
Abstract
This study aimed at a better understanding of the wood-water interaction, in particular the role of the hydroxyl accessibility during the humidity-dependent change in moisture content. Thin sections (80 µm) of never-dried Norway spruce sapwood that contained early- and latewood were used for the experiments. Sorption isotherm measurements confirmed the humidity-dependent moisture content changes and the effect of the first drying of the wood sections. Changes in hydroxyl accessibility were then determined by deuteration of the sections using deuterium oxide, followed by their re-protonation in water (H2O) vapor at different relative humidity: 15, 55 or 95%. The deuteration and re-protonation of the wood sections were quantified by dry mass changes as well as by changes in the OH and OD stretching vibrations in the Fourier transform infrared spectra. The results showed that the deuterated sections could be almost completely re-protonated in H2O vapor, nearly irrespective of the applied relative humidity. Therefore, changes in hydroxyl accessibility were not the driving force for the humidity-dependent changes in moisture content. However, a slow re-protonation rate at low relative humidity had to be considered. Nonetheless, a small quantity of OD groups persisted the re-protonation in H2O vapor and liquid H2O, which was not related to the drying of the wood.
Graphic abstract

Similar content being viewed by others
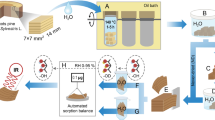
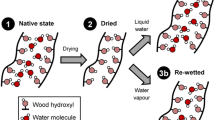
Introduction
Lignocelluloses, such as wood, interact with water vapor from the surroundings because they contain a large number of polar groups that attract water molecules, in particular hydroxyl (OH) groups (Berthold et al. 1996). Water accessible OH groups in lignocelluloses can be studied using hydrogen–deuterium exchange that occurs when deuterium oxide (D2O) forms hydrogen bonds with OH groups in wood or other cellulosic materials. The exchanged hydrogen can be quantified either gravimetrically by a dry mass increase of 1 g per mol exchanged hydrogen (Pönni et al. 2014; Uimonen et al. 2020), or spectroscopically by the shift of the OH stretching band to lower wavenumbers (Mann and Marrinan 1956; Hofstetter et al. 2006; Salmén and Stevanic 2018). This provides an estimate for the number of accessible OH groups in lignocelluloses, which has been applied to study the relationship between sorption sites and absorbed water (Gibbons 1953; Rouselle and Nelson 1971; Guthrie and Heinzelman 1974; Popescu et al. 2014; Altgen et al. 2018; Salmén and Stevanic 2018; Thybring et al. 2020). Correlations between the concentration of accessible OH groups and the amount of absorbed water have been found for cellulosic fibers (Gibbons 1953; Jeffries 1964; Rouselle and Nelson 1971; Guthrie and Heinzelman 1974) and for solid wood (Popescu et al. 2014; Altgen et al. 2018). Nonetheless, there is still uncertainty about the exact relationship between accessible OH groups and absorbed water. Several studies have shown that the amount of absorbed water can change independently from the OH accessibility (Stevens and Smith 1970; Rautkari et al. 2013; Salmén and Stevanic 2018). There is evidence that additional factors have a strong impact on the amount of absorbed water without affecting the sorption site density, such as the degree of cross-linking in heat-treated wood (Altgen et al. 2018; Willems et al. 2020) or the spatial availability of wood cell walls in modified wood (Thybring et al. 2020; Altgen et al. 2020). Furthermore, Lindh et al. (2016) showed that OH(3) groups in cellulose that are associated with the C(3) atom of the glucose units are unreactive to H–D exchange, but these OH groups may still form hydrogen bonds with water molecules.
