
Abstract
Blended cements, where Portland cement clinker is partially replaced by supplementary cementitious materials (SCMs), provide the most feasible route for reducing carbon dioxide emissions associated with concrete production. However, lowering the clinker content can lead to an increasing risk of neutralisation of the concrete pore solution and potential reinforcement corrosion due to carbonation. carbonation of concrete with SCMs differs from carbonation of concrete solely based on Portland cement (PC). This is a consequence of the differences in the hydrate phase assemblage and pore solution chemistry, as well as the pore structure and transport properties, when varying the binder composition, age and curing conditions of the concretes. The carbonation mechanism and kinetics also depend on the saturation degree of the concrete and CO2 partial pressure which in turn depends on exposure conditions (e.g. relative humidity, volume, and duration of water in contact with the concrete surface and temperature conditions). This in turn influence the microstructural changes identified upon carbonation. This literature review, prepared by members of RILEM technical committee 281-CCC carbonation of concrete with supplementary cementitious materials, working groups 1 and 2, elucidates the effect of numerous SCM characteristics, exposure environments and curing conditions on the carbonation mechanism, kinetics and structural alterations in cementitious systems containing SCMs.
Similar content being viewed by others

1 Mechanisms of carbonation in cementitious hydrates, and CO2 sequestration
This section summarises the chemical reactions occurring during carbonation of cementitious hydrate phases, and the effects of these reactions on hydrated reaction products and pore solution chemistry. In Portland cement-based systems, the term carbonation describes the reaction of dissolved carbonates with cementitious products, mainly through interaction with calcium ions that are extracted from hydrate phases. Once CO2 enters the material, at pH > 10, gaseous CO2 rapidly dissolves into the alkaline pore solution and subsequently hydrolyses to bicarbonate (HCO3−) and carbonate ions (CO32−) via a multi-step reaction sequence that can be summarised as: CO2 + OH− → HCO3− → CO32− + H+ [1, 2]. At pH < 8, direct hydration of CO2 occurs and results in carbonic acid H2CO3 (via CO2 + H2O → H2CO3), which can then dissociate into HCO3− and CO32− ions [3] at higher pH. At pH 8-10, both mechanisms are important. In the pore solution the carbonate ions react with calcium ions that are re-solubilised from the solid products, and precipitate as calcium carbonate. Carbonation is a reactive transport process. The presence of water lowers the diffusion of gaseous CO2. At the same time, the presence of water is essential for calcium and carbon dioxide to dissolve and react.
The main carbonation reaction product is calcium carbonate, which can precipitate in three crystalline polymorphs: calcite, aragonite and vaterite, depending on the internal concrete conditions (e.g. pH, temperature and supersaturation) [4], and the presence of impurities or additives [5,6,7]. Calcite is the most stable polymorph under ambient conditions, while metastable phases including amorphous calcium carbonate, monohydrocalcite, vaterite and aragonite can form prior to or along with calcite precipitation (Ostwald’s rule of stages) [8, 9]. Amorphous calcium carbonate and vaterite form in the early stages of atmospheric carbonation and also during carbonation in a CO2-rich atmosphere [10, 11]. Calcite and vaterite are the most common forms of CaCO3 found in carbonated cement paste [12, 13] at ambient or close to ambient conditions.
The formation of calcium carbonate through carbonation leads to the release of some of the water that was bound in portlandite and other hydrates, and results in changes in the total solid volume as illustrated in Fig. 1. These changes might be positive or negative dependent on the cement chemistry and can have a major impact on porosity and hence on the transport properties of the carbonated cement paste. Thermodynamic modelling of the carbonation of hydrated cement can be used to understand the pH changes and the destabilisation sequence of the different Portland cement hydrated products. The thermodynamic model depicted in Fig. 1 shows the phase assemblage of a white Portland cement as a function of the amount of CO2 that has reacted with the cement paste, and the corresponding decrease of pH and of the Ca/Si ratio of the calcium silicate hydrate (C–S–H). Carbonation proceeds according to the following steps:

Thermodynamic modelling of the phase assemblage during carbonation of a white PC w/b = 0.5 and degree of hydration 90% (a), and changes of pH and Ca/Si of the C–S–H during carbonation (b). In both graphics, the undamaged cement paste is shown on the right-hand side, moving to the left as more CO2 reacts with the hydrates. Adapted from [16]
-
Initially any monosulfate- and hemicarbonate-AFm phases, if present, will destabilise to monocarbonate-AFm [14], leading to small changes of the solid volume (molar volume of monosulphate: 332 cm3/mol; hemicarbonate: 285 cm3/mol and monocarbonate-AFm: 262 cm3/mol) (not shown in Fig. 1).
-
Portlandite is the first major hydrate that decomposes to calcium carbonate during carbonation, leading to a moderate volume increase (ΔV = 12% of portlandite) as the molar volume of calcite (36.9 cm3/mol) is greater than that of portlandite (33.0 cm3/mol). The pH remains stable at around ~ 12.5 [12].
-
After all accessible portlandite is consumed, C–S–H starts to decalcify down to a Ca/Si ratio of ~ 1.3. The pH decreases only marginally during this step. This decalcification is not accompanied by significant volume change.
-
Monocarbonate decomposes to strätlingite, which accommodates the released aluminium and silicon from the C–S–H; C–S–H continues to be consumed without change of its Ca/Si ratio. This reaction also leads to some changes of the solid volume. The molar volume of monocarbonte (262 cm3/mol) is greater than that of strätlingite (216 cm3/mol), which is compensated by the formation of calcium carbonate and consumption of C–S–H.
-
After monocarbonate is consumed, the decalcification and decomposition of the C–S–H continues down to Ca/Si of ~ 0.75 and pH of ~ 11. This step is accompanied by the largest drop in pH.
-
Upon further carbonation strätlingite decomposes, followed by ettringite at pH around 10. The released aluminium and sulphur are bound in aluminium hydroxide or zeolites [15] and gypsum, respectively. In parallel, the C–S–H decalcifies to Ca/Si of around 0.67 while the pH is lowered to 10.
-
Upon further carbonation, C–S–H decomposed into calcite and hydrated amorphous silica and the pH drops rapidly below 10 to 8.5.
-
At the final stage, hydrotalcite decomposes at pH around 8.5.
-
The total CO2 bound is around 40 to 50 g CO2/100 g Portland cement.
The same sequence of destabilisation is also observed in PC blended with fly ash [15], metakaolin [16] and in calcium sulfoaluminate cements [14]. The consumption of calcium during the carbonation reaction lowers the pH in pore solution from initially 13.5 to 14 (in the absence of carbonation) to below 8, when the cement hydrates have completely carbonated. This decrease happens stepwise, as the pH is buffered by different hydrate assemblages, which are destabilised [17,18,19,20]. This buffering capacity, i.e. the amount of calcium oxide available, as well as the buffered pH depend on the type of the hydrates [14, 21].
It is worth noting, however, that under real carbonation exposure conditions different steps might occur simultaneously, and/or may halt before completion for kinetic or microstructural reasons, although the general sequence will be the same as indicated by thermodynamic modelling. For example, it has often been observed that not all portlandite and C–S–H are carbonated, particularly at low RH [13, 22, 23]. From a thermodynamic point of view initially no C–S–H should decalcify, which does not agree with what has been reported in real carbonated pastes [24, 25].
Portlandite carbonation occurs as a consequence of a dissolution–precipitation reaction and the reaction kinetic is initially more rapid than the carbonation of C–S–H [13]. Calcium carbonate grows on the portlandite surfaces as illustrated in Fig. 2, which slows down the further carbonation by limiting the transport of water and CO2 [22, 23]. CH carbonation releases water, while C–S–H loses water mainly upon complete carbonation [27].

The most abundant calcium-bearing phase in hydrated cement paste is C–S–H, and this is also the phase that displays the most complex carbonation behaviour. The carbonation process of C–S–H depends on its original Ca/Si ratio, CO2 concentration, and on the presence of portlandite [28]. The C–S–H carbonation proceeds in steps; first CaO is removed from the interlayer space. The initially short silicate chains present in high-Ca C–S–H become longer until a C–S–H with a very low Ca/Si ratio of around 0.7 and a pH value of 10 is reached. Only then is the main CaO in C–S–H also consumed, leading to the formation of amorphous silica [11, 29] and further reduction of pH to around 8.5. The destabilisation of C–S–H to silica gel, however, often remains incomplete, particularly at lower relative humidity [27, 30].
The extent of C–S–H carbonation also depends on the CO2 concentration [31]. Above 3% of CO2 in the gas phase, C–S–H can fully decompose into calcium-modified silica gel, while below this concentration, partially decalcified C–S–H was found after the “completion” of carbonation [29,30,31,32]. The behaviour of C–S–H gels also varies depending on Ca/Si, due to the composition dependence of the thermodynamic stability of C–S–H [33].
Initial C–S–H decalcification, involving interlayer calcium, is not accompanied by significant volume changes (see Fig. 1), and C–S–H carbonation can thus contribute significantly to the densification of cement paste microstructure by calcite precipitation. However, below a Ca/Si ratio of ~ 1.3, significant decalcification shrinkage has been observed [34]. The fine-textured initial microstructure of C–S–H is preserved after the initial decalcification [35], and both inner and outer C–S–H undergo a comparable decalcification although only outer C–S–H shows morphological changes upon decalcification [35]. These morphological changes involve coarsening of the porosity and precipitation of calcium carbonate at the tips of the C–S–H fibrils. Carbonation shrinkage has been observed to increase with CO2 concentration and with decreasing Ca/Si ratio, and shows a maximum at high to moderate relative humidity [36], mainly attributed to the polymerisation of C–S–H.
The total amount of CO2 that can be bound (binding or buffering capacity of a cement) depends directly on the amount of CaO available to form CaCO3 [9, 12, 23, 37], i.e., all CaO in the hydrates minus calcium bound in calcium carbonate or calcium sulfates. Thus, the CO2 binding capacity of blended cements and calcium sulfoaluminate cements is generally lower than that of plain PC, since the available CaO content is lower [14, 16,17,18,19,20,21]. Additionally, a lower degree of carbonation of portlandite and calcium silicate hydrate in specific conditions alters the buffering capacity. These differences mean that a direct application of the understanding of the carbonation mechanism and CO2 binding capacity for plain PC cannot be used to infer how concretes with SCMs will perform when exposed to different environments.
2 Carbonation kinetics and carbonation coefficient
The dependence of carbonation resistance on CO2 diffusion and the amount of carbonatable matter is also clear from the well-known square-root-time relation (Eq. 1) for carbonation rate, mentioned in various literature including [38,39,40], which is essentially a solution to Fick’s first law of diffusion.
Here xc is the carbonation depth (m), Dc is the diffusion coefficient of CO2 (m2/s), cs is the CO2 concentration at the concrete surface (kg/m3), ac is the amount of carbonatable material per unit volume (kg/m3), t is the time (s), Rcarb (= ac/Dc) is the carbonation resistance ((kg/m3)/(m2/s)), and k (= √(2·s/Rcarb) is the carbonation coefficient (m/√s). Equation (1) assumes a linear decline of CO2 concentration across the surface layer, from the CO2 concentration at the concrete surface (cs), down to the concentration at the carbonation front, which has a negligible value. The carbonation progress is accelerated with increasing CO2 concentration (cs) and increasing CO2 diffusion coefficient (Dc) but slows down with increasing amount of carbonatable material (ac).
The formula containing the carbonation coefficient k in Eq. (1) is commonly used in experimental studies. When plotting carbonation depths as a function of the square-root of the exposure time, a more or less linear relationship is obtained, the slope of which is taken as the carbonation coefficient.
It should be noted, though, that Eq. 1 assumes a steady-state condition (i.e. constant carbonation coefficient). However, since carbonation reaction products tend to block the pores in concrete and cause a reduction in gas diffusivity, and because cementitious materials continue to hydrate and refine the pores inside the material, the carbonation coefficient should decrease with time (or depth). When carbonation coefficient becomes time dependent, Eq. 2 can be applied as used in [41, 42].
Here the exponent n should be negative, to account for pore blockage by reaction products, wetting events, increased moisture content and ageing effects; a value of zero would define pure diffusion control and an unchanging material, neither with depth nor time. A positive value indicates that the material dries out and/or cracks due to carbonation shrinkage. The empirical exponent n was found to be between 0.0 and − 0.2 in [42]. They observed that n depends on curing duration and binder type. Hunkeler and Greve-Dierfeld [43] showed for three days water cured samples a decrease in n with decreasing w/b ratio and increasing relative humidity.
The carbonation resistance of a cement paste or a concrete—which is usually expressed in terms of the depth to which atmospheric CO2 has penetrated the material at a given time, or as a coefficient describing this rate of ingress—depends not only on its CO2 binding capacity, but also on its porosity and pore size distribution [16, 21, 44]. Changes in the effective permeability due to hydrate/carbonate volume changes and microcracking can be a significant consequence of carbonation [45]. In concretes with conventional SCMs, an increase in porosity upon carbonation is typically reported, in contrast to plain PC systems where carbonation decreases their permeability due to pore blocking as discussed above [46]. However, the correlation between the hydrate products in blended PCs and the associated volume and permeability changes is not yet clear. Of more concern, it is not clear what mechanism is leading to microcracking, and to what extent these microcracks might modify the transport properties of the carbonated layer, reducing the resistance to ingress of CO2. Due to the large variety of SCMs used in the production of modern concretes and the broad range of properties exhibited by concretes produced with them, in the following sections the main effects of the type and amount of SCM added are discussed, as well as the influence of concrete mix design parameters and other factors on the carbonation process of SCM-containing materials.
3 Concrete mix design and its effect on carbonation
3.1 Effect of SCM type
When PC is blended with SCMs, the phase assemblages and chemistry of the pore solution change significantly. In PC blended with siliceous SCMs, a reduced content of portlandite is present, as consequence of pozzolanic or latent-hydraulic reactions. The main reaction product observed is an Al-substituted C–S–H type phase, with lower Ca/Si than in plain PC systems [47]. Formation of different secondary reaction products such as layered double hydroxides (e.g. AFm phases and Mg–Al hydroxides) is also reported when using Al-rich SCMs [48].
The effect of SCMs on the chemistry and properties of cementitious materials has been extensively evaluated, including by recent RILEM technical committees, and the reader can find more detailed information in [49,50,51]. In this review a brief description of the chemistry of SCM-blended cements is presented to explain potential correlations between material properties and carbonation performance.
Addition of SCMs can increase the early reactivity of PC clinker due to its (physical) filler effect promoting nucleation and growth of the hydrates, and the higher effective water/cement (w/c) ratio [52, 53]. In the pozzolanic reaction SiO2 and Al2O3 react with water and CH to form additional C–S–H with a reduced Ca/Si ratio, an increased silicate mean chain length, and some incorporation of Al [54,55,56]. Thermodynamic calculations indicate that for complete consumption of portlandite, the required ratio of SCM to total binder content is approximately 75 wt% for blast furnace slag (BFS) [44, 53], 35 wt% for Class F/siliceous fly ash (FA), 18 wt% for silica fume (SF) [47, 57, 58] and 6–18 wt% for metakaolin (MK) [59], although microstructural (kinetic) constraints can lead to the persistence of portlandite at higher SCM contents than would be indicated from thermodynamics alone [60]. The consumption of portlandite leads to a reduced pH value in the pore solution, which is in the range of pHPC,LS ≈ 12.7–13.8, pHBFS,FA,MK ≈ 12–13, pHSF ≈ 11.0–12.5 at high clinker replacement levels, according to [57, 61, 62] and is accompanied by reduced K+ and Na+ concentration [15, 57]. Furthermore, a reduction of coarse porosity and an improvement in quality of the interfacial transition zone are generally noticed [61, 63].
Limestone (LS) provides CaO mainly in form of CaCO3. The replacement of a small amount of PC by limestone is beneficial as it promotes nucleation and increases the effective w/c ratio for hydration [64]. In addition, CaCO3 can react with C3A to form monocarboaluminate, which indirectly stabilises ettringite by increasing sulfate availability, leading to an increase of the total volume of hydrate phases [16, 64,65,66]. According to [67], 15 wt% LS addition with a similar size distribution to PC, and used as an addition instead of interground, increases porosity of the hydrated binder, while a smaller size distribution and intergrinding decreases porosity.
Figure 3a shows a Ca–Si–Al ternary diagram, highlighting the regions corresponding to the major SCM groups and Portland cement [68]. Figure 3b shows cement hydrate phases that commonly form from Portland cement—SCM blends in the H2O-CaO-Al2O3-SiO2 system [47]. The precise extent of the C–S–H and C–A–S–H domains is the subject of ongoing research, but these graphics do highlight the broad range of compositions of the main binding phases forming in SCM blended cementitious systems, as a function of the type of SCM used.

