C [47]. Additionally, it has been demonstrated that exopolysaccharides can be used to solubilize P. As Cyanobacteria are well known as exopolysaccharide producers, it might be an advantage in MT soils where more reaction sites are available [49]. An additional advantage of some Cyanobacteria could be their potential capability to fix nitrogen and CO2 [50].
In contrast, the relative abundance of predicted genes involved in P mineralization did not significantly differ in the two soils. This was further accompanied by comparable Po concentrations. This included genes coding for alkaline phosphatases (ALP), namely phoA and phoD, and ugpQ, which coded for a glycerophosphoryl diester phosphodiesterase. While PhoA includes mainly phosphomonoesterase, PhoD shows phosphodiesterase activity as well [51]. UgpQ has phosphodiesterase activity and hydrolyzes glycerophosphoryl diesters to glycerol-3-phosphate (G3P). In line with that, we also detected reads, which were assigned to all four subunits of the glycerol-3-phosphate transporter system (ugpBAEC), whose relative abundance exceeded those detected in other studies [44, 45]. This could allow the utilization of organic P sources in nutrient-depleted systems. The detection of predicted ALP genes was higher than that of the predicted acid phosphatase genes in both soils. However, this was not surprising as both soils had a pH of 7.7 at the beginning of the experiment [17] and of 7.2 (MT) and 7.6 (IL) after 842 days of incubation [4], which is still above the optimal pH for acid phosphatase activities [52]. Moreover, Fraser et al. [53] demonstrated that the addition of manure might increase the abundance of the phoD community as well as the ALP activity. Genes coding for the Pho regulon [54, 55] that controls phoA and phoD gene expression were also highly abundant in both soils, which was in line with the high abundance of pstS, underlining again the need for a strict control of P utilization.
Interestingly, the predicted quinoprotein glucose dehydrogenase (PQQGDH), potentially involved in P solubilization, was among the top three abundant genes in both soils. Its relative abundance was even higher compared to a study from agricultural soils [44], which regularly obtained organic fertilizers. Solubilization processes so far have been mostly described for P-rich systems [45]. However, the PNaHCO3 concentration and the total Pi concentration (Table 1) suggest that there is still a high potential to solubilize P from the stable inorganic fraction.
Conclusion
The predicted key players involved in P transformation in our study were among the dominant taxa, which underlines the importance of P acquisition in both artificial soil mixtures. The dominant families predicted to be involved in P turnover like Bacillaceae and Microchaetaceae are perfectly adapted to harsh environments with the ability to rest as spores or acquire additional nutrients like C and N by fixation. Our data indicate that organic P is an important source in both artificial soil mixtures, as many reads were assigned to genes potentially involved in the effective use of organic P sources, including different alkaline phosphatases and glycerophosphoryl diester phosphodiesterase. These might be involved in the mineralization of still available Po introduced with the manure, or Po released from the microbial biomass, whose composition significantly changed in more than 300 days after the last manure addition. In MT, which has a larger soil surface area, the relative abundance of the predicted pstC gene was significantly increased compared to IL. This gene belongs to the high affinity phosphate-specific transporter (pstSCAB) system acting under P starvation. Moreover, the potential to use internal P sources, by the breakdown of poly-P species, was greater in MT compared to IL.
However, the data represent the system’s potential, which was predicted on the basis of metagenomic assignments for certain processes and do not confirm the actual turnover. In order to deepen the insight into P turnover mechanisms and to identify active microbial communities involved in the processes, RNA and proteome-based studies and additional experiments should be employed.
Data Availability
The metagenomic sequencing data have been uploaded to the sequencing read archive (SRA) under the bioproject number PRJNA556907. All other data generated or analyzed during this study have been included in this published article (and its supplementary information files).
