

Abstract
Decline of protein quality control in neurons contributes to age-related neurodegenerative disorders caused by misfolded proteins. 4E-BP1 is a key node in the regulation of protein synthesis, as activated 4E-BP1 represses global protein translation. Overexpression of 4E-BP1 mediates the benefits of dietary restriction and can counter metabolic stress, and 4E-BP1 disinhibition on mTORC1 repression may be neuroprotective; however, whether 4E-BP1 overexpression is neuroprotective in mammalian neurons is yet to be fully explored. To address this question, we generated 4E-BP1-overexpressing transgenic mice and confirmed marked reductions in protein translation in 4E-BP1-overexpressing primary neurons. After documenting that 4E-BP1-overexpressing neurons are resistant to proteotoxic stress elicited by brefeldin A treatment, we exposed primary neurons to three different Parkinson's disease (PD)-linked toxins (rotenone, maneb, or paraquat) and documented significant protection in neurons from newborn male and female 4E-BP1-OE transgenic mice. We observed 4E-BP1-dependent upregulation of genes encoding proteins that comprise the mitochondrial unfolded protein response, and noted 4E-BP1 overexpression required activation of the mitochondrial unfolded protein response for neuroprotection against rotenone toxicity. We also tested whether 4E-BP1 could prevent α-synuclein neurotoxicity by treating 4E-BP1-overexpressing primary neurons with α-synuclein preformed fibrils, and we observed marked reductions in α-synuclein aggregation and neurotoxicity, thus validating that 4E-BP1 is a powerful suppressor of PD-linked pathogenic insults. Our results indicate that increasing 4E-BP1 expression or enhancing 4E-BP1 activation can robustly induce the mitochondrial unfolded protein response and thus could be an appealing strategy for treating a variety of neurodegenerative diseases, including especially PD.
SIGNIFICANCE STATEMENT In neurodegenerative disease, misfolded proteins accumulate and overwhelm normal systems of homeostasis and quality control. One mechanism for improving protein quality control is to reduce protein translation. Here we investigated whether neuronal overexpression of 4E-BP1, a key repressor of protein translation, can protect against misfolded protein stress and toxicities linked to Parkinson's disease, and found that 4E-BP1 overexpression prevented cell death in neurons treated with brefeldin A, rotenone, maneb, paraquat, or preformed fibrils of α-synuclein. When we sought the basis for 4E-BP1 neuroprotection, we discovered that 4E-BP1 activation promoted the mitochondrial unfolded protein response. Our findings highlight 4E-BP1 as a therapeutic target in neurodegenerative disease and underscore the importance of the mitochondrial unfolded protein response in neuroprotection against various insults.
- 4E-BP1
- alpha-synuclein
- mitochondrial unfolded protein response
- neuroprotection
- Parkinson's disease
- protein translation
Introduction
More than two decades ago, research into the pathogenesis of inherited neurodegenerative disorders and sporadic neurodegenerative diseases revealed a common theme of protein misfolding as the mechanistic basis of almost all such disorders (Taylor et al., 2002). The importance of protein quality control in maintaining homeostasis in neurons and other CNS cells was further reinforced by the realization that autophagy is critically important for normal CNS function, as conditional deletion of an essential autophagy pathway gene from neurons is sufficient to produce rapidly progressive neurodegeneration in mice, accompanied by accumulation of ubiquitinated protein aggregates (Hara et al., 2006; Komatsu et al., 2006). In CNS cells subjected to the accumulation of a toxic disease-causing misfolded protein, as occurs in neurodegenerative disorders, such as Alzheimer's disease, Parkinson's disease (PD), or amyotrophic lateral sclerosis, the proteotoxic stress causes a disruption of proteostasis, resulting in altered solubility of the proteome in neurons and glia (Pace et al., 2018). While numerous studies have shown that boosting proteostasis can effectively counter such CNS dyshomeostasis, another approach would be to reduce protein translation.
The mechanistic target of rapamycin (mTOR) is a serine/threonine protein kinase that regulates cellular growth, cytoskeletal organization, differentiation, development, survival, and aging (Maiese et al., 2013). mTOR is ubiquitously expressed (including in the CNS) and serves as the central component of two major complexes in multicellular organisms, mTORC1 and mTORC2. While mTORC2 regulates the organization of the actin cytoskeleton, activation of mTORC1 increases protein translation through activation of S6 kinase 1 and inhibition of 4E binding protein 1 (4E-BP1) (Lipton and Sahin, 2014). As 4E-BP1 represses the activity of eukaryotic initiation factor 4E (eIF4E), increased 4E-BP1 function results in reduced protein translation. Numerous studies have documented that rapamycin inhibition of mTORC1 is neuroprotective; but although this effect has often been attributed to autophagy activation, rapamycin can inhibit protein aggregation in Huntington's disease cellular models independently of autophagy by reducing protein synthesis (King et al., 2008). Furthermore, activation of 4E-BP1 alone has been shown to prevent dopaminergic neurodegeneration in a Drosophila model of PD (Tain et al., 2009). Whether 4E-BP1 overexpression is neuroprotective in mammalian neurons is yet to be fully explored.
Primers for PCR Analysis
Here we studied a line of transgenic mice that overexpress WT 4E-BP1, and observed marked neuroprotection against protein misfolding stress, PD-linked toxin insult, and preformed fibrils (PFFs) of α-synuclein (α-syn) in primary neurons overexpressing 4E-BP1. When we examined the expression of metabolic regulators and mitochondrial stress response genes, we noted that 4E-BP1 overexpression activated the mitochondrial unfolded protein response (UPRmt), which is a mitochondrial stress response documented to be neuroprotective in cell culture, worm, and mouse models of Alzheimer's disease (Jovaisaite et al., 2014). We confirmed that neuroprotection in neurons overexpressing 4E-BP1 depends on activation of the UPRmt. These findings underscore the importance of 4E-BP1 as a therapeutic target in neurodegenerative proteinopathies.
Materials and Methods
4E-BP1 transgenic mice
All animal experimentation adhered to National Institutes of Health guidelines and was approved by, and performed in accordance with, the University of California, San Diego and University of California, Irvine Institutional Animal Care and Use Committees and the Duke University Committee on the Use and Care of Animals. 4E-BP1-OE transgenic lines were maintained on the C57BL/6J strain background.
