
Abstract
The disruption of apoptotic cell death process is closely associated with the etiology of various diseases, including cancer. Permanent viral infections can cause different types of cancers. Oncogenic viruses manipulate both external and internal apoptosis pathways, and inhibit the activity of proapoptotic proteins and signaling pathways, which facilitates carcinogenesis. Ineffective immune surveillance or immune response suppression can induce uncontrolled virus propagation and host cell proliferation. In this review, we discuss current data that provide insights into mechanisms of apoptotic death suppression by viruses and their role in oncogenesis.
Similar content being viewed by others
INTRODUCTION
In multicellular organisms apoptosis plays an important role in the maintenance of cellular homeostasis, morphogenesis, and is also involved in various stages of development, immune system functioning, and the removal of damaged or infected cells [1]. The initiation of apoptosis is triggered by extracellular or intracellular factors, such as activation of death receptors, disturbance in the cell cycle signals, accumulation of misfolded proteins, DNA damage, metabolic disorders, and various bacterial and viral infections.
There are two main apoptosis signaling pathways: the extrinsic (receptor-dependent signaling pathway involving death receptors) and the intrinsic (mitochondrial) pathway. The extrinsic pathway is initiated by interactions between specific death ligands and death receptors [CD95, TRAIL-R1/2, and tumor necrosis factor receptor 1 (TNFR1)] on the cell surface. Ligands interacting with the receptors promote receptor oligomerization and conformational changes that facilitate binding to the adaptor proteins [Fas-associated protein with death domain (FADD); TNFR1-associated death domain protein (TRADD)] and to the inactive precursor of the cysteine protease-initiator caspases (pro-caspase-8, -10) [2]. Within the Death Inducing Signaling Complex (DISC) initiator caspases are activated and subsequently activate effector caspases (caspase-3, -6, -7) [3]. This caspase cascade leads to hydrolysis of various proteins, such as nuclear lamins, cytoskeletal proteins, protein kinases, and DNA repair proteins and inactivation of the apoptosis inhibitor proteins. As a result of these processes, morphological and biochemical changes occur that are incompatible with normal cellular functioning.
Intracellular signals, such as cellular DNA damage, oxidative stress, and disturbance of the cell cycle and other signaling pathways initiate apoptosis mainly via the intrinsic pathway. A key feature of the intrinsic pathway is permeabilization of the mitochondrial membrane, which is mediated by proapoptotic members of the family of proteins that regulate apoptosis (Bcl-2 family), such as Bax and Bak. Formation of a mitochondrial pore leads to the release of proapoptotic proteins [cytochrome c, apoptosis inducing factor (AIF), and endonuclease G (EndoG), etc.] from the mitochondria through the high permeability channels of the outer membrane to the cytoplasm. Cytochrome c binds with apoptotic protease activating factor 1 (Apaf-1) protein in the presence of 2′-deoxynucleoside-5′-triphosphate (dATP) which causes its oligomerization, and leads to the recruitment of procaspase-9. As a result, a protein complex known as apoptosome is formed in the cytoplasm. Activation of the initiator caspase-9 occurs within the complex, which then activates effector caspases leading to cell death [4].
Some viral infections are able to inhibit apoptotic cell death to facilitate viral replication and survival. Many oncogenic viruses modulate the oncosuppressive function of p53, activate antiapoptotic signaling pathways, and inhibit receptor-dependent and intrinsic apoptotic pathways, which shifts the balance between pro- and anti-apoptotic factors. Such changes in apoptotic pathways disturb the homeostatic balance of the cell, and facilitate the progression of infection and virus-induced pathologies (table). In subsequent sections of this work, various mechanisms of viral infection that suppress apoptosis and promote cellular transformation are described.
REGULATION OF THE ONCOSUPPRESSIVE PROTEIN p53
One of the central players within the apoptotic signaling pathways is the oncosuppressive protein p53. p53 is a transcription factor that regulates expression of ~500 target genes, and thereby controls a wide range of cellular processes including the cell cycle arrest, cell aging, DNA repair, metabolic adaptation, and cell death [18]. Many viral infections use different approaches to manipulate the function of p53 to successfully replicate and spread. For example, some viruses cause the p53-mediated apoptotic death of the cell host in order to spread; however, other viruses stimulate cell proliferation, which weakens the function of p53 as a regulator of the cell cycle, to promote oncogenic processes [19].
An example of a virus that promotes oncogenesis is hepatitis B. The study of this virus showed that the viral-encoded protein HBx (hepatitis B virus X protein) negatively affects induction of the p53-induced apoptosis [20]. Association between HBx and cytoplasmic p53 and p73 proteins was demonstrated in the human hepatocellular carcinoma cell lines HepG2 and Hep3B. This interaction prevented nuclear translocation and initiation of proapoptotic factors. In addition, HBx enhances expression of the N-terminal isoforms of p73, which is a trans-dominant inhibitor of p53 [21]. Also, HBx has been shown to enhance expression level of HURP (hepatoma upregulated protein), which promotes p53 degradation and suppression of apoptosis induced by chemotherapeutic DNA damaging agents [22]. Thus, reactivation of hepatitis B virus, which is often observed as a result of therapy for hematologic malignancies, breast cancer, and other malignant tumors, diminishes response to the treatment [23].
Along with studies of the hepatitis B virus, numerous investigations have assessed carcinogenicity of the hepatitis C virus. It was shown that increased expression levels of NS2, NS3/4A, and NS5A viral proteins affect p53-dependent apoptosis via induction of its translocation from the nucleus to the cytoplasmic/perinuclear region. In particular, the nonstructural protein NS5A directly interacts with p53 and suppresses p53-induced apoptosis [24]. Elevation of the NS5A protein levels is also associated with generation of reactive oxygen species (ROS) in hepatocytes, which causes oxidative stress and contributes to the development of hepatocellular carcinoma. Along with nonstructural components, viral capsid proteins, E1 and E2, promote severe oxidative stress, which accelerates tumor development. Interestingly, expression of the NS3-NS5B proteins is sufficient for creating two-membrane vesicles, which resemble autophagosomes. The NS4B protein induces lipidation of LC3 and the accumulation of autophagosomes [24]. Thus, hepatitis C could use the autophagy pathway to enhance viral replication and suppress antiviral interferon-dependent signaling by inhibiting apoptotic death.
DNA viruses, like adenovirus and SV40, use DNA replication mechanisms of host cells to enhance viral propagation. Viral T antigen, or adenovirus E1B proteins bind and inactivate p53, which allows the cell to avoid cell cycle arrest and promotes its transition to S-phase [25]. Moreover, viral T-antigens are capable of “DNA mimicry” [25] by imitating the charge and contour of the DNA duplex. This feature allows T-antigens to bind p53 and thereby disrupt transcriptional regulation of the p53 target genes that induce host cell transformation [26]. Connection between p53 and SV40 not only suppresses normal functioning of p53, but also stimulates “auxiliary” activity that enhances replication of SV40. The N-terminal domain of p53 recruits transcriptional regulators such as p300/CBP and MDM2, which alters expression levels of the cellular genes necessary for the spread of the virus [27].