References
Kögel-Knabner I, Guggenberger G, Kleber M, Kandeler E, Kalbitz K, Scheu S, Eusterhues K, Leinweber P (2008) Organo-mineral associations in temperate soils: integrating biology, mineralogy, and organic matter chemistry. J Plant Nutr Soil Sc 171:61–82. https://doi.org/10.1002/jpln.200700048
Totsche KU, Rennert T, Gerzabek MH, Kögel-Knabner I, Smalla K, Spiteller M, Vogel H-J (2010) Biogeochemical interfaces in soil: the interdisciplinary challenge for soil science. J Plant Nutr Soil Sc 173:88–99. https://doi.org/10.1002/jpln.200900105
Stotzky G (1997) Soil as an environment for microbial life. In: van Elsas JD, Trevors JT, Wellington EMH (eds) Modern soil microbiology. Marcel Dekker Inc, New York, pp 1–20
Vogel C, Babin D, Pronk GJ, Heister K, Smalla K, Kogel-Knabner I (2014) Establishment of macro-aggregates and organic matter turnover by microbial communities in long-term incubated artificial soils. Soil Biol Biochem 79:57–67. https://doi.org/10.1016/j.soilbio.2014.07.012
Gerard F (2016) Clay minerals, iron/aluminum oxides, and their contribution to phosphate sorption in soils-a myth revisited. Geoderma 262:213–226. https://doi.org/10.1016/j.geoderma.2015.08.036
Pronk GJ, Heister K, Vogel C, Babin D, Bachmann J, Ding G-C, Ditterich F, Gerzabek MH, Giebler J, Hemkemeyer M, Kandeler E, Kunhi Mouvenchery Y, Miltner A, Poll C, Schaumann GE, Smalla K, Steinbach A, Tanuwidjaja I, Tebbe CC, Wick LY, Woche SK, Totsche KU, Schloter M, Kögel-Knabner I (2017) Interaction of minerals, organic matter, and microorganisms during biogeochemical interface formation as shown by a series of artificial soil experiments. Biol Fertil Soils 53:9–22. https://doi.org/10.1007/s00374-016-1161-1
Vogel C, Heister K, Buegger F, Tanuwidjaja I, Haug S, Schloter M, Kogel-Knabner I (2015) Clay mineral composition modifies decomposition and sequestration of organic carbon and nitrogen in fine soil fractions. Biol Fertil Soils 51:427–442. https://doi.org/10.1007/s00374-014-0987-7
Seeling B, Zasoski RJ (1993) Microbial effects in maintaining organic and inorganic solution phosphorus concentrations in a grassland topsoil. Plant Soil 148:277–284. https://doi.org/10.1007/BF00012865
Olander LP, Vitousek PM (2004) Biological and geochemical sinks for phosphorus in soil from a wet tropical forest. Ecosystems 7:404–419. https://doi.org/10.1007/s10021-004-0264-y
Rodriguez H, Fraga R, Gonzalez T, Bashan Y (2006) Genetics of phosphate solubilization and its potential applications for improving plant growth-promoting bacteria. Plant Soil 287:15–21. https://doi.org/10.1007/s11104-006-9056-9
Illmer P, Schinner F (1992) Solubilization of inorganic phosphates by microorganisms isolated from forest soils. Soil Biol Biochem 24:389–395. https://doi.org/10.1016/0038-0717(92)90199-8
Kumar V, Behl RK, Narula N (2001) Establishment of phosphate-solubilizing strains of Azotobacter chroococcum in the rhizosphere and their effect on wheat cultivars under green house conditions. Microbiol Res 156:87–93. https://doi.org/10.1078/0944-5013-00081
Afzal A, Bano A (2008) Rhizobium and phosphate solubilizing bacteria improve the yield and phosphorus uptake in wheat (Triticum aestivum). Int J Agri Biol 10:1560–8530
Barreto TR, da Silva ACM, Soares ACF, de Souza JT (2008) Population densities and genetic diversity of actinomycetes associated to the rhizosphere of Theobroma cacao. Braz J Microbiol 39:464–470. https://doi.org/10.1590/S1517-838220080003000010