Primary cortical neuron culture experiments
Primary cortical neurons were cultured from dissociated cortex of postnatal day 0 (P0) to day 1 (P1) mice, as described previously (Dubinsky et al., 2014). The dissociation was done using trypsin (T9935), trypsin inhibitor (T6522), and DNase I (10104159001, Roche Diagnostics). Primary neurons were seeded onto plates coated with 0.1 mg/ml poly-D-lysine hydrobromide (P1024-50MG) and grown in complete media (CM) consisting of Neurobasal-A medium (Thermo Fisher Scientific, 10888022) supplemented with 0.5 mm L-glutamine (Thermo Fisher Scientific, 25030149), 0.25% penicillin-streptomycin (Thermo Fisher Scientific, 15140148), and 0.25% B-27 supplement (Thermo Fisher Scientific, 17504001). Primary neurons were maintained until treatment by removing half of the CM and replacing it with fresh CM on day 3 (where day 0 is the day of cell seeding), day 5, and every second day after day 5. For SUnSET assay, neurons were treated with puromycin for 10 min (10 µg/ml), followed by incubation in puromycin-free media for 20 min. On day 14, primary neurons were treated for 5 h with 50 µg/ml brefeldin A (B5936). Treatment with 10 μm paraquat, 1 μm maneb, or 0.1 μm rotenone was done on day 14 (DIV 14) followed by immunoblot, propidium iodide staining, or LDH assay. Primary hippocampal neurons were prepared in a similar manner as the cortical neurons; and in both cases, we used comparable numbers of males and females between 4E-BP1 transgenic and WT littermate control mice. Briefly, the hippocampus from P0 mice was dissected, meninges removed, dissociated into single cells, and maintained in the same manner as primary cortical neurons. The primary hippocampal neurons were seeded with preformed α-syn fibrils at a concentration of 100 ng/ml on day 10 and maintained for an additional 7 d, before immunocytochemistry was conducted with purified anti-α-syn phospho (Ser129) antibody 81A (825701, BioLegend; previously Covance catalog #MMS-5091) as described previously (Luk et al., 2009, 2012).
LDH and propidium iodide assays
The LDH assay was conducted using LDH-Cytotoxicity Assay Kit II (BioVision, K313-500-2) according to the manufacturer's instructions. Briefly, LDH Assay Buffer and WST Substrate Mix were added to media harvested from primary neurons. Plate readings were taken at four time points: 30, 60, 90, and 120 min. The percent of LDH released was calculated as the percent cytotoxicity over the amount of protein (μg/μl). Amount of protein was separately determined using BCA assay using the Pierce BCA Protein Assay Kit (catalog #23225). For the High Control, cells were treated with 10 μl of the cell lysis buffer provided with the LDH assay kit and kept in the incubator for 20 min before the media was harvested for LHD assay. For the Low Control, the cells/neurons were treated with complete media for neuronal growth. After harvesting media, cells were treated with propidium iodide (P4864, 1:3000) and Hoechst (33342, 1:10,000) and kept in the incubator for 15 min before imaging each well under a 780 LSM confocal microscope (Carl Zeiss) at 10× or Keyence BZ-X710. Briefly, three images were taken per well, and each experiment was done in triplicate and repeated 3 times independently, unless otherwise mentioned.
JC-1 assay
Primary neurons were treated with 50 μg/μl brefeldin A (B5936) in CM, or CM alone for 5 h. Primary neurons were also incubated with 30 μm carbonyl cyanide 3-chlorophenylhydrazone (C2759, Sigma Millipore) for 20-30 min at 37°C. This was followed by incubation with 100 μm JC-1 dye (Calbiochem, catalog #420200) at 37°C for 20-30 min. Following this, the cells were washed carefully 3 times with warm PBS buffer before the fluorimetric reading was taken. Red to green ratio was measured using Tecan infinite M200 pro. JC-1 dye exhibits potential-dependent accumulation in mitochondria, indicated by a fluorescence emission shift from green (∼529 nm) to red (∼590 nm). A decrease in the red/green ratio indicates mitochondrial depolarization.
Lentivirus preparation and transduction
Validated lentiviral pLKO.1 vectors harboring shRNA against Chop (SHCLNG NM_007837), Cebpβ (SHCLNG NM_009883), and Hspd1 (SHCLNG NM_010477) specific against mouse gene targets were ordered from Sigma Millipore; 7.5 µg of shRNA plasmids was purified and cotransfected with 7.5 µg LP1, LP2, and VSVG plasmids in a 15 cm dish with 70% confluent HEK293FT using Lipofectamine 2000 according to the manufacturer's protocol. Six hours later, the media was changed to media without antibiotics and the virus was harvested 48 h after transfection from the media. Briefly, the media was filter sterilized (0.45 μm filter) and subjected to 20% sucrose gradient centrifugation (the media was put in the ultracentrifuge tube, followed by addition of 2 ml of sucrose at the bottom of the well) at 20,000 rpm at 4°C for 2 h. The media was removed along with the sucrose solution and a light pellet was observed which was solubilized in ice-cold PBS. Lentivirus was used at 30-50 multiplicity of infection. Prepared lentivirus was infected in primary cortical neurons 7 d after plating. On day 14, the neurons were treated with rotenone before subjecting them to propidium iodide staining or LDH assay. For knockdown efficiency testing, the infected neurons were harvested on day 14 by Trizol and subjected to RT-PCR analysis.
RT-PCR
RNA was harvested from cultured primary neurons using TRIzol Reagent (Thermo Fisher Scientific, 15596018) according to the manufacturer's instructions; 1 μg of RNA was used to prepare cDNA using USB First-Strand cDNA Synthesis Kit (USB products, catalog #75780) or Superscript II First strand (Invitrogen, catalog #1701598) for Real-Time PCR according to the manufacturer's instructions. PCR was performed using SYBR Green (Thermo Fisher Scientific, 4367659). A complete list of primers follows is given in Table 1.
Immunoblot analysis and immunohistochemistry
Western blotting was done as previously described (Dubinsky et al., 2014). Briefly, cell lysates were prepared using 1× RIPA buffer (Thermo Scientific Pierce, PI89901) plus protease inhibitor tablets (4693124001) and phosphatase inhibitor tablets (4906837001). Equivalent amounts of protein (10-25 μg) from each sample were mixed with 4× LDS sample buffer (Thermo Fisher Scientific, NP0007), 10× Reducing agent (Thermo Fisher Scientific, NP0009), and 1× RIPA buffer. The sample mixtures were heated for 10 min at 70°C, subjected to SDS-PAGE (sodium dodecyl sulphate–polyacrylamide gel electrophoresis), and transferred onto a PVDF membrane. The membranes were blocked for 1 h with 5% nonfat dry milk in PBS plus 0.1% Tween 20 (Thermo Fisher Scientific, BP337500) and then incubated with primary antibodies overnight at 4°C. This was followed by incubation with secondary antibodies for 1 h at room temperature. Chemiluminescent substrate was added to the blot, and chemiluminescence was detected using autoradiography.