It also remains unclear how changes in the amount of absorbed water across the hygroscopic range are related to changes in OH accessibility. At nearly saturated water vapor pressure (≥ 92% RH), it is estimated that 1–2 water molecules are attached to each sorption site on average (Berthold et al. 1996). However, this number decreases at lower relative humidity (RH) and water molecules may bind to two nearby OH groups simultaneously when the moisture content is low (Joly et al. 1996; Willems 2018). However, while moisture content changes within the hygroscopic range have been studied extensively, only Taniguchi et al. (1978) measured the OH accessibility of wood in dependence on the relative D2O vapor pressure applied during the H–D exchange. They suggested that new water sorption sites are formed in wood until reaching a RH of 60%, above which their number remains constant. This RH threshold is particularly interesting in view of the steep upward bend in the sorption isotherm of wood above ca. 60% RH that has been associated with the plasticizing effect of water on the glassy wood polymer matrix (Engelund et al. 2013; Hill and Beck 2017). Unfortunately, only few experimental details are given by Taniguchi et al. (1978) and one concern is the duration of the H–D exchange experiments. Different deuterium exchange rates of OH groups in cellulosic materials have been found depending on the structure (Frilette et al. 1948; Mann and Marrinan 1956; Jeffries 1964; Hishikawa et al. 1999). A fast deuteration occurs in regions where the β-glucan molecules are gathered relatively loose, but a tighter packing slows down the deuterium exchange reaction (Frilette et al. 1948; Hishikawa et al. 1999). OH groups in cellulose crystallites are accessible for H–D exchange only at their surface or at irregularities, but not at their core, as shown in a number of studies (Mann and Marrinan 1956; Hofstetter et al. 2006; Salmén and Bergström 2009; Lindh et al. 2016). Uimonen et al. (2020) showed that the complete deuteration of all accessible OH groups in a wood piece of ca. 20 mg requires about ten hours in D2O vapor at 95% target RH. However, when the RH is reduced, the amount of D2O that is provided to the wood is lower. Thereby, fewer D2O molecules are simultaneously adsorbed to the accessible OH groups to initiate H–D exchange. Furthermore, since the deuterium atoms are randomly exchanged between OH groups and bound water molecules, not all of the absorbed D2O molecules cause an H–D exchange and the wood needs to be supplied with a certain amount of D2O molecules until (nearly) full deuteration is reached. Lastly, the exchanged protium (1H) needs to be removed from the wood, which occurs faster if the concentration of D2O in the wood and the air flow is high. All of these factors should prolong the time required for the complete deuteration of accessible OH groups at low RH. For cellulose materials, a humidity-dependent reaction time has indeed been observed during tritium exchange of accessible OH groups in tritiated water (HTO) vapor by Sepall and Mason (1961). They showed that the reaction is slower at a RH of 20% compared to 50 and 75%, but that very similar hydroxyl accessibilities are reached when given sufficient time. The importance of sufficient reaction times has also been observed in re-protonation experiments on deuterated wood. Suchy et al. (2010) interpreted the presence of residual OD groups in deuterated and then dried wood after re-protonation in liquid H2O for 60 min as evidence for a loss in hydroxyl accessibility. However, Thybring et al. (2017) showed that a nearly complete re-protonation of deuterated and then dried wood can be achieved by prolonged soaking in H2O for 29–30 h, which disconfirmed an effect of drying on the accessibility of sorption sites.
In this study, the approach of Suchy et al. (2010) and Thybring et al. (2017) was adapted to study the re-protonation of deuterated and then dried Norway spruce wood in water vapor at 95, 55 or 15% RH. Thereby, a lower OH accessibility would be observed by the incomplete re-protonation of the deuterated wood, which was determined gravimetrically and by the OD stretching vibrations in the Fourier-transform infrared (FT-IR) spectrum. The aim of the study was to determine how changes in moisture content within the hygroscopic range are linked to the OH accessibility. In particular, it was evaluated if a decrease in RH causes a reduction in the OH accessibility of wood.
Materials and methods
Wood samples
Stem disks of never-dried Norway spruce (Picea abies (L.) Karst.) sapwood were collected from a tree in southern Finland. Within one hour after felling, the stem disks were sealed into plastic bags and stored at 7 °C the same day. Two blocks of ca. 6 × 10 × 20 mm3 (R × L × T) were cut from the sapwood and stored in H2O at 7 °C. Sapwood was differentiated from the heartwood by the large difference in moisture content. From the blocks, radial sections with a thickness of 80 µm were cut on a sledge microtome (Fig. 1). All sections contained equal proportions of early- and latewood and were separated into three groups: One group of sections was kept in H2O at 7 °C (“never-dried and soaked in H2O”). Another group was dried in an automated sorption apparatus (DVS intrinsic, Surface Measurement Systems, UK) at 0% target RH, 25 °C and a nitrogen flow (grade 5.0; ≤ 3 ppm H2O) of 200 sccm for five days, which was followed by re-soaking in D2O at room temperature with two exchanges of the D2O (“dried and re-soaked in D2O”). The last group of sections was transferred into D2O without any prior drying step and the D2O was exchanged three times prior to any measurements to remove all residual H2O before the samples were stored at room temperature (“never-dried and soaked in D2O”). The deuteration and re-protonation procedures for the different analytical methods are displayed in Fig. 2.

Sample preparation from never-dried Norway spruce (Picea abies (L.) Karst.) sapwood

Flow chart illustrating the drying and soaking procedures for the different analytical methods
Sorption isotherms
Four sections of never-dried Norway spruce (stored in H2O) were combined, shortly wiped with a wet cloth to remove excess water and placed on the sample pan of the automated sorption apparatus (DVS intrinsic, Surface Measurement Systems, UK). Temperature and nitrogen flow (grade 5.0, ≤ 3 ppm H2O) were kept constant at ca. 25 °C and 200 sccm, respectively. The desorption isotherm from the water-saturated state was determined by conditioning the sample to the following RH steps: 95% for 60 h, 80% for 24 h, 65% for 12 h, 50% for 12 h, 35% for 12 h; 20% for 12 h and 0% for 24 h. The durations at the different RH steps were adapted from Fredriksson and Thybring (2018). The absorption isotherm from the dry state and the scanning isotherm from the conditioned (95% RH) state were then determined using the same RH steps (Fig. 3a). After the measurements, the dry samples were removed from the automated sorption apparatus and soaked in H2O for ca. 72 h. The re-soaked samples were then wiped with a wet cloth and placed again in the automated sorption apparatus to measure the initial desorption isotherm from water-saturated state, the absorption isotherm and the scanning isotherm from conditioned state using the same RH sequence as before. The conditioned mass of each RH step was determined as the average mass over the final 10 min. The moisture content (in g g− 1) was determined by relating the water mass to the dry sample mass that was determined in the first 0% RH step. The mass change per minute (dm/dt, in µg g− 1 min− 1) was determined as the slope of a linear regression line over the final 60 min of each RH step.