Human papilloma virus (HPV) is one of the best-known oncogenic viruses associated with benign prostate hyperplasia and anogenital carcinomas. HPV uses special properties of E6 and E7 viral proteins to inhibit p53 activity. The E6 viral protein promotes 26S proteasome-dependent p53 degradation via association with the E3 ubiquitin ligase E6AP, which reduces levels of p53 in infected cells. Recently, it was shown that the E7 viral protein affects functioning of the DREAM complex (dimerization partner, RB-like, E2F, and multi-vulval class B), a transcriptional repressor complex. The p53-p21-DREAM pathway is a key regulator of the cell cycle checkpoint control via p53. By triggering the p53-p21-DREAM pathway, p53 suppresses a number of genes involved in regulation of the DREAM complex. However, the E7 oncoprotein can directly bind the DREAM complex, which enhances expression levels of most cell cycle genes, and inhibits p53 functioning [28]. Thus, the E6 and E7 HPV proteins modulate p53 level and its signaling pathway, alter host cell cycle progression, promote unregulated cell division, and suppress apoptosis (figure).

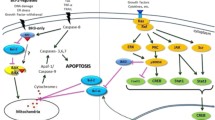
Inhibition of the extrinsic and intrinsic apoptotic pathway and modulation of the cell signaling by viral infection (see explanation in the text). Red arrows indicate viral suppression of the corresponding target, and blue arrows indicate activation of the selected pathway or proteins. Genes associated with the inhibition of apoptotic death are marked in red italics, genes involved in the initiation of apoptosis are indicated with blue italics. (Color version of the figure is available in online version of the article and can be accessed at: https://www.springer.com/journal/10541)
Non-oncogenic viruses, such as smallpox virus, which have large genomes, reduce p53 levels and destabilize the protein to facilitate viral replication [29]. It has been found, that the kinase B1R, which is encoded by the early viral genome, hyper-phosphorylates p53 at Ser-15 and Thr-18 residues to enhance p53 ubiquitination and degradation [30].
Human immunodeficiency virus type 1 (HIV-1), which reduces levels of CD4-lymphocytes and causes immune system dysfunction, also interacts with p53 protein during the infection process. Depending on the stage of infection, the virus either inhibits or activates p53. In the early stage of infection, HIV-1 proteins, such as Nef and LTR, directly interact with p53 through its N-terminal domain and inactivate the protein via inactivation of its transcriptional activity and, as a consequence, impede apoptosis [31]. In the later stages of infection, HIV-1 Tat proteins induce p53 activity, and stimulate T-cell apoptosis to promote HIV-1 spread [32].
Recent studies of the Epstein–Barr virus (EBV) within the herpesvirus family revealed that the viral oncogene EBNA1 affects the p53 pathway via viral mRNA. The viral EBNA1 protein inhibits translation its own mRNA, which promotes activation of the PI3Kδ kinase and stabilizes the p53-specific ubiquitin ligase MDM2, promoting p53 monoubiquitination and degradation via nuclear and cytoplasmic proteasomes [33]. Taken together, these data reveal that various types of oncogenic viruses, using their own proteins or RNA, modulate endogenous level of p53 or inhibit its transcriptional activity, which eventually suppresses apoptotic pathway and stimulates host cell proliferation.
INHIBITION OF THE RECEPTOR-DEPENDENT, APOPTOTIC DEATH PATHWAY
Many viruses have developed a variety of strategies for interfering with both internal cellular pathways and external pathways of apoptosis. Death receptor-mediated apoptosis plays an important role in pathogenesis of viral infection and antiviral response of the host. Many viruses have acquired the ability to suppress receptor-dependent apoptosis and evade the host immune response using viral-encoded anti-apoptotic factors (figure) [34].
Some viruses, such as poxviruses, encode death receptor homologs (T2 protein, CrmE). Free forms of the proteins neutralize tumor necrosis factor (TNF) ligand, which is required for induction of the external apoptotic pathway [35]. The hepatitis B protein HBc is a strong inhibitor of the TRAIL-induced apoptosis in human hepatoma cells. Resistance of the HBc-expressing cells to TRAIL-induced apoptosis is associated with significant decrease of the death receptor 5 (DR5 or TRAILR2) expression and is observed in the patients with chronic hepatitis [36]. EBV, in contrast, prevents TNF pathway activation by decreasing expression levels of the TNFR1 receptor [37]. It should be noted that adenoviruses and human papilloma virus suppress the expression of the death receptors Fas and TNFR1/2 [38, 39].
In addition to regulating cell surface receptors and death ligands concentrations, viruses influence the assembly of primary apoptotic complexes that function as a part of the extrinsic apoptotic pathway, including DISC and complex I. It has been shown that many viruses of the herpesvirus and poxvirus families encode a vFLIP protein that is homologous to the cell death regulator cFLIP. The vFLIP protein structure includes two death effector domains (DEDs), which allow the protein to interact with FADD adapter protein in the DISC, which prevents activation of the initiator caspase-8 and apoptotic pathway initiation [40]. The hepatitis C virus, in turn, regulates expression of the endogenous cFLIP protein and suppresses activation of caspase-8, thereby shifting the balance toward necroptotic cell death [41, 42].
The role of HPV in suppression of the TNF-mediated apoptosis should be emphasized. HPV encodes two oncoproteins, E6 and E7, which are directly responsible for the development of HPV-induced carcinogenesis. Studies have shown that the E6 oncoprotein interacts with the death domain of TNFR1 and blocks binding of the TNFR1 to the TRADD adapter protein suppressing initiation of apoptosis [43]. The E6 protein can also protect cells from TRAIL-induced apoptosis facilitating degradation of FADD protein and caspase-8 [44]. The E7 HPV oncoprotein, in turn, inhibits TNF-mediated apoptosis via modulation of the level of E3-ubiquitin ligase cellular inhibitor of apoptosis proteins (cIAP2), which is involved in degradation of caspases and DISC proteins [45].
It has been reported that viral proteins may directly interact with caspases and suppress their activity. Herpesvirus family members, herpes simplex virus and cytomegalovirus, encode caspase inhibitors, ribonucleotide reductase R1, and vICA, respectively. These proteins suppress CD95-mediated apoptosis by binding to the prodomain of procaspase-8 through their death effector domain, which prevents caspase activation [46, 47]. Poxviruses (vaccinia, rabbitpox, variola, etc.) also encode conservative inhibitors of serine proteases called serpins, which suppress activity of caspase-8 and -10 when the external pathway of apoptosis is triggered [48].
In this way, many oncogenic viruses have acquired the ability to inhibit the extrinsic apoptotic pathway and avoid the host immune response. This mainly occurs as a result of the directional suppression of the host death receptors and their ligands, as well by modulating activities of the key components of the receptor complexes.
INHIBITION OF THE MITOCHONDRIAL APOPTOSIS PATHWAY
Throughout the viral evolution, a mechanism for regulating apoptosis via modulation of the Bcl-2 family proteins was revealed. Today, more than 30 mammalian proteins that are members or are associated with the family have been described. These proteins have been classified into two main groups: pro- and antiapoptotic [49]. Antiapoptotic proteins of the Bcl-2 family include Bcl-2, Bcl-xL, Bcl-W, Mcl-1, and Bfl-1/A1 and are structurally homologous. These antiapoptotic proteins may interact directly with proapoptotic proteins Bim, Puma, Bad, Bid, Bik, Bmf, Hrk, Bax, Bak, and Noxa to suppress their activity. Activation of the apoptotic pathway shifts the balance toward proapoptotic proteins of the Bcl-2 family and leads to the assembly of the Bax-Bak multimeric pores on the outer mitochondrial membrane (OMM), its permeabilization and, as mentioned above, release of cytochrome c and other intramitochondrial factors to the cytosol [50].