The significant difference in Al2O3 contained in SCMs influences the composition and structure of the C-A-S–H phases and secondary reaction products forming in these materials [44]. Therefore, the role of Al in defining the carbonation rates and mechanisms of C(-A)-S–H has received some attention in the recent literature. Irbe [69] found faster carbonation of C–A–S–H than a comparable C–S–H gel when testing synthetic gels of molar ratio Ca/(Al + Si) = 0.96-0.97, under 0.04%, 2% and 4% CO2 conditions. The thermogravimetry data of [70] show a significant increase in CaCO3 formation after 28 days of carbonation of synthetic gels of molar ratio Ca/(Al + Si) ~ 1 when moving from zero Al content to Al/Si = 0.02, but no notable trend when increasing Al content further up to Al/Si = 0.14. In this study and other publications [29, 71] the formation of an additional Al-enriched silicate gel as a carbonation product is described when C-A-S–H gels are decalcified. This phase (and/or the formation of additional potential Al-containing reaction products) deserves further attention in characterisation, as its formation may alter the energetics of the carbonation process sufficiently to induce changes in its rate or mechanism.
According to several studies [42, 72,73,74], PC carbonation under moderate environmental conditions is governed by the reaction of portlandite to mainly form calcite, yielding a denser microstructure due to the positive difference of molar volume of calcite compared to CH (+4 cm3/mol as mentioned in Sect. 1), which more than compensates the shrinkage induced by C–S–H decomposition and subsequent microcracking e.g. [74, 75].
In the case of BFS-blended cements, with increasing C-A-S–H carbonation the percentage of the CaCO3 that is present as calcite is found to be reduced, while the metastable aragonite and vaterite tend to increase [21, 75,76,77]. The microstructural changes identified in these materials lead to higher permeability [75] and diffusivity [78] after carbonation, compared to PC with the same water to binder (w/b) ratio. The reduced calcium and alkali contents lead to a lower buffering capacity in BFS blended cements, and polymerisation shrinkage may lead to increased porosity. Experimental results from [79, 80] indicate that at a replacement level ≥ 25%, the carbonation resistance is decreased compared to plain PC. Several researchers have determined that, under moderately accelerated carbonation conditions (3-5 vol% CO2), not all portlandite and C–A–S–H were consumed during carbonation of BFS-blended cements [21, 26]. However, especially at high BFS replacement levels, the degree of portlandite and C-A-S–H carbonation is higher compared to plain Portland cement systems [21, 74]. This may compensate for the lowered buffering capacity to some extent, because if the degree of portlandite and C–A–S–H carbonation is increased, the actual available Ca is increased. However, if the maximum possible degree of portlandite carbonation is reduced, for example because portlandite is covered by calcium carbonate crystals and therefore the release of Ca from portlandite into the pore solution is hindered, the available Ca to buffer carbonation is reduced.
Compared to BFS, low calcium fly ash (FA) provides less initial CaO to blended systems. At the same FA replacement level (30 wt%), portlandite reduction compared to PC is higher for FA than for BFS [15, 64, 65, 76, 81]. A higher degree of C-A-S–H carbonation (full polymerisation) was identified at > 20 wt% FA replacement levels [73]. A less pronounced decrease in mercury and water intruded porosity upon carbonation has been found in FA blended systems compared to plain PC and BFS-containing concretes [24, 27, 77, 82, 83] and an increase in pore size threshold upon carbonation [82]. The slow pozzolanic reaction of FA compared with slag hydration, and the higher degree of C-A-S–H carbonation accompanied by a release of physical water [73] may be a reason for the deviation from Fick´s first law that has been observed in these systems [42, 84, 85]. There is an increase in the partly carbonated zone with increasing FA replacement, determined through depth dependent pH measurements by the ex situ leaching method [86], indicating divergence from the assumption of pure diffusion-controlled kinetics required for Fickian diffusion (and formation of a sharp carbonation front). In several studies [87,88,89,90], a significant increase in carbonation progress compared to PC was found with increasing FA replacement levels between 30 and 50 wt% for the same w/b ratio. The difference was more pronounced for longer water curing durations (> 7 days) and high relative humidity during carbonation exposure. Thomas and Matthews [91] found that concretes with the same strength grade containing 15–30 wt% FA in the cementitious fraction carbonated to only a slightly greater extent than plain PC. However, concretes containing 50 wt% FA carbonated at a significantly higher rate than PC concrete.
From a thermodynamic point of view, less CO2 can be bound if less calcium is available in the liquid and solid phases (except CaO already bound in CaCO3 or CaSO4) and if less Na+ or K+ are available in the pore solution. When using highly siliceous SCMs such as SF, where Al2O3 and CaO are solely provided by the Portland clinker, the carbonation reaction process is comparable to that identified in plain Portland cement systems, as the main reaction product forming is C–S–H. For synthetic C–S–H it has been reported [33, 92] that with an increasing molar ratio of C–S–H versus portlandite, i.e. with increasing C–S–H carbonation, decalcification shrinkage may be dominant, leading to a coarsening of the pore structure. The carbonation (decalcification) shrinkage seems to increase with the degree of C–S–H polymerisation, and with a decrease in Ca/Si molar ratio. Carbonation shrinkage may reverse the beneficial, experimentally determined, porosity reduction that is related to portlandite carbonation to CaCO3 [27].
The C–S–H amount in SF blended paste is high and the amounts of ettringite and AFm are reduced compared to PC or blends containing FA or BFS. However, the higher extents of self-desiccation and autogenous shrinkage leading to high internal stresses and micro-cracking, a pronounced reduction of pH, challenges of early-age workability, and high increase in strength provided by small amounts of SF addition usually lead to its application at low replacement levels. The number of published research papers on carbonation of SF-blended binary [78, 93, 96] and ternary [74] cement pastes is limited compared to BFS or FA blends. According to Leemann et al. [96], the reduction in total porosity of SF blended cement paste is less pronounced and coarse porosity is increased compared to PC upon carbonation. Consequently, higher oxygen diffusion coefficients are expected for carbonated SF blended cementitious materials than for PC and BFS blended cements for similar replacement levels. A decreasing carbonation resistance compared to PC was found for constant w/b [96] and for constant strength grade [93].
MK provides less CaO than FA but makes available more Al2O3 into the cementitious system than other SCMs. Carbonation when using MK or other calcined clays as SCMs has been investigated in an increasing number of studies [16, 25, 83, 93, 94]. At moderate replacement levels, a negligible amount of portlandite was found prior to carbonation due to pozzolanic reaction, and the C-A-S–H carbonation degree was higher than for PC (polymerised C-A-S–H for MK, decalcified C–S–H for PC) [16]. Consequently, the accessible porosity was on a similar level [16, 83] and the threshold pore radius was increased compared to PC upon carbonation [16]. Higher carbonation rates were found for MK blended cementitious materials either for a constant strength grade [93] or for constant w/b [16] upon carbonation compared to PC, but lower than for SF blends [93] or for LS blends [16] at the same replacement level.
Initial portlandite contents in LS blended cement pastes are slightly lower than in PC pastes [16, 64, 66]. The LS-containing pastes were found to carbonate to a higher degree [16, 95]. Both the initial porosity and the porosity upon carbonation were increased compared to PC. A similar reduction in accessible porosity compared to plain PC was found after carbonation [16, 96], leading to a reduced diffusivity.
Figure 4 shows some effects of clinker replacement levels on the change in carbonation coefficient relative to PC mixes as reported by different authors—all samples have been cured for 28 days. According to these analyses, at the same replacement level the reduction in carbonation resistance is more pronounced for FA and SF blended concretes and mortars, than for LS or BFS blended concretes and mortars. It should be stated that differences in w/b, aggregate type and grading, execution and carbonation conditions can affect these results.

In order to assess the carbonation resistance of a specific cementitious material from its composition three main approaches have been developed:
-
(a)
estimating carbonation resistance based on the clinker content and water to binder ratio (w/b) [41, 97].
-
(b)
k-value concept (EN 206), using the type of SCM and water/(cement + k·addition) (w/ceq) [99].
-
(c)
assessing buffering capacity based on the ratio of mixing water to reactive calcium oxide (w/CaOreactive), see Sect. 1 [44, 96, 100].
Figure 5 a, b, c illustrate the approaches (a), (b) and (c), respectively.

approach a carbonation resistance in dependency of w/b and clinker content for 7-day water cured concretes (20 °C/ 65% RH/ 0.04 vol%CO2) [41, 97]. Error bars indicate ± standard deviation; b) approach b carbonation resistance in dependency of w/ceq of concretes [99] c) approach c carbonation resistance in dependency of w/CaOreactive of samples tested according to SIA262-1 [44] and samples cured for 91-day [100] and 28-day [96] carbonated at 1% CO2 and related to 0.04% CO2
The approach (a), which uses the clinker content and w/b [41, 97], shows carbonation rates of concretes water cured for 7-day and carbonated at 65% RH, 20 °C under natural CO2 concentration. The lines show the potential regression curves to the mean carbonation rates of groups of concretes summarized in dependency of the ranges of clinker content according to EN197-1, including CEM I (95-100% clinker), CEM II/A (80–94% clinker), CEM II/B (65–79% clinker), CEM III/A (35-64% clinker) and CEM III/B (20–34% clinker). In addition the effect of maximum grain size (4 - 32 mm), admixtures (retarder, water reducers, air-entrainment agent), binder content, cement strength grade and clinker replacement level by specific types of SCM has been evaluated for mixes with the same w/b ratio. No systematic effect has been found for the maximum grain size, binder content and the admixture except for the air-entrainment admixture. With air-entrainment admixture the carbonation rate increased at fresh concrete air content > 8%. A small decrease in carbonation rate has been found with increasing cement strength grade in this curing protocol. The increase in carbonation rate with increasing replacement level was higher for LS than for BFS.
The k-value concept, approach (b) according to [99], shows carbonation rates of concretes water cured for 7-day and carbonated at 65% RH, 20 °C under natural CO2 concentration. Based on the equivalent performance concept for type II additions in CEN/TR 16639 the efficiency factor kc has been determined for the concrete property carbonation rate. The efficiency factor is used to reduce the creditable amount of mineral addition when calculating the w/ceq with ceq = clinker + kc · addition. Using the w/ceq the same performance shall be reached as if pure Portland cement would have been used with the same w/c = w/ceq ratio. Hence, plain PC concrete has the efficiency factor kc = 1.0, which is named category 0. For BFS and FA blended cements 3 and 2 categories have been built, respectively. Increasing category represents increasing amount of type II addition. For example, CEM III/B concretes are allocated to category 3 BFS. Efficiency factors were in the range of 0.8-0.6 for BFS blended cement concretes and 0.3–0.35 for FA blended cement concretes. For BFS blends the efficiency factors where in the range of those determined in [101] with 0.65–0.81 and higher than determined in [98] with 0.2–0.4. For FA blends the efficiency factor was similar with those determined in [101] with 0.25–0.58 and [98] with 0.05–0.3.
Approach (c) [44, 96, 100] replaces the clinker content in approach (a) by the amount of all CaO available to react with CO2 -, i.e. w/CaOreactive (w/CaOreactive). In order to determine the CaOreactive Papadakis et al. [38] suggested a simplified mass balance equation to calculate CaO: [Ca(OH)2] + 3[C–S–H] + 3[C3S] + 2[C2S], this expression however, neglects CaO present in aluminate or ferrite phases. For modern cements, which can contain significant amounts of calcium carbonate as an SCM or minor additional constituent, the fraction of CaO already bound by CaCO3 has also to be considered as well as the amount of SO3, as SO3 will be present as CaSO4 in completely carbonated cements [14, 21]. Also, the amount of CaO present in unreacted cement clinker and supplementary cementitious material must be accounted for [14, 21, 44]. Thus, the CaO available for carbonation can be calculated according to: \({\text{CaO}}_{\mathrm{reactive}} = \, {\text{CaO}}_{\mathrm{total}} - {\text{CaO}}_{CaCO3\_init} - {\text{CaO}}_{CaSO4} - {\text{CaO}}_{\mathrm{unreacted clinker}} - {\text{CaO}}_{\mathrm{unreacted SCM}}\) where each term can be determined using thermodynamic modelling (see Fig. 1) or mass balance calculations.
The simplified approaches (a-c) used to describe the carbonation resistance in dependency of parameters of the mix design have their benefits and drawbacks. Approach (a), clinker content and w/b, is simple to apply, but it neglects the buffering capacity of the specific type of SCM and their differences regarding alteration in pore size distribution upon carbonation. The K-value concept, approach (b) is less simple to apply because it requires the allocation of the specific concretes to efficiency factor required for each type of SCM. Approach (c), is the most scientific approach. It also accounts for the contribution of SCM on CO2 binding capacity. But for its application, the reactive CaO content has to be known or assessed and the effect of specific types of SCM on pore size distribution are little accounted for. Approach (a) and (b) are restrictive regarding the implementation of new materials, while in approach (c) new materials can be simply included. All three approaches are highly dependent on degree of hydration. In all three approaches, the mixing water w accounts for the diffusivity i.e. the volume of capillary pores. Alteration of porosity prior and upon carbonation due to the use of alternative materials and SCM`s is little accounted for.
In addition to those three approaches, which describe the carbonation resistance based on the mix design, other approaches have been developed, which describe carbonation resistance based on compressive strength [101, 102] or permeability [103]. For a 7 day compressive strength between 20 and 25 MPa the carbonation rate was found to be in the range between 4 to 7 mm/year0.5 in [102] for concrete samples water cured for 7 day and carbonated at 65% RH, 20 °C and natural CO2. The concretes where prepared with CEM I, CEM III/A, CEM II/B-M and CEM IV as well as CEM II/C-M cements and the carbonation rate was the highest for concretes with high clinker replacement levels within the same strength grade. Similar were the results from [101] for samples carbonated in 2% CO2 converted to 0.04 vol%with k(0.04% CO2) = k(2% CO2)·√(0.04/2). Imamoto et al. [103] used the correlation between air permeability and carbonation rate determined from existing structures, and thus under variable environmental conditions (moisture, temperature, CO2 concentration) in Japan, Switzerland and Portugal to rate the quality of the placed concrete. For example, for a coefficient of air permeability between 0.1 and 1 10-16 m2 the concrete was classified moderate and the carbonation rate was found in the broad range between > 0 and 5 mm/year0.5. Compressive strength as well as the air permeability are indicators for the air tightness of a concrete and hence also its resistance against CO2 diffusion; however compressive strength does not account for the CO2 binding capacity of the specific type of binder and the differences in alteration of pore size distribution upon carbonation. This may explain to some extend the high scatter observed for this correlation. The benefit of both approaches is that the compressive strength is usually available and the air permeability [103] may be determined non-destructively.
To summarize, the carbonation resistance is depending on more factors than just the clinker replacement level, FA or BFS, the calcium oxide content, compressive strength or air permeability. In addition, the binder specific alteration of porosity, the reaction products formed, the composition dependent reaction kinetics, the degree of hydration, the water retention and hence the testing conditions are important parameters, which leads to a limited precision in all the correlations discussed above.
3.2 Aggregate volume and grading: paste-mortar-concrete
The carbonation coefficients reported in [38, 39] for plain Portland cement concrete samples carbonated at 50 vol% CO2, 30 °C and 65% RH increased by a factor of 1.1 and 1.5 when the aggregate to cement ratio (a/c) increased by a factor of 1.7 and 2.3, respectively. Similar results have been reported at 5 vol% CO2 [104]. If a coarser aggregate was used (unchanged total volume of aggregate), the measured carbonation depth was similar or slightly higher [38, 39]. In contrast, Jung et al. [105] found no significant differences in the effective CO2 diffusion coefficients between concrete and mortar, but a significant increase in diffusivity was observed for cement paste systems upon carbonation (i.e. if the a/c ratio decreases). The increase of gas diffusion coefficient for paste systems was explained by the larger porosity per unit of volume hydrated paste compared to hydrated concrete. This is in line with investigations of plain Portland cement pastes and mortars from [27, 105, 106].
Carbonation rates of cement paste, mortar and concrete systems have been compared in [107]. Figure 6 plots the carbonation coefficients obtained after 7 weeks in 2 and 10 vol%CO2 for cement paste, mortar and concrete, manufactured with either 30 wt% PC replacement by FA (70PC30FA), or 70 wt% PC replacement by BFS (30PC70S). Carbonation coefficients of mortar and cement paste (w/b = 0.5) were higher by a factor of 1.2–1.3 and 1.6–2.3, respectively (depending on binder type and CO2 concentration), compared to the carbonation coefficients of concrete. The increase in carbonation coefficient for cement paste compared to concrete is more pronounced for FA than for slag systems, and more pronounced at higher CO2 concentration. A better correlation has been observed between mortar and concrete (R2 = 0.96) than between cement paste and concrete (R2 = 0.82 for w/b ratio of 0.50).