Exemplary relative humidity sequences during a sorption isotherm measurements and b deuteration in D2O vapor (step 2) followed by re-protonation in H2O vapor (step 4)
Re-protonation after deuteration in D2O vapor
The measurements were performed on never-dried wood sections (stored in H2O). Four sections were combined, shortly pressed on a dry cloth to remove excess water and placed on the sample pan of the DVS apparatus (DVS ET, Surface Measurement Systems, UK). During all DVS runs, temperature and nitrogen flow (grade 6.0, ≤ 0.5 ppm H2O) were kept constant at ca. 25 °C and 200 sccm, respectively. Samples stored in H2O were exposed to the following RH sequence: (1) 0% RH for 24 h; (2) 95% target RH using D2O vapor for 12 h; (3) 0% RH for 24 h; (4) 15, 55 or 95% RH using H2O vapor for 24 h; and finally (5) 0% RH for 24 h (see example in Fig. 3b). The measured RH during step (4) was ca. 16.0, 59, or 95.8%, thus there were small deviations from the target RH. Additional measurements were performed by prolonging the exposure of the samples to 15% RH (H2O) during step (4) to 120 h (5 days). Exchanged OH groups after H–D exchange, remaining OD groups after re-protonation, absorbed D2O in step (2) and absorbed H2O in step (4) were calculated according to Eqs. 1–4:
where m1, m2, m3, m4 and m5 are the samples masses in steps (1), (2), (3), (4) and (5), respectively. The concentration of re-protonated OD groups (in mmol g− 1) was calculated by subtracting the amount of remaining OD groups from the amount of exchanged OH groups.
All measurements were taken in duplicate. After each measurement, three FT-IR spectra were collected from random positions of the sections as described below.
An additional sequence in the automated sorption apparatus, during which H2O vapor was used in step (2) instead of D2O vapor, resulted in very small changes in dry sample mass that were equivalent to a loss of 0.04 mmol g− 1 OD groups when applying Eq. 2 (Supplementary Fig. S.1). Furthermore, an additional deuteration and re-protonation sequence was applied during which the intermediate drying step (3) was omitted (Supplementary Fig. S.2).
Re-protonation after deuteration in liquid D2O
The measurements were taken on never-dried as well as on dried and re-soaked sections that were stored in D2O. Four sections were combined, shortly pressed on a dry cloth to remove excess water and placed on the sample pan of the automated sorption apparatus (DVS intrinsic, Surface Measurement Systems, UK). During the sequence, temperature and nitrogen flow (grade 5.0, ≤ 3 ppm H2O) were kept constant at ca. 25 °C and 200 sccm, respectively. The sections were first dried at 0% RH for 24 h and then exposed to a target RH (H2O) of either 15, 55 or 95% for one week. During the RH exposure, FT-IR spectra of the sections were taken at different time intervals. This required to remove the sections from the automated sorption apparatus for several minutes (< 10 min. in total) during which the sections were exposed to ambient laboratory conditions. Finally, the sections were soaked in liquid H2O for ca. 48 h with one water change after ca. 24 h and then dried in the automated sorption apparatus under dry nitrogen flow for ca. 24 h, before their FT-IR spectra were recorded again. In order to confirm the effect of the intermediate drying of the deuterated sections before the re-protonation, four deuterated sections per sample group were immediately washed in liquid H2O without prior drying and soaked for 48 h with five water changes. Their FT-IR spectra were recorded after drying under nitrogen flow as described above.
Infrared spectroscopy
FT-IR spectra were measured on a FT-IR spectrometer (SpectrumTwo, PerkinElmer, USA) equipped with an ATR unit and a diamond crystal. The time between removing the sections from the automated sorption apparatus and measuring the FT-IR spectra was limited as much as possible. The FT-IR spectra were collected within the wavenumber region 4000–600 cm− 1 using a resolution of 4 cm− 1 and 10 accumulation. The spectra were ATR and baseline corrected using the Spectrum 10 software (PerkinElmer, USA). They were normalized to the maximum absorbance of the CH/CH2 stretching band at ca. 2890 cm− 1 (± 4 cm− 1), which is not affected by absorbed water or deuteration (Hofstetter et al. 2006). The intensities of the OH and OD stretching bands were quantified by integration of the normalized spectra in the wavenumber regions 3020–3680 cm− 1 and 2330–2700 cm− 1, respectively, with the absorbance being set to zero at these thresholds. The amount of OD groups in a sample was estimated by the OD band area and by the relative OH accessibility (in %), which was calculated as the ratio of the OD band area to the sum of the OH and OD band areas.