Many large DNA viruses mimic Bcl-2 protein (vBcl-2), which prevents the accumulation and oligomerization of proapoptotic Bax and Bak proteins and blocks induction of the internal apoptotic pathway. The blockage of the premature host cell death in the early stages of viral infection is critical for successful infection [51] (figure). Adenovirus was one of the first viruses in which a homologue of the Bcl-2 protein was identified. The E1B 19K protein is homologous to the BH1 and BH2 domains of the Bcl-2 protein. It was shown that E1B 19K can interact with Bax, Bak, and Bik proteins, is functionally interchangeable with antiapoptotic Bcl-2 protein upon adenoviral infection, and acts as a potent inhibitor of apoptosis [52, 53].
Many members of the herpesvirus family also encode Bcl-2 homologues: BHRF1 and Ks-Bcl-2 are expressed in EBV and herpesvirus associated with Kaposi’s sarcoma, respectively [54, 55]. Studies have shown that BHRF1 and Ks-Bcl-2 proteins are able to bind such proapoptotic proteins as Bad, Bik, Bmf, Hrk, Noxa, Bax with high affinity, suppressing their activity and blocking apoptosis [56, 57]. A protein expressed during the cytomegalovirus infection, vMIA, while not displaying any homology to Bcl-2, is similar to Bcl-xL in its tertiary structure. vMIA binds proapoptotic proteins Bax and Bak preventing their oligomerization and blocking mitochondrial pore opening [58, 59].
It should also be noted that the herpes virus associated with Kaposi’s sarcoma encodes an additional anti-apoptotic protein, which is homologous to the cellular protein survivin, a member of a family of inhibitor of apoptosis proteins (IAPs). This protein is called K7 or viral inhibitor of apoptosis (vIAP) and it inhibits caspase activity and suppresses cell death [60]. vIAP was shown to bind the cellular Bcl-2 protein, as well as active caspase-3, inhibiting its proteolytic activity and suppressing proapoptotic cellular signaling [60].
The poxvirus family contains large DNA viruses that have apoptosis inhibitors within their structures. The F1L protein of the vaccinia virus has no sequence similarity with the Bcl-2 protein, but is able to mimic the unusual topology of the Bcl-2 tertiary structure. This allows F1L to bind proapoptotic proteins, such as Bim [61] and Bak [62], and prevent intrinsic apoptotic pathway induction. Notably, many members of the poxvirus family encode inhibitors of proapoptotic BH3-containing proteins that are highly adaptable to the structure of Bcl-2 and are able to modulate mitochondria-dependent apoptosis signaling.
Several poxvirus family strains encode another anti-apoptotic protein vGAAP (viral Golgi anti-apoptotic protein). vGAAP is not required for virus replication; however, it affects virulence. Interestingly, vGAAP demonstrates extremely high conservatism with the human hGAAP protein, which is localized in the Golgi complex, and forms a cation channel to regulate calcium ion (Ca2+) flux [63]. Increased levels of vGAAP or hGAAP expression leads to cellular resistance to apoptotic death, which is probably due to reduction in the release of Ca2+ from intracellular stores and decrease in penetration of Ca2+ ions into mitochondria [63]. However, the detailed mechanism by which GAAP controls apoptosis remains unknown.
Almost all apoptosis-inhibiting viral vBcl-2 proteins contain transmembrane anchor domains, which are required for their localization on the OMM. Various vBcl-2 proteins inhibit different stages of Bax activation and translocation to the OMM, which facilitates modulation of the internal apoptotic pathway activity [64].
Some viruses, such as hepatitis B and C viruses, affect apoptosis events that occur after the opening of mitochondrial pores. The hepatitis C virus, using non-structural proteins NS5A/B, suppresses caspase-3 activation, presumably by inhibiting caspase-9 [65]. The hepatitis B HBx protein interacts with AIF, an apoptotic factor, which affects emergence of the high molecular weight DNA fragments and chromatin condensation, and prevents apoptosis induction [66]. Notably, HBx expression affects localization and activity of the Drp1 and Parkin proteins, shifting cellular balance toward mitophagy [67].
Nevertheless, principal targets of the internal apoptotic pathway for many oncogenic viruses are members of the Bcl-2 family. Viral-encoded proteins show high degree of adaptability to the structure of Bcl-2 proteins and modulate signaling through various mechanisms.
THE PI3K-Akt SIGNALING PATHWAY AS A TARGET FOR VIRAL INFECTION
Phosphatidylinositol-3-kinase (PI3K) and protein kinase B (Akt) play an important role in regulation of the cell cycle and apoptosis [68]. The ability of Akt to prevent apoptotic death in the cell lines is accomplished by phosphorylation and inhibition of proapoptotic mediators such as Bad, Bax and caspase-9 [69, 70]. Additionally, Akt activates the transcription factor cAMP response element-binding protein (CREB), or IkB-kinase (IKK), which is a positive regulator of nuclear transcription factor kappa-B (NF-κB), to alter expression levels of genes with antiapoptotic activity [71]. Therefore, the PI3K-Akt signaling pathway is highly active in many tumor tissues [72].
Some oncogenic viruses such as HPV, herpesvirus family members [EBV; herpesvirus associated with Kaposi’s sarcoma (KSHV)], and human T-lymphotropic retrovirus (HTLV-1) have developed mechanisms for activating PI3K-Akt signaling pathway to inhibit apoptosis or autophagy, which can hinder viral replication [73]. The most widely studied virus is HPV, wherein each of the viral oncoproteins E5, E6, and E7 either directly or indirectly target the PI3K-Akt pathway and promote cell survival and proliferation as well as progression of the malignant formations [74].
LMP2A, an EBV herpesvirus membrane protein, also induces phosphorylation of Akt and activates PI3K-Akt to prevent the removal of infected cells. LMP2A provides a selective advantage to LMP2A-expressing cells during the development of EBV-associated malignant neoplasms [75]. LMP2A-mediated activation of the PI3K-AKT pathway also inhibits differentiation of the EBV-infected epithelial cells, thereby facilitating progression of the EBV-associated carcinomas and lymphomas [76]. Kaposi virus, a member of the herpesvirus family, encodes a viral G-protein-coupled receptor (vGPCR), which promotes Akt phosphorylation and induces sarcomogenesis in the allograft mouse model [77]. Moreover, increased activation of Akt was also observed in the biopsies of human Kaposi’s sarcoma taken from the patients with immunodeficiency virus [77]. Expression of the Kaposi virus K1 protein in B-lymphocytes, resulted in the PI3P-Akt pathway activation, inhibition of PTEN phosphatase and members of the Forkhead transcription factor family (FKHR), which are the key regulators of cell cycle and apoptosis. The viral protein K1 expression promotes cell survival and viral pathogenesis, and prevents premature apoptosis in the virus-infected cells (figure) [78].
HTLV-1, a retrovirus, in turn, modulates Akt in CD4+ T cells, which produces a long latency phase [79]. It was found that the HTLV-1 oncoprotein Tax activated the Akt pathway and induced the Akt-dependent inactivation of the FOXO3 (Forkhead box O3) transcription factor. This promoted removal of CD4+ T cells via induction of the proapoptotic and antiproliferative target genes [79]. Thus, FOXO3 inhibition promotes CD4+ T cells survival and proliferation, which retain the ability to spread infectious HTLV-1 particles [79].