For immunohistochemistry, all reagents were prepared in PBS. Coverslips with primary neurons were fixed with 4% PFA (Electron Microscopy Sciences, 15714) for 20 min, followed by addition of 0.2% Triton (T8787) for 5 min. Blocking was done in a solution of 5% BSA (A7906) plus 5% goat serum (Jackson ImmunoResearch Laboratories, 005-000-121) for 1 h at room temperature. Primary antibodies were diluted with 5% BSA plus 5% goat serum. Incubation with primary antibodies was done overnight at 4°C. Secondary antibodies were prepared in 5% BSA plus 5% goat serum. Incubation with secondary antibodies was for 1 h at room temperature. Coverslips with primary neurons were incubated in Hoechst 33 342 (Thermo Fisher Scientific, H3570) at a 1:10,000 dilution for 10 min at room temperature before mounting them on Fluoromount G (Electron Microscopy Sciences, catalog #17984-25). The fixed cells were imaged using a 63× objective in an LSM 780 confocal microscope (Carl Zeiss). For immunocytochemistry of hippocampal neurons treated with 100 ng/ml of α-syn PFFs, neurons were fixed with 4% PFA, 4% sucrose, and 1% Triton X-100. Following fixation, neurons were blocked with 5% BSA in PBS, and incubated in primary antibody followed by AlexaFluor-488-conjugated secondary antibodies.
Unless indicated otherwise, all antibodies were purchased from Sigma Millipore. Primary antibodies used in this study were as follows: β-actin (Abcam, ab8226, 1:5000), 4E-BP1 (Cell Signaling Technology, 9644S, 1:1000), Hsp60 (Cell Signaling Technology, 12165S, 1:7500), cleaved caspase-3 (Cell Signaling Technology, 9664S, 1:1000), α-syn (BioLegend, previously Covance catalog #MMS-5091, 1:400 for IC), cleaved caspase-3 (Cell Signaling Technology, 9664S, 1:400 for IC), microtubule-associated protein 2 (MAP2 [microtubule-associated protein 2], Millipore, MAB3418, 1:400 for IC), puromycin (Millipore, MABE343, 1:1000), and 4EBP1 (Cell Signaling Technology, 9644S,1:400). For Western blot analysis, primary antibodies were used at a dilution of 1:1000 and secondary antibodies (from Santa Cruz Biotechnology) were used at concentrations ranging from 1:5000 to 1:10,000. For immunocytochemistry, primary antibodies were used at a concentration of 1:400 and secondary antibodies (from Thermo Fisher Scientific) were used at a 1:100 dilution.
Experimental design and statistical analysis
All the graphs in this study were generated using GraphPad Prism 6 software. Statistical analysis was done using Microsoft Excel, Prism 6.0 (GraphPad), and SigmaPlot (Systat Software). Statistical significance was defined at p < 0.05. All t tests were two-tailed Student's t tests, and level of significance (α) was always set to 0.05. For one-way and two-way ANOVA, if statistical significance (p < 0.05) was achieved, then we performed post hoc analysis, as specified, to account for multiple comparisons. Details of each experiment, including the number of biological replicates and technical replicates, as well as the number of neurons analyzed, are provided in each figure legend. In addition to exact p values, figure legends include the degrees of freedom and effect size (EF) for each statistical comparison.
Results
4E-BP1 overexpression in neurons yields a marked reduction in protein translation
To examine the role of protein translation status as a modifier of disease, we generated 4E-BP1 transgenic mice in which a loxP-flanked STOP codon cassette is placed 5′ to the 4E-BP1 transgene and 3′ to the CAGGS promoter-enhancer (Tsai et al., 2015). These 4E-BP1 conditional transgenic mice were bred to CMV-Cre transgenic mice to yield a line of 4E-BP1-OE mice that display whole-body fourfold to fivefold overexpression of 4E-BP1, which was confirmed by immunoblot analysis of various tissues (Tsai et al., 2015, 2016). To determine the effect of 4E-BP1 overexpression on protein translation in neurons of 4E-BP1-OE mice, we derived primary cortical neurons from 4E-BP1-OE mice and nontransgenic WT littermate controls, and treated the cultured cortical neurons with a brief pulse of puromycin to monitor protein translation via SUnSET assay (Schmidt et al., 2009). As puromycin is a structural analog of aminoacyl tRNAs, it gets incorporated into nascent polypeptide chains, prevents further elongation of the translating protein, and can be detected by antibody staining. Immunohistochemistry analysis of puromycin-treated primary cortical neurons revealed visibly decreased puromycin immunostaining in primary cortical neurons from 4E-BP1-OE mice (Fig. 1A), and quantification of puromycin immunoreactivity confirmed a marked reduction in cortical neurons overexpressing 4E-BP1 (Fig. 1B). To corroborate these findings, we also performed immunoblot analysis of protein lysates from puromycin-treated primary cortical neurons from 4E-BP1-OE mice and WT littermate controls, and observed decreased puromycin-labeled protein in neurons overexpressing 4E-BP1 (Fig. 1C). Densitometry analysis of puromycin immunoblots similarly confirmed a significant reduction in protein translation in primary cortical neurons from 4E-BP1-OE mice (Fig. 1D).

Protein translation is markedly reduced in 4E-BP1-overexpressing neurons. A, DIV14 primary cortical neurons from control and 4E-BP1 transgenic mice were treated with puromycin (10 µg/ml for 10 min), immunostained with antipuromycin antibody, and counterstained with DAPI. Puromycin immunoreactivity is visibly decreased in primary neurons from 4E-BP1 transgenic mice. Scale bar, 10 µm. B, Densitometry analysis of puromycin immunostaining analysis shown in A. n = 3 biological replicates; n = 4 technical replicates/culture. *p = 0.020 (two-tailed t test; df = 21; EF = 1.1). C, DIV14 primary cortical neurons were treated with puromycin (10 µg/ml for 10 min) and then harvested for protein isolation followed by SDS-PAGE and immunoblotting with antipuromycin antibody. Puromycin immunoreactivity is visibly decreased in primary neurons from 4E-BP1 transgenic mice. β-actin served as a loading control, and transgene status was confirmed by immunoblotting for 4E-BP1. D, Densitometry analysis of puromycin immunoblotting analysis shown in C. n = 3 biological replicates. *p = 0.014 (two-tailed t test; df = 4; EF = 5.7). Error bars indicate SEM.