Oliveira CA, Alves VMC, Marriel IE, Gomes EA, Scotti MR, Carneiro NP, Guimarães CT, Schaffert RE, Sá NMH (2009) Phosphate solubilizing microorganisms isolated from rhizosphere of maize cultivated in an oxisol of the Brazilian Cerrado Biome. Soil Biol Biochem 41:1782–1787. https://doi.org/10.1016/j.soilbio.2008.01.012
Richardson AE, Barea J-M, McNeill AM, Prigent-Combaret C (2009) Acquisition of phosphorus and nitrogen in the rhizosphere and plant growth promotion by microorganisms. Plant Soil 321:305–339. https://doi.org/10.1007/s11104-009-9895-2
Pronk GJ, Heister K, Ding GC, Smalla K, Kogel-Knabner I (2012) Development of biogeochemical interfaces in an artificial soil incubation experiment; aggregation and formation of organo-mineral associations. Geoderma 189:585–594. https://doi.org/10.1016/j.geoderma.2012.05.020
SaundersS WMH, Williams EG (1955) Observations on the determination of total organic phosphorus in soils. J Soil Sc 6:254–267. https://doi.org/10.1111/j.1365-2389.1955.tb00849.x
Schubert M, Lindgreen S, Orlando L (2016) AdapterRemoval v2: rapid adapter trimming, identification, and read merging. BMC Research Notes 9:88. https://doi.org/10.1186/s13104-016-1900-2
MacManes M (2014) On the optimal trimming of high-throughput mRNA sequence data. Front Genet 5:13. https://doi.org/10.3389/fgene.2014.00013
Schmieder R, Edwards R (2011) Fast identification and removal of sequence contamination from genomic and metagenomic datasets. PLoS ONE 6:e17288. https://doi.org/10.1371/journal.pone.0017288
Buchfink B, Xie C, Huson DH (2015) Fast and sensitive protein alignment using DIAMOND. Nat Methods 12:59–60. https://doi.org/10.1038/nmeth.3176
Kanehisa M, Goto S (2000) KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28:27–30. https://doi.org/10.1093/nar/28.1.27
Huson DH, Mitra S, Ruscheweyh H-J, Weber N, Schuster SC (2011) Integrative analysis of environmental sequences using MEGAN4. Genome Res 21:1552–1560. https://doi.org/10.1101/gr.120618.111
Team RC (2008) A language and environment for statistical computing. R foundation for statistical computing. https://www.R-project.org. Accessed 22 Nov 2015
Oksanen J, Blanchet FG, Kindt R, Legendre P, Minchin PR, O’Hara RB, Simpson GL, Solymos P, Henry M, Stevens H, Wagner H (2015) vegan: community ecology package. R package version 22-1. http://CRAN.R-project.org/package=vegan. Accessed 22 Nov 2015
Chen J (2012) GUniFrac: generalized UniFrac distances. R package version 10. https://cran.r-project.org/packages=GUniFrac. Accessed 22 Nov 2015
Auguie B (2015) gridExtra: miscellaneous functions for “Grid” graphics. R package version 200. http://CRAN.R-project.org/package=gridExtra. Accessed 22 Nov 2015
Maechler M, Roussseeuw P, Struyf A et al. (2015) cluster: “Finding Groups in Data”: cluster analysis extended Rousseeuw et al. R package version 2.0.3. http://CRAN.R-project.org/package=cluster. Accessed 22 Nov 2015
Graffelman J (2013) calibrate: calibration of scatterplot and biplot axes. R package version 1.7.2. http://CRAN.R-project.org/package=calibrate. Accessed 22 Nov 2015
Neuwirth E (2014) RColorBrewer: ColorBrewer palettes. R package version 1.1-2. http://CRAN.R-project.org/package=RColorBrewer. Accessed 22 Nov 2015
Warnes G, Bolker B, Bonebakker L et al. (2013) gplots: Various R programming tools for plotting data. R package version 2.12.1. http://CRAN.R-project.org/package=gplots. Accessed 22 Nov 2015
Meyer D, Zeileis A, Hornik K et al (2014) vcd: visualizing categorical data. R package version 1.3-2. http://CRAN.R-project.org/package=vcd. Accessed 22 Nov 2015