THE NF-κB SIGNALING PATHWAY AS AN ANTAGONIST OF APOPTOTIC DEATH
Activation of the PI3K-Akt pathway can trigger the NF-κB signaling pathway, which is often activated in many types of cancer cells promoting tumorigenesis [80]. There are several mechanisms by which NF-κB antagonizes cell death. First, the activation of NF-κB increases levels of antiapoptotic genes such as cIAP1/2, Bcl-2, Bcl-xL, TRAF1/2, survivin, and p21. The pathway also induces the expression of some pro-oncogenic genes and a number of pro-inflammatory and lymphatic cytokines [81]. Moreover, NF-κB signaling promotes tumor progression, facilitates transition of epithelial cells to mesenchymal and metastasis, and contributes to vascularization of the tumors [82].
NF-κB is also activated in response to acute viral infection. However, some viruses use the constitutive activation of NF-κB to promote viral spread. For example, the EBV transmembrane protein LMP1 stimulates development of lymphoma by activating the NF-κB pathway [83]. LMP1 is able to interact with TRAF, a TNFR-family cellular receptors, to transmit intracellular signals. Thus, LMP1-induced activation of NF-κB promotes proliferation and survival of the infected cells [84].
NF-κB is also constitutively activated in most primary effusion lymphoma (PEL) cells, a disease induced by the Kaposi virus [85]. In these cells, the viral protein vFLIP activates NF-κB by directly binding with the regulatory subunit of the IKK-NEMO complex (also known as IKK-gamma). This results in activation of this complex and release of the DNA-binding transcription factors [86]. In transgenic mice expressing the viral vFLIP protein, NF-κB pathway activation enhanced lymphocyte proliferation and increased lymphoma incidence [87].
The HTLV-1 retrovirus activates the NF-κB pathway in the similar manner. The viral oncoprotein Tax modulates cellular signaling pathways to enhance T-cell proliferation and survival. Recent studies have shown that the Tax protein affects activation of the ubiquitin-dependent kinases, and undergoes K63-dependent polyubiquitination. This modification is crucial for the Tax interaction with NEMO and NF-κB activation (figure) [88].
It should be noted that some oncogenic viruses, such as hepatitis B and C viruses, and EBV, increase the level of cellular ROS due to mitochondrial dysfunction and response to protein misfolding. Such viral-induced oxidative stress triggers not only metabolic changes, but also activation of NF-κB, which promotes oncogenesis in liver tissues and blood [89]. Non-oncogenic viruses, such as hepatitis delta virus, which co-infects host cells together with the hepatitis B virus, enhance ROS production and activate NF-κB and STAT3 pathways. This accelerates development of the liver pathologies and onset of hepatocellular carcinoma [90].
NF-κB-mediated inflammation plays an important role in the proper functioning of the innate immune response to acute infection. Nevertheless, the effects of viral proteins on the key targets of this pathway facilitate induction of the cell transformation. NF-κB pathway activation not only promotes tumor cells proliferation and suppresses apoptosis, but also induces the epithelial to mesenchymal transition and metastasis [91]. Thus, suppression of the NF-κB pathway in infected cells may be a promising therapeutic strategy.
CONCLUSION AND PERSPECTIVES
When normal mechanisms for controlling cell growth and death are disrupted, some cells exhibit uncontrolled proliferation and cease to perform their tissue-specific functions, which leads to the development of cancer. It is assumed that infection with oncogenic viruses causes ~15% of all human cancers [92]. To date, the most well-known cancer-causing viruses are EBV, hepatitis B and C, human T lymphotropic virus 1 (HTLV-1), human papilloma virus, herpesvirus associated with Kaposi’s sarcoma, and Merkel cell polyomavirus (MCPyV) (table).
Another type of viruses that should also be noted is the most studied and relevant virus encountered in recent years – severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) of the coronavirus family. This virus belongs to the family of non-retroviral RNA viruses and has a single-stranded RNA genome. Many studies have demonstrated the ability of SARS-CoV-2 to trigger both the external and internal apoptotic pathways of the host cell death which facilitates the spread of the virus and enhances its pathogenicity [93, 94]. However, some authors have suggested that SARS-CoV-2 may participate in the persistent infection of the host, which may cause pulmonary fibrosis over time. For example, after a few years, viral infection could result in the growth of neoplasms [95]. One mechanism that has been suggested by the authors on the basis of protein homology with SARS-CoV-1, is destruction of the p53 tumor suppressor protein. Other mechanisms that have potential to promote carcinogenesis are cytokine storm and oxidative stress. The latter acts both as initiator and promoter of carcinogenesis due to the direct mutagenic effect of ROS and the ability of ROS to promote cell proliferation and invasion. However, these hypotheses require experimental verification.
Human oncogenic viruses have diverse genomes, cell tropisms, and oncological pathologies; however, they have many common properties that lead to oncogenesis. Most oncogenic viruses are transmitted between people and cause chronic infections that last for years without obvious symptoms. During this time, oncogenic viruses adapt to the host cell by altering cellular processes and disrupting immune recognition. Viral oncoproteins are able to manipulate both extrinsic and intrinsic apoptotic pathways, which affects the expression of death receptors, the assembly of apoptotic complexes, and suppress activities of caspases and proapoptotic proteins. However, suppression of the immune system and inadequate immune surveillance can induce uncontrolled viral replication and activate expression of the viral proteins that disrupt regulation of the host cell proliferation and stimulate tumorigenesis [96]. Although malignant transformation is a unifying pathological feature of oncogenic viruses, it is not evolutionarily beneficial for the virus nor is it necessary for viral spread.
A logical approach for the prevention or treatment of the cancer of viral etiology is targeted suppression of the virus. This notion was confirmed by the advances in clinical practice, which reduced dramatically the number of virus-associated tumor diseases [97]. The emergence of antiviral therapy for hepatitis C virus significantly improved effectiveness of the treatment of the vast majority of patients. This approach is proved to be an effective way to prevent hepatocellular carcinoma [98]. To date, the use of vaccines against HPV and the hepatitis B virus in the developed and some developing countries helped to reduce significantly the incidence of cervical cancer, liver cancer, and other viral associated diseases. Also, the vaccine trials and immunotherapy against EBV are currently underway [99]. Prevention or treatment of EBV can reduce the incidence of lymphoproliferative diseases and certain lymphomas and carcinomas of the nasopharynx [100].
Thus, the development of antiviral drugs and immunotherapy targeting tumor antigens are the priority goals to be achieved to combat cancer.
Abbreviations
- IF:
-
apoptosis inducing factor
- Akt:
-
protein kinase B
- CD95/Fas/Apo-1:
-
death receptor
- cIAP:
-
cellular inhibitor of apoptosis proteins
- DISC:
-
death inducing signaling complex
- EBV:
-
Epstein–Barr virus
- FADD:
-
Fas-associated protein with death domain
- HBx:
-
hepatitis B virus protein
- HIV-1:
-
Human Immunodeficiency Virus
- HPV:
-
human papillomavirus
- HTLV-1:
-
retrovirus of human T-cell leukemia type 1
- KSHV:
-
herpesvirus associated with Kaposi’s sarcoma
- NF-κB:
-
nuclear transcription factor kappa-B
- PI3K:
-
phosphatidylinositol 3-kinase
- SARS-CoV:
-
positive-sense single-stranded RNA virus of the coronavirus family
- TNF:
-
tumor necrosis factor
- TNFR1:
-
tumor necrosis factor receptor 1
- TRADD:
-
TNFR1-associated death domain protein
- ROS:
-
reactive oxygen species
References
Jin, Z., and El-Deiry, W. S. (2005) Overview of cell death signaling pathways, Cancer Biol. Ther., 4, 139-163, doi: https://doi.org/10.4161/cbt.4.2.1508.