4E-BP1-overexpressing neurons are protected against brefeldin A-mediated proteotoxicity and are resistant to treatment with PD-linked environmental toxins
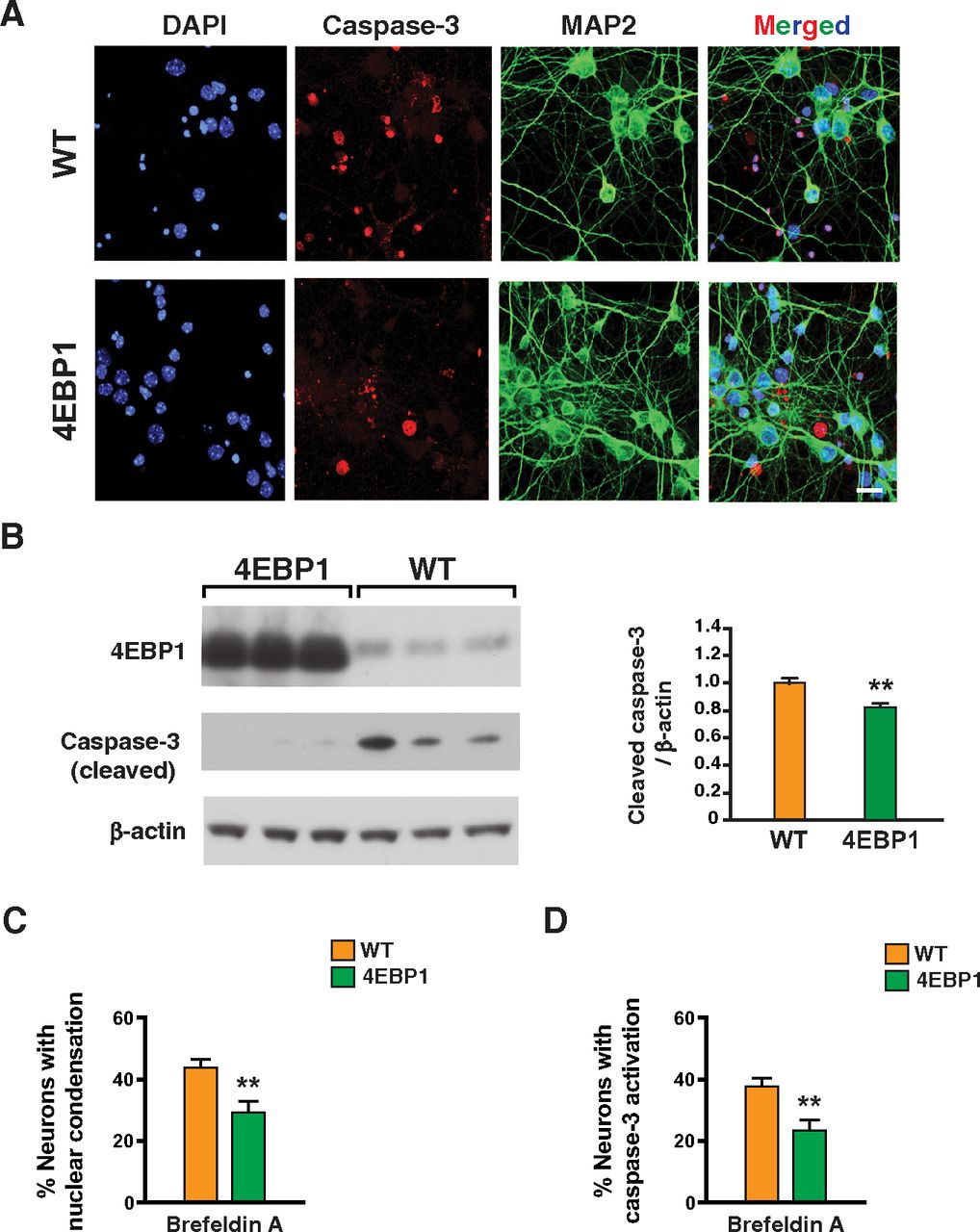
Proteostasis is essential for optimal cell growth and survival, dysregulation of which causes protein aggregation and is associated with age-related neurodegenerative disease (Labbadia and Morimoto, 2015). One possible remedy for proteostasis decompensation in neurodegenerative proteinopathies would be to reduce protein synthesis and promote protein refolding capacity. To determine whether 4E-BP1 overexpression can counter impaired protein quality control, we treated primary cortical neurons from 4E-BP1-OE mice and WT littermate controls with brefeldin A, a fungal metabolite known to inhibit protein secretion in eukaryotic cells by interfering with the function of the Golgi apparatus leading to proteotoxic endoplasmic reticulum stress (Fujiwara et al., 1988). Immunostaining of brefeldin A-treated cortical neurons for activated caspase-3 and MAP2 revealed obviously reduced neurotoxicity in 4E-BP1-overexpressing neurons (Fig. 2A,B). Quantification of neurotoxicity by the presence of nuclear condensation and by detection of activated caspase-3 in individual neurons confirmed that 4E-BP1 overexpression can significantly protect against general proteotoxic stress induced by brefeldin A treatment (Fig. 2C,D). In addition to promoting apoptotic activation, brefeldin A treatment also resulted in significant mitochondrial membrane depolarization, which was rescued in primary cortical neurons overexpressing 4E-BP1 (Fig. 3).
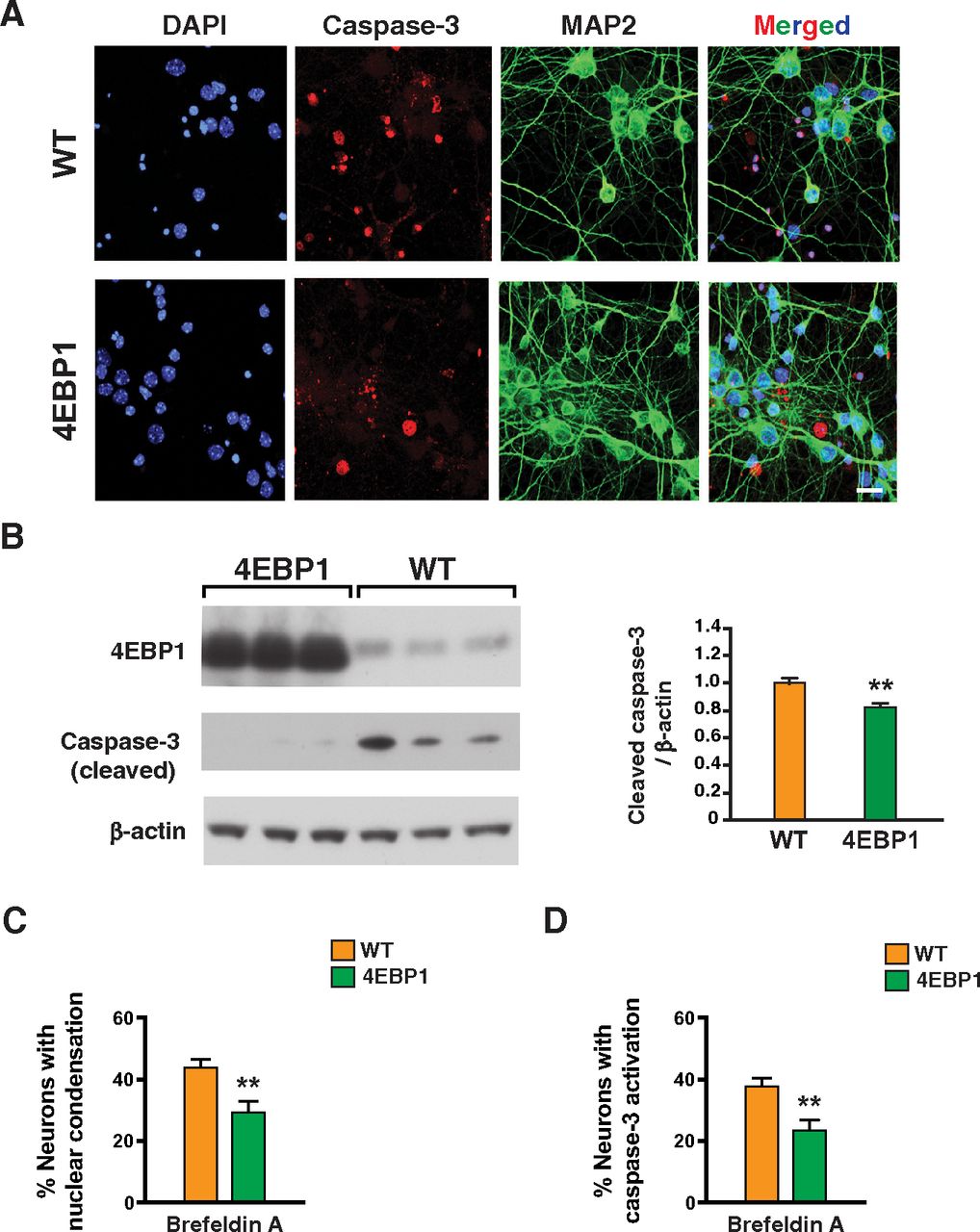
4E-BP1-overexpressing neurons are protected against brefeldin A proteotoxicity. A, DIV14 primary cortical neurons from control and 4E-BP1 transgenic mice were treated with brefeldin A (50 µg/ml), immunostained with antibodies against activated caspase-3 and MAP2, and counterstained with DAPI (4′,6-diamidino-2-phenylindole). Cell death is visibly decreased in primary neurons from 4E-BP1 transgenic mice, based on decreased activated caspase-3 nuclear staining and increased MAP2 neurite staining. Scale bar, 10 µm. B, DIV14 primary cortical neurons from control and 4E-BP1 transgenic mice were treated with brefeldin A (50 µg/ml), and then harvested for protein isolation followed by SDS-PAGE and immunoblotting with anti-activated caspase-3 antibody. Caspase-3 activation is visibly decreased in primary neurons from 4E-BP1 transgenic mice. β-Actin served as a loading control, and transgene status was confirmed by immunoblotting for 4E-BP1. Right, Densitometry analysis of activated caspase-3 immunoblotting. n = 3 biological replicates; n = 2 technical replicates. **p = 0.007 (two-tailed t test; df = 12; EF =1.9). C, DIV14 primary cortical neurons from control and 4E-BP1 transgenic mice were treated with brefeldin A (50 µg/ml), and counterstained with DAPI as in A. We then counted the number of neurons showing nuclear condensation. n = 4 biological replicates; n ≥ 50 neurons. **p = 0.008 (two-tailed t test; df = 8; EF = 2.6). D, DIV14 primary cortical neurons from control and 4E-BP1 transgenic mice were treated with brefeldin A (50 µg/ml), immunostained with antibodies against activated caspase-3 and MAP2, and counterstained with DAPI as in A. We then counted the number of neurons showing activated caspase-3 nuclear staining. n = 4 biological replicates; n ≥ 50 neurons. **p = 0.001 (two-tailed t test; df = 8; EF = 3.7). Error bars indicate SEM.