The increase in carbonation coefficient with increasing a/c for PC concretes reported in [38, 39, 104] may be attributed to the increasing fraction of more porous portlandite-rich interfacial transition zone (ITZ) providing paths for fast CO2 diffusion [77]. In contrast, the faster carbonation in the presence of less aggregate reported in [107] could be related to more autogenous and drying shrinkage in case of cement paste system and subsequent increase of porosity [106,107,108,109].
The majority of studies evaluating carbonation resistance when using SCMs are conducted in paste and mortars, with the assumption that the observations for these systems can be directly translated into concrete. However, this is not the case for the data shown here due to the differences in CO2 diffusivity, consistent with variations in pore structure based on the different paste contents and ITZ zones; differences in the degree of water saturation in materials with diverse permeability; and the different type and amount of carbonation products expected to be forming at a given time. The porosity of carbonated and partly carbonated regions as well as the amount of cement phases buffering the reactions involving CO2, will modify the carbonation reaction kinetics. This has important implications for the development of modelling tools for prediction of the long-term performance of concretes with SCMs, and for the determination of the real carbonation resistance of these materials.
3.3 Effect of recycled and lightweight aggregate
Independent of the SCM used, concretes produced with recycled and lightweight aggregate show to some extent different carbonation performance from those produced with conventional dense aggregates. Recycled aggregate (RA) may contain mortar, unbound aggregate, ceramics, floating particles and bituminous matter. Its different physical (e.g. water absorption, porosity) and mechanical properties (e.g. Los Angeles coefficient) compared with natural aggregate may impact concrete performance. In recent reviews on the durability of concretes with recycled aggregates, Guo et al. [110] and Silva et al. [111] reported that concrete carbonation resistance is affected adversely by the aggregate replacement ratio, w/c ratio, adhered mortar, aggregate size, and exposure time, particularly in concretes with SCMs. Carbonation resistance improves with addition of superplasticisers, consistent with a reduced water content in the concrete, and can also be improved by RA pre-treatment.
Even in the absence of major RA contaminants, due to the heterogeneity and wide variability in the properties of recycled concrete aggregate (RCA), which depend on its source and service history, it is impossible to draw general conclusions about its potential effect on carbonation performance of concretes with SCMs. However, numerous authors [112,113,114,115,116,117,118,119,120,121,122,123,124] studying the effect of the RCA replacement ratio on carbonation resistance of concretes with and without SCMs found that in the case of coarse aggregate replacement the carbonation depth, relative to normal concrete, increased 1.06 to 2.00-fold. However, some studies report higher carbonation resistance in RCA concrete [119, 125, 126], which can be explained by two mutually opposing effects, as the adverse effect of larger porosity counteracts the benefits of greater amounts of (alkaline) carbonatable matter (uncarbonated mortar). Using recycled aggregate as fine rather than as coarse aggregates seems to increase the carbonation depth, so that concrete with RCA showed carbonation depths up to 3.75 to 12.25 times greater than for concrete with natural aggregate [127,128,129,130].
The use of porous and gas-permeable lightweight aggregate (LWA) requires a concrete cover at least 5 mm thicker than the maximum particle size [131]. LWA nonetheless lowers concrete gas permeability, as it absorbs the water in fresh concrete which can form an adhesive water layer on a `dense` aggregate surface. The water adsorption may reduce the water/binder ratio around the LWA in the aggregate-matrix interface, and thus eliminates the interfacial transition zone (ITZ) typically identified when using natural aggregates (Fig. 7). As cement hydration reactions proceed, the absorbed water migrates from the LWA to the hydrating cement paste, favouring internal curing. The outcome is a larger high-quality paste-aggregate transition zone (larger grey zone in Fig. 7 bottom left), less permeable in lightweight than in normal concrete [132, 133]. Research on existing structures confirms the beneficial impact of LWA on the carbonation coefficient [134,135,136].

4 Effect of curing on carbonation resistance
Curing has an important effect on the carbonation resistance of concrete as it influences the pore structure and the degree of hydration of the cement, and consequently the amount of carbonatable material. The effect is less pronounced for plain PC concrete than for concrete with SCMs due to their slower reaction. Nevertheless, if adequate precautions are taken, e.g. sufficiently long curing and reduced w/c ratio, the depth of carbonation of concrete with binders containing SCMs may be the same as for concrete with PC [138]. Thus, the type of curing, its duration and temperature as well as other environmental and geometrical conditions like wind speed, size of structural element, and type of formwork need to be considered when estimating the impact on the carbonation resistance.
Typical curing methods in practice are in air, moist, sealed, heat or steam curing, or the use of a chemical curing compound. The type of curing drastically affects the carbonation rate as shown by [139], who reported that the carbonation rate of water-cured samples was only 17% of the carbonation rate of air-cured samples (28 days), in a chamber filled continuously with 100% CO2 (carbonation measurements between 1 and 28 days).
In the laboratory, curing at > 95% RH is common practice but is not representative for on-site hydration circumstances. The duration of curing on construction sites is typically a few days. For varying replacement levels by FA and BFS, researchers have reported that the carbonation coefficient increases dramatically when shortening the curing periods from 7 to 1 day [140,141,142]. In [143], it is shown for concretes (grades C16/20 to C30/37) with blended cements (CEM II A-M and CEM IV A-M) based on natural carbonation tests for 1 year, how the period of wet curing, and the type of curing (wet curing vs. use of a membrane), affect the carbonation rate and therefore the service life of reinforced concrete structures. In that study, an extension of the wet curing period from 1 to 3 days increased the predicted service life by a factor of more than 2, and a factor of 4 improvement is gained by increasing from 1 to 7 days of wet curing. It was found that 3 days of curing should be enough for replacement levels of less than 30 wt% for FA or 50 wt% for BFS [141, 142]. For higher replacement levels, longer curing periods are desirable.
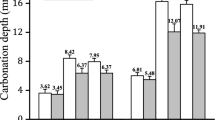
For accelerated carbonation testing in the laboratory, longer curing periods are in most cases recommended to obtain a more realistic and representative microstructure before subjecting the concrete to high CO2 levels. Continuous curing over periods longer than 1 month can significantly increase the durability of BFS concrete (50–85 wt% cement replacement), but periods longer than 3 months were found not to considerably affect the resistance to carbonation anymore [52]. In contrast, Sailio [144] showed that the carbonation depth of systems containing slag cement, or 30 wt% FA, seems to decrease progressively and continuously also with curing times exceeding 3 months. However, no significant effect of wet curing beyond 3 months was observed for Portland cement systems containing 10 wt% or 25 wt% MK [143,144,145]. Atis [146] reported for concrete with FA (50 wt% and 70 wt% cement replacement) the importance of a longer initial curing before testing at 5 vol% CO2 (testing from 3 days up to 3 months of moist curing), which resulted in lower carbonation depths. Burdon [87] found that the carbonation rate significantly increases with increasing FA replacement between 30 and 50 wt% for the same w/b ratio, but that the relative difference to PC concrete decreases with moist curing time. Parrott [79] examined the effect of 15–25 wt% LS replacement (at the samew/b = 0.59) on carbonation rate on samples water cured for 1, 3 and 28 days prior to natural carbonation exposure. Compared to plain cement, the carbonation rate increased with increasing LS replacement; the difference was smallest for 1-day curing for both exposure times under temporal variable outdoor conditions, see Fig. 8. With increasing exposure time and therefore decreasing moisture and temperature variations at the carbonation front, the differences decrease compared to plain PC after 1.5 years of exposure.

Effect of sealed curing duration on carbonation rate for different limestone replacement levels, compared to PC [79] concretes for the same w/b ratio
Also, the temperature during curing has an impact on the carbonation resistance. Borges et al. [75] concluded that, for a curing period of 90 days, raising temperature from 20 °C to 60 °C reduced the carbonation rate by 10–30% for cement pastes with high replacement levels by BFS. Li et al. [148] investigated the effect of high temperature curing (water curing at 20 °C, 40 °C, 60 °C, 80 °C until equal strength grade) of concrete made of PC and with FA, FA and BFS, and SF. The minimal carbonation depth was found for samples cured at 60 °C, pronounced for blended cements and less pronounced for PC.
In different models e.g. fib MC 2006 bulletin 34 [149] an execution transfer parameter is introduced to take into account the influence of curing on the effective carbonation resistance. It should be noted that all actions preventing premature desiccation of the concrete close to the surface are here considered as curing measures, and that no distinction is made for the type of curing and type of binder. However, Van den Heede et al. [85] emphasise the importance of a binder-dependent curing parameter. The effect of curing on carbonation rate as obtained from [42, 79, 91, 147, 150,151,152] has been evaluated in Fig. 9. It can be seen that for slag and LS blended cements, increasing curing time from 7 to 28 days is less effective with regard to improving carbonation resistance, despite the expected further reaction processes involving LS and slag with time [66]. In contrast, the FA blended samples showed a pronounced reduction of carbonation rate when increasing curing from 7 to 28 days, especially for the samples exposed outside under sheltered conditions with variable RH and temperature (exposed samples). It should be mentioned that moisture conditions in sheltered small samples differ from large structural elements; i.e., more homogeneous micro- and macro-climates in small samples than in structural elements may lead to different moisture transport processes.

Increase in carbonation resistance in dependency of moist curing time relative to 7 days curing for a PC, b PC with LS addition c PC with BFS addition d PC with FA addition. Data from [42, 79, 91, 147, 150,151,152]; i.e. 20/65/3 indicates T [°C]/ RH [%]/ CO2 [vol. %] where the climate conditions are given in the original literature source
5 Effect of relative humidity, temperature and CO2 concentration on carbonation
5.1 Relative humidity and temperature
Carbonation of cementitious materials occurs when CO2 diffuses through the material and dissolves in the pore solution where it reacts with the solid phases. At very low RH, there is not enough water in the pores to dissolve CO2 for the chemical reaction to happen. At very high RH, the pores become saturated and consequently the diffusion of CO2 is slowed down significantly. For Portland cement-based materials, the carbonation rate at 20 °C reaches maximum values at RH between 40% and 80% [38, 39, 153,154,155]. This RH range is quite large because the literature results relate to different CO2 concentrations with various preconditioning conditions (temperature and relative humidity, duration) and different curing times, as well as different material compositions (paste vs. mortar, water-to-cement ratio). In recent studies, the fastest carbonation has been observed at approximately 50–55% RH for plain cementitious materials; the carbonation coefficient of plain cementitious material is reduced by a factor of 0.6 ± 0.4, 0.5 ± 0.4 and 0.4 ± 0.4 if the relative humidity increases to 70%, 80% and 90% respectively [43, 44, 72]. The carbonation coefficient has been observed to be reduced to zero in the range 10 to 30% RH [72]. These broad ranges and various conditions involve varying degrees of saturation of the test specimens. For example, in the accelerated carbonation tests for both French standard XP P 18-458 [156] and European standard EN 12390-12 [157], the RH during carbonation is fixed at 57 ± 3%, while conditions specified for sample preconditioning are different.
For materials containing SCMs, the RH range at which carbonation proceeds the fastest may differ from the values determined for Portland cement systems (Fig. 10). Drouet et al. [72] found the most rapid carbonation RH around 33% for CEM V/A paste (containing slag and fly ash) carbonated at a CO2 concentration of 50% at 20 °C, which is a much lower RH value than was obtained for plain Portland cement paste (CEM I) in the same study, which was around 50%. Leemann and Moro [44] studied mixtures with different SCMs: BFS, SF, FA and LS. They found that increasing RH from 57 to 70% or 80%, at a CO2 concentration of 4% not only decreases the carbonation coefficient but also changes the ranking of the different concrete mixtures. The authors explained these differences via the effect of pore size effect on capillary condensation. For cementitious materials with a higher amount of fine pores, such as those containing blast furnace slag and pozzolans, the pore volume filled with water due to condensation is higher at a fixed value of RH [72, 158, 159]. De Ceukelaire et al. [160] found a maximum carbonation progress at 50% RH for concretes containing BFS, at different CO2 concentrations. A decrease of carbonation coefficient with increasing RH (40% to 60% to 80%) for different binders containing SCMs was also reported in [25].