Results and discussion
Sorption isotherms and hysteresis
The sorption isotherms measured in desorption from the water-saturated state, in absorption from the dry state and in desorption from the conditioned (95% RH) state (scanning desorption) are shown in Fig. 4a. The measurements were taken on never-dried Norway spruce sections and then repeated using the same sections after re-soaking in liquid H2O. The mass change per minute at the end of each RH step did not exceed 5 µg min− 1; hence, the sections were close to reaching equilibrium moisture content at every RH step (see Supplementary Table S.1).

Sorption isotherms (a) and hysteresis (b) measured by DVS. Closed symbols and solid lines show the sequence that started with never-dried spruce; Open symbols and dashed lines show the sequence that started after soaking the dried spruce samples in water
Although the measurements were limited to seven RH steps, all isotherms showed the typical sigmoidal shape with an upwards bend for relative humidities above 65% RH (Fig. 4a). In line with previous studies (Hoffmeyer et al. 2011; Fredriksson and Thybring 2018), the moisture content at 95% RH, obtained by desorption from the water-saturated state, exceeded the moisture content reached by absorption from the dry state. The desorption isotherm initiated from water-saturated state and the scanning isotherm that started from the conditioned state began to merge below 50% RH. The sorption behavior measured after re-soaking in H2O was very similar to the sorption behavior measured in never-dried state. Nonetheless, re-soaking the sections did not restore the equilibrium moisture content at 95% RH completely; the moisture content at 95% RH measured from the never-dried state was ca. 2%-points higher compared to the measurement after re-soaking. The absorption and scanning isotherms were almost identical for never-dried and re-soaked sections, except that the measurement after re-soaking resulted in a slightly lower moisture content at 95% RH in absorption.
From the sorption isotherms, absolute hysteresis was calculated from the desorption and the scanning isotherms (Fig. 4b). In line with Fredriksson and Thybring (2018), desorption isotherms resulted in a nearly linear hysteresis, while scanning isotherms gave non-linear hysteresis curves with a maximum at 80% RH. Desorption after re-soaking the sections also resulted in a nearly linear hysteresis curve, but the slope was lower when compared to the desorption from the never-dried state. A slightly lower hysteresis of the re-soaked sections was also noticed for the hysteresis based on the scanning isotherm. Therefore, soaking the sections in H2O for ca. 72 h could not restore the initial hygroscopicity in the never-dried state completely. This was in line with the observations by Penttilä et al. (2020), who found that drying and re-soaking wood samples in liquid water resulted in lower moisture contents at ca. 89% RH compared to never-dried samples for different soft- and hardwood species. Using small-angle neutron scattering, they showed that the initial drying of the wood samples caused changes of the cell wall structure on the microfibril level that were not fully recovered when the samples were re-soaked in water.
Re-protonation after deuteration in D2O vapor
Initiating the deuteration and re-protonation in a single measurement sequence using the automated sorption apparatus enabled the quantification of the number of OH groups that were deuterated in D2O vapor and the number of deuterated OH groups that remained after exposure to H2O vapor. This was based on the changes in dry mass compared to the initial dry mass. An additional measurement using an identical sequence, except that D2O vapor was replaced by H2O vapor, showed very little changes in dry mass between the different steps (equivalent to |0.04| mmol g− 1 OD groups or less, see Supplementary Fig. S.1). Therefore, it is reasonable to assume that dry mass changes during the actual measurements were only caused by deuteration and re-protonation of accessible OH groups.
The mass changes caused by deuteration and re-protonation are shown in Fig. 5a for an exemplary sequence with a re-protonation step at 15% RH for 24 h. The sample mass first decreased during the initial drying step and the mass at the end of this step was used as a reference mass (= 0 mmol g− 1 OD groups) for the calculation of the concentration of OD groups within the sample. The sample was then exposed to nearly saturated D2O vapor, which caused a steep mass increase due to the absorption of D2O and the hydrogen–deuterium exchange in accessible OH groups. The latter also resulted in an increased dry mass at the end of the subsequent drying step and this mass increase was used to quantify the concentration of accessible OH groups (= 9.8 mmol g− 1 in the example). In the next step, the dry and fully deuterated sample was exposed to H2O vapor, which caused another mass increase due to the absorption of H2O. This absorption also caused the re-protonation of OD groups that were accessible to H2O. At 15% RH, this re-protonation during the H2O vapor exposure could be observed by the slow decrease in mass after it had increased initially. The final dry mass of the sample was then used to determine the remaining OD groups after the re-protonation (= 1.7 mmol g− 1 in the example in Fig. 5a).

Gravimetric changes caused by deuteration in D2O vapor and re-protonation in H2O vapor: a example of mass changes (% of initial dry mass) caused by deuteration in D2O vapor and re-protonation in H2O vapor at 15% RH, b absorbed water at the end of the deuteration and the re-protonation steps, with each bar representing the average of two replicates and error bars show the data range, c concentration of remaining OD groups after re-protonation at different RH, d ratio of absorbed H2O per re-exchanged OD group in dependence on the target RH during the re-protonation
As to be expected, the RH had a large effect on the concentration absorbed water molecules during the exposure to water vapor. At the end of the H2O vapor exposure, the concentration of absorbed water molecules ranged between 2.3 (15% RH) and 13.4 (95% RH) mmol g− 1 (Fig. 5b). Furthermore, it was also noticed that the concentration of adsorbed D2O molecules in D2O vapor at a target RH of 95% was ca. 0.7 mmol g− 1 lower than the concentration of H2O molecules that were absorbed in H2O vapor at the same target RH. However, it should be noted that the exposure to D2O vapor was shorter than the exposure to H2O vapor, and that the RH control and measurement of the sorption apparatus was calibrated for H2O and not for D2O. This gave false readings for the RH of D2O vapor and potentially increased the deviation from the target RH.