Schleich, K., Warnken, U., Fricker, N., Öztürk, S., Richter, P., et al. (2012) Stoichiometry of the CD95 death-inducing signaling complex: experimental and modeling evidence for a death effector domain chain model, Mol. Cell, 47, 306-319, doi: https://doi.org/10.1016/j.molcel.2012.05.006.
Ashkenazi, A., and Dixit, V. M. (1998) Death receptors: signaling and modulation, Science, 281, 1305-1308, doi: https://doi.org/10.1126/science.281.5381.1305.
Zamaraev, A. V., Kopeina, G. S., Zhivotovsky, B., and Lavrik, I. N. (2014) Cell death controlling complexes and their potential therapeutic role, Cell. Mol. Life Sci., 72, 505-517, doi: https://doi.org/10.1007/s00018-014-1757-2.
Raab-Traub, N. (2012) Novel mechanisms of EBV-induced oncogenesis, Curr. Opin. Virol., 2, 453-458, doi: https://doi.org/10.1016/j.coviro.2012.07.001.
Young, L. S., and Rickinson, A. B. (2004) Epstein–Barr virus: 40 years on, Nat. Rev. Cancer, 4, 757-768, doi: https://doi.org/10.1038/nrc1452.
Levrero, M., and Zucman-Rossi, J. (2016) Mechanisms of HBV-induced hepatocellular carcinoma, J. Hepatol., 64, 84-101, doi: https://doi.org/10.1016/j.jhep.2016.02.021.
Gessain, A., and Cassar, O. (2012) Epidemiological aspects and world distribution of HTLV-1 infection, Front. Microbiol., 3, doi: https://doi.org/10.3389/fmicb.2012.00388.
Matsuoka, M., and Jeang, K. T. (2007) Human T-cell leukaemia virus type 1 (HTLV-1) infectivity and cellular transformation, Nat. Rev. Cancer, 7, 270-280, doi: https://doi.org/10.1038/nrc2111.
Schiffman, M., Clifford, G., and Buonaguro, F. M. (2009) Classification of weakly carcinogenic human papillomavirus types: addressing the limits of epidemiology at the borderline, Infect. Agent. Cancer, 4, doi: https://doi.org/10.1186/1750-9378-4-8.
Harper, D. M., and DeMars, L. R. (2017) HPV vaccines – a review of the first decade, Gynecol. Oncol., 146, 196-204, doi: https://doi.org/10.1016/j.ygyno.2017.04.004.
Mitchell, J. K., Lemon, S. M., and McGivern, D. R. (2015) How do persistent infections with hepatitis C virus cause liver cancer? Curr. Opin. Virol., 14, 101-108, doi: https://doi.org/10.1016/j.coviro.2015.09.003.
Goossens, N., and Hoshida, Y. (2015) Hepatitis C virus-induced hepatocellular carcinoma, Clin. Mol. Hepatol., 21, 105-114, doi: https://doi.org/10.3350/cmh.2015.21.2.105.
Schulz, T. F., and Cesarman, E. (2015) Kaposi Sarcoma-associated Herpesvirus: mechanisms of oncogenesis, Curr. Opin. Virol., 14, 116-128, doi: https://doi.org/10.1016/j.coviro.2015.08.016.
Wendzicki, J. A., Moore, P. S., and Chang, Y. (2015) Large T and small T antigens of Merkel cell polyomavirus, Curr. Opin. Virol., 11, 38-43, doi: https://doi.org/10.1016/j.coviro.2015.01.009.
Liu, W., MacDonald, M., and You, J. (2016) Merkel cell polyomavirus infection and Merkel cell carcinoma, Curr. Opin. Virol., 20, 20-27, doi: https://doi.org/10.1016/j.coviro.2016.07.011.
Krump, N. A., and You, J. (2018) Molecular mechanisms of viral oncogenesis in humans, Nat. Rev. Microbiol., 16, 684-698, doi: https://doi.org/10.1038/s41579-018-0064-6.
Vousden, K. H., and Lane, D. P. (2007) p53 in health and disease, Nat. Rev. Mol. Cell Biol., 8, 275-283, doi: https://doi.org/10.1038/nrm2147.
Kaminskyy, V., and Zhivotovsky, B. (2010) To kill or be killed: how viruses interact with the cell death machinery: symposium, J. Int. Med., 267, 473-482, doi: https://doi.org/10.1111/j.1365-2796.2010.02222.x.
Wang, X. W., Gibson, M. K., Yeh, H., Forrester, K., Harris, C. C., et al. (1995) Abrogation of p53-induced apoptosis by the Hepatitis B virus X gene, Cancer Res., 55, 6012-6016, doi: https://doi.org/10.1385/1-59259-079-9:57.
Knoll, S., Fürst, K., Thomas, S., Baselga, S. V., Stoll, A., Schaefer, S., and Pützer, B. M. (2011) Dissection of cell context-dependent interactions between HBx and p53 family members in regulation of apoptosis: a role for HBV-induced HCC, Cell Cycle, 10, 3554-3565, doi: https://doi.org/10.4161/cc.10.20.17856.
Chao, C. C. K. (2016) Inhibition of apoptosis by oncogenic hepatitis B virus X protein: Implications for the treatment of hepatocellular carcinoma, World J. Hepatol., 8, 1061-1066, doi: https://doi.org/10.4254/wjh.v8.i25.1061.
Voican, C. S., Mir, O., Loulergue, P., Dhooge, M., Brezault, C., et al. (2016) Hepatitis B virus reactivation in patients with solid tumors receiving systemic anticancer treatment, Ann. Oncol., 27, 2172-2184, doi: https://doi.org/10.1093/annonc/mdw414.
Vescovo, T., Refolo, G., Vitagliano, G., Fimia, G. M., and Piacentini, M. (2016) Molecular mechanisms of hepatitis C virus-induced hepatocellular carcinoma, Clin. Microbiol. Infect., 22, 853-861, doi: https://doi.org/10.1016/j.cmi.2016.07.019.
Levine, A. J., and Oren, M. (2009) The first 30 years of p53: Growing ever more complex, Nat. Rev. Cancer, 9, 749-758, doi: https://doi.org/10.1038/nrc2723.
Liu, X., and Marmorstein, R. (2006) When viral oncoprotein meets tumor suppressor: a structural view, Genes Dev., 20, 2332-2337, doi: https://doi.org/10.1101/gad.1471706.
Hermannstadter, A., Ziegler, C., Kuhl, M., Deppert, W., and Tolstonog, G. V. (2009) Wild-type p53 enhances efficiency of Simian virus 40 large-T-antigen-induced cellular transformation, J. Virol., 83, 10106-10118, doi: https://doi.org/10.1128/jvi.00174-09.
Engeland, K. (2018) Cell cycle arrest through indirect transcriptional repression by p53: I have a DREAM, Cell Death Differ., 25, 114-132, doi: https://doi.org/10.1038/cdd.2017.172.
Moss, B. (1990) Regulation of vaccinia virus transcription, Annu. Rev. Biochem., 59, 661-688, doi: https://doi.org/10.1146/annurev.bi.59.070190.003305.
Santos, C. R., Vega, F. M., Blanco, S., Barcia, R., and Lazo, P. A. (2004) The vaccinia virus B1R kinase induces p53 downregulation by an Mdm2-dependent mechanism, Virology, 328, 254-265, doi: https://doi.org/10.1016/j.virol.2004.08.013.
