
Abstract
A series of heterometallic [TiIV7MIII] rings are reported which contain a single paramagnetic metal centre, either FeIII or CrIII. The structures contain an octagon of metals bridged by oxides within the ring and pivalate ligands outside the ring. The structures encapsulate a single cation, either a secondary ammonium cation or a cesium. Multi-frequency EPR spectroscopy was used to measure the zero-field splitting parameters for the paramagnetic metal sites. The FeIII sites have rhombic anisotropy, with D around − 0.18 cm−1 and λ (= D/E) approximately 0.25. The CrIII have a more axial anisotropy, with D around − 0.5 cm−1 and λ around 0.05. Attempts to find a magneto-structural correlation are vitiated by the difficulty of refining the challenging crystal structures.
Similar content being viewed by others


1 Introduction
When the spin, s, of a system is greater than ½ the sub-levels of that spin are not degenerate in zero-field. The splitting of the sub-levels, called the zero-field splitting (ZFS), is vital in determining the magnetic behaviour of the system and is key in area such as single-molecule magnets [1] and in interpreting the spectroscopic behaviour of metalloproteins [2]. The relationship between structure and ZFS is reasonably well understood for the first coordination sphere of the 3d-metal, e.g., for five-coordinate nickel(II) complexes [3], and Bencini and Gatteschi have shown how the single ion ZFS can be used to calculate the ZFS of a polymetallic cage assuming that the strong exchange limit applies [4].
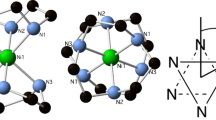
We have been studying cyclic octametallic rings for a number of years [5]. They have allowed very attractive experiments, such as the report of 4D-inelastic neutron scattering which allows an understanding of magnetic behaviour without the need for a spin Hamiltonian [6]. We have carried out more detailed studies of heterometallic octametallic rings [7]; these studies include an examination of whether the strong exchange limit is valid in such spin systems [8]. More recently, we have synthesized a series of octametallic rings of general formula [CAT][TiIV7MIIIO8(O2CtBu)16] (where CAT = a cation; MIII = FeIII 1 or CrIII 2] [9] (Fig. 1a). We observed that the EPR spectra of these compounds depend on the cation present. This allows us to examine how the ZFS of s = 5/2 and 3/2 centres depends on subtle changes in the coordination around the paramagnetic centres; the central cation interacts with the two bridging oxide ligands (Fig. 1b). We report the results of these studies here.

Structure of the heterometallic rings studied in the crystal. a The complete octametallic ring anion; the cation sits at the centre of the ring. b The interaction between a cation, di-n-propylammonium, and the two bridging oxides attached to a trivalent metal centre. Color scheme: Fe/Cr, brown; Ti, grey-blue; O, red; N, blue; C, grey. Methyl groups of pivalates omitted for clarity. H-atoms omitted for clarity except those involved in N–H…O hydrogen bonds (color figure online)
2 Experimental Details
2.1 General Information
Unless stated otherwise, all reagents and solvents were purchased from commercial sources and used without further purification. The compounds were synthesized and crystallised under a nitrogen atmosphere in anhydrous solvents, using standard Schlenk line techniques. Analytical data were obtained by the microanalysis laboratory at the University of Manchester—carbon, hydrogen, nitrogen analysis (CHN) by a Flash 2000 elemental analyser and metals analysis by Thermo iCap 6300 inductively coupled plasma optical emission spectroscopy (ICP-OES). A Thermo Fisher Scientific “Exactive Plus EMR Orbitrap” mass spectrometer was used for mass spectrometric analysis. The samples were ionised in the electrospray ion source operated in both positive and negative modes, ~ 3.5 kV was applied to the capillary. The ions were detected using an electrostatic trap with mass resolution of ~ 140,000 and stability sufficient to achieve a precision of < 5 ppm for mass measurements at the region of interest.
2.2 Starting Materials
{[Fe3O(O2CtBu)6(H2O)3](O2CtBu)}.tBuCO2H [10] and cesium pivalate were prepared as reported in literature [11].