The total concentration of accessible OH groups was quantified by the dry mass increase after the exposure of the sections to D2O vapor, which is shown in Fig. 5c as the remaining OD groups at a re-protonation RH of 0%. On average, an OH accessibility of ca. 9.6 mmol g− 1 was determined gravimetrically before the deuterated sections were re-protonated in H2O vapor (Fig. 5c). This value is slightly higher than the OH accessibilities of 8.4 and 9.3 mmol g− 1 reported by Thybring et al. (2017) for air-dried Norway spruce early- and latewood, respectively. However, it is slightly lower than the OH accessibility of 10.0 mmol g− 1 measured on kiln-dried Norway spruce by Lillqvist et al. (2019).
The exposure of the deuterated sections to H2O vapor resulted in the nearly complete re-protonation of the OD groups, because the final dry mass after exposure to H2O vapor was very close to the initial dry mass before the deuterium exchange. The concentration of remaining OD groups after the re-protonation was estimated by the mass difference between the initial and final drying step and the results are shown in Fig. 5c. When the re-protonation step was performed for 24 h, a higher concentration of remaining OD groups was recorded for a RH of 15%. However, when the re-protonation at 15% RH was prolonged to 120 h (five days), the effect of the RH during re-protonation on the remaining OD groups was very small and average concentrations of 0.51, 0.43 and 0.25 mmol OD groups per gram dry wood were recorded after re-protonation at 15, 55 and 95% RH, respectively. This showed that the exposure time had a larger effect on the re-protonation of OD groups than the RH level. It also showed that, when given enough time, the difference in OH accessibility between 15 and 95% is almost negligible and, therefore, not the cause for the difference in the amount of absorbed water molecules. Instead, the main difference between the RH levels was the average quantity of absorbed water molecules per accessible OH groups. When relating the amount of water molecules at the end of the re-protonation step to the amount of re-protonated OD groups, ratios between ca. 0.3 at 15% and ca. 1.5 at 95% RH were calculated (Fig. 5d).
The presence of remaining OD groups after re-protonation was verified by FT-IR spectroscopy using the areas of the OH and OD stretching bands (Fig. 6a). As expected, deuteration gave rise to an OD stretching band, while the OH stretching band decreased. Re-protonation in H2O vapor canceled the effect of deuteration nearly completely. Especially after the re-protonation at 95%, the OD stretching band could barely be detected in the FT-IR spectra. However, it needs be noted that the FT-IR spectroscopic measurements were taken ex-situ, since they required the removal of the sections from the automated sorption apparatus. The short exposure of deuterated wood to laboratory conditions caused uncertainties in the OH accessibility determination due to the rapid re-protonation, as described in detail by Tarmian et al. (2017). The relative OH accessibility of deuterated sections without re-protonation was ca. 31% (Fig. 6b), which was lower than the ratio of ca. 40% that was measured on fully deuterated wood (Thybring et al. 2017) and pulp (Salmén and Stevanic 2018), but similar to the ratio measured by Tarmian et al. (2017) on a freshly cut surface. Presumably, the short exposure of the fully deuterated sections to ambient laboratory conditions caused the re-protonation of some easily accessible OD groups. However, the re-protonation during the FT-IR measurements was less likely when the samples were already exposed to H2O vapor, since one would assume that most accessible OD groups were already re-protonated. Indeed, the results of the relative OH accessibility and of the OD band area for re-protonated sections were well in line with the gravimetrically determined concentrations of remaining OD groups. Even at 15% RH, the relative OH accessibility decreased to less than 3% when the exposure was prolonged to 120 h and only very small differences between the different RH levels were observed.

FT-IR spectroscopic changes after deuteration and re-protonation in water vapor. a Exemplary ATR and baseline corrected FT-IR spectra of dry wood sections in the wavenumber range 3650–2300 cm− 1. The areas of the OH and OD stretching bands are highlighted, b relative OH accessibility (%) and OD band area after re-protonation in H2O vapor and drying; Each column represents an average of six replicates and the error bars show the standard deviation
The nearly complete re-protonation of the deuterated wood sections disconfirmed a significant humidity dependence of the OH accessibility of wood that was suggested by Taniguchi et al. (1978). Instead, the present results were in line with the findings of Sepall and Mason (1961), who showed that the RH had only a small effect on the OH accessibility of cellophane and wood cellulose. When given sufficient time, even a low concentration of absorbed water molecules can re-protonate the majority of OD groups in fully deuterated wood. Since water sorption is a dynamic process that involves simultaneous ad- and desorption of water molecules, water molecules may “migrate” between different OH groups. Thereby, OH (or OD) groups may become accessible by hydrogen bond breaking at one position, while other OH groups become momentarily inaccessible by water desorption and hydrogen bond formation with adjacent OH groups. Presumably, the total concentration of accessible OH groups remains constant, while the number OH groups that simultaneously interact with water molecules decreases when reducing the RH. However, the re-protonation rate may not only depend on the number of simultaneously water-interacting OH groups. Re-protonation may not be initiated by the first H2O molecule that bonds to an OD group, because deuterium atoms are exchanged randomly between hydroxyls and water molecules. Furthermore, exchanged deuterium atoms need to be removed from the system to prevent re-deuteration. These two factors are not only affected by the number of OH groups that are simultaneously interacting with water molecules, but also by the amount of water molecules provided to the system over time.