Greenway, A. L., McPhee, D. A., Allen, K., Johnstone, R., et al. (2002) Human immunodeficiency virus type 1 Nef binds to tumor suppressor p53 and protects cells against p53-mediated apoptosis, J. Virol., 76, 2692-2702, doi: https://doi.org/10.1128/jvi.76.6.2692-2702.2002.
Thakur, B. K., Chandra, A., Dittrich, T., Welte, K., and Chandra, P. (2012) Inhibition of SIRT1 by HIV-1 viral protein Tat results in activation of p53 pathway, Biochem. Biophys. Res. Commun., 424, 245-250, doi: https://doi.org/10.1016/j.bbrc.2012.06.084.
Gnanasundram, S., Malbert-Colas, L., Chen, S., Fusée, L., Daskalogianni, C., et al. (2020) MDM2’s dual mRNA binding domains co-ordinate its oncogenic and tumour suppressor activities, Nucleic Acids Res., 48, 6775-6787, doi: https://doi.org/10.1093/nar/gkaa431.
Benedict, C. A., Norris, P. S., and Ware, C. F. (2002) To kill or be killed: viral evasion of apoptosis, Nat. Immunol., 3, 1013-1018, doi: https://doi.org/10.1038/ni1102-1013.
Reading, P. C., Khanna, A., and Smith, G. L. (2002) Vaccinia virus CrmE encodes a soluble and cell surface tumor necrosis factor receptor that contributes to virus virulence, Virology, 292, 285-298, doi: https://doi.org/10.1006/viro.2001.1236.
Du, J., Liang, X., Liu, Y., Qu, Z., Gao, L., et al. (2009) Hepatitis B virus core protein inhibits TRAIL-induced apoptosis of hepatocytes by blocking DR5 expression, Cell Death Differ., 16, 219-229, doi: https://doi.org/10.1038/cdd.2008.144.
Morrison, T. E., Mauser, A., Klingelhutz, A., and Kenney, S. C. (2004) Epstein–Barr virus immediate-early protein BZLF1 inhibits tumor necrosis factor alpha-induced signaling and apoptosis by downregulating tumor necrosis factor receptor 1, J. Virol., 78, 544-549, doi: https://doi.org/10.1128/jvi.78.1.544-549.2004.
Benedict C. A., Norris P. S., Prigozy T. I., Bodmer J. L., Mahr J. A., et al. (2001) Three adenovirus E3 proteins cooperate to evade apoptosis by tumor necrosis factor-related apoptosis-inducing ligand receptor-1 and -2, J. Biol. Chem., 276, 3270-3278, doi: https://doi.org/10.1074/jbc.m008218200.
Kabsch, K., and Alonso, A. (2002) The Human Papillomavirus type 16 E5 protein impairs TRAIL- and FasL-mediated apoptosis in HaCaT cells by different mechanisms, J. Virol., 76, 12162-12172, doi: https://doi.org/10.1128/jvi.76.23.12162-12172.2002.
Thome, M., Schneider, P., Hofmann, K., Fickenscher, H., Meinl, E., et al. (1997) Viral FLICE-inhibitory proteins (FLIPs) prevent apoptosis induced by death receptors, Nature, 386, 517-521, doi: https://doi.org/10.1038/386517a0.
Kim, H., and Ray, R. (2014) Evasion of TNF-α-mediated apoptosis by hepatitis C virus, Methods Mol. Biol., 1155, 125-132, doi: https://doi.org/10.1007/978-1-4939-0669-7_11.
Nailwal, H., and Chan, F. K. M. (2019) Necroptosis in anti-viral inflammation, Cell Death Differ., 26, 4-13, doi: https://doi.org/10.1038/s41418-018-0172-x.
Filippova, M., Filippov, V. A., Kagoda, M., Garnett, T., Fodor, N., and Duerksen-Hughes, P. J. (2009) Complexes of Human Papillomavirus type 16 E6 proteins form pseudo-death-inducing signaling complex structures during tumor necrosis factor-mediated apoptosis, J. Virol., 83, 210-227, doi: https://doi.org/10.1128/jvi.01365-08.
Garnett, T. O., Filippova, M., and Duerksen-Hughes, P. J. (2006) Accelerated degradation of FADD and procaspase 8 in cells expressing human papilloma virus 16 E6 impairs TRAIL-mediated apoptosis, Cell Death Differ., 13, 1915-1926, doi: https://doi.org/10.1038/sj.cdd.4401886.
Yuan, H., Fu, F., Zhuo, J., Wang, W., Nishitani, J., An, D. S., Chen, I. S. Y., and Liu, X. (2005) Human papillomavirus type 16 E6 and E7 oncoproteins upregulate c-IAP2 gene expression and confer resistance to apoptosis, Oncogene, 24, 5069-5078, doi: https://doi.org/10.1038/sj.onc.1208691.
Dufour, F., Sasseville, A. M. J., Chabaud, S., Massie, B., Siegel, R. M., and Langelier, Y. (2011) The ribonucleotide reductase R1 subunits of herpes simplex virus types 1 and 2 protect cells against TNFα- and FasL-induced apoptosis by interacting with caspase-8, Apoptosis, 16, 256-271, doi: https://doi.org/10.1007/s10495-010-0560-2.
McCormick, A. L., Skaletskaya, A., Barry, P. A., Mocarski, E. S., and Goldmacher, V. S. (2003) Differential function and expression of the viral inhibitor of caspase 8-induced apoptosis (vICA) and the viral mitochondria-localized inhibitor of apoptosis (vMIA) cell death suppressors conserved in primate and rodent cytomegaloviruses, Virology, 316, 221-233, doi: https://doi.org/10.1016/j.virol.2003.07.003.
Veyer, D. L., Carrara, G., Maluquer de Motes, C., and Smith, G. L. (2017) Vaccinia virus evasion of regulated cell death, Immunol. Lett., 186, 68-80, doi: https://doi.org/10.1016/j.imlet.2017.03.015.
Chipuk, J. E., Moldoveanu, T., Llambi, F., Parsons, M. J., and Green, D. R. (2010) The BCL-2 family reunion, Mol. Cell, 37, 299-310, doi: https://doi.org/10.1016/j.molcel.2010.01.025.
Danial, N. N., and Korsmeyer, S. J. (2004) Cell death: critical control points, Cell, 116, 205-219, doi: https://doi.org/10.1016/s0092-8674(04)00046-7.
Altmann, M., and Hammerschmidt, W. (2005) Epstein–Barr virus provides a new paradigm: a requirement for the immediate inhibition of apoptosis, PLoS Biol., 3, 1-10, doi: https://doi.org/10.1371/journal.pbio.0030404.
Han, J., Wallen, H. D., Nuñez, G., and White, E. (1998) E1B 19,000-molecular-weight protein interacts with and inhibits CED-4-dependent, FLICE-mediated apoptosis, Mol. Cell. Biol., 18, 6052-6062, doi: https://doi.org/10.1128/mcb.18.10.6052.
Farrow, S. N., White, J. H. M., Martinou, I., Raven, T., Pun, K. T., Grinham, C. J., Martinou, J. C., and Brown, R. (1995) Cloning of a bcl-2 homologue by interaction with adenovirus E1B 19K, Nature, 374, 731-733, doi: https://doi.org/10.1038/374731a0.
Sarid, R., Sato, T., Bohenzky, R. A., Russo, J. J., and Chang, Y. (1997) Kaposi’s sarcoma-associated herpesvirus encodes a functional Bcl-2 homologue, Nat. Med., 3, 293-298, doi: https://doi.org/10.1038/nm0397-293.
