
Abstract
The organically modified titanium alkoxides Ti2(Oi-Pr)4(OOCCMe2O)2(i-PrOH)2 and Ti4(Oi-Pr)4(SA)6 were obtained from the reaction of Ti(Oi-Pr)4 with 2-hydroxy-isobutyric acid and salicyladoxime (SA-H2), respectively. Reaction of 1,3-dibenzoyl acetone (DBA-H) did not result in a substituted titanium alkoxide derivative, but instead in the oxo cluster Ti4O2(Oi-Pr)8(DBA)2 after allowing moisture to diffuse into the reaction mixture. The three titanium compounds show common structural features which are different to derivatives void of ligand OH groups. The latter play a decisive role in coordinating the ligands to the titanium centers.
Graphic abstract

Similar content being viewed by others

Introduction
Metal alkoxides, M(OR)n, are usually not employed as such for sol–gel processing, but instead modified by organic ligands. Reasons for doing so are manifold (control of reactivity, creation of porosity, functionalization, etc.) and have been discussed elsewhere [1]. Bidentate monoanionic groups (X ∩ X or X ∩ Y) are preferred, because they are more strongly bonded than monodentate ligands. The organically modified derivatives are prepared by reacting M(OR)n with the corresponding protonated compounds.
The best-investigated (structural) chemistry of organically modified metal alkoxides is that of titanium. Substitution of titanium alkoxides, Ti(OR)4, with X ∩ X or X ∩ Y in a 1:1 ratio commonly results in derivatives of the composition Ti2(OR)6(X ∩ X/Y)2. In the compounds obtained by reaction of β-diketones, β-ketoesters, oximes, or aminoalcohols each titanium atom is chelated by one X ∩ X or X ∩ Y ligand (structure type A, Scheme 1) [2]. Two bridging bidentate ligands (structure type B, Scheme 1) were observed in Ti2(OMe)6[(CH2)2PMe2]2 [3] and, with a slight variation (one carboxylate ligand bridging and the second hydrogen-bonded to a coordinated ROH), in the rare derivatives Ti2(OR)6(OOCRʹ)2(ROH) [4, 5]. Reactions with carboxylic acids normally give oxo clusters of the type TiaOb(OR)c(OOCRʹ)d, with bridging carboxylate ligands, because water is generated by reaction of the eliminated ROH with the carboxylic acid [2]. The dimeric nature of A and B is due to the preference of the Ti atoms for octahedral coordination, which is achieved by a pair of bridging OR groups.

When Ti(OEt)4 was reacted with the amino acid glycine, a compound with the composition Ti2(OEt)6(glycinate)2 (1, Scheme 2) was obtained (and not an oxo cluster), but the structure was different to B [6]. Instead of bridging carboxylate groups, five-membered chelate rings were formed with the α-amino group and only one COO oxygen coordinated to Ti. Thus, the structure of Ti2(OEt)6(glycinate)2 is of the A type, with X = O and Y = N.

The structure of 1 shows that a substituent in the chelating/bridging ligand capable of also coordinating to the metal center (as the amino group in 1) might change the coordination of the ligand in the modified titanium alkoxides compared to the corresponding unsubstituted ligands. We now tested whether and how a hydroxy substituent would influence the structures. Derivatives Ti2(OR)6(X ∩ X/Y)2 with such ligands were previously unknown; they were now obtained by the reaction of Ti(Oi-Pr)4 with an OH-substituted carboxylic acid and an OH-substituted oxime as well as a 1,3,5-triketone, which can formally be considered an OH-substituted β-diketone. We concentrate on the solid-state structures of the derivatives, and elaborate on some common structural features. The solution chemistry of the compounds is disregarded, because it is well known that IR and NMR spectra of metal alkoxide derivatives in most cases do not provide meaningful structural information; the structures are often dynamic so that the NMR spectra just show averaged signals.
Results and discussion
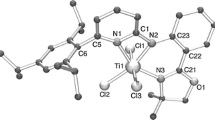
No oxo cluster was formed when Ti(Oi-Pr)4 was reacted with an equimolar amount of 2-hydroxy-isobutyric acid, as in the reaction with α-amino acids, but instead a centrosymmetric dimer 2 with again only one COO oxygen and the α-hydroxy group of the hydroxycarboxylate ligands coordinated to titanium (Fig. 1). The hydroxycarboxylate ligands, however, were chelating-bridging contrary to the chelating amino carboxylate ligands in 1. The central Ti2O2 unit and the two chelate rings of 2 are nearly coplanar.