Nonetheless, the gravimetric and spectroscopic results indicated a small number of remaining OD groups that persisted the re-protonation. One may assume that the intermediate drying step between deuteration and re-protonation resulted in the entrapment of some D2O or OD groups within the cell wall. Drying has been shown to trap solvents within the wood structure (Uimonen et al. 2020). However, an additional measurement sequence during which the intermediate drying step was omitted, so that the re-protonation in H2O vapor for 24 h at 95% RH directly followed after the deuteration in D2O vapor, resulted in nearly the same number of remaining OD groups (see Supplementary Fig. S.2). OD groups that resisted the re-protonation in H2O vapor were also observed by Hofstetter et al. (2006) as a small OD band in the FT-IR spectra of cellulose samples. The reason for the small but measureable amount of remaining OD groups is still unclear. Similar to the different deuteration rates of cellulosic materials (Frilette et al. 1948; Mann and Marrinan 1956; Jeffries 1964; Hishikawa et al. 1999), some of the OD groups within the tightly packed cell wall structures may be re-protonated at a very slow rate and require even longer exposure to H2O vapor. Furthermore, there is a possible isotope effect on the hydrogen- and deuterium-bonding due to the mass difference between hydrogen (1H) and deuterium (2H) atoms (Buckingham and Fan-Chen 1981). If deuterium bonds in wood were stronger than hydrogen bonds, absorption of water may preferentially result in the breaking of hydrogen bonds, which could delay or even prevent the complete re-protonation of OD groups.
Re-protonation after deuteration in liquid D2O
In another set of re-protonation experiments, never-dried sections were deuterated by soaking in liquid D2O and this was compared to sections that were first dried and then soaked in liquid D2O. Both set of samples were dried in the automated sorption apparatus followed by the re-protonation in H2O vapor for one week and finally in liquid H2O for 48 h.
As exemplarily shown in Fig. 7a and b for deuterated sections that were exposed to H2O vapor at 15% RH, a rapid decrease in the OD stretching band occurred already after the first day and this decrease continued at a lower rate during the following exposure. We also noticed a shift in the maximum of the OD stretching band towards lower wavenumbers. The time-dependent decrease in the OD stretching band during the H2O vapor exposure was quantified by the OD band area, because the increase in the OH stretching band by absorbed H2O during re-protonation did not allow the calculation of the relative OH accessibility (Fig. 7c). A time-dependent decrease was especially notable during the first four days of exposure to 15% RH, which further emphasized the need for adequate durations of re-protonation experiments. As explained above, the lower amount of water molecules that were supplied to the wood and the lower amount of simultaneously water-interacting sorption sites at 15% RH reduced the rate at which the re-protonation of the OD groups occurred compared to the exposure to 55 or 95% RH. By tendency, a very small decrease in the OD band area in the course of one week was also observed during the re-protonation at higher RH. However, a small but noticeable OD stretching band was still observed at the end of the one-week H2O vapor exposure. Similar findings were reported by Hofstetter et al. (2006) who found that re-protonation of deuterated cellulose in H2O vapor continued even when the diffusion of water into the structure reached an equilibrium. They also observed that some OD groups persisted in the cellulose structure even after long exchange times.

Spectroscopic changes during the re-protonation of wood sections that were deuterated in liquid D2O: changes in absorbance at 2700–2330 cm− 1 during re-protonation at 15% RH and soaking in H2O for sections that were deuterated in never-dried state (a) and after drying and re-soaking (b). c Band area changes during a 1-week exposure to H2O vapor; (d) OD band area after 1-week exposure to H2O vapor and after additional soaking in liquid H2O
In agreement with our previous re-protonation results, the differences in the OD band area between the different RH levels were very small after 1 week (Fig. 7d). Furthermore, larger OD band areas were recorded for sections that were deuterated in never-dried state compared to sections that were first dried and then deuterated. Soaking the sections in liquid H2O for 48 h after the re-protonation in H2O diminished the small RH-dependent differences in the OD band area. However, a small OD stretching band was still detected after soaking and the difference between never-dried and dried sections remained. One might initially assume that this was the result of the drying of the deuterated wood. The closed the cell wall pores in the dried wood may have not fully re-opened during re-soaking, particularly after the first drying of never-dried wood. This could have resulted in the inaccessibility of some OD groups (Suchy et al. 2010) or an entrapment of D2O within the cell wall structure (Uimonen et al. 2020). However, when the deuterated sections were not dried, but immediately washed in liquid H2O for ca. 48 h with five water changes and then dried under nitrogen flow, the same OD band areas were observed. Therefore, the remaining OD stretching vibrations after soaking in H2O were not an indication of a loss in OH accessibility or entrapped D2O in the cell wall structure caused by drying. Instead, there may have been a small quantity of OD groups in the deuterated wood that re-protonated at an extremely slow rate, as discussed above. Further studies are required to understand the persistence of these OD groups and its possible link to the cell wall structure.