4E-BP1-overexpressing neurons maintain normal mitochondrial membrane depolarization on brefeldin A-mediated proteotoxicity. DIV14 primary cortical neurons from control and 4E-BP1 transgenic mice were treated with brefeldin A (50 mg/ml), treated with JC-1 dye, and then fluorescence emission measured to determine the extent of mitochondrial membrane depolarization. Results were normalized to nontransgenic (WT) neurons at baseline (untreated). Carbonyl cyanide m-chlorophenyl hydrazine (CCCP) treatment served as a positive control for depolarization. ANOVA with post hoc Tukey test: **p = 0.001 for WT untreated versus WT brefeldin A-treated (df = 4, EF = 0.9). *p = 0.012 for WT brefeldin A treated versus 4EBP1 brefeldin A-treated (df = 4, EF = 2.6). Error bars indicate SEM.
As proteotoxic stress in neurodegenerative proteinopathies may result in impaired general proteostasis, we reasoned that a reduced burden of newly synthesized proteins, as occurs on 4E-BP1 overexpression, could be neuroprotective in neurodegenerative disorders involving protein misfolding. PD is a progressive neurodegenerative disorder characterized by slowness of movement, rigidity, and tremor because of the selective loss of dopaminergic neurons in the substantia nigra, and in most cases likely results from α-syn misfolding (Dawson and Dawson, 2003). Rotenone is an insecticide and has been shown to produce parkinsonism in rodents in association with the formation of α-syn aggregate inclusions, which may reflect rotenone's ability to alter α-syn protein conformation (Betarbet et al., 2000; Sherer et al., 2003). In addition to rotenone, other environmental toxins prominently implicated in PD pathogenesis in rodent modeling include maneb and paraquat, which produce motor abnormalities in treated rodents, presumably because of altered dopamine metabolism, but do not elicit α-syn inclusion formation (Roede and Jones, 2014). To determine whether 4E-BP1 overexpression can protect against these PD-linked toxins, we treated primary cortical neurons from 4E-BP1-OE mice and WT littermate controls with rotenone, maneb, or paraquat, and we observed a dramatic increase in neurotoxicity in cortical neurons treated with each of these toxins (Fig. 4A). In all 3 cases, 4E-BP1 overexpression significantly blunted the neurotoxicity based on propidium iodide exclusion and LDH release assays (Fig. 4A,B). As rotenone can promote α-syn inclusion formation, we studied rotenone treatment of primary cortical neurons further by performing immunostaining for activated caspase-3 and MAP2, and observed visibly reduced neurotoxicity in 4E-BP1-overexpressing neurons (Fig. 4C). Quantification of neurotoxicity by the presence of nuclear condensation and detection of activated caspase-3 in individual neurons confirmed that 4E-BP1 overexpression can protect against rotenone toxicity (Fig. 4D,E).