2.3 Synthesis of Compounds
The syntheses of [nPr2NH2][Ti7FeO8(O2CtBu)16], 1.nPr2NH2 and [nPr2NH2][Ti7CrO8(O2CtBu)16], 2.nPr2NH2 have been previously reported [9].
2.4 [PrNH3][Ti7FeO8(O2CtBu)16], 1.PrNH3
Pivalic acid (36.0 g, 353 mmol), n-propylamine (nPrNH2) (0.69 ml, 8.4 mmol), [Fe3O(O2CtBu)6(H2O)3](O2CtBu).tBuCO2H (1.2 g, 1.2 mmol), pivalic anhydride (1 ml, 0.92 g, 4.93 mmol) in toluene (24 ml) were refluxed for 30 min, and a clear orange solution was obtained. This was then cooled to RT, and titanium(IV) isopropoxide (6.0 mL, 5.76 g, 20.3 mmol) was added. The reaction mixture was then stirred while refluxing for 36 h (T bath 140 °C). The toluene was removed by distillation, and the residue cooled to 70 °C and MeCN (100 ml) was added. The solution was left to cool to room temperature while stirring. The precipitate was collected by filtration and washed with MeCN (4 × 24 ml), and acetone (2 × 12 ml) and then extracted into hexane (3 × 24 ml). The combined extract was filtered, and the solvent removed under reduced pressure. Yield: 5.7 g (88% based on Ti). Elemental analysis (%) calculated for C83H154FeNO40Ti7: C: 45.37; H: 7.06; N: 0.64; Fe: 2.54; Ti: 15.26. Found: C, 45.09; H, 7.12; N, 0.64; Fe, 2.82; Ti, 14.64. ESI–MS (Sample dissolved in toluene, MeCN mobile phase), m/z: 2198 [M + H]+.
Pale yellow crystals suitable for single crystal X-ray diffraction studies were grown from hexane.
2.5 [Me2NH2][Ti7FeO8(O2CtBu)16], 1.Me2NH2
Pivalic acid (36.0 g, 353 mmol), Dimethylammonium dimethylcarbamate ((CH3)2NH·(CH3)2NCOOH) (2 ml, 15.6 mmol), [Fe3O(O2CtBu)6(H2O)3](O2CtBu).tBuCO2H (1.2 g, 1.2 mmol) and pivalic anhydride (2 ml, 1.84 g, 9.86 mmol) in toluene (24 ml) were refluxed for 30 min, and a clear orange solution was obtained. This was then cooled to RT, and titanium(IV) isopropoxide (6.0 ml, 5.76 g, 20.3 mmol) was added. The reaction mixture was then stirred while refluxing for 24 h (T bath 140 °C). The toluene was removed by distillation, pivalic acid was also partially removed after 2 h and after the residue cooled to 80 °C, MeCN (100 ml) was added. The solution was left to cool to room temperature while stirring. The precipitate was collected by filtration and washed with MeCN (4 × 25 ml), and acetone (2 × 30 ml) and then extracted into pentane (2 × 25 ml). The combined extract was filtered, and the solvent removed under reduced pressure. Yield: 6.4 g (98% based on Ti). Elemental analysis (%) calculated for C82H152FeNO40Ti7: C: 45.11; H: 7.02; N: 0.64; Fe: 2.56; Ti: 15.36. Found: C, 45.83; H, 7.19; N, 0.67; Fe, 2.82; Ti, 15.12. ESI–MS (Sample dissolved in toluene, MeCN mobile phase), m/z: 2184 [M + H]+.
Pale yellow crystals suitable for single crystal X-ray diffraction studies were grown from Et2O/MeCN.