Structure of Ti2(Oi-Pr)4(OOCCMe2O)2(i-PrOH)2 (2) (red: O, light grey: Ti). Hydrogen atoms, except that of the hydrogen bond, were omitted for clarity. Dashed lines represent the hydrogen bonds (color figure online)
For electroneutrality, one of the coordinated groups must be uncharged; i.e. the question was whether the composition of the compound was Ti2(Oi-Pr)6(OOCCMe2OH)2 (analogous to 1) or Ti2(Oi-Pr)4(OOCCMe2O)2(i-PrOH)2. This question was clearly answered by the Ti–O bond lengths, since Ti–O6 was much longer (221.56(6) pm) than Ti–O1 (184.48(6) pm), the other Ti–OR group perpendicular to the plane of the chelate rings (Table 1). O6 is, therefore, the oxygen atom of a coordinated i-PrOH molecule and O1 that of an i-PrO− group. Furthermore, the sum of angles around the bridging oxygen O2 is exactly 360° (it would by pyramidal if protonated). Thus, the composition of 2 is Ti2(Oi-Pr)4(OOCCMe2O)2(i-PrOH)2, i.e. the hydroxy group of the hydroxycarboxylate ligand is deprotonated and takes the position of the bridging OR group. The coordinated i-PrOH ligand is stabilized by a hydrogen bond to the neighboring Oi-Pr group: the O1···O6* distance of 277.7 pm is a clear indication of an O–H···O arrangement. The bridging hydrogen atom was identified in the X-ray structure analysis. The i-PrOH ligand is bent towards the hydrogen-accepting O1* (Ti1–Ti1–O6 77.14(2) pm) to facilitate a hydrogen bond interaction. The same stabilization of a neutral ligand has been observed in dimeric titanium alkoxides with fluorinated alkoxo groups (RF), Ti2(ORF)8(ROH)2 (R = Et, i-Pr) [7, 8], in dimeric carboxylate derivatives Ti2(OR)6(OOCR’)2(ROH) (see above) and in amine adducts Ti2(OR)6(NH2R)2. In the latter, the amino group interacts with a neighboring OR group through an N–H···O interaction [9, 10].
The Ti–O2–Ti* bridge is strongly asymmetric, as can be seen from the Ti-O2 distances (196.40(5) and 205.72(5) pm) as well as the C4-O2-Ti angles (137.13(4) and 117.98(4)).
The structure of 2 can be generalized to C, where Oa and Ob are the oxygen atoms of the chelating-bridging ligand and L a neutral ligand. A combination of the structural motifs A and C (Scheme 3) was found in Ti4(Oi-Pr)4(SA)6 (3), which was obtained from the reaction of Ti(Oi-Pr)4 with variable proportions of salicylaldoxime (H2SA) (molar ratio 1:1–1:5). The chelating-bridging entity in 3, however, is not a simple ligand, but instead a building block of the composition Ti(SA)2(Oi-Pr), schematically shown at the bottom of Scheme 3 (where Oa and Ob are the oxygen atoms coordinating to the central Ti2 unit according to D, Oc is an oxygen atom of the SA ligand bound to the central Ti2 unit). Thus, the six-membered chelating unit is O–N–Ti–N–O.