4E-BP1-overexpressing neurons are protected against PD-associated environmental toxins. A, DIV14 primary cortical neurons from control and 4E-BP1 transgenic mice were treated with rotenone (10 nm), maneb (1 μm), or paraquat (10 μm), or left untreated. We then measured neurotoxicity by propidium iodide exclusion. n ≥ 3 biological replicates. ANOVA with post hoc Tukey test: ***p = 0.0001 for WT untreated versus WT rotenone-treated (df = 63, EF = 3.1). ***p = 0.0001 for WT untreated versus WT maneb-treated (df = 63, EF = 20.4). ***p = 0.0001 for WT untreated versus WT paraquat-treated (df = 63, EF = 22.1). ***p = 0.0001 for WT rotenone-treated versus 4EBP1 rotenone-treated (df = 69, EF = 1.8). ***p = 0.0001 for WT maneb-treated versus 4EBP1 maneb-treated (df = 71, EF = 11.6). ***p = 0.0001 for WT paraquat-treated versus 4EBP1 paraquat-treated (df = 70, EF = 10.7). B, DIV14 primary cortical neurons from control and 4E-BP1 transgenic mice were treated with rotenone (10 nm), maneb (1 μm), or paraquat (10 μm). We then measured neurotoxicity by quantifying LDH release into the culture media. n ≥ 3 biological replicates. Two-tailed t test: *p = 0.041 for WT rotenone-treated versus 4EBP1 rotenone-treated (df = 7, EF = 1.7). *p = 0.027 for WT maneb-treated versus 4EBP1 maneb-treated (df = 7, EF = 1.0). *p = 0.018 for WT paraquat-treated versus 4EBP1 paraquat-treated (df = 7, EF = 0.4). C, DIV14 primary cortical neurons from control and 4E-BP1 transgenic mice were treated with rotenone (10 nm), immunostained with antibodies against activated caspase-3 and MAP2, and counterstained with DAPI. Cell death is visibly decreased in primary neurons from 4E-BP1 transgenic mice, based on decreased activated caspase-3 nuclear staining and increased MAP2 neurite staining. Scale bar, 10 µm. D, DIV14 primary cortical neurons from control and 4E-BP1 transgenic mice were treated with rotenone (10 nm), and counterstained with DAPI as in C. We then counted the number of neurons showing nuclear condensation. n = 3 biological replicates; n ≥ 50 neurons. *p = 0.048 (two-tailed t test; df = 4; EF = 2.8). E, DIV14 primary cortical neurons from control and 4E-BP1 transgenic mice were treated with rotenone (10 nm), immunostained with antibodies against activated caspase-3 and MAP2, and counterstained with DAPI as in C. We then counted the number of neurons showing activated caspase-3 nuclear staining. n = 3 biological replicates; n ≥ 50 neurons. p = 0.083 (two-tailed t test; df = 4; EF = 2.4). Error bars indicate SEM.
4E-BP1 induces the expression of genes required for metabolism, antioxidant defense, and the UPRmt, as well as their transcription factor regulators
While reduction of protein translation by 4E-BP1 overexpression is a key factor in achieving neuroprotection, this mechanism alone is unlikely to account for protection against rotenone, maneb, and paraquat, where altered dopamine metabolism, increased ROS production, and impaired energy production have all been implicated (Drechsel and Patel, 2008). To assess whether 4E-BP1 neuroprotection has a transcriptional component affecting these pathways, we measured the expression of key regulators from these pathways, beginning with metabolic enzymes involved in glycolysis and TCA cycle function. We observed significant increases in the expression levels of citrate synthase, hexokinase 1, and pyruvate carboxylase in 4E-BP1-overexpressing primary cortical neurons compared with WT littermate control neurons (Fig. 5A). Importantly, these 4E-BP1-dependent expression changes persisted in rotenone-treated neurons (Fig. 5A). We next considered antioxidant factors and noted significant expression increases in the glutathione-S-transferase subunit gene, superoxide dismutase 1, and superoxide dismutase 2 in 4E-BP1-overexpressing neurons, and confirmed that these expression increases were also present in 4E-BP1-overexpressing neurons exposed to rotenone (Fig. 5B). As recent work has highlighted the importance of the UPRmt in promoting proteostasis in stress situations and in neurodegenerative proteinopathies (Nargund et al., 2012; Sorrentino et al., 2017), we then analyzed the expression levels of factors involved in the UPRmt, and documented marked increases in the expression of heat shock protein d1 (HSP60), mitochondrial ATP-dependent Clp protease proteolytic subunit (ClpP), and mitochondrial ATP-dependent Clp protease ATP-binding subunit clpX-like (ClpX) in primary cortical neurons from 4E-BP1-OE mice compared with WT littermate control mice, and documented increased expression of UPRmt mediators, which was sustained in 4E-BP1-overexpressing neurons exposed to rotenone (Fig. 5C). As the genes that encode factors comprising the UPRmt contain a response element corresponding to the consensus binding site of a transcription factor known as the CCAAT/enhancer-binding protein homologous protein (CHOP), and previous studies have documented that CHOP expression itself is upregulated during the UPRmt (Zhao et al., 2002), we measured CHOP expression in 4E-BP1-overexpressing neurons, and observed significant induction compared with WT neurons (Fig. 5D). We also found that 4E-BP1-overexpressing neurons display elevated expression of ATF4, another transcriptional regulator that itself is upregulated during the UPRmt (Quiros et al., 2017), and the master regulator PPARγ coactivator 1α (PGC-1α), which promotes mitochondrial biogenesis by upregulating expression of mitochondrial genes (Fig. 5D). Rotenone treatment yielded increased expression of these master regulators in 4E-BP1-overexpressing neurons (Fig. 5D), consistent with the observed expression increases documented for their target genes in 4E-BP1-overexpressing neurons (Fig. 5A,C). Immunoblot analysis of HSP60 confirmed a significant increase at the protein level in 4E-BP1-overexpressing neurons compared with control neurons (Fig. 6).

4E-BP1 induces the expression of genes required for metabolism, antioxidant defense, the UPRmt, and positive transcription factor regulators of these pathways. A-D, We isolated RNA from DIV14 primary cortical neurons from control and 4E-BP1 transgenic mice that were left untreated or treated with rotenone (10 nm). We performed qRT-PCR analysis on isolated RNAs for various genes as indicated, comprising four categories: (A) metabolism; (B) antioxidant defense; (C) UPRmt; and (D) positive transcription factors regulators of the UPRmt and mitochondria biogenesis. n = 3 biological replicates. *p < 0.05; **p < 0.01; ***p < 0.001; ANOVA with post hoc Tukey test. Error bars indicate SEM. For degrees of freedom, EFs, and exact p values for Figure 5, see Extended Data Figure 5-1.
Figure 5-1
Summary of statistical data for expression analysis of genes required for metabolism, anti-oxidant defense, the UPRmt, and positive transcription factor regulators of these pathways in 4E-BP1-overexpressing neurons. We performed qRT-PCR analysis on isolated RNAs from DIV14 primary cortical neurons from control and 4E-BP1 transgenic mice for genes comprising four categories: A) metabolism; B) anti-oxidant defense; C) UPRmt; and D) positive transcription factors regulators of the UPRmt and mitochondria biogenesis. Here we present the degrees of freedom, EFs, and exact p values for each experimental result shown in Figure 5. Download Figure 5-1, DOC file.

4E-BP1-overexpressing neurons display increased protein expression of Hsp60. A, Immunoblot analysis of DIV14 primary cortical neurons from control and 4E-BP1 transgenic mice. β-actin served as a loading control, and transgene status was confirmed by immunoblotting for 4E-BP1. B, Densitometry analysis of Hsp60 immunoblotting analysis shown in A. **p = 0.007 (two-tailed t test; df = 13; EF = 1.9). Error bars indicate SEM.