Conclusions
Deuterated wood sections were nearly completely re-protonated in H2O vapor at 15–95% RH, which showed that RH-dependent changes in moisture content were not caused by changes in the number of accessible OH groups in wood. The RH only affected the rate of the re-protonation reactions. These results support the assumption that individual sorption sites in wood become momentarily inaccessible and re-accessible due to the dynamic ab- and desorption of water molecules in interaction with the surrounding water vapor. However, a very small amount of OD groups persisted the re-protonation in H2O vapor and liquid H2O within the period monitored in our experiments. Although the origin of these persistent OH groups remains unknown, it is clear that they are not caused by the drying of deuterated wood.
References
Altgen M, Willems W, Hosseinpourpia R, Rautkari L (2018) Hydroxyl accessibility and dimensional changes of Scots pine sapwood affected by alterations in the cell wall ultrastructure during heat-treatment. Polym Degrad Stab 152:244–252. https://doi.org/10.1016/j.polymdegradstab.2018.05.005
Altgen M, Altgen D, Klüppel A, Rautkari L (2020) Effect of curing conditions on the water vapor sorption behavior of melamine formaldehyde resin and resin-modified wood. J Mater Sci 55:11253–11266. https://doi.org/10.1007/s10853-020-04814-0
Berthold J, Rinaudo M, Salmeń L (1996) Association of water to polar groups; estimations by an adsorption model for ligno-cellulosic materials. Colloids Surf Physicochem Eng Asp 112:117–129. https://doi.org/10.1016/0927-7757(95)03419-6
Buckingham AD, Fan-Chen L (1981) Differences in the hydrogen and deuterium bonds. Int Rev Phys Chem 1:253–269. https://doi.org/10.1080/01442358109353322
Engelund ET, Thygesen LG, Svensson S, Hill CAS (2013) A critical discussion of the physics of wood–water interactions. Wood Sci Technol 47:141–161. https://doi.org/10.1007/s00226-012-0514-7
Fredriksson M, Thybring EE (2018) Scanning or desorption isotherms? Characterising sorption hysteresis of wood. Cellulose 25:4477–4485. https://doi.org/10.1007/s10570-018-1898-9
Frilette VJ, Hanle J, Mark H (1948) Rate of exchange of cellulose with heavy water. J Am Chem Soc 70:1107–1113. https://doi.org/10.1021/ja01183a071
Gibbons GC (1953) The moisture regain of methylcellulose and cellulose acetate. J Text Inst Trans 44:T201–T208
Guthrie JD, Heinzelman DC (1974) Deuterium–hydrogen-exchange accessibility of cellulose by use of D218O and mass-spectroscopy. Text Res J 44:981–985
Hill C, Beck G (2017) On the applicability of the Flory–Huggins and Vrentas models for describing the sorption isotherms of wood. Int Wood Prod J 8:50–55. https://doi.org/10.1080/20426445.2016.1275094
Hishikawa Y, Togawa E, Kataoka Y, Kondo T (1999) Characterization of amorphous domains in cellulosic materials using a FTIR deuteration monitoring analysis. Polymer 40:7117–7124. https://doi.org/10.1016/S0032-3861(99)00120-2
Hoffmeyer P, Engelund ET, Thygesen LG (2011) Equilibrium moisture content (EMC) in Norway spruce during the first and second desorptions. Holzforschung 65:875–882. https://doi.org/10.1515/hf.2011.112
Hofstetter K, Hinterstoisser B, Salmén L (2006) Moisture uptake in native cellulose—the roles of different hydrogen bonds: a dynamic FT-IR study using deuterium exchange. Cellulose 13:131–145. https://doi.org/10.1007/s10570-006-9055-2
Jeffries R (1964) The amorphous fraction of cellulose and its relation to moisture sorption. J Appl Polym Sci 8:1213–1220. https://doi.org/10.1002/app.1964.070080314
Joly C, Gauthier R, Escoubes M (1996) Partial masking of cellulosic fiber hydrophilicity for composite applications. Water sorption by chemical modified fibers. J Appl Polym Sci 61:57–69
Lillqvist K, Källbom S, Altgen M, Belt T, Rautkari L (2019) Water vapour sorption properties of thermally modified and pressurised hot-water-extracted wood powder. Holzforschung 73:1059–1068. https://doi.org/10.1515/hf-2018-0301
Lindh EL, Bergenstråhle-Wohlert M, Terenzi C, Salmén L, Furó I (2016) Non-exchanging hydroxyl groups on the surface of cellulose fibrils: the role of interaction with water. Carbohydr Res 434:136–142. https://doi.org/10.1016/j.carres.2016.09.006