Babin D, Ding GC, Pronk GJ, Heister K, Kogel-Knabner I, Smalla K (2013) Metal oxides, clay minerals and charcoal determine the composition of microbial communities in matured artificial soils and their response to phenanthrene. FEMS Microbiol Ecol 86:3–14. https://doi.org/10.1111/1574-6941.12058
Ding GC, Pronk GJ, Babin D, Heuer H, Heister K, Kogel-Knabner I, Smalla K (2013) Mineral composition and charcoal determine the bacterial community structure in artificial soils. FEMS Microbiol Ecol 86:15–25. https://doi.org/10.1111/1574-6941.12070
Mandic-Mulec I, Stefanic P, Elsas J (2015) Ecology of Bacillaceae. Microbiol Spect. 3:TBS-0017-2013. https://doi.org/10.1128/microbiolspec
Su C, Zhang M, Lin L, Yu G, Zhong H, Chong Y (2020) Reduction of iron oxides and microbial community composition in iron-rich soils with different organic carbon as electron donors. Int Biodeter Biodegr 148:104881. https://doi.org/10.1016/j.ibiod.2019.104881
Fierer N, Bradford MA, Jackson RB (2007) Toward an ecological classification of soil bacteria. Ecology 88:1354–1364
Lang F, Krüger J, Amelung W, Willbold S, Frossard E, Bünemann EK, Bauhus J, Nitschke R, Kandeler E, Marhan S, Schulz S, Bergkemper F, Schloter M, Luster J, Guggisberg F, Kaiser K, Mikutta R, Guggenberger G, Polle A, Pena R, Prietzel J, Rodionov A, Talkner U, Meesenburg H, von Wilpert K, Hölscher A, Dietrich HP, Chmara I (2017) Soil phosphorus supply controls P nutrition strategies of beech forest ecosystems in Central Europe. Biogeochemistry 136:5–29. https://doi.org/10.1007/s10533-017-0375-0
Cleveland CC, Liptzin D (2007) C:N:P stoichiometry in soil: is there a “Redfield ratio” for the microbial biomass? Biogeochemistry 85:235–252. https://doi.org/10.1007/s10533-007-9132-0
Heuck C, Weig A, Spohn M (2015) Soil microbial biomass C:N:P stoichiometry and microbial use of organic phosphorus. Soil Biol Biochem 85:119–129. https://doi.org/10.1016/j.soilbio.2015.02.029
Santos-Beneit F (2015) The Pho regulon: a huge regulatory network in bacteria. Front Microbiol 6:402. https://doi.org/10.3389/fmicb.2015.00402
Richardson AE, Simpson RJ (2011) Soil microorganisms mediating phosphorus availability. Plant Physiol 156:989–996. https://doi.org/10.1104/pp.111.175448
Grafe M, Goers M, von Tucher S, Baum C, Zimmer D, Leinweber P, Vestergaard G, Kublik S, Schloter M, Schulz S (2018) Bacterial potentials for uptake, solubilization and mineralization of extracellular phosphorus in agricultural soils are highly stable under different fertilization regimes. Environ Microbiol Rep 10:320–327. https://doi.org/10.1111/1758-2229.12651
Bergkemper F, Schöler A, Engel M, Lang F, Krüger J, Schloter M, Schulz S (2016) Phosphorus depletion in forest soils shapes bacterial communities towards phosphorus recycling systems. Environ Microbiol 18:1988–2000. https://doi.org/10.1111/1462-2920.13188
Keasling JD, Bertsch L, Kornberg A (1993) Guanosine pentaphosphate phosphohydrolase of Escherichia coli is a long-chain exopolyphosphatase. Proc Natl Acad Sci U S A 90:7029–7033. https://doi.org/10.1073/pnas.90.15.7029
Adams MM, Gómez-García MR, Grossman AR, Bhaya D (2008) Phosphorus deprivation responses and phosphonate utilization in a thermophilic Synechococcus sp. from microbial mats. J Bacteriol 190:8171–8184. https://doi.org/10.1128/jb.01011-08
Docampo R, de Souza W, Miranda K, Rohloff P, Moreno SNJ (2005) Acidocalcisomes? Conserved from bacteria to man. Nat Rev Microbiol 3:251–261. https://doi.org/10.1038/nrmicro1097