7-day carbonation depth as a function of RH, for CEM I and CEM V/A pastes with w/b = 0.40, at a CO2 concentration of 50% [72]
It should be noted that apart from the externally imposed humidity conditions, the applied CO2 concentration during carbonation testing could also affect the internal humidity and saturation degree of cementitious binders. According to [161], excessive water production during carbonation at high CO2 levels could have a pore blocking effect. Hence, the humidity range at which carbonation is most rapid is most probably dependent on CO2 concentration for any concentrations above the natural atmospheric levels.
In addition to RH, temperature is a main external parameter that affects carbonation of cementitious materials. Higher temperature accelerates carbonation as chemical reactions and diffusion are faster. However, higher temperature also decreases the solubility of portlandite and CO2 in water [2, 162, 163].
The carbonation coefficient increases by a factor of approximately 1.1 from 20 to 30 °C and from 30 to 40 °C for plain and for blended cements (e.g. limestone and low calcium FA). For calcined clay blended cements, a higher increase of the carbonation coefficient was determined (by approximately a factor of 1.3) [25]. The decrease in carbonation coefficient when the temperature decreases from 20 to 10 °C is less pronounced (multiplicative factor of 0.95) [25, 38, 164].
Temperature also affects the stability of the calcium carbonate polymorphs that precipitate during carbonation. Tai and Chen [165] have shown that the formation of these polymorphs by precipitation from a CaCl2/Na2CO3 solution is a function of pH and temperature (Fig. 11). At high pH, calcite is the main polymorph regardless of temperature (24 °C and 58 °C). At lower pH, the dominant polymorph changes as a function of temperature; for high temperature (58 °C), aragonite will be the main polymorph to precipitate while for ambient temperature (24 °C) vaterite has been observed. Drouet et al. [72] have studied the effect of both temperature and RH on the stability of calcium carbonate polymorphs. They found that the two metastable forms of calcium carbonate (aragonite and vaterite) were observed in varying amounts for both Portland cement and CEM V pastes. The amount of aragonite and vaterite increased when the RH decreased. This increase is more significant at higher temperature.

Polymorphs of calcium carbonate observed to precipitate from a 1:1 molar mix of CaCl2 and Na2CO3 at a supersaturation of 5.5, according to pH (controlled by NaOH or HCl addition) and temperature [165]
One key aspect is that if the temperature increases without maintaining a fixed RH, a part of the water from the specimen evaporates and the carbonation could be lower in spite of the increased temperature. The literature results reflect these contradictory effects. Drouet et al. [72] observed that the carbonation depth of Portland cement paste increased continuously with temperature at a fixed RH, whereas the carbonation depth of CEM V/A reached a maximum at around 50 °C.
These findings are particularly important considering that when evaluating carbonation resistance of concretes according to accelerated carbonation standardised testing methodologies, the exposure temperature is generally set to be between 20 and 25 °C, which is not representative of all climates, and can yield carbonation results that can be far from being a realistic representation of what can be expected of concretes with SCMs under natural carbonation conditions in the field.
5.2 CO2 concentration
Increasing the CO2 concentration is a common approach to accelerate carbonation for testing purposes, and the results of accelerated tests are often used as performance criteria for validation of new concrete mixes and service life prediction models. Different factors and equations have been proposed to transform the results from accelerated carbonation testing to predict natural carbonation [151, 166, 167]. However, there is still need for research to define relations applicable for different concrete compositions [142].
From a thermodynamic viewpoint, increasing the CO2 concentration should not change the expected phase formation in plain Portland cement [28, 168]. Nonetheless, experiments have shown that:
-
The C–S–H carbonation rate increases with the applied CO2 concentration [32]. The proportion of CaCO3 originating from portlandite and C–S–H carbonation changes significantly from 1% CO2 onwards, in favor of C–S–H carbonation [169].
-
A CO2 concentration above 3% favours formation of aragonite and vaterite [26, 170] due to pH reduction because of increased amounts of dissolved CO2, [171], (Fig. 11).
-
A CO2 concentration above 10% leads to incomplete reaction of portlandite, e.g. due to a fast surface reaction causing an overgrowth of portlandite with calcite, which permanently prevents further portlandite carbonation [28, 170, 172]. A high CO2 concentration also lowers the pH and thus the solubility of CO2 in water or in solutions containing alkali hydroxides (such as cement pore fluids), leading to the formation of more HCO -3 and less CO 2-3 [2, 171].
-
However, in cementitious materials with limestone fillers only calcite is formed [168, 172, 173]. In high-volume fly ash binder systems, the preferred precipitation of aragonite and vaterite over calcite could not be confirmed for 1% and 10% CO2, but was evident at 0.03–0.04% CO2 [169].
-
For a high CO2 level, the amount of water produced during carbonation could be more than the porous matrix is capable of expelling in the same time interval. The time needed to establish a condition of equilibrium again is believed to slow down further carbonation [161, 174].
-
The correlation between accelerated and natural carbonation coefficients decreased from laboratory to sheltered (R2 = 0.69) and finally to unsheltered exposure (R2 = 0.15). Key parameters to consider for natural carbonation are temperature, relative humidity and amount of precipitation [44, 96]. The correlation strongly depends on the microclimatic conditions (i.e. RH, orientation, wind, precipitation).
Rozière et al. [175] found correlation coefficients of only 0.45-0.67 when correlating natural to accelerated carbonation at 50% CO2, although the ranking of the concrete types considered remained similar. This is in line with other studies [38, 39, 176]. On the other hand, several researchers [175,176,177] noticed small changes in ranking between mixes when carbonated at 1%, 4% or 10% CO2. Increasing the RH at 4% CO2 from 57% to 70-80% RH also affected the ranking for carbonation resistance, probably as the result of a different w/c and pore size distributions of the concrete types tested [44].
As the pore structure and the ratio w/CaOreactive are cement-specific, the correlation between accelerated and natural carbonation depends strongly on the binder [150, 179]. Hainer 150 points out that clinker-reduced concrete shows relatively higher carbonation rates under field than under accelerated conditions. In contrast, Leemann et al. [96] and Bernal et al. [171] noted that accelerated carbonation is more aggressive than natural carbonation for concrete with SCMs. A detailed comparison of literature data is challenging due to the many different storage and exposure conditions assessed by different authors, and is beyond the scope of this review.
The increase in carbonation coefficient with increasing CO2 concentration (CO2acc) compared to natural CO2 concentration (CO2nat) is in most experimental investigations lower than what is expected from the theoretical relationship √(CO2acc/CO2nat) [38, 96, 151]. According to these investigations, the deviation from the theoretical relationship is more pronounced for plain cements and cements with LS than for slag blended cementitious materials. It has been indicated [42] that a formula in which the carbonation depth relies on the square-root of the CO2 concentration in play should be reliable up to 3% CO2. Yet, even from 1% CO2, the relationship does not seem to hold [169]. Given that this is the case, the assumption of pure diffusion control of carbonation kinetics appears to break down at higher CO2 concentrations, and this needs more detailed investigation to determine what is the true rate-controlling step if accelerated test results are to be used to predict natural carbonation rates in standards and codes.
6 Effect of carbonation on porosity and transport properties
6.1 Porosity and pore size distribution
Porosity of cementitious mixtures plays an important role in relation to the carbonation resistance, as it is directly linked to the ingress of CO2. When investigating porosity, different aspects need to be considered, for instance pore volume, pore size distribution, tortuosity and interconnectivity. Castellote et al. [31] reported pore clogging and formation of dense, carbonated areas as the main cause for limited accelerated carbonation rates in plain PC pastes. Hyvert et al. [32] and Anstice et al. [180] confirmed a decreasing total porosity with an increasing CO2 concentration, and Knöfel et al. [173] recorded a refinement in pore size distributions using mercury intrusion porosimetry. Mortars with plain PC showed increased amounts of gel pores, while the fraction of capillary pores decreased to around 10%. The decrease in total porosity, and pore clogging, are attributed to the formation of calcium carbonate which takes up a larger volume than the initial hydration product Ca(OH)2. Several studies [24, 25, 181] also identified a decrease in the total porosity of plain cement pastes due to carbonation in combination with a coarsening of the capillary pores. For BFS-containing binders a decrease in gel porosity but an increase in capillary pores (coarsening of the pore structure) has been reported in [26, 83, 182]. Similar are the findings at high FA or SF replacement levels [26, 27, 182, 183]. This coarsening is usually associated with C–S–H/C-A-S–H carbonation. The higher the amount of reactive alumina provided by the SCM, the larger the increase in porosity on carbonation, due to the decomposition of AFm and AFt phases [75].
According to [106] the decrease in micro-pores is attributed to clogging by CaCO3 from C–S–H carbonation, while the reduction of macro-pores is mainly attributed to pore clogging by CaCO3 from portlandite carbonation. However, the increase in meso-pores has been interpreted differently by various authors, and the potential causes are listed in [106] as follows:
-
(i)
porous structure of silica gel formed during carbonation.
-
(ii)
micro-cracks in the CaCO3 layer surrounding portlandite crystals due to the hydric gradient, and.
-
(iii)
micro-cracks induced by carbonation shrinkage.
The more pronounced increase in meso- and macro-pore volumes for FA, SF and slag blended cements upon carbonation can also be attributed to the lower Ca/Si ratio of C–S–H/C-A-S–H formed in these blended cements, by enabling the entrapment of alkalis reducing the amount of hydroxyl ions and also the increased carbonation shrinkage for Ca/Si less than 1.3 [34].
Also Shah et al. [25] reported that because the replacement of PC with Si-rich SCMs results in less portlandite, the decalcification of the C–S–H present leads to an increase of the total porosity in combination with a coarsening of the pore structure. Consequently, the extent of pore coarsening and the increase of total porosity during carbonation depend on both the amount and the type of SCMs.
Prior to carbonation, SCMs can increase the porosity of cementitious materials at early ages but contribute to the development of a more refined pore structure over the time [184]. These two opposing effects will affect the resistance to carbonation of concretes depending on the type of SCM used and its ability to cause pore refinement, the amount of cement replacement, and the reaction degree as a function of time. Upon carbonation it is observed that the total porosity increases with increasing SCM replacement ratios [75, 82]. For example, when SF is used in concrete, Kulakowski et al. [185] found a ‘critical threshold’ related to the amount of replacement and w/b ratio: cement replacement up to 10 wt% by SF will result in an increased carbonation resistance. However, if SF replacement is higher than 10%, this will lead to more carbonation.
The data collected in Table 1, from [82], show an increasing total porosity prior to carbonation when the replacement level by FA is increased from 0 to 30 wt% and 60 wt% substitution. Upon carbonation, the total pore volume decreases in all cases. The decrease of the pore volume in the MIP-accessible range (3 nm to 500 µm) during carbonation seems to be lower at higher replacement levels for the MIP measurements, and similar in the case of GRAM (gamma ray attenuation method) measurements (accessing coarse pores size 200 μm to 1000 μm [186]).
For ternary systems with 50% clinker, 31% calcined clay, 15% limestone and 4% gypsum, a clear increase of the total porosity was observed in Fig. 12a [24], under both natural and accelerated (3%) carbonation conditions. An experimental study on the use of slag in pastes [24] showed that an increase in total porosity is detected when the clinker replacement is 70 wt%. The increase in capillary porosity due to carbonation further accelerates diffusion of detrimental substances and will therefore make concrete with SCMs more vulnerable to deterioration by coupled mechanisms.

Mercury intrusion curve of uncarbonated cylindrical paste samples (solid line) compared with cylindrical paste samples carbonated in natural (dotted line) and accelerated conditions at 3% CO2 (dashed line) cured in lime water for 120 day and preconditioned for 15 day at 60% RH and 27 °C; a) Cumulative intrusion curve of PC, b) Cumulative intrusion curve of 70% PC + 30% FA, c) Cumulative intrusion curve of limestone calcined clay cement (50% clinker, 31% calcined clay, 15% LS, 4% gypsum) from [24]
6.2 Transport properties
The alteration of the pore structure upon carbonation of plain and blended cementitious materials affects the vapour sorption/desorption isotherms, gas and moisture transport. The transport of CO2 through cementitious materials is one of the governing parameters affecting carbonation rate and is strongly affected by moisture conditions as was discussed above. The degree of water saturation under steady-state and isothermal conditions has been determined by means of water vapour sorption/desorption tests [38, 74, 187]. Figure 13 shows sorption isotherms for plain and blended cement pastes. A benefit of adsorption/desorption isotherms is the provision of information about changes in micropores < 10 nm, which are less well reflected by MIP (molecule radius of H2O is 0.1 nm, of nitrogen N2 is 0.215 nm and both are smaller than that of mercury; it avoids also the risk of microcracking in MIP [106]).

Effect of carbonation on degree of pore saturation (adsorption (Ads.)/desorption (Des.) isotherms) of plain PC mortars with w/c = 0.5 from [38, 187] a and desorption isotherms with 22% slag + 22% FA (circle) or 61% slag (rhombus) blended mortars with w/c = 0.4 from [74] b for non-carbonated (NC) and carbonated C samples
Papadakis et al. [38] and Houst et al. [187] observed no alteration in water saturation degree at > 50 vol%CO2 for plain cementitious materials. Borges et al. [75] found a small alteration at low RH when applying 3 vol%CO2 to plain PC, indicating an increase in micro-porosity, compared to a pronounced alteration of pore saturation in FA and BFS blended cementitious materials upon carbonation, indicating an increase in micro-, meso- and macro porosity. Hyvert [188] determined a notable alteration of the water sorption isotherm for plain PC, and an even more evident alteration for blended cementitious materials. The reduced degree of saturation in carbonated samples at 50% ≤ RH ≤ 85% affects effective CO2 diffusion coefficients in two ways: (i) the coarsened pore structure opens the path for CO2 diffusion, as the capillary pores are the decisive pores for high diffusion rates; and (ii) the degree of saturation is reduced in the relative humidity ranges expected under common site conditions.
Effective CO2 diffusion coefficients in non- and fully carbonated pastes, mortars and concretes have been determined experimentally under different conditions [21, 38, 39, 44, 78, 105, 182, 187, 189,190,191].
Prior to carbonation it was found that the effective CO2 diffusion coefficient decreases with:
-
Decreasing w/b (decreasing capillary porosity) [21, 39, 78, 105].
-
Increasing relative humidity above 40% RH, which increases the degree of water saturation [38, 105, 182, 187, 189].
-
Increasing PC replacement level by FA, SF or BFS, respectively increasing the fraction of the binder that is C(-A)-S–H [21, 78, 182, 189].
-
Increasing degree of hydration [182].
Upon carbonation the effective diffusion coefficient was found to:
-
Decrease in the case of plain cementitious materials and blends with portlandite or LS, with increasing ratio of portlandite carbonation [21, 38, 39, 44, 78, 105, 182, 187, 189,190,191] for RH > 30% [182].
-
Increase with increasing cement replacement level by SF, FA, calcined clay, or mostly for BFS, due to increasing C(-A)-S–H carbonation [21, 78, 182, 190, 191]. The difference becomes more pronounced with increasing degree of water saturation.
-
Increase with carbonation degree at constant high RH (93%) for all mix design unless the water to cement ratio is very low (< 0.4) [191].
In water CO2 diffusion coefficients have been found to be < 10-9 m2/s [192]. Water vapour diffusion coefficients are up to two orders of magnitude higher and increase with the difference in relative humidity, increasing w/b ratio and decrease with increasing replacement by SF or BFS [193]. As CO2 is consumed by the carbonation reaction, CO2 diffusion coefficients are often derived from O2, N2 or He diffusion coefficients [182, 187, 189]. The value of the corresponding CO2 diffusion coefficient depends on whether the process is limited by the molecular mass (molecular diffusion dominating in larger pores) or by the Knudsen diffusion (i.e. diffusion in small pores, where the diffusion resistance is dominated by collisions with the pore walls). Furthermore, transport may also occur as surface diffusion, if the gas interacts by a succession of adsorption–desorption reactions with the surfaces [187, 194]. According to [187, 189] the CO2 diffusion through non-carbonated cementitious materials is both a Knudsen and molecular diffusion process over the entire range of water saturation degree. However, upon carbonation this only holds for plain cements. For slag (approximately 80% replacement level) or FA and SF blended cements (replacement level > 44%) the pores can coarsen sufficiently that Knudsen processes involving pore walls are less important, and so transport instead becomes molecular diffusion controlled [182, 192].
Figure 14 shows effective O2 diffusion coefficients (DCO2/DO2 ≈ 1.4) determined by Leemann et al. [21, 78] after oven drying. The determined effective diffusion coefficients are smaller or similar after carbonation for plain PC, portlandite-blended and limestone-blended cements, but higher for silica fume and slag blends, consistent with the preceding discussion for the rates of carbonation of cements containing these SCMs.