The solid-state structure of 3 contains two independent centrosymmetric molecules with only minor structural differences; only one is reproduced in Fig. 2 and Table 2. While the coordination octahedra of Ti3 and Ti3* are condensed with each other through the bridging oxygen atoms O23 and O23* and form the central unit D, Ti2 and Ti2* are part of the “chelating unit” and are connected to the central unit through bridging oximate groups. They are coordinated by a terminal Oi-Pr group and by two chelating SA ligands, which interact with the metal atom though the phenolate oxygen and the nitrogen atom (the two nitrogen and oxygen atoms being cis to each other). The coordination octahedron is completed by an oxygen atom from the SA ligand of the central unit D (see below). Ti3 of the central unit is coordinated by one chelating SA ligand, one Oi-Pr group and three oximate oxygens from the “chelating unit”.

Structure of Ti4(Oi-Pr)4(SA)6 (3) (red: O, blue: N, light grey: Ti). Only one molecule in the asymmetric unit is shown. Hydrogen atoms were omitted for clarity (color figure online)
Each SA ligand in 3 coordinates through the phenolic oxygen and the oximate nitrogen, but there are three different SA ligands with regard to the metal coordination (Scheme 4; the numbers in refer to the Harris notation of multidentate ligands [11]): the two SA ligands chelated to Ti2 differ in a way that the oximate oxygen of one of them (with O6 and N9) is coordinated to only one Ti3 atom (2.111, left in Scheme 4), while that of the second (with O3 and N11) is coordinated to both Ti3 and Ti3* (3.211). The third type of SA ligand (2.111, right in Scheme 4) chelates to Ti3 and additionally coordinates to Ti2 through the oximate oxygen. The 3.211 coordination mode was also found in another SA-substituted titanium alkoxide, viz. Ti3(Oi-Pr)8(SA)2 [12] and the SA-substituted oxo derivative Ti4(OMe)6(SA)4, both being structurally unrelated to 3 [13]. This coordination of SA is in contrast to titanium alkoxide derivatives with aliphatic or aromatic aldoximate ligands without the OH group in ß-position, where the oximate NO group is always side-on coordinated to the same titanium atom. Representative examples with aromatic oximate ligands are Ti2(Oi-Pr)4(benzaldoximate)4 and Ti2(Oi-Pr)4(anisaldoximate)4 [14].

The O23 bridge in 3 is symmetric (205.2(5) and 207.8(5) pm), probably because the “chelating unit” in 3 is less strained as the chelating hydroxycarboxylate ligand in 2. The shortest Ti–O distances are those trans to the coordinated nitrogen atoms (Ti3–O2 174.8(5) and Ti2–O11 176.9(5) pm) thus compensating the relatively weak Ti–N interactions (Ti2–N9 223.5(7) and Ti2–N11 218.5(6) pm). For comparison, the Ti–N distances in Ti2(Oi-Pr)4(ON = CHPh)4, with side-on coordinated NO groups, are in the range 206.5–211.0 pm and that of Ti2(Oi-Pr)8(NH2R)2 in the range 229–231 pm [14].
The titanium alkoxide derivatives 2 and 3 demonstrate the great impact of OH substituents on the structures formed upon reaction of Ti(OR)4 with carboxylic acids or oximes. In both cases, the C–OH group was deprotonated and bridged two titanium atoms. It thus had a structure-determining role and resulted in completely different structures compared with the compounds with unsubstituted carboxylate or oximate ligands. Along this line, we also tested 1,3,5-triketones, which can be considered OH substituted β-diketones. We were unable to isolate a substituted titanium alkoxide derivative when 1,5-diphenyl-pentane-1,3,5-trione (1,3-dibenzoyl acetone, DBA-H) was reacted with Ti(Oi-Pr)4 in dichloromethane. However, when ambient moisture was allowed to diffuse slowly in the reaction solution, the crystalline titanium oxo cluster Ti4O2(Oi-Pr)8(DBA)2 (4) (Fig. 3 and Table 3) was reproducibly obtained (Scheme 5).