Sharma SB, Sayyed RZ, Trivedi MH, Gobi TA (2013) Phosphate solubilizing microbes: sustainable approach for managing phosphorus deficiency in agricultural soils. SpringerPlus 2:587–587. https://doi.org/10.1186/2193-1801-2-587
Rai AN, Borthakur M, Bergman B (1992) Nitrogenase derepresssion, its regulation and metabolic changes associated with diazotrophy in the non-heterocystous cyanobacterium Plectonema boryanum PCC 73110. Microbiology 138:481–491. https://doi.org/10.1099/00221287-138-3-481
Rodriguez F, Lillington J, Johnson S, Timmel CR, Lea SM, Berks BC (2014) Crystal structure of the Bacillus subtilis phosphodiesterase PhoD reveals an iron and calcium-containing active site. J Biol Chem 289:30889–30899. https://doi.org/10.1074/jbc.M114.604892
Nannipieri P, Giagnoni L, Landi L, Renella G (2011) Role of phosphatase enzymes in soil. In: Bünemann E, Oberson A, Frossard E (eds) Phosphorus in action, Soil Biology vol 26. Springer, Berlin, pp 215–241
Fraser TD, Lynch DH, Bent E, Entz MH, Dunfield KE (2015) Soil bacterial phoD gene abundance and expression in response to applied phosphorus and long-term management. Soil Biol Biochem 88:137–147. https://doi.org/10.1016/j.soilbio.2015.04.014
Eder S, Shi L, Jensen K, Yamane K, Hulett FM (1996) A Bacillus subtilis secreted phosphodiesterase/alkaline phosphatase is the product of a Pho regulon gene, phoD. Microbiology 142:2041–2047. https://doi.org/10.1099/13500872-142-8-2041
Wanner BL (1990) Phosphorus assimilation and its control of gene expression in Escherichia coli. In: Hauska G, Thauer RK (eds) The molecular basis of bacterial metabolism 41. Colloquium der Gesellschaft für Biologische Chemie 5–7 April 1990 in Mosbach/Baden. Springer, Berlin, pp 152–163
Acknowledgments
This project was carried out within the framework of the priority program 1315 “Biogeochemical Interfaces in Soil” founded by the Deutsche Forschungsgemeinschaft (DFG): KO 1035/33-1, KO 1035/33-2, and SCHL 446/4-2. Moreover, the data analysis was supported by the Federal Ministry of Education and research (BMBF), funding the BonaRes project “Innovative solutions to sustainable Soil Phosphorus management” (InnoSoilPhos) (No. 031B0509 B). We further thank Juliette Ohan for the English proofreading.
Funding
Open Access funding enabled and organized by Projekt DEAL. This research was funded by the Deutsche Forschungsgemeinschaft (DFG): KO 1035/33-1, KO 1035/33-2, and SCHL 446/4-2 and the Federal Ministry of Education and research (BMBF) (No. 031B0509 B).
Author information
Authors and Affiliations
Contributions
CV, GP, and IKK designed and conducted the experiment. CV performed P measurements. GV, AS, and SK performed sequencing and assisted in bioinformatics data analysis. IT analyzed the metagenomics data, wrote the original draft of the manuscript, and performed the lab work. SS, MS, and MMF supervised IT, and reviewed and edited the manuscript. MS and IKK obtained the funding. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Consent to Participate
Not applicable.
Consent for Publication
The authors declare that consent is given for publication.
Code Availability
Not applicable.
Supplementary Information
ESM 1
(PDF 829 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tanuwidjaja, I., Vogel, C., Pronk, G.J. et al. Microbial Key Players Involved in P Turnover Differ in Artificial Soil Mixtures Depending on Clay Mineral Composition. Microb Ecol 81, 897–907 (2021). https://doi.org/10.1007/s00248-020-01635-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00248-020-01635-1