In some carbonation exposure conditions involving high pressure differences, both diffusive and convective transport coexist, and permeability may become dominant [195]. Gas permeability of carbonated and non-carbonated cementitious materials has been reported e.g. in [75, 104, 196,197,198,199,200,201,202] under the assumption of laminar flow. Gas permeability was in the range of 0.1 × 10− 9 m/s < ke < 10 × 10-9 m/s. The gas permeability decreased with decreasing w/b ratio and increasing relative humidity or increasing cement replacement by SF, FA or BFS. Upon carbonation, gas permeability was increased or at least similar as prior to carbonation. But the increase was more pronounced for BFS blended cement systems with replacement levels of 75 wt% to 90 wt% [75, 198].
Water transport properties of cementitious materials have been reported e.g. in [55, 177, 178]. The coefficient of water permeability is usually up to four orders of magnitude lower than the coefficient of gas permeability and may be related to gas permeability in dependency of the applied pressure head according to Klinkenberg explained in [203]. Upon carbonation the water permeability decreased in the case of plain and LS blended cementitious materials but increased for slag, FA and SF blended cementitious materials with moderate to high replacement levels [55, 177, 178]. Similarly, capillary sorption describes permeability of concrete where the pressure head is replaced by the capillary forces including water and water vapour transport [204, 205].
Figure 15 shows coefficients of water sorptivity determined by Jansson et al. [101] for non- and partly-carbonated cementitious materials. He observed for non-carbonated samples a decrease in sorptivity from plain PC over blends with 20 or 35 wt% FA, or 35, 50, or 65 wt% BFS. After carbonation, the sorptivity was significantly reduced for PC samples, slightly reduced for samples containing 20 wt% or 35 wt% FA, or 35 wt% or 50 wt% BFS and increased for samples containing 65 wt% BFS where a pronounced C-A-S–H carbonation can be expected. Similarly, a decrease in capillary sorptivity with carbonation has been observed for plain cement materials [204].