4E-BP1 neuroprotection depends on induction of the UPRmt
To determine whether neuroprotection against rotenone toxicity afforded by 4E-BP1 overexpression depends on induction of the UPRmt, we treated primary cortical neurons from 4E-BP1-OE transgenic mice and WT littermate controls with rotenone, and performed shRNA knockdown of a transcription factor activator of the UPRmt, CHOP, or CCAAT/enhancer-binding protein-β (C/EBPβ), or heat shock protein d1 (Hspd1/HSP60), a principal mitochondrial chaperone protein required for an effective UPRmt, after validating robust knockdown with shRNA-expressing constructs (Fig. 7). Propidium iodide staining revealed that 4E-BP1 overexpression rescued toxicity in untreated neurons, in mock-transfected neurons, and in neurons transfected with a scrambled shRNA, compared with WT primary cortical neurons; however, shRNA knockdown of CHOP, Hspd1, or C/EBPβ prevented neuroprotection in 4E-BP1-overexpressing neurons (Fig. 8A). Evaluation of neurotoxicity by quantification of LDH release independently corroborated that 4E-BP1 overexpression in rotenone-treated neurons only yielded increased neuron survival if expression levels of CHOP, Hspd1, or C/EBPβ were not reduced by shRNA knockdown (Fig. 8B). These findings reveal a central role of the UPRmt in protecting against rotenone toxicity, and demonstrate that 4E-BP1 overexpression relies on UPRmt induction for such neuroprotection.

Validation of robust knockdown by lentivirus shRNA constructs. DIV6 primary cortical neurons from control mice were transduced with lentivirus-expressing shRNA, as indicated, for 1 week, after which we isolated RNA and performed qRT-PCR analysis for the gene indicated.

Activation of the UPRmt is required for 4E-BP1 neuroprotection. A, DIV14 primary cortical neurons from control and 4E-BP1 transgenic mice were left untreated or were treated with rotenone (10 nm) after being subjected to shRNA lentiviral transduction for 1 week, as indicated. We then measured neurotoxicity by propidium iodide exclusion. n = 3 biological replicates; n = 4 technical replicates; n ≥ 5000 neurons. Two-tailed t test: **p = 0.001 for WT rotenone-treated versus 4EBP1 rotenone-treated [no shRNA], df = 12, EF = 2.5. ***p = 0.0001 for WT rotenone-treated versus 4EBP1 rotenone-treated [scrambled shRNA], df = 16, EF = 3.2. B, DIV14 primary cortical neurons from control and 4E-BP1 transgenic mice were treated with rotenone (10 nm) after being subjected to shRNA lentiviral transduction for 1 week, as indicated. We then measured neurotoxicity by quantifying LDH release into the culture media. Two-tailed t test: *p = 0.034 for WT rotenone-treated versus 4EBP1 rotenone-treated [no shRNA], df = 6, EF = 2.3. *p = 0.015 for WT rotenone-treated versus 4EBP1 rotenone-treated [scrambled shRNA], df = 6, EF = 2.9. Error bars indicate SEM.
4E-BP1 protects neurons against α-syn fibril proteotoxicity
It is well established that α-syn (α-syn), a presynaptic protein of uncertain function, plays a central role in the pathogenesis of PD (Polymeropoulos et al., 1997). In the PD brain, the normally soluble α-syn protein is converted into insoluble filamentous assemblies, leading to its intraneuronal deposition in Lewy bodies and Lewy neurites, which are histopathological hallmarks of PD and related disorders (Rochet and Lansbury, 2000). Neurotoxicity in PD is closely correlated with the levels of protofibrils of α-syn, rather than mature fibrils. These oligomeric forms of α-syn may target intracellular organelles and cellular pathways, including mitochondria, the ubiquitin-proteasome system, and the autophagy pathway, leading to neuron dysfunction and cell death. Evidence strongly points to α-syn misfolding and accumulation in the pathogenesis of PD because of impaired proteostasis and defective mitochondrial quality control (Trojanowski et al., 1998; Singleton et al., 2003; Samii et al., 2004). To model α-syn proteotoxicity, a protocol for the production of α-syn PFFs has been developed (Volpicelli-Daley et al., 2011), and a method for recapitulating α-syn proteotoxicity based on treatment of primary neurons with α-syn PFFs has been established (Volpicelli-Daley et al., 2014). To determine whether 4E-BP1 overexpression is capable of countering α-syn PFF toxicity, we exposed primary hippocampal neurons from 4E-BP1-OE transgenic mice and WT littermate controls to α-syn PFFs. Immunostaining for α-syn revealed visibly decreased accumulation of α-syn inclusions in 4E-BP1-overexpressing neurons (Fig. 9A,B). When we quantified hippocampal neurotoxicity by evaluating nuclear condensation and measuring caspase-3 activation in individual neurons, we observed markedly reduced toxicity in hippocampal neurons from 4E-BP1-OE transgenic mice (Fig. 9C,D). Hence, 4E-BP1 overexpression can protect neurons against both PD-relevant chemical toxins and misfolded α-syn.

4E-BP1-overexpressing neurons are protected against α-syn PFFs. A, DIV10 primary hippocampal neurons from control and 4E-BP1 transgenic mice were treated with α-syn PFFs (100 ng/ml) for 1 week, immunostained with an antibody against phosphorylated α-syn, and stained with DAPI. Accumulation of α-syn aggregates is visibly reduced in 4E-BP1-overexpressing neurons. Scale bar, 10 µm. B, Densitometry analysis of phospho-α-syn immunostaining analysis shown in A. n = 5 biological replicates. **p = 0.001 (two-tailed t test; df = 66; EF = 1.0). C, DIV10 primary hippocampal neurons from control and 4E-BP1 transgenic mice were treated with α-syn PFFs (100 ng/ml) for 1 week and counterstained with DAPI. We then counted the number of neurons showing nuclear condensation. n = 5 biological replicates; n ≥ 50 neurons. ***p = 0.0001 (two-tailed t test; df = 66; EF = 1.9). D, DIV10 primary hippocampal neurons from control and 4E-BP1 transgenic mice were treated with α-syn PFFs (100 ng/ml) for 1 week, immunostained with an antibody against activated caspase-3, and counterstained with DAPI. We then counted the number of neurons showing activated caspase-3 nuclear staining. n = 5 biological replicates; n ≥ 50 neurons. ***p = 0.0001 (two-tailed t test; df = 66; EF = 1.7). Error bars indicate SEM.