Molecular structure of 4

In the centrosymmetric tetramer 4, each titanium atom is octahedrally coordinated; the four [TiO6] polyhedra share edges (Fig. 4). The same cluster core structure was observed for Ti4O4(Oi-Pr)4(OCHPhCHMeNHMe)4 [15]. The comparison of 4 with Ti4O2(Oi-Pr)10(acac)2 (acac = acetylacetonate) [16], one of the few titanium oxo clusters with β-diketonate ligands, is interesting (Fig. 4), where—formally—the DBA is replaced by (acac + Oi-Pr). While all titanium atoms in 4 are octahedrally coordinated, two of the titanium atoms in Ti4O2(Oi-Pr)10(acac)2 (the ones without acac ligands) are only 5-coordinate. Furthermore, the polyhedra are differently connected. The reason for this difference is that the central oxygen atoms of the (planar) DBA ligands bridge two Ti atoms. This supplementary coordination renders the inner titanium atoms also octahedrally coordinated. The highly condensed cluster core of 4 results from three different bridging groups: DBA linking Ti1 and Ti2, μ3-O20 connecting Ti2, Ti2* and Ti1, and a µ2-Oi-Pr bridge (O7) between Ti2 and Ti1*

Comparison of the cluster core structure in 4 (left) with that of Ti4O2(Oi-Pr)10(acac)2 (right)
The Ti–Oacac distances in Ti2(Oi-Pr)6(acac)2 (with chelating acac ligands) are 207.3(4) and 203.0(3) pm [17]. They are typical Ti–O bond lengths of coordinated β-diketonates. In comparison, the Ti–O2 distance of the bridging DBA oxygen (O2) in 4 is much longer (Ti1–O2 215.6(1) and Ti2–O2 211.9(1) pm) and that of the “outer” oxygen atoms O1 and O3 (Ti1–O1 197.9(1) and Ti2–O3 197.3(1) pm) much shorter. The corresponding C-O distances in 4 are not significantly different from each other (129.1–130.9 pm). As a matter of fact, the Ti–O2 distance in 4 is also much longer than the corresponding distances of the bridging oxygen atoms in 2 and 3. A possible interpretation could be that a mesomeric form of the coordinated DBA prevails, in which the oxygen atoms in the 1- and 5-position of the DBA ligand carry a higher negative charge, while that in the 3-position is more ketone-like. This is supported by the bond distances trans to O2: the bridging Oi-Pr ligand O7 is very unsymmetrical (Ti2–O7 191.0(1) and Ti1–O7* 218.7(1) pm), the shorter distance being trans to O2. Likewise, the Ti1–O5 distance (177.2(1) pm), also being trans to O2, is shorter than typical distances of Ti–OR groups.
Conclusion
The structures reported in this article clearly show that an OH-substituent in bidentate organic compounds commonly used for the modification of titanium alkoxides (or metal alkoxides in general), such as carboxylic acids, oximes, or β-diketones, has a decisive influence on the structural outcome of the modified titanium alkoxides. In each case, an OR group exchange took place with the Rʹ–OH group of the ligand replacing a Ti–OR group, in addition to coordination of the bidentate group. This additional coordination also changed the coordination mode of the carboxylate, oximate or β-diketonate group. Nevertheless, common structural features between compounds 2–4 were identified, which can also be traced back to the structures of the derivatives with OH-free ligands.
Experimental
Bis(2-oxoisobutyrato)tetrakis(isopropoxy)bis(isopropanol)tetratitanium(IV) (2, C26H56O12Ti2)
An amount of 1.42 g of Ti(Oi-Pr)4 (5 mmol) was added to a solution of 520 mg 2-hydroxy-isobutyric acid (5 mmol) in 2 cm3 of water-free 2-propanol. Compound 2 started to crystallize after one day. The crystals were washed with 2-propanol and stored under moisture-free conditions.
Hexakis([(oxyimino)methyl]phenolato)tetrakis(isopropoxy)-tetratitanium(IV) (3, C54H58N6O16Ti4)