Coefficients of sorptivity reported in [101] for non- carbonated and partly carbonated concretes along with the carbonation depth (small squares), the notation shows the cement type respectively the amount and type of addition and the w/b ratio
Gas permeability data shows an increase upon carbonation for most binder types, while both the effective diffusion coefficients and the sorptivity coefficients show a decrease upon carbonation except for SF or BFS blended cementitious materials. Hence it can be assumed, that the induced pressure in gas permeability experiments alters the pore structure and mirrors mainly the transport in larger pores. Moisture transport (sorption, capillary suction and water permeability) strongly depends on the initial moisture conditions. Additionally, moisture transport may be altered by chemical interaction of water with the cement, such as re-hydration of unreacted cement and dehydrated cement pastes, changes of microstructure of C–A–S–H and secondary reaction products (depending on the type of SCM used) during moisture transport, drying shrinkage induced by the microscopic damage to pores due to capillary forces, and swelling associated with the water uptake [204,205,206,207,208,209].
7 General remarks and conclusions
It is clear that the carbonation mechanism in cementitious systems containing SCMs differs from that of plain Portland cement systems, due to the differences in the phase assemblage evolution, pore structure, and pore solution chemistry. The main difference between these systems is the role of portlandite carbonation, as it is expected that the amount of portlandite decreases at higher SCM replacement levels. This is a consequence of the lower amount of Portland clinker in the cement (which produces portlandite during its hydration) and the consumption of portlandite during the pozzolanic reaction. Portlandite carbonation is the main contributor of released water during carbonation, and usually leads to a reduction in pore size and total volume of meso- and macro- pores due to CaCO3 precipitation. However, the literature results are to some extent contradictory, which may be due to the different measurement methods along with the different pore sizes investigated. Portlandite carbonation may furthermore be incomplete due to the covering of portlandite crystals by calcium carbonate.
With the reduced content of portlandite in SCM containing systems, carbonation will happen more rapidly in the main CO2-binding phases, C–S–H in the case of using SF, and C–A–S–H phases in the case of using BFS, FA, MK and other Al-containing SCMs. Carbonation of these hydrates seems to be the main contributor to carbonation shrinkage (polymerisation shrinkage), especially for low Ca/Si C(-A)-S–H and induces coarsening of pore structure upon carbonation and reduction of mechanical strength. This is particularly critical when evaluating carbonation of concretes with SCMs, as carbonation not only reduces the overall pH, but can also lead to a coarsening in pore structure, consequently reducing the ability of these materials to withstand other forms of degradation by chemical or physical attack.
Carbonation of C(-A)-S–H does not seem to contribute to the release of water prior to complete polymerisation, but there is little investigation regarding structural changes and water released in these hydrates upon carbonation, despite their role as the dominant binding phase present in modern cements. There is an urgent need to determine the carbonation mechanism of Al substituted C–S–H phases, and those containing alkalis (C-(N)-A-S–H), as a function of exposure conditions (e.g. temperature, RH and CO2 concentration), as these are the main CO2-binding phases identified in SCM-containing cements, particularly those produced with BFS. The degree of crosslinking in these gels and the reduced Ca/Si ratio compared with the C–S–H formed in plain PC systems will strongly influence how carbonation proceeds.
There is little understanding in the role of secondary hydration products in the carbonation resistance of systems with SCMs. In the case of using BFS, there is a consensus that Mg–Al layered double hydroxides (LDHs) (e.g. hydrotalcite), whose formation is favoured when using BFS with > 5 wt% MgO, seem to play a key role in improving carbonation resistance in systems where higher quantities of these phases are formed. Understanding of the roles of other phases such as AFm and/or AFt phases, as well as zeolites present in some natural SCMs, in the carbonation process is imperative if next-generation binders are to be designed to favour formation of specific microstructural features to maximise their carbonation resistance.
There is no direct correlation between the carbonation rates identified in pastes, mortars and concretes with a given binder type, independent of the SCM used, which is consistent with the differences in the pore structure induced by the addition of aggregates and reduction of paste, and variation in degrees of saturations that can be achieved in these materials. All these factors will influence gas diffusivity and dissolution, and consequently the mechanism and kinetics of carbonation. Many studies centred on evaluating carbonation of SCM containing materials, particularly for new SCMs, are limited to the assessment of mortars. The carbonation coefficients obtained in those studies cannot directly be used to predict performance of concrete made with similar materials. It is important to understand the limitations of the tests conducted, which in the case of mortars can only give an indication of concrete performance but not a full description of the behaviour of the material.
It is well known that the carbonation exposure conditions (e.g. CO2 concentration, RH and temperature) will strongly influence the kinetics of carbonation and the type and amount of carbonation products forming in different cementitious systems. In the case of plain PC systems this is well understood, and it has been suggested that the carbonation products forming when using > 3% CO2 are not identical to those observed in naturally carbonated materials. This is of particular importance as several national accelerated carbonation standards prescribe the use of CO2 concentration beyond this value, which can lead to misleading results.
The effect of temperature on carbonation is usually overlooked, particularly when evaluating carbonation under accelerated testing methods, as the majority of standard test methods have similar ranges of exposure temperatures. It is evident that temperature will play a key role in reaction kinetics and gas transport, which means that the results of accelerated carbonation tests using temperature conditions with no relevance to a specific region may not provide meaningful results to predict how these concretes will perform under real exposure conditions at a temperature very different from the testing temperature.
An overview of the effect of various parameters on the carbonation coefficient, as found in current literature as summarised in this review, is given in Table 2. It should be noted that these factors are strongly dependent on the composition of the material (type and replacement level of SCM) as well as on the carbonation conditions (i.e. CO2 concentration and RH) and the preconditioning of the material prior to testing.
The preparation of this literature review enabled the members of RILEM TC 281-CCC to elucidate that there is a large number of open questions regarding carbonation of concretes with SCMs, although these materials are extensively, if not exclusively, used in modern infrastructure development. There is great scope for future research in this area, as questions at all scales need to be resolved to determine the best strategies to be implemented to increase the longevity of modern and future infrastructure concretes.
In an effort to answer some of these questions, the RILEM TC 281-CCC (WG1&2) is currently performing a round robin test to assess the effects on carbonation resistance in pastes, mortars and concretes produced with commercial SCM containing cements, when adopting different carbonation exposure conditions outlined in currently applied standards and test specifications.
Change history
31 March 2021
A Correction to this paper has been published: https://doi.org/10.1617/s11527-021-01658-1
16 December 2020
A Correction to this paper has been published: https://doi.org/10.1617/s11527-020-01599-1
References
Kern M (1960) The hydration of carbon dioxide. J Chem Educ 39:1–14. https://doi.org/10.1021/ed037p14
Stumm W, Morgan J (1995) Aquatic chemistry: chemical equilibria and rates in natural waters. Wiley, New York
Adamczyk K, Prémont-Schwarz M, Pines D, Pines E, Nibbering ETJ (2009) Real-time observation of carbonic acid formation in aqueous solution. Science 326:1690–1694
Dubina E, Korat L, Black L, Strupi-Šuput J, Plank J (2013) Influence of water vapour and carbon dioxide on free lime during storage at 80 & #xB0;C, studied by Raman spectroscopy. Spectrochimica Acta Part A Mol Biomol Spectrosc 111:299–303. https://doi.org/10.1016/j.saa.2013.04.033
Tai C, Chen FB (2004) Polymorphism of CaCO3, precipitated in a constant-composition environment. AIChE J. https://doi.org/10.1002/aic.690440810
Fernández-Díaz L, Prieto M (2010) The role of sulfate groups in controlling CaCO3 polymorphism. Geochim Cosmochim Acta 74(21):6064–6076. https://doi.org/10.1016/j.gca.2010.08.010
De Choudens-Sánchez V, González LA (2009) Calcite and Aragonite Precipitation Under Controlled Instantaneous Supersaturation: elucidating the Role of CaCO3 Saturation State and Mg/Ca Ratio on Calcium Carbonate Polymorphism. J Sediment Res 79(6):363–376. https://doi.org/10.2110/jsr.2009.043
Rodriguez-Navarro C, Kudłacz K, Cizer Ö, Ruiz-Agudo E (2015) Formation of amorphous calcium carbonate and its transformation into mesostructured calcite. Cryst Eng Comm 17:58–72. https://doi.org/10.1039/c4ce01562b
Ostwald WZ (1987) Studien über die Bildung und Umwandlung fester Körper. Z Phys Chem 22:289–330. https://doi.org/10.1515/zpch-1897-2233
Cizer Ö, Rodriguez-Navarro C, Ruiz-Agudo E, Elsen J, Gemert DV, Balen KV (2012) Phase and morphology evolution of calcium carbonate precipitated by carbonation of hydrated lime. J Mater Sci 47:6151–6165. https://doi.org/10.1007/s10853-012-6535-7
Black L, Breen C, Yarwood J, Garbev K, Stemmermann P, Gasharova B (2007) Structural features of C–S-H(I) and its carbonation in air—A Raman spectroscopic study. Part II: carbonated phases. J Am Ceram Soc 90(2):908–917. https://doi.org/10.1111/j.1551-2916.2006.01429.x
Lagerblad B (2005) Carbon dioxide uptake during concrete life cycle - state of the art. CBI Report 2, Swedish Cement and Concrete Research Institute, Stockholm Sweden
Groves GW, Brough A, Richardson IG, Dobson CM (1991) Progressive changes in the structure of hardened C3S cement pastes due to carbonation. J Am Ceram Soc 74(11):2891–2896. https://doi.org/10.1111/j.1151-2916.1991.tb06859.x
Hargis CW, Lothenbach B, Müller CJ, Winnefeld F (2017) Carbonation of calcium sulfoaluminate mortars. Cement Concr Compos 80:123–134. https://doi.org/10.1016/j.cemconcomp.2017.03.003
De Weerdt K, Plusquellec G, Revert AB, Geiker MR, Lothenbach B (2019) Effect of carbonation on the pore solution of mortar. Cement Concr Res 118:38–56. https://doi.org/10.1016/j.cemconres.2019.02.004
Shi Z, Lothenbach B, Geiker MR, Kaufmann J, Leemann A, Ferreiro S, Skibsted J (2016) Experimental studies and thermodynamic modeling of the carbonation of Portland cement—metakaolin—limestone mortars. Cement Concr Res 88:60–72. https://doi.org/10.1016/j.cemconres.2016.06.006
Femenias YS, Angst U, Elsener B (2017) pH-monitoring in mortar with thermally-oxidized iridium electrodes. RILEM Tech Lett 2:59–66
Houst Y, Wittmann F (2002) Depth profiles of carbonates formed during natural carbonation. Cement Concr Res 32:1923–1930
Grengg C, Müller B, Staudinger C, Mittermayr F, Breininger J, Ungerböck B, Borisov S, Mayr T, Dietzel M (2019) High-resolution optical pH imaging of concrete exposed to chemically corrosive environments. Cement Concr Res 116:231–237
Khan MSH, Noushini A, Castel A (2017) Carbonation of a low-calcium fly ash geopolymer concrete. Mag Concr Res 69(1):24–34. https://doi.org/10.1680/jmacr.15.00486
Leemann A, Pahlke H, Loser R, Winnefeld F (2018) Carbonation resistance of mortar produced with alternative cements. Mater Struct 51:114. https://doi.org/10.1617/s11527-018-1239-3
Galan I, Glasser FP, Baza D, Andrade C (2015) Assessment of the protective effect of carbonation on portlandite crystals. Cement Concr Res 74:68–77. https://doi.org/10.1016/j.cemconres.2015.04.001
Cizer Ö, Van Balen K, Elsen J, Van Gemert D (2012) Real-time investigation of reaction rate and mineral phase modifications of lime carbonation. Constr Build Mater 35:741–751. https://doi.org/10.1016/j.conbuildmat.2012.04.036
Shah V, Scrivener K, Bhattacharjee B, Bishnoi S (2018) Changes in microstructure characteristics of cement paste on carbonation. Cement Concr Res 109:184–197. https://doi.org/10.1016/j.cemconres.2018.04.016
Shah V, Bishnoi S (2018) Carbonation resistance of cements containing supplementary cementitious materials and its relation to various parameter of concrete. Constr Build Mater 178:219–232. https://doi.org/10.1016/j.conbuildmat.2018.05.162
Auroy M, Poyet S, Le Bescop P, Torrenti JM, Charpentier T, Moskura M, Bourbon X (2018) Comparison between natural and accelerated carbonation (3% CO2): impact on mineralogy, microstructure, water retention and cracking. Cement Concr Res 109:64–80. https://doi.org/10.1016/j.cemconres.2018.04.012¨
Morandeau A, Thiéry M, Dangla P (2014) Investigation of the carbonation mechanism of CH and C–S-H in terms of kinetics, microstructure changes and moisture properties. Cement Concr Res 56:153–170. https://doi.org/10.1016/j.cemconres.2013.11.015
Visser JHM (2014) Influence of the carbon dioxide concentration on the resistance to carbonation of concrete. Constr Build Mater 67:8–13. https://doi.org/10.1016/j.conbuildmat.2013.11.005
Sevelsted TF, Skibsted J (2015) Carbonation of C–S–H and C–A–S–H samples studied by 13C, 27Al and 29Si MAS NMR spectroscopy. Cement Concr Res 71:56–65. https://doi.org/10.1016/j.cemconres.2015.01.019
Steiner S, Lothenbach B, Borgschulte A, Proske T, Winnefeld F (2018) Effect of relative humidity on the carbonation rate of portlandite, calcium silicate hydrates and ettringite. 20. In: Internationale Baustofftagung (ibausil), 12.—14. September 2018, Weimar Germany
Castellote M, Fernandez L, Andrade C, Alonso C (2009) Chemical changes and phase analysis of OPC pastes carbonated at different CO2 concentrations. Mater Struct 42:515–525. https://doi.org/10.1617/s11527-008-9399-1
Hyvert N, Sellier A, Duprat F, Rougeau P, Francisco P (2010) Dependency of C–S–H carbonation rate on CO2 pressure to explain transition from accelerated tests to natural carbonation. Cement Concr Res 40:1582–1589. https://doi.org/10.1016/j.cemconres.2010.06.010
Wu B, Ye G (2019) Study of carbonation rate of synthetic C–S-H by XRD, NMR and FTIR. HERON 64(1–2):21–38
Chen JJ, Thomas JJ, Jennings HM (2006) Decalcification shrinkage of cement paste. Cement Concr Res 36:801–809. https://doi.org/10.1016/j.cemconres.2005.11.003
Groves GW, Rodway DI, Richardson IG (1990) The carbonation of hardened cement pastes. Advances in Cement Research 3(11):117–125. https://doi.org/10.1680/adcr.1990.3.11.117
Swenson EG, Sereda PJ (1968) Mechanism of the carbonation shrinkage of lime and hydrated cement. J Appl Chem 18:111–117
Galan I, Andrade C, Mora P, Sanjuan MA (2010) Sequestration of CO2 by concrete carbonation. Environ Sci Technol 44:3181–3186. https://doi.org/10.1021/es903581d
Papadakis VG, Vayenas CG, Fardis MN (1991) Fundamental modeling and experimental investigation of concrete carbonation. ACI Mater J 88:363–373
Papadakis VG, Vayenas CG, Fardis MN (1991) Physical and chemical characteristics affecting the durability of concrete. ACI Mater J 88:186–196
Visser JHM (2012) Accelerated carbonation testing of mortar with supplementary cementing materials: limitation of the acceleration due to drying. Heron 57(3):231–246
von Greve-Dierfeld S (2015) Bemessungsregeln zur Sicherstellung der Dauerhaftigkeit von XC exponierten Stahlbetonbauteilen (in German). Dissertation, Technical University Munich
Sisomphon K, Franke L (2007) Carbonation rates of concretes containing high volume of pozzolanic materials. Cement Concr Res 37(12):1647–1653. https://doi.org/10.1016/j.cemconres.2007.08.014
Hunkeler F, Greve-Dierfeld S (2019) Karbonatisierung von Beton und Korrosionsgeschwindigkeit der Bewehrung im karbonatisierten Beton (Carbonation of concrete and reinforcement corrosion rate in carbonated concrete). Research report published by the Swiss Federal Roads Office VSS No. 696
Leemann A, Moro F (2017) Carbonation of concrete: the role of CO2 concentration, relative humidity and CO2 buffer capacity. Mater Struct 50:30. https://doi.org/10.1617/s11527-016-0917-2
Johannesson B, Utgenannt P (2001) Microstructural changes caused by carbonation of cement mortar. Cement Concr Res 31(6):925–931. https://doi.org/10.1016/S0008-8846(01)00498-7
Taylor HF (1997) Cement chemistry, vol 2. Thomas Telford, London
Lothenbach B, Scrivener K, Hooton RD (2011) Supplementary cementitious materials. Cement Concr Res 41(12):1244–1256. https://doi.org/10.1016/j.cemconres.2010.12.001
Juenger M, Snellings R, Bernal SA (2019) Supplementary cementitious materials: new sources, characterization, and performance insights. Cement Concr Res 122:257–273. https://doi.org/10.1016/j.cemconres.2019.05.008