Henderson, S., Huen, D., Rowe, M., Dawson, C., Johnson, G., and Rickinson, A. (1993) Epstein–Barr virus-coded BHRF1 protein, a viral homologue of Bcl-2, protects human B cells from programmed cell death, Proc. Natl. Acad. Sci. USA, 90, 8479-8483, doi: https://doi.org/10.1073/pnas.90.18.8479.
Kvansakul, M., Yang, H., Fairlie, W. D., Czabotar, P. E., Fischer, S. F., et al. (2008) Vaccinia virus anti-apoptotic F1L is a novel Bcl-2-like domain-swapped dimer that binds a highly selective subset of BH3-containing death ligands, Cell Death Differ., 15, 1564-1571, doi: https://doi.org/10.1038/cdd.2008.83.
Flanagan, A. M., and Letai, A. (2008) BH3 domains define selective inhibitory interactions with BHRF-1 and KSHV BCL-2, Cell Death Differ., 15, 580-588, doi: https://doi.org/10.1038/sj.cdd.4402292.
Karbowski, M., Norris, K. L., Cleland, M. M., Jeong, S. Y., and Youle, R. J. (2006) Role of Bax and Bak in mitochondrial morphogenesis, Nature, 443, 658-662, doi: https://doi.org/10.1038/nature05111.
Norris, K. L., and Youle, R. J. (2008) Cytomegalovirus proteins vMIA and m38.5 link mitochondrial morphogenesis to Bcl-2 family proteins, J. Virol., 82, 6232-6243, doi: https://doi.org/10.1128/jvi.02710-07.
Wang, H. W., Sharp, T. V., Koumi, A., Koentges, G., and Boshoff, C. (2002) Characterization of an anti-apoptotic glycoprotein encoded by Kaposi’s sarcoma-associated herpesvirus which resembles a spliced variant of human survivin, EMBO J., 21, 2602-2615, doi: https://doi.org/10.1093/emboj/21.11.2602.
Taylor, J. M., Quilty, D., Banadyga, L., and Barry, M. (2006) The vaccinia virus protein F1L interacts with Bim and inhibits activation of the pro-apoptotic protein Bax, J. Biol. Chem., 281, 39728-39739, doi: https://doi.org/10.1074/jbc.M607465200.
Postigo, A., Cross, J. R., Downward, J., and Way, M. (2006) Interaction of F1L with the BH3 domain of Bak is responsible for inhibiting vaccinia-induced apoptosis, Cell Death Differ., 13, 1651-1662, doi: https://doi.org/10.1038/sj.cdd.4401853.
Carrara, G., Parsons, M., Saraiva, N., and Smith, G. L. (2017) Golgi anti-apoptotic protein: a tale of camels, calcium, channels and cancer, Open Biol., 7, 170045, doi: https://doi.org/10.1098/rsob.170045.
Cross, J. R., Postigo, A., Blight, K., and Downward, J. (2008) Viral pro-survival proteins block separate stages in Bax activation but changes in mitochondrial ultrastructure still occur, Cell Death Differ., 15, 997-1008, doi: https://doi.org/10.1038/cdd.2008.14.
Masalova, O., Lesnova, E., Solyev, P., Zakirova, N., Prassolov, V., et al. (2017) Modulation of cell death pathways by Hepatitis C virus proteins in Huh7.5 hepatoma cells, Int. J. Mol. Sci., 18, 2346, doi: https://doi.org/10.3390/ijms18112346.
Liu, H., Yuan, Y., Guo, H., Mitchelson, K., Zhang, K., et al. (2012) Hepatitis B virus encoded X protein suppresses apoptosis by inhibition of the caspase-independent pathway, J. Proteome Res., 11, 4803-4813, doi: https://doi.org/10.1021/pr2012297.
Kim, S. J., Khan, M., Quan, J., Till, A., Subramani, S., and Siddiqui, A. (2013) Hepatitis B virus disrupts mitochondrial dynamics: induces fission and mitophagy to attenuate apoptosis, PLoS Pathog., 9, 1-12, doi: https://doi.org/10.1371/journal.ppat.1003722.
Brazil, D. P., Yang, Z. Z., and Hemmings, B. A. (2004) Advances in protein kinase B signalling: AKTion on multiple fronts, Trends Biochem. Sci., 29, 233-242, doi: https://doi.org/10.1016/j.tibs.2004.03.006.
Datta, S. R., Brunet, A., and Greenberg, M. E. (1999) Cellular survival: a play in three akts, Genes Dev., 13, 2905-2927, doi: https://doi.org/10.1101/gad.13.22.2905.
Takino, J. I., Sato, T., Nagamine, K., and Hori, T. (2019) The inhibition of Bax activation-induced apoptosis by RasGRP2 via R-Ras-PI3K-Akt signaling pathway in the endothelial cells, Sci. Rep., 9, 16717, doi: https://doi.org/10.1038/s41598-019-53419-4.
Fresno Vara, J. Á., Casado, E., de Castro, J., Cejas, P., Belda-Iniesta, C., and González-Barón, M. (2004) P13K/Akt signalling pathway and cancer, Cancer Treat. Rev., 30, 193-204, doi: https://doi.org/10.1016/j.ctrv.2003.07.007.
Zhao, H. F., Wang, J., Shao, W., Wu, C. P., Chen, Z. P., To, S. T., and Li, W. P. (2017) Recent advances in the use of PI3K inhibitors for glioblastoma multiforme: current preclinical and clinical development, Mol. Cancer, 16, 100, doi: https://doi.org/10.1186/s12943-017-0670-3.
Surviladze, Z., Sterk, R. T., DeHaro, S. A., and Ozbun, M. A. (2013) Cellular entry of Human Papillomavirus type 16 involves activation of the phosphatidylinositol 3-kinase/Akt/mTOR pathway and inhibition of autophagy, J. Virol., 87, 2508-2517, doi: https://doi.org/10.1128/jvi.02319-12.
Zhang, L., Wu, J., Ling, M. T., Zhao, L., and Zhao, K. N. (2015) The role of the PI3K/Akt/mTOR signalling pathway in human cancers induced by infection with human papillomaviruses, Mol. Cancer, 14, doi: https://doi.org/10.1186/s12943-015-0361-x.
Fukuda, M., and Longnecker, R. (2004) Latent membrane protein 2A inhibits transforming growth factor-1-induced apoptosis through the phosphatidylinositol 3-kinase/Akt pathway, J. Virol., 78, 1697-1705, doi: https://doi.org/10.1128/jvi.78.4.1697-1705.2004.
Scholle, F., Bendt, K. M., and Raab-Traub, N. (2000) Epstein–Barr virus LMP2A transforms epithelial cells, inhibits cell differentiation, and activates Akt, J. Virol., 74, 10681-10689, doi: https://doi.org/10.1128/jvi.74.22.10681-10689.2000.
Sodhi, A., Montaner, S., Patel, V., Gómez-Román, J. J., Li, Y., Sausville, E. A., Sawait, E. T., and Gutkind, J. S. (2004) Akt plays a central role in sarcomagenesis induced by Kaposi’s sarcoma herpesvirus-encoded G protein-coupled receptor, Proc. Natl. Acad. Sci. USA, 101, 4821-4826, doi: https://doi.org/10.1073/pnas.0400835101.
Tomlinson, C. C., and Damania, B. (2004) The K1 protein of Kaposi’s sarcoma-associated Herpesvirus activates the Akt signaling pathway, J. Virol., 78, 1918-1927, doi: https://doi.org/10.1128/jvi.78.4.1918-1927.2004.