The reaction between Ti(Oi-Pr)4 and various proportions of salicylaldoxime (SA-H2) (1:1–1:5) was carried under argon. The corresponding amount of Ti(Oi-Pr)4 was added to the oxime solution in water-free CH2Cl2. Compound 3 crystallized rapidly from the solution at room temperature. They were collected and stored under moisture-free conditions.
Bis(1,5-diphenylpentane-1,3,5-trionato)-di(oxido)octakis-(isopropoxy)tetratitanium(IV) (4, C58H80O16Ti4)
An amount of 176 mg (0.66 mmol) of 1,5-diphenyl-pentane-1,3,5-trione was dissolved in 4 cm3 of CH2Cl2, and 0.4 cm3 (1.32 mmol) of Ti(Oi-Pr)4 was added dropwise. The mixture was stirred for 10 min and then kept at room temperature at ambient conditions to allow slow diffusion of water into the reaction mixture. Compound 4 started to crystallize from the solution after 24 h.
X-ray structure analyses
All measurements were performed using Mo Kα (λ = 71.073 pm) radiation. Data were collected on a Bruker AXS SMART APEX II four-circle diffractometer with κ-geometry. Data were collected with φ and ω-scans and 0.5° frame width. The data were corrected for polarization and Lorentz effects, and an empirical absorption correction (SADABS) was employed. The cell dimensions were refined with all unique reflections. SAINT PLUS software (Bruker Analytical X-ray Instruments, 2007) was used to integrate the frames. Symmetry was then checked with the program PLATON. Details of the X-ray investigations are given in Table 4.
The structures were solved by the Patterson method (SHELXS97). Refinement was performed by the full-matrix least-squares method based on F2 (SHELXL97) with anisotropic thermal parameters for all non-hydrogen atoms. Hydrogen atoms were inserted in calculated positions, if not otherwise stated, and refined riding with the corresponding atom.
CCDC-2017662 (for 2), -2017663 (for 3), and -2017664 (for 4) contain the supplementary crystallographic data. These data can be obtained free of charge from The Cambridge Crystallographic Data Center via www.ccdc.cam.ac.uk/structures/.
References
Schubert U, Hüsing N, Lorenz A (1995) Chem Mater 7:2010
Schubert U (2005) J Mater Chem 15:3701
Scharf W, Neugebauer D, Schubert U, Schmidbaur H (1978) Angew Chem Int Ed 17:601
Boyle TJ, Tyner RP, Alam TM, Scott BL, Ziller JW, Potter BG Jr (1999) J Am Chem Soc 121:12104
Czakler M, Artner C, Schubert U (2012) Eur J Inorg Chem 2012:3485
Schubert U, Tewinkel S, Möller F (1995) Inorg Chem 34:995
Fisher J, van der Sluys WG, Huffman JC, Sears J (1993) Synth React Inorg Met Org Chem 23:479
Campbell C, Bott SG, Larsen R, van der Sluys WG (1994) Inorg Chem 33:4950
Fric H, Schubert U (2005) New J Chem 29:232
Fric H, Puchberger M, Schubert U (2006) J Sol-Gel SciTechnol 40:155
Coxall RA, Harris SG, Henderson DK, Parsons S, Tasker PA, Winpenny REP (2000) J Chem Soc Dalton Trans 2000:2349
Davidson MG, Johnson AL, Jones MD, Lunn MD, Mahon MF (2007) Polyhedron 26:975
Chen S, Fang WH, Zhang L, Zhang J (2018) Inorg Chem 57:8850
Baumann SO, Bendova M, Puchberger M, Schubert U (2011) Eur J Inorg Chem 2011:573
Fric H, Puchberger M, Schubert U (2008) Eur J Inorg Chem 2008:1452
Moran PD, Rickard CEF, Bowmaker GA, Cooney RP, Bartlett JR, Woolfrey JL (1998) Inorg Chem 37:1417
Errington RJ, Ridland J, Clegg W, Coxall RA, Sherwood JM (1998) Polyhedron 17:659
Funding
Open access funding provided by TU Wien (TUW).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Schubert, U., Bendova, M., Czakler, M. et al. The structural chemistry of titanium alkoxide derivatives with OH-substituted bidentate ligands. Monatsh Chem 151, 1697–1703 (2020). https://doi.org/10.1007/s00706-020-02698-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00706-020-02698-z