De Belie N, Soutos M, Gruyaert E (2018) Properties of fresh and hardened concrete containing supplementary cementitious materials, State-of-the-Art Report of the RILEM Technical Committee 238-SCM Working Group 4. Springer, Berlin
Scrivener K, Lothenbach B, De Belie N, Gruyaert E, Skibsted J, Snellings R, Vollpracht A (2015) TC 238-SCM: hydration and microstructure of concrete with SCMs. Mater Struct 48:835–862. https://doi.org/10.1617/s11527-015-0527-4
Villagrán-Zaccardi Y, Vollpracht A, Gruyaert E, De Belie N (2018) Recommendation of RILEM TC 238-SCM: determination of the degree of reaction of siliceous fly ash and slag in hydrated cement paste by the selective dissolution method. Mater Struct. https://doi.org/10.1617/s11527-017-1134-3
Gruyaert E, Van Den Heede P, De Belie N (2013) Carbonation of slag concrete: effect of the cement replacement level and curing on the carbonation coefficient—effect of carbonation on the pore structure. Cement Concr Compos 35(1):39–48. https://doi.org/10.1016/j.cemconcomp.2012.08.024
Gruyaert E. (2010) Effect of blast furnace slag as cement replacement on hydration, microstructure, strength and durability of concrete. Dissertation, Ghent University
Herterich J (2017) Microstructure and Phase Assemblage of Low-Clinker Cements during Early Stages of Carbonation. Dissertation, School of Civil Engineering at the University of Leeds
Deschner F, Winnefeld F, Lothenbach B, Seufert S, Schwesig P, Dittrich S, Goetz-Neunhoeffer F, Neubauer J (2012) Hydration of Portland cement with high replacement by siliceous fly ash. Cement Concr Res 42(10):1389–1400. https://doi.org/10.1016/j.cemconres.2012.06.009
Prentice D, Bernal S, Bankhead M, Hayes M, Provis J (2018) Phase evolution of slag-rich cementitious grouts for immobilisation of nuclear wastes. Adv Cement Res 30(8):345–360. https://doi.org/10.1680/jadcr.17.00198
Vollpracht A, Lothenbach B, Snellings R, Haufe J (2016) The pore solution of blended cements: a review. Mater Struct 46:3341–3367. https://doi.org/10.1617/s11527-015-0724-1
Azad VJ, Suraneni P, Trejo D, Weiss WJ, Isgor B (2018) Thermodynamic investigation of allowable admixed chloride limits in concrete. ACI Mater J 115(5):727–738
Kunther W, Zhuo D, Skibsted J (2015) Thermodynamic modelling of Portland cement—metakaolin—limestone blends. In: 1st international conference on calcined clays for sustainable concrete doi.org/https://doi.org/10.1007/978-94-017-9939-3
Skocek J, Zajac M, Stabler C, Ben Haha M (2017) Predictive modelling of hydration and mechanical performance of low Ca composite cements: possibilities and limitations from industrial perspective. Cement Concr Res 100:68–83. https://doi.org/10.1016/j.cemconres.2017.05.020
Lothenbach B, Rentsch D, Wieland E (2014) Hydration of a silica fume blended low-alkali shotcrete cement. Phys Chem Earth Parts A/B/C 70–71:3–16. https://doi.org/10.1016/j.pce.2013.09.007
McPolin DO, Basheer PAM, Long AE (2009) Carbonation and pH in mortars manufactured with supplementary cementitious materials. J Mater Civil Eng 21(5):217–225
Sadrmomtazi A, Khoshkbijari R, Tahmouresi B (2018) Effect of fly ash and silica fume on transition zone, pore structure and permeability of concrete. Mag Concrete Res 70(10):519–532. https://doi.org/10.1680/jmacr.16.00537
De Weerdt K, Ben Haha M, Saout G, Kjellsen KO, Justnes H, Lothenbach B (2011) Hydration mechanism of ternary Portland cements containing limestone powder and fly ash. Cement Concr Res 41(3):279–291. https://doi.org/10.1016/j.cemconres.2010.11.014
De Weerdt K, Ben Haha M, Le Saout G, Kjellsen KO, Justness H, Lothenbach B (2012) The effect of temperature on the hydration of composite cements containing limestone powder and fly ash. Mater Struct 45:1101–1114. https://doi.org/10.1617/s11527-011-9819-5
Lothenbach B, Le Saout G, Galucci E, Scrivener K (2008) Influence of limestone on the hydration of Portland cements. Cement Concr Res 38(6):848–860. https://doi.org/10.1016/j.cemconres.2008.01.002
Tennis PE, Thomas MDA, Weiss WJ (2011) State-of-the-Art Report on use of limestone in cements at levels up to 15%. Report of the Portland Cement Association, Illinois
Snellings R, Mertens G, Elsen J (2012) Supplementary cementitious materials. Rev Miner Geochem 74:211–278. https://doi.org/10.2138/rmg.2012.74.6
Irbe L (2019) The role of aluminium in C-A-S-H during chemical attack on concrete. Dissertation TU München, Germany
Li J, Yu Q, Huang H, Yin S (2019) Effects of Ca/Si ratio, aluminum and magnesium on the carbonation behavior of calcium silicate hydrate. Materials 12:1268. https://doi.org/10.3390/ma12081268
Ortaboy S, Li J, Geng G, Myers R, Monteiro PJM, Maboudian R, Carraro C (2017) Effects of CO2 and temperature on the structure and chemistry of C–(A–)S–H investigated by Raman spectroscopy. RSC Adv 7:48925–48933. https://doi.org/10.1039/c7ra07266j
Drouet E, Poyet S, Le Bescop P, Torrenti JM, Bourbon X (2019) Carbonation of hardened cement pastes: influence of temperature. Cement Concr Res 115:445–459. https://doi.org/10.1016/j.cemconres.2018.09.019
Thiery M, Faure P, Morandeau A, Platre G, Boutelou J, Dangl P, Baroghel-Bouney V (2011) Effect of carbonation on the microstructure and moisture properties of cement-based materials. In: 12th International conference on durability of building materials and components, 12th–15th April 2011, Porto Portugal
Auroy M, Poyet S, Le Bescop P, Torrenti JM (2015) Impact of carbonation on unsaturated water transport properties of cement-based materials. Cement Concr Res 74:44–58. https://doi.org/10.1016/j.cemconres.2015.04.002
Borges P, Costa J, Milestone N, Lynsdale C, Streatfield R (2010) Carbonation of CH and C–S-H in composite cement pastes containing high amounts of BFS. Cement Concr Res 40(2):284–292. https://doi.org/10.1016/j.cemconres.2009.10.020
Van den Heede P, De Schepper M, De Belie N (2019) Accelerated and natural carbonation of concrete with high volumes of fly ash: chemical, mineralogical and microstructural effects. R Soc Open Sci 6:181665. https://doi.org/10.1098/rsos.181665
Branch JL, Epps R, Kosson DS (2018) The impact of carbonation on bulk and ITZ porosity in microconcrete materials with fly ash replacement. Cement Concr Res 103:170–178. https://doi.org/10.1016/j.cemconres.2017.10.012
Leemann A, Loser R, Münch B, Lura P (2017) Steady-state O2 and CO2 diffusion in carbonated mortars produced with blended cements. Mater Struct 50:247. https://doi.org/10.1617/s11527-017-1118-3
Parrott L (1996) Some effects of cement and curing upon carbonation and reinforcement corrosion in concrete. Mater Struct 29:164–173. https://doi.org/10.1007/BF02486162
Wierig H (1985) Longtime studies on the carbonation on concrete under normal outdoor exposure. RILEM Seminar on the Durability of Concrete Structures under normal outdoor exposure. Mater Struct 18:323. https://doi.org/10.1007/BF02472928
Younsy A, Turcry P, Ait-Mokhtar A, Staquet S (2013) Accelerated carbonation of concrete with high content of mineral additions: effect of interactions between hydration and drying. Cement Concr Res 43:25–33. https://doi.org/10.1016/j.cemconres.2012.10.008
Morandeau A, Thiery M, Dangla P (2015) Impact of accelerated carbonation on OPC cement paste blended with fly ash. Cement Concr Res 67:226–236. https://doi.org/10.1016/j.cemconres.2015.10.003
Saillio M, Bertin M, Baroghel-Bouny V (2019) Coupling effect and durability in cement pastes and concretes with supplementary cementitious materials. In: International conference on sustainable materials, system and structures (SMSS2019), pp 20–22 March 2019, Rovinj Croatia
Hussain S, Bhunia D, Singh S (2017) Comparative study of accelerated carbonation of plain cement andfly-ash concrete. J Build Eng 10:26–31. https://doi.org/10.1016/j.jobe.2017.02.001
Van den Heede P, De Belie N (2015) Durability Based Life Cycle Assessment of Concrete with Supplementary Cementitious Materials Exposed. In: International Conference on Sustainable Structural Concrete, La Plata Argentina, (1):13–24, LEMIT (Laboratorio de Entrenamiento Multidisciplinario para la Investigación Tecnológica)
Lu C, Wang W, Li Q, Hao M, Xu Y (2018) Effects of micro-environmental climate on the carbonation depth and the pH value in fly ash. J Clean Prod 181:309–317. https://doi.org/10.1016/j.jclepro.2018.01.155
Burdon D (2006) The durability of concrete containing high levels of fly ash. PCA R&D Serial No. 2989. Portland Cement Association, Skokie US
Sideris K, Savva A (2001) Resistance of fly ash and natural pozzolans blended cement mortars and concrete to carbonation, sulfate attack and chloride ion penetration.In: Seventh CANMET/ACI international conference on fly ash, silica fume, slag and natural pozzolans in concrete, Mandras India
Papadakis V (1999) Effect of fly ash on Portland cement systems: part I. Low-calcium fly ash. Cement Concr Res 29(11):1727–1736. https://doi.org/10.1016/S0008-8846(99)00153-2
Papadakis V (2000) Effect of fly ash on Portland cement systems. Part II: high calcium fly ash. Cement Concr Res 30(10):1647–1654. https://doi.org/10.1016/S0008-8846(00)00388-4
Thomas M, Matthews J (1992) Carbonation of fly ash concrete. Mag Concr Res 44(160):217–228. https://doi.org/10.1680/macr.1992.44.160.217
Wu B, Ye G (2019) X-ray diffraction study of carbonation rate of C–S-H with different Ca/Si ratio. International conference on sustainable materials, system and structures (SMSS2019), 20-22 March 2019, Rovinj Croatia
Newlands M (2001) Development of a simulated natural carbonation test of selected CEM II concretes. Dissertation, University Dundee
Thienel KC, Beuntner N (2012) Effects of calcined clay as low carbon cementing materials on the properties of concrete. Concrete in the Low Carbon Era, 9.-11. July, Dundee UK
Shao Y, Rostami V, He Z, Boyd A (2014) Accelerated carbonation of Portland limestone cement. J Mater Civil Eng 26(1):117–124. https://doi.org/10.1061/(ASCE)MT.1943-5533.0000773
Leemann A, Nygaard P, Kaufmann J, Loser R (2015) Relation between carbonation resistance, mix design and exposure of mortar and concrete. Cement Concr Compos 62:33–43. https://doi.org/10.1016/j.cemconcomp.2015.04.020
von Greve-Dierfeld S, Gehlen C (2016) Performance-based durability design, carbonation, part 2—classification of concrete. Struct Concr 17:523–532. https://doi.org/10.1002/suco.201600067
Ribeiro A, Santos T, Goncalves A (2018) Performance of concrete exposed to natural carbonation: use of the k-value concept. Constr Build Mater 175:360–370. https://doi.org/10.1016/j.conbuildmat.2018.04.206
Nobis C, Vollpracht A (2015) k-Value for Carbonation of Concretes with Supplementary Cementitious Materials. In: 27th Biennial National Conference of the Concrete Institute of Australia in Conjunction with the 69th RILEM Week—Concrete 2015, 30 Aug.—2 Sept. 2015, Melbourne Australia
Shi Z (2016) Durability of Portland Cement - Calcined Clay - Limestone Blends. Dissertation. Aarhus University Denmark
Jansson H, Snibb R, Bohlin K, Löfgren I (2017) Carbonation of concrete with mineral additions. XXIII Nordic Concrete Research Symposium, Aalborg Denmark
Müller C (2019) Konzepte zur Herstellung von ressourceneffizienten Betonen am Beispiel Zement (Concepts for the production of resource efficient concretes with the example cement). Beton 69(9):311–314
Imamoto K, Neves R, Torrent R (2016) Carbonation rate in old structures assessed with air-permeability site NDT. IABMAS 2016 Paper 426, Foz do Iguacu, Brazil, 26–30 June 2016
Russel D, Basheer PAM, Rankin IB, Long AE (2001) Effect of relative humidity and air permeability on prediction of the rate of carbonation of concrete. Struct Build 146(3):319–526
Jung SH, Lee MK, Oh BH (2011) Measurement device and characteristics of diffusion coefficient of carbon dioxide in concrete. ACI Mater J 108(6):589–595
Pham S, Prince W (2014) Effects of carbonation on the microstructure of cement materials: influence of measuring methods and types of cement. Int J Concr Struct Mater 8:327–333. https://doi.org/10.1007/s40069-014-0079-y
Cortvriendt M, Gruyaert E, Vanoutrive H (2019) Comparison between the carbonation of cement paste, mortar and concrete with supplementary cementitious materials. Master Thesis KU Leuven
Schwartzentruber A, Catherine C (2000) Method of the concrete equivalent mortar (CEM)—A new tool to design concrete containing admixture (in French). Mater Struct 33(232):475–482. https://doi.org/10.1007/BF02480524
Akcay B (2017) Aggregate restraining effect on autogenous shrinkage of cementitious materials. KSCE J Civil Eng 22(6):3102–3111. https://doi.org/10.1007/s12205-017-0635-5
Guo H, Shi C, Guan X, Zhu J, Ding Y, Ling TC, Zhang H, Wang Y (2018) Durability of recycled aggregate concrete—A review. Cement Concr Compos 89:251–259. https://doi.org/10.1016/j.cemconcomp.2018.03.008
Silva RV, Neves R, de Brito J, Dhir RK (2015) Carbonation behaviour of recycled aggregate concrete. Cement Concr Compos 62:22–32. https://doi.org/10.1016/j.cemconcomp.2015.04.017
Singh N, Singh SP (2016) Carbonation and electrical resistance of self-compacting concrete made with recycled concrete aggregates and metakaolin. Constr Build Mater 121:400–409. https://doi.org/10.1016/j.conbuildmat.2016.06.009
de Brito J, Ferreira J, Pacheco J, Soares D, Guerreiro M (2016) Structural, material, mechanical and durability properties and behaviour of recycled aggregates concrete. J Build Eng 6:1–16. https://doi.org/10.1016/j.jobe.2016.02.003
Kou SC, Poon CS (2012) Enhancing the durability properties of concrete prepared with coarse recycled aggregate. Constr Build Mater 35:69–76. https://doi.org/10.1016/j.conbuildmat.2012.02.032
Buyle-Bodin F, Hadjieva-Zaharieva R (2002) Influence of industrially produced recycled aggregates on flow properties of concrete. Mater Struct 35:504–509. https://doi.org/10.1007/BF02483138
Amorim P, de Brito J, Evangelista L (2012) Concrete made with coarse concrete aggregate: influence of curing on durability. ACI Mater J 109:195–204
Jiménez LF, Moreno EI (2015) Durability indicators in high absorption recycled aggregate concrete. Adv Mater Sci Eng ID505423:8. https://doi.org/10.1155/2015/505423
Limbachiya M, Meddah MS, Ouchagour Y (2012) Performance of portland/silica fume cement concrete produced with recycled concrete aggregate. ACI Mater J 109:91–100
Xiao J, Lei B, Zhang C (2012) On carbonation behavior of recycled aggregate concrete. Sci China Technol Sci 55:2609–2616. https://doi.org/10.1007/s11431-012-4798-5
Pedro D, de Brito J, Evangelista L (2015) Performance of concrete made with aggregates recycled from precasting industry waste: influence of the crushing process. Mater Struct 48:3965–3978. https://doi.org/10.1617/s11527-014-0456-7
Otsuki N, Miyazato S, Yodsudjai W (2003) Influence of recycled aggregate on interfacial transition zone, strength, chloride penetration and carbonation of concrete. J Mater Civil Eng 15(5):443–451. https://doi.org/10.1061/(ASCE)0899-1561(2003)15:5(443)
Zhang K, Xiao J (2018) Prediction model of carbonation depth for recycled aggregate concrete. Cement Concr Compos 88:86–99. https://doi.org/10.1016/j.cemconcomp.2018.01.013
Kisku N, Joshi H, Ansari M, Panda SK, Nayak S, Dutta SC (2017) A critical review and assessment for usage of recycled aggregate as sustainable construction material. Constr Build Mater 131:721–740. https://doi.org/10.1016/j.conbuildmat.2016.11.029
Xiao J, Lu D, Ying J (2013) Durability of recycled aggregate concrete: an overview. J Adv Concr Technol 11:347–359. https://doi.org/10.3151/jact.11.347
Levy SM, Helene P (2004) Durability of recycled aggregates concrete: a safe way to sustainable development. Cement Concr Res 34:1975–1980. https://doi.org/10.1016/j.cemconres.2004.02.009
Medina C, Frías M, Sánchez de Rojas MI, Thomas C, Polanco JA (2012) Gas permeability in concrete containing recycled ceramic sanitary ware aggregate. Constr Build Mater 37:597–605. https://doi.org/10.1016/j.conbuildmat.2012.08.023
Evangelista L, de Brito J (2010) Durability performance of concrete made with fine recycled concrete aggregates. Cement Concr Compos 32:9–14. https://doi.org/10.1016/j.cemconcomp.2009.09.005
Zega CJ, Di Maio ÁA (2011) Use of recycled fine aggregate in concretes with durable requirements. Waste Manag 31:2336–2340. https://doi.org/10.1016/j.wasman.2011.06.011
Geng J, Sun J (2013) Characteristics of the carbonation resistance of recycled fine aggregate concrete. Constr Build Mater 49:814–820. https://doi.org/10.1016/j.conbuildmat.2013.08.090
Vieira T, Alves A, de Brito J, Correia JR, Silva RV (2016) Durability-related performance of concrete containing fine recycled aggregates from crushed bricks and sanitary ware. Mater Des 90:767–776. https://doi.org/10.1016/j.matdes.2015.11.023
Sveindottir EL, Maage M, Poot S, Hansen EA, Bennenk HW, Helland S, Norden G, Kwint E, Milencovic A, Smeplass S, Tarquis F, Holand I, Price B, van Breugel K, Thienel KC (1998) LWAC Material Properties - State-of-the-Art. The European Union – Brite EuRam III, Gouda Netherlands
Hossain KMA, Ahmed S, Lachemi M (2011) Lightweight concrete incorporating pumice based blended cement and aggregate: mechanical and durability characteristics. Constr Build Mater 25:1186–1195. https://doi.org/10.1016/j.conbuildmat.2010.09.036
Vaysburd A (1992) Durability of lightweight concrete and its connections with the composition of concrete, design, and construction methods. ACI Spec Publ 136:295–318
Holm TA, Bremner TW, Vaysburd A (1988) Carbonation of marine structural lightweight concretes. ACI Spec Publ 109:667–676
Thienel KC, Schmidt-Döhl F, Feldrappe V (2000) In-Situ Tests on Existing LWAC Structures. In: Second International Conference on Structural Lightweight Aggregate Concrete, Kristiansand Norway
Swamy RN, Jiang ED (1992) Pore structure and carbonation of lightweight concrete after years exposure. Mater Sci. https://doi.org/10.14359/4012