Discussion
Dietary restriction has been shown to extend lifespan and be neuroprotective in worms, flies, and mice (Mattson, 2005), and numerous studies have linked the beneficial effects of dietary restriction to inhibition of mTOR (Kapahi et al., 2004; Kaeberlein et al., 2005). As previous studies of 4E-BP1 have documented that 4E-BP1 is required for lifespan extension on dietary restriction in Drosophila (Zid et al., 2009), and we found that 4E-BP1 overexpression can counter metabolic decline and obesity in mice (Tsai et al., 2016), 4E-BP1 activation is likely a major contributor to lifespan extension and antiaging neuroprotection. Consistent with this model, hyperactivation of mTOR has been implicated in the pathogenesis of many neurodegenerative disorders (An et al., 2003; Ravikumar et al., 2004; Zemke et al., 2007; Gkogkas et al., 2013; Santini et al., 2013; Wong, 2013; Pryor et al., 2014; Tang et al., 2015). To determine whether 4E-BP1 can prevent misfolded protein stress and protect against neurodegenerative insults, we derived primary neurons from 4E-BP1-OE mice. To directly test whether 4E-BP1 overexpression can confer neuroprotection against various protein misfolding and neurodegenerative stresses, we compared the health and survival of primary cortical neurons from 4E-BP1-OE transgenic mice and WT littermate control mice, and documented markedly reduced toxicity in 4E-BP1-overexpressing neurons treated with brefeldin A, rotenone, maneb, or paraquat. While brefeldin A causes accumulation of proteins destined for secretion in the endoplasmic reticulum and thus elicits a general proteostasis stress (Fujiwara et al., 1988), rotenone, maneb, and paraquat promote mitochondrial dysfunction by inhibiting complex I, triggering oxidative stress, and increasing levels of ROS (Drechsel and Patel, 2008), although other effects on mitochondrial quality control have been proposed for these PD-linked toxins (Desplats et al., 2012). Interestingly, 4E-BP1 overexpression also prevented mitochondrial dysfunction that developed in neurons exposed to brefeldin A. These findings indicate that 4E-BP1 overexpression must somehow engage a mitochondrial stress response pathway in neurons to promote mitochondrial function and sustain cellular homeostasis.
To determine the basis for 4E-BP1 activation of a mitochondrial stress response pathway, we evaluated the transcription levels of various mitochondrial metabolic, antioxidant, and chaperone factors in primary neurons overexpressing 4E-BP1, and we observed significant expression increases. In particular, we documented 4E-BP1-dependent upregulation of the expression of genes encoding proteins that comprise the UPRmt, which was initially described as a mitochondrial-to-nuclear signaling pathway that is activated when misfolded proteins accumulate within mitochondria (Pellegrino et al., 2013). However, beyond this classical model of UPRmt activation, further research has demonstrated that the UPRmt is activated in response to a diverse array of both mitochondrial and cellular stresses. Indeed, recent studies have shown that activation of the integrated stress response, which turns off global protein translation on regulatory phosphorylation of eIF2α, culminates with activation of the UPRmt (Fiorese et al., 2016; Munch and Harper, 2016; Quiros et al., 2017; Samluk et al., 2019). To test whether 4E-BP1 overexpression, which also reduces global protein synthesis, could be activating the UPRmt, we measured the transcription levels of two master UPRmt regulators, CHOP and ATF4, whose transcriptional upregulation has been shown to yield UPRmt activation (Zhao et al., 2002; Wang et al., 2018). When we observed significant increases in the transcription of CHOP and ATF4 in primary neurons from 4E-BP1-OE transgenic mice, we repeated our studies of 4E-BP1 neuroprotection in combination with shRNA knockdown of CHOP, shRNA knockdown of the CHOP cofactor C/EBPβ, or shRNA knockdown of HSP60, an essential mitochondrial chaperone for the UPRmt, and we confirmed that primary neurons overexpressing 4E-BP1 are not rescued from rotenone treatment unless these factors are expressed at normal levels. The mechanism by which 4E-BP1 promotes the transactivation of CHOP and ATF4 is yet to be defined, but may involve engagement with the MEK/JNK pathway, as c-Jun phosphorylation can increase CHOP expression (Horibe and Hoogenraad, 2007). Nonetheless, our findings reveal that 4E-BP1 is capable of turning on the UPRmt in neurons.
A number of studies have found that upregulation of 4E-BP1 could be an effective treatment in models of neurodegenerative diseases, including especially PD. For example, increased dosage of the Drosophila ortholog of mammalian 4E-BP1 strongly suppressed motor phenotypes and dopaminergic neuron degeneration in Drosophila Parkin and PINK1 loss-of-function models of PD (Tain et al., 2009). Furthermore, 4E-BP1 has been shown to be a substrate of LRRK2 (Imai et al., 2008), mutations of which account for the most common genetic form of familial PD. As mutant G2019S LRRK2, the most common PD-causing allele in the human population, results in increased phosphorylation of 4E-BP1 (Imai et al., 2008), reduced 4E-BP1 function may contribute to PD pathogenesis. To further examine the connection between 4E-BP1 function and PD, we exposed primary neurons to three different PD-linked toxins (rotenone, maneb, or paraquat), and we documented significant neuroprotection in neurons from 4E-BP1-OE transgenic mice. We also tested whether 4E-BP1 could prevent α-syn neurotoxicity by treating 4E-BP1-overexpressing primary hippocampal neurons with α-syn PFFs, and we observed marked reductions in α-syn aggregation and neurotoxicity, thus validating that 4E-BP1 is also a powerful suppressor of the central player in the PD pathogenic cascade. While previous studies have attributed 4E-BP1 neuroprotection to reduced protein synthesis, we have now extended the understanding of 4E-BP1 neuroprotection by discovering that 4E-BP1 activation induces the UPRmt in neurons. This finding underscores the broad neurotherapeutic potential for 4E-BP1 activation, as activation of the UPRmt can potently reduce Alzheimer's disease-associated amyloid-β proteotoxicity (Sorrentino et al., 2017). Although much effort has been focused on developing compounds to inhibit mTORC1, as mTORC1 inhibition can activate autophagy and derepress 4E-BP1, this therapeutic strategy has been complicated by concern over off-target effects of broad mTOR inhibition and by unease regarding the potential deleterious effects of inducing autophagy when the autophagy pathway is impaired and autophagic flux is blocked in neurodegeneration because of misfolded protein stress in diseased neurons. For these reasons, increasing the expression of 4E-BP1 or enhancing 4E-BP1 activation by maintaining abundant levels of dephosphorylated 4E-BP1 may represent an appealing therapeutic opportunity for treating a variety of neurodegenerative diseases, including especially PD.
Footnotes
The authors declare no competing financial interests.
This work was supported by National Institutes of Health Grants R01 AG033082 and R01 NS065874 to A.R.L.S.; and Department of Biotechnology, India RLS fellowship to S.G.D. We thank K. Luk and V. Lee for providing α-synuclein preformed fibrils; E. Lopez and A. Foley for mouse colony management assistance; and N.J. Arboleda, M.S. Shrilaxmi, J. Oh, O. Puckett, D. Sanders, J. Hsu, A. Risbud, M. Gue, and M. Wadhwa for technical and statistical assistance.
- Correspondence should be addressed to Albert R. La Spada at alaspada{at}uci.edu or Somasish Ghosh Dastidar at somasish.gd{at}manipal.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}