Mann J, Marrinan HJ (1956) The reaction between cellulose and heavy water. Part 1. A qualitative study by infra-red spectroscopy. Trans Faraday Soc 52:481–487. https://doi.org/10.1039/TF9565200481
Penttilä PA, Altgen M, Carl N, van der Linden P, Morfin I, Österberg M, Schweins R, Rautkari L (2020) Moisture-related changes in the nanostructure of woods studied with X-ray and neutron scattering. Cellulose 27:71–87. https://doi.org/10.1007/s10570-019-02781-7
Pönni R, Rautkari L, Hill CAS, Vuorinen T (2014) Accessibility of hydroxyl groups in birch kraft pulps quantified by deuterium exchange in D2O vapour. Cellulose 21:2117–1226. https://doi.org/10.1007/s10570-014-0166-x
Popescu C-M, Hill CAS, Curling S, Ormondroyd G, Xie Y (2014) The water vapour sorption behaviour of acetylated birch wood: how acetylation affects the sorption isotherm and accessible hydroxyl content. J Mater Sci 49:2362–2371. https://doi.org/10.1007/s10853-013-7937-x
Rautkari L, Hill CAS, Curling S, Jalaludin Z, Ormondroyd G (2013) What is the role of the accessibility of wood hydroxyl groups in controlling moisture content? J Mater Sci 48:6352–6356. https://doi.org/10.1007/s10853-013-7434-2
Rouselle M-A, Nelson ML (1971) Accessibility of cotton cellulose by deuterium exchange. Text Res J 41:599–604. https://doi.org/10.1177/004051757104100708
Salmén L, Bergström E (2009) Cellulose structural arrangement in relation to spectral changes in tensile loading FTIR. Cellulose 16:975–982. https://doi.org/10.1007/s10570-009-9331-z
Salmén L, Stevanic JS (2018) Effect of drying conditions on cellulose microfibril aggregation and “hornification”. Cellulose 25:6333–6344. https://doi.org/10.1007/s10570-018-2039-1
Sepall O, Mason SG (1961) Hydrogen exchange between cellulose and water: I. Measurement of accessibility. Can J Chem 39:1934–1943. https://doi.org/10.1139/v61-260
Stevens CV, Smith BF (1970) Crosslinking cotton cellulose with ethyleneurea derivatives having varying hydrogen-bonding capabilities. II. Accessibility determinations. J Appl Polym Sci 14:1691–1700. https://doi.org/10.1002/app.1970.070140704
Suchy M, Virtanen J, Kontturi E, Vuorinen T (2010) Impact of drying on wood ultrastructure observed by deuterium exchange and photoacoustic FT-IR spectroscopy. Biomacromolecules 11:515–520. https://doi.org/10.1021/bm901268j
Taniguchi T, Harada H, Nakato K (1978) Determination of water adsorption sites in wood by a hydrogen–deuterium exchange. Nature 272:230–231. https://doi.org/10.1038/272230a0
Tarmian A, Burgert I, Thybring EE (2017) Hydroxyl accessibility in wood by deuterium exchange and ATR-FTIR spectroscopy: methodological uncertainties. Wood Sci Technol 51:845–853. https://doi.org/10.1007/s00226-017-0922-9
Thybring EE, Thygesen LG, Burgert I (2017) Hydroxyl accessibility in wood cell walls as affected by drying and re-wetting procedures. Cellulose 24:2375–2384. https://doi.org/10.1007/s10570-017-1278-x
Thybring EE, Piqueras S, Tarmian A, Burgert I (2020) Water accessibility to hydroxyls confined in solid wood cell walls. Cellulose 27:5617–5627. https://doi.org/10.1007/s10570-020-03182-x
Uimonen T, Hautamäki S, Altgen M, Kymäläinen M, Rautkari L (2020) Dynamic vapour sorption protocols for the quantification of accessible hydroxyl groups in wood. Holzforschung 74:412–419. https://doi.org/10.1515/hf-2019-0058
Willems W (2018) Hygroscopic wood moisture: single and dimerized water molecules at hydroxyl-pair sites? Wood Sci Technol 52:777–791. https://doi.org/10.1007/s00226-018-0998-x
Willems W, Altgen M, Rautkari L (2020) A molecular model for reversible and irreversible hygroscopicity changes by thermal wood modification. Holzforschung 74:420–425. https://doi.org/10.1515/hf-2019-0057
Acknowledgments
The financial support by the Academy of Finland (Grant No. 309881) is acknowledged. We thank Dr. Daniela Altgen for creating the illustrations, Dr. Paavo Penttilä for providing the wood material and Dr. Wim Willems for the interesting discussions that led up to this study.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Altgen, M., Rautkari, L. Humidity-dependence of the hydroxyl accessibility in Norway spruce wood. Cellulose 28, 45–58 (2021). https://doi.org/10.1007/s10570-020-03535-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10570-020-03535-6