Olagnier, D., Sze, A., Bel Hadj, S., Chiang, C., Steel, C., et al. (2014) HTLV-1 Tax-mediated inhibition of FOXO3a activity is critical for the persistence of terminally differentiated CD4+ T cells, PLoS Pathog., 10, e1004575, doi: https://doi.org/10.1371/journal.ppat.1004575.
Bai, D., Ueno, L., and Vogt, P. K. (2009) Akt-mediated regulation of NF-κB and the essentialness of NF-κB for the oncogenicity of PI3K and Akt, Int. J. Cancer, 125, 2863-2870, doi: https://doi.org/10.1002/ijc.24748.
Feng, C., Wu, B., Fan, H., Li, C., and Meng, S. (2014) NF-kappaB-induced gp96 up-regulation promotes hepatocyte growth, cell cycle progression and transition, Acta Microbiol. Sinica, 54, 1212-1220.
Huber, M. A., Azoitei, N., Baumann, B., Grünert, S., Sommer, A., et al. (2004) NF-κB is essential for epithelial-mesenchymal transition and metastasis in a model of breast cancer progression, J. Clin. Invest., 114, 569-581, doi: https://doi.org/10.1172/jci21358.
Kulwichit, W., Edwards, R. H., Davenport, E. M., Baskar, J. F., Godfrey, V., and Raab-Traub, N. (1998) Expression of the Epstein–Barr virus latent membrane protein 1 induces B cell lymphoma in transgenic mice, Proc. Natl. Acad. Sci. USA, 95, 11963-11968, doi: https://doi.org/10.1073/pnas.95.20.11963.
Wang, L. W., Jiang, S., and Gewurz, B. E. (2017) Epstein–Barr virus LMP1-mediated oncogenicity, J. Virol., 91, e01718-16, doi: https://doi.org/10.1128/jvi.01718-16.
Gopalakrishnan, R., Matta, H., and Chaudhary, P. M. (2013) A purine scaffold HSP90 inhibitor BIIB021 has selective activity against KSHV-associated primary effusion lymphoma and blocks vFLIP k13-induced NF-κB, Clin. Cancer Res., 19, 5016-5026, doi: https://doi.org/10.1158/1078-0432.ccr-12-3510.
Briggs, L. C., Chan, A. W. E., Davis, C. A., Whitelock, N., Hotiana, H. A., et al. (2017) IKKγ-mimetic peptides block the resistance to apoptosis associated with Kaposi’s sarcoma-associated Herpesvirus infection, J. Virol., 91, e01170-17, doi: https://doi.org/10.1128/jvi.01170-17.
Chugh, P., Matta, H., Schamus, S., Zachariah, S., Kumar, A., Richardson, J. A., Smith, A. L., and Chaudhary, P. M. (2005) Constitutive NF-κB activation, normal Fas-induced apoptosis, and increased incidence of lymphoma in human herpes virus 8 K13 transgenic mice, Proc. Natl. Acad. Sci. USA, 102, 12885-12890, doi: https://doi.org/10.1073/pnas.0408577102.
Lavorgna, A., and Harhaj, E. W. (2014) Regulation of HTLV-1 tax stability, cellular trafficking and NF-κB activation by the ubiquitin-proteasome pathway, Viruses, 6, 3925-3943, doi: https://doi.org/10.3390/v6103925.
Kgatle, M. M., Spearman, C. W., Kalla, A. A., and Hairwadzi, H. N. (2017) DNA oncogenic virus-induced oxidative stress, genomic damage, and aberrant epigenetic alterations, Oxid. Med. Cell. Longev., 2017, 1-16, doi: https://doi.org/10.1155/2017/3179421.
Williams, V., Brichler, S., Khan, E., Chami, M., Dény, P., Kremsdorf, D., and Gordien, E. (2012) Large hepatitis delta antigen activates STAT-3 and NF-κB via oxidative stress, J. Viral Hepat., 19, 744-753, doi: https://doi.org/10.1111/j.1365-2893.2012.01597.x.
Xia, Y., Shen, S., and Verma, I. M. (2014) NF-κB, an active player in human cancers, Cancer Immunol. Res., 2, 823-830, doi: https://doi.org/10.1158/2326-6066.cir-14-0112.
Zur Hausen, H., and de Villiers, E. M. (2014) Cancer “causation” by infections – individual contributions and synergistic networks, Semin. Oncol., 41, 860-875, doi: https://doi.org/10.1053/j.seminoncol.2014.10.003.
Ren, Y., Shu, T., Wu, D., Mu, J., Wang, C., et al. (2020) The ORF3a protein of SARS-CoV-2 induces apoptosis in cells, Cell. Mol. Immunol., 17, 881-883, doi: https://doi.org/10.1038/s41423-020-0485-9.
Varga, Z., Flammer, A. J., Steiger, P., Haberecker, M., Andermatt, R., et al. (2020) Endothelial cell infection and endotheliitis in COVID-19, Lancet, 395, 1417-1418, doi: https://doi.org/10.1016/S0140-6736(20)30937-5.
Alpalhão, M., Ferreira, J. A., and Filipe, P. (2020) Persistent SARS-CoV-2 infection and the risk for cancer, Med. Hypotheses, 143, 109882, doi: https://doi.org/10.1016/j.mehy.2020.109882.
Mesri, E. A., Feitelson, M. A., and Munger, K. (2014) Human viral oncogenesis: a cancer hallmarks analysis, Cell Host Microbe, 15, 266-282, doi: https://doi.org/10.1016/j.chom.2014.02.011.
Van Kriekinge, G., Castellsagué, X., Cibula, D., and Demarteau, N. (2014) Estimation of the potential overall impact of human papillomavirus vaccination on cervical cancer cases and deaths, Vaccine, 32, 733-739, doi: https://doi.org/10.1016/j.vaccine.2013.11.049.
McQuaid, T., Savini, C., and Seyedkazemi, S. (2015) Sofosbuvir, a significant paradigm change in HCV treatment, J. Clin. Transl. Hepatol., 3, 27-35, doi: https://doi.org/10.14218/jcth.2014.00041.
Schiller, J. T., and Lowy, D. R. (2010) Vaccines to prevent infections by oncoviruses, Annu. Rev. Microbiol., 64, 23-41, doi: https://doi.org/10.1146/annurev.micro.112408.134019.
Bu, W., Joyce, M. G., Nguyen, H., Banh, D. V., Aguilar, F., et al. (2019) Immunization with components of the viral fusion apparatus elicits antibodies that neutralize Epstein–Barr Virus in B cells and epithelial cells, Immunity, 50, 1305-1316, doi: https://doi.org/10.1016/j.immuni.2019.03.010.
Funding
This work was supported by the Russian Science Foundation (project no. 19-15-00125). Work in our laboratory was also supported by the Russian Foundation for Basic Research (projects nos. 18-29-09005, 20-015-00157), and by the Swedish (project no. 190345) and the Stockholm (project no. 181301) Cancer Societies.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare no conflicts of interest in financial or any other sphere. This article does not contain any studies with human participants or animals performed by any of the authors.
Rights and permissions
About this article

Cite this article
Zamaraev, A.V., Zhivotovsky, B. & Kopeina, G.S. Viral Infections: Negative Regulators of Apoptosis and Oncogenic Factors. Biochemistry Moscow 85, 1191–1201 (2020). https://doi.org/10.1134/S0006297920100077
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0006297920100077