Expanded Shale, Clay & Slate Institute (2006). Internal Curing. Using Expansed Shale, Clay and Slate Lightweight Aggregate. Publication nº 4362 (https://www.escsi.org/internal-curing/technical-docs/- Accesed date: 2019-06-03)
Sanjuan A, Andrade C, Cheyrezy M (2003) Concrete carbonation tests in natural and in accelerated conditions. Adv Cement Res 15(4):171–180. https://doi.org/10.1680/adcr.2003.15.4.171
Fattuhi NI (1988) Concrete carbonation as influenced by curing regime. Cement Concr Res 18(3):426–430. https://doi.org/10.1016/0008-8846(88)90076-2
Sanjuán MÁ, Estévez E, Argiz C, del Barrio D (2018) Effect of curing time on granulated blast-furnace slag cement mortars carbonation. Cement Concr Compos 90(2):257–265. https://doi.org/10.1016/j.cemconcomp.2018.04.006
Bier T (1986) Influence of type of cement and curing on carbonation progress and pore structure of hydrated cement pastes. Symp M Microstruct Dev During Hydr Cement 85:123. https://doi.org/10.1557/PROC-85-123
Ekolu SO (2016) A review on effects of curing, sheltering, and CO2 concentration upon natural carbonation of concrete. Constr Build Mater 127:306–320. https://doi.org/10.1016/j.conbuildmat.2016.09.056
Sideris KK, Zarras SC (2006) Influence of curing time on the durability of concrete against carbonation induced corrosion. In: 16th Hellenic Concrete Conference, October 25-27, Alexandroupolis Greece
Saillio M (2012) Interactions physico-chimiques ions-matrice dans les bétons sains ou carbonatés: influence sur le transport ionique. Dissertation (in French), University Paris-Est
Bucher R, Diederich P, Escadeilla G, Cyr M (2017) Service life of metakaolin-based concrete exposed to carbonation - Comparison with blended cement containing fly ash, blast furnace slag and limestone filler. Cement Concr Res 99:18–29. https://doi.org/10.1016/j.cemconres.2017.04.013
Atis CD (2003) Accelerated carbonation and testing of concrete made with fly ash. Constr Build Mater 17:147–152. https://doi.org/10.1016/S0950-0618(02)00116-2
Thomas M (2004) The effect of curing and concrete quality on the durability of concretes with high-volumes of supplementary cementing materials. University of New Brunswick Fredericton, Canada
Li G, Yao F, Li P, Yan C (2016) Long-term carbonation resistance of concrete under initial high-temperature curing. Mater Struct 49(7):2799–2806. https://doi.org/10.1617/s11527-015-0686-3
Schiessl P, Gehlen C et. al. (2006) fib Bulletin 34: Model Code for service life design. ISBN: 2-88394-074-6
Hainer S, Prosk T, Graubner C (2015) Effect of curing on the carbonation of concrete (Einfluss der Nachbehandlung auf das Karbonatisierungsverhalten von Beton aus klinkerarmen Zementen, in German). Beton- und Stahlbetonbau 110:1. https://doi.org/10.1002/best.201400042
Hunkeler F, Lammer A (2012) Requirements for the carbonation resistance of concrete (in German). AGB2008/012. Federal Roads Office FEDRO, Switzerland
Von Greve-Dierfeld S, Gehlen C (2016) Performance-based durability design, carbonation, part 1—benchmarking of deemed-to-satisfy rules. Struct Concr 17:309–328. https://doi.org/10.1002/suco.201600066
Verbeck G (1958) Carbonation of hydrated Portland cement. ASTM Spec Tech Publ 205:17–36. https://doi.org/10.1520/STP39460S
Thiery M (2005) Modélisation de la carbonatation atmosphérique des matériaux cimentaires (in French). Dissertation, Ecole Nationale des Ponts et Chaussées (ENPC) Paris
Galan I, Andrade C, Castellote M (2013) Natural and accelerated CO2 binding kinetics in cement paste at different relative humidities. Cement Concr Res 49:21–28. https://doi.org/10.1016/j.cemconres.2013.03.009
Essai pour béton durci—Essai de carbonatation accélérée—Mesure de l’épaisseur de béton carbonaté. XP P 18-458, French standard, AFNOR 2008
Testing hardened concrete—Part 12: Determination of the potential carbonation resistance of concrete: Accelerated carbonation method. EN 12390-12:2020
Diamond S (1971) A critical comparison of mercury porosimetry and capillary condensation pore size distributions of portland cement pastes. Cement Concr Res 1:531–545. https://doi.org/10.1016/0008-8846(71)90058-5
Baroghel-Bouny V (2007) Water vapour sorption experiments on hardened cementitious materials part I: essential tool for analysis of hygral behaviour and its relation to pore structure. Cement Concr Res 37:414–437. https://doi.org/10.1016/j.cemconres.2006.11.019
De Ceukelaire L, Van Nieuwenburg D (1993) Accelerated carbonation of a blast-furnace cement concrete. Cement Concr Res 23(2):442–452. https://doi.org/10.1016/0008-8846(93)90109-M
Saetta AV, Vitaliani RV (2004) Experimental investigation and numerical modeling of carbonation process in reinforced concrete structures: part I: theoretical formulation. Cement Concr Res 34:571–579. https://doi.org/10.1016/j.cemconres.2003.09.009
Glasser FP, Pedesen J, Goldthorpe K, Atkins M (2005) Solubility reactions of cement components with NaCl solutions: ca(OH)2 and CSH. Adv Cement Res 17(2):57–64. https://doi.org/10.1680/adcr.2005.17.2.57
Chaussadent T (1999) Etats des lieux et réflexions sur la carbonatation du béton armé. Laboratoire Central des Ponts et Chausees (LCPC). https://www.ifsttar.fr/fileadmin/user_upload/editions/lcpc/ERLPC/ERLPC-OA-LCPC-OA29.pdf
He R (2010) A study on carbonation for low calcium fly ash concrete under different temperature and relative humidity. EJGE 15(Q):1871–1877
Tai CY, Chen FB (1998) Polymorphism of CaCO3, precipitated in a constant-composition environment. AIChE J 44:1790–1798. https://doi.org/10.1002/aic.690440810
Gehlen C (2000) Probabilistische Lebensdauerbemessung von Stahlbetonbauwerken: Zuverlässigkeitsbetrachtungen zur wirksamen Vermeidung von Bewehrungskorrosion (in German). Dissertation, RWTH Aachen
Uomoto T, Takada Y (1993) Factors affecting concrete carbonation ratio. Doboku Gakkai Ronbunshu 1992 451:119–128. https://doi.org/10.2208/jscej.1992.451_119
Thiel C (2019) Einfluss von CO2-Druck auf das Porengefüge zementgebundener Werkstoffe während der Carbonatisierung. Dissertation, Technische Universität München
Van den Heede P, De Belie N (2019) Difference in carbonation behavior at 0.04%, 1% and 10% CO2 for high-volume fly ash mortar: effect on internal humidity, and resistivity. In: International conference on sustainable materials, systems and structures (SMSS2019), March 18–22, Rovinj, Croatia, vol. 3, pp 317-324
Thiery M, Villain G, Dangla P, Platret G (2007) Investigation of the carbonation front shape on cementitious materials: effects of the chemical kinetics. Cement Concr Res 37(7):1047–1058. https://doi.org/10.1016/j.cemconres.2007.04.002
Bernal SA, Provis JL, Brice DG, Kilcullen A, Duxson P, van Deventer JSJ (2012) Accelerated carbonation testing of alkali-activated binders significantly underestimate the real service life: the role of the pore solution. Cement Concr Res 42:1317–1326
Thiel C, Schön A, Gehlen C (2018) Einfluss der CO2-Permeation auf die Carbonatisierung zementgebundener Baustoffe. 20. In: Internationale Baustofftagung (ibausil), 12.-14. September 2018, Weimar Germany
Knöfel D, Eßer G (1993) Einfluss unterschiedlicher Kohlendioxidkonzentrationen auf Zementmörtel (in German). Werkstoffwissenschaften und Bausanierung, Wittmann F H, Bartz W J (eds), Tagungsbericht des 3. Int. Koll. pp 1408–1418
Da Silva FG, Helene P, Castro-Borges P, Liborio J (2009) Sources of variations when comparing concrete carbonation results. J Mater Civil Eng 21(7):333. https://doi.org/10.1061/(ASCE)0899-1561(2009)21:7(333)
Rozière E, Loukili A, Cussigh F (2009) A performance based approach for durability of concrete exposed to carbonation. Constr Build Mater 23(1):190–199. https://doi.org/10.1016/j.conbuildmat.2008.01.006
Durán-Herrera A, Mendoza-Rangel J, De-Los-Santos E, Vazquez P, Bentz D (2013) Accelerated and natural carbonation of concretes with internal curing and shrinkage/viscosity modifiers. Mater Struct 48:1207–1214. https://doi.org/10.1617/s11527-013-0226-y
Harrison TA, Jones MR, Newlands MD, Kandasami S, Khanna G (2012) Experience of using the prTS 12390-12 accelerated carbonation test to assess the relative performance of concrete. Mag Concr Res 64(8):737–747. https://doi.org/10.1680/macr.11.00162
Dunster AM (2000) Accelerated carbonation testing of concrete. Ihs Bre Press, Bracknell UK. ISBN 13 9781860814549
Neves R, Branco F, de Brito J (2013) Field assessment of the relationship between natural and accelerated concrete carbonation resistance. Cement Concr Compos 41:9–15. https://doi.org/10.1016/j.cemconcomp.2013.04.006
Anstice DJ, Page CL, Page MM (2005) The pore solution phase of carbonated cement pastes. Cement Concr Res 35:377–383. https://doi.org/10.1016/j.cemconres.2004.06.041
Ngala VT, Page CL (1997) Effects of carbonation on pore structure and diffusional properties of hydrated cement pastes. Cement Concr Res 27(7):995–1007. https://doi.org/10.1016/S0008-8846(97)00102-6
Boumaaza M, Turcry P, Huet B, Aït-Mokhtar A (2020) Influence of carbonation on the microstructure and the gas diffusivity of hardened cement pastes. Constr Build Mater 253:119227. https://doi.org/10.1016/j.conbuildmat.2020.119227
Justness H, Skocek J, Øsnor TA, Engelsen CJ, SkjØlsvold O (2020) Microstructural changes of hydrated cement blended with fly ash upon carbonation. Cement Concr Res 137:106192. https://doi.org/10.1016/j.cemconres.2020.106192
Berodier E, Scrivener K (2015) Evolution of pore structure in blended systems. Cement Concr Res 73:25–35. https://doi.org/10.1016/j.cemconres.2015.02.025
Kulakowski M, Pereira F, Dal Molin D (2009) Carbonation-induced reinforcement corrosion in silica fume concrete. Constr Build Mater 23(3):1189–1195. https://doi.org/10.1016/j.conbuildmat.2008.08.005
Moreira AC, Appoloni CR, Rocha WRD, Oliveira LF, Fernandes CP, Lopes RT (2010) Determination of the porosity and pore size distribution of SiC ceramic foams by nuclear methodologies. Adv Appl Ceram 109(7):416–420. https://doi.org/10.1179/174367510x12722693956158
Houst Y, Wittmann F (1994) Influence of porosity and water content on the diffusivity of CO2 and O2 through hydrated cement paste. Cement Concr Res 24(6):1165–1176. https://doi.org/10.1016/0008-8846(94)90040-X
Hyvert N (2009) Application de l’approche probabiliste à la durabilité des produits préfabriqués en béton (in French). Dissertation, University of Toulouse
Dutzer V, Dridi W, Poyet S, Le Bescop P, Bourbon X (2019) The link between gas diffusion and carbonation in hardened cement pastes. Cement Concr Res 123(9):105795. https://doi.org/10.1016/j.cemconres.2019.105795
Soja W, Maraghechi H, Georget F, Scrivener K (2018) Changes of microstructure and diffusivity in blended cement pastes to natural carbonation. In: 5th International Conference on Concrete Repair, Rehabilitation, and Retrofitting (ICCRRR 2018), Cape Town, South Africa
Boumaaza M, Huet B, Pham G, Ait-Mokhtar A, Gehlen C (2018) A new test method to determine the gaseous oxygen diffusion coefficient of cement pastes as a function of hydration duration, microstructure and relative humidity. Mater Struct 51:51. https://doi.org/10.1617/s11527-018-1178-z
Zeebe R (2011) On the molecular diffusion of dissolved CO2, HCO -3 and CO 2-3 and their dependence on isotopic mass. Geochim Cosmochim Acta 75:2483–2498. https://doi.org/10.1016/j.gca.2011.02.010
Olsson N, Nilsson LO, Ahs M, Baroghel-Bouney V (2018) Moisture transport and sorption in cement based materials containing slag or silica fume. Cement Concr Res 106:23–32. https://doi.org/10.1016/j.cemconres.2018.01.018
Secombe J, Vidal R, Galle C, Adenot F (2006) Experimental study of gas diffusion in cement paste. Cement Concr Res 37(4):579–588. https://doi.org/10.1016/j.cemconres.2006.12.003.cea-02360113
Zou D, Liu T, Du C, Teng J (2015) Influence of wind pressure on the carbonation of concrete. Materials 8:4652–4667. https://doi.org/10.3390/ma8084652
Villain G, Thiery M (2005) Impact of carbonation on microstructure and transport properties of concrete. In: 10th International conference on durability of building materials and components, pp 17–20 2005, Lyon France
Dinku A, Reinhardt HW (1997) Gas permeability coefficient of cover concrete as a performance control. Mater Struct 30:387–393. https://doi.org/10.1007/BF02498560
Amsler D, Bur M (2018) Einfluss der Kapillaren Wasseraufnahme auf die Karbonatisierung von Beton. CAS Arbeit der Fachhochschule Nordwestschweiz, Switzerland
Pham S, Prince W (2013) Effects of carbonation on the microstructure and macro physical properties of cement mortars. World Acad Sci Technol Int J Civil Environ Eng 7(6):434–437
Juhart J, Bregar R, David G, Krüger M (2019) Air permeability, water penetration and water absorption to specify durability of eco-efficient concrete. RILEM Tech Lett 4:67–73. https://doi.org/10.21809/rilemtechlett.2019.86
Claisse P, Elsayad H, Shaaban I (1999) Permeability and pore volume of carbonated concrete. Mater J 96(3):378–381
Tracz T, Zdeb T (2019) Effect of hydration and carbonation progress on the porosity and permeability of cement pastes. Materials. https://doi.org/10.3390/ma12010192
Claisse PA, Elsayad HI, Ganjian E (2009) Water vapour and liquid permeability measurements in cementitious samples. Adv Cement Res 21(2):83–89. https://doi.org/10.1680/adcr.8.00046
Dias WPS (2000) Reduction of concrete sorptivity with age through carbonation. Cement Concr Res 30(8):1255–1261. https://doi.org/10.1016/s0008-8846(00)00311-2
Zhang SP, Zong I (2014) Evaluation of relationship between water absorption and durability of concrete materials. Adv Mater Sci Eng. https://doi.org/10.1155/2014/650373
Zhang Z, Thierry M, Baroghel-Bouny V (2016) Investigation of moisture transport properties of cementitious materials. Cement Concr Res. https://doi.org/10.1016/j.cemconres.2016.08.013
Alderete N, Villagran Zaccardi Y, Snoeck D, Van Belleghem B, Van den Heede P, Van Tittelboom K, De Belie N (2019) Capillary imbibition in mortars with natural pozzolan, limestone powder and slag evaluated through neutron radiography, electrical conductivity, and gravimetric analysis. Cement Concr Res 118:57–68. https://doi.org/10.1016/j.cemconres.2019.02.011
Edvardsen C (1999) Water permeability and autogenous healing of cracks in concrete. ACI Mater J 96(4):448–454
Hall C (2019) Capillary imbibition in cement-based materials with time-dependent permeability. Cement Concr Res 124:105835. https://doi.org/10.1016/j.cemconres.2019.105835
Acknowledgements
The authors would like to thank all members of RILEM TC-281 CCC, for the valuable discussions regarding carbonation of concretes with SCMs, and their insight during the preparation and review of this paper.
Funding
Participation of S.A. Bernal was partially sponsored by EPSRC through grants EP/R001642/1 and EP/T008407/1, and by the National Science Foundation (NSF) through award 1903457. The participation of J.L. Provis was sponsored by EPSRC through grant EP/T008407/1. N. De Belie and P. Van den Heede acknowledge the support of the Research Foundation Flanders (FWO) for funding a Bilateral Scientific Cooperation project with China (NSFC) - G0F3619N and for funding the postdoctoral fellowship of Philip Van den Heede (project No. 3E013917). Kosmas Sideris acknowledge TITAN S.A. Cement Company GREECE and Nordia S.A. Greece are thanked for providing materials. Charlotte Thiel acknowledges the support of the German research institution DFG. Elke Gruyaert and Hanne Vanoutrive would like to thank KU Leuven for the internal funding. Natalia Alderete would like to thank the Research Foundation-Flanders (FWO-Vlaanderen) for the financial support (12ZG820N).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article has been prepared by work group 1 and 2 within RILEM TC 281. The recommendation has been reviewed and approved by all members of the TC.
TC Members:
Chair: Nele De Belie.
Deputy Chair: Susan Bernal.
Members: Nele De Belie, Elke Gruyaert, Philip Van den Heede, Natalia Alderete, Ueli Angst, Deepankar Kumar Ashish, Veronique Baroghel-Bouny, Muhammed Basheer, Leon Black, Patrick Dangla, Susan Bernal, Aires Camões, Özlem Cizer, Martin Cyr, Vilma Ducman, Ana Fernández Jiménez, Christoph Gehlen, Mette Geiker, Gregor Gluth, R. Doug Hooton, Bruno Huet, Ivan Ignjatović, Siham Kamali-Bernard, Sylvia Kessler, Olonade Kolawole A, Tung-Chai Ling, Barbara Lothenbach, Marija Nedeljkovic, César Medina Martínez, José Fernando Martirena Hernández, Jose Pacheco, Angel Palomo, Solmoi Park, Ravi Patel, John Provis, Francisca Puertas, Javier Sanchez, Karen Scrivener, Marijana Serdar, Kosmas Sideris, Ruben Snellings, Charlotte Thiel, Yury Villagrán Zaccardi, Anya Vollpracht, Stefanie von Greve-Dierfeld, Brant Walkley, Fazhou Wang, Ling Wang, Li Juan, Yan Yao, Guang Ye, Christian Thienel, Kei-ichi Imamoto, Fabrizio Moro, Jan Elsen, Hanne Vanoutrive, Xinyuan Ke, Andres Idiart, Luca Valentini, Maciej Zajac, Shiju Joseph, Antonis Kanellopoulos, Shishir Mundra, Quoc Tri Phung, Janez Perko, Bei Wu, Carmen Andrade, Isabel Fuencisla Saez del Bosque, Zhenguo Shi, Heejeong Kim, Katja Dombrowski-Daube, Cyrill Grengg, Anna Varzina, Bahman Ghiassi, Yogarajah Elakneswaran, Qing-feng Liu, Miren Etxeberria, Zhiyuan Liu, Zengfeng Zhao, Hans Beushausen, Cassandre Le Galliard, Ning Li, Daniel Geddes, Semion Zhutovsky, Isabel Martins.
The original online version of this article was revised: Dr. Isabel Martins was missing from the RILEM membership list that was published in the original article.
The original version of this article was revised due to a retrospective Open Access order.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
von Greve-Dierfeld, S., Lothenbach, B., Vollpracht, A. et al. Understanding the carbonation of concrete with supplementary cementitious materials: a critical review by RILEM TC 281-CCC. Mater Struct 53, 136 (2020). https://doi.org/10.1617/s11527-020-01558-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1617/s11527-020-01558-w