2.6 [Cs][Ti7FeO8(O2CtBu)16], 1.Cs
Pivalic acid (36.0 g, 353 mmol), caesium pivalate (1 g, 4.27 mmol) were stirred at 100 °C for 10 min. Then [Fe3O(O2CtBu)6(H2O)3] O2CtBu}. tBuCO2H (1.2 g, 1.2 mmol), pivalic anhydride (1 ml, 0.92 g, 4.93 mmol) in toluene (25 ml) were refluxed for 15 min, and a clear orange solution was obtained. This was then cooled to RT, and titanium(IV) isopropoxide (6.0 mL, 5.76 g, 20.3 mmol) was added. The reaction mixture was then stirred while refluxing for 48 h (T bath 140 °C). Toluene was removed by distillation under N2, and the residue cooled to 40 °C and MeCN (120 ml) was added. The solution was left to cool to room temperature while stirring. The precipitate was collected by filtration and washed with MeCN (5 × 20 ml) and dried with a flow of N2. Then the solid was extracted into hexane (30 ml). The combined extract was filtered and the solvent removed under reduced pressure. Yield: 6.2 g (95% based on Ti).
Elemental analysis (%) calculated for C80H144FeO40Ti7Cs: C: 42.31; H: 6.44; Fe: 2.46; Ti: 14.76; Cs: 5.85. Found: C, 43.17; H, 6.76; N, none found; Fe, 2.30; Ti, 14.33. ESI–MS (Sample dissolved in toluene, MeCN mobile phase), m/z: 2271 [M + H]+.
Pale yellow crystals suitable for single crystal X-ray diffraction studies were grown from toluene.
2.7 [Cs][Ti7CrIIIO8(O2CtBu)16], 2.Cs
Pivalic acid (36.0 g, 353 mmol) and Cr(NO3)3 9H2O (3.0 g, 7.5 mmol) were heated with stirring at 160 °C for 4 h. Then the flask was cooled to RT and pivalic anhydride (1 ml, 0.92 g, 4.93 mmol), pivalic acid (4.0 g, 39 mmol), caesium pivalate (1 g, 4.27 mmol) and toluene (25 ml) were added and stirred for 10 min at 140 °C. After cooling at RT, titanium(IV) isopropoxide (6.0 ml, 5.76 g, 20.3 mmol) was added and the solution was refluxed at 140 °C under N2 atmosphere for up to 7 days. Toluene was removed by distillation and the solution was left to cool to RT. MeCN (50 ml) was added, stirred for 1 h then the precipitate was collected by filtration and washed with acetone (5 × 20 ml), dried and then extracted into hexane (3 × 24 ml). The combined extract was filtered and the solvent removed under reduced pressure. Yield: 5.34 g (85% based on Ti).
Elemental analysis (%) calculated for C80H144CrO40Ti7Cs: C: 42.40; H: 6.40; Cr: 2.29; Ti: 14.79. Found: C, 41.97; H, 6.45; N, none found; Cr, 2.28; Ti, 15.06. ESI–MS (Sample dissolved in tert-butyl methyl ether, acetone mobile phase), m/z: 2237 [M + H]+
Pale green crystals suitable for single crystal X-ray diffraction studies were grown from toluene.
2.8 X-ray Crystallography
2.8.1 Data Collection
X-ray data for compound 1.nPr2NH2 were collected at a temperature of 150 K on an Oxford X’calibur 2 with an Atlas CCD detector with MoKα radiation, (λ = 0.71073 Å). X-ray data for compound 2.nPr2NH2 were collected at a temperature of 150 K on an Agilent Technologies Supernova diffractometer equipped with an Eos CCD detector with MoKα radiation, (λ = 0.71073 Å) X-ray data for compound 1.Me2NH2 were collected at a temperature of 150 K on a Bruker Prospector diffractometer with an Apex II CCD detector with CuKα radiation, (λ = 1.54184 Å). X-ray data for compounds 1.nPrNH3 and 2.Cs were collected at a temperature of 150 K and 100 K, respectively, on a Rigaku FR-X diffractometer with a HyPix HP6000 detector with MoKα radiation, (λ = 0.71073 Å). X-ray data for compound 1.Cs were collected at a temperature of 100 K on a Rigaku FR-X diffractometer with a Hypix HP6000 detector with CuKα radiation, (λ = 1.54184 Å). All the diffractometers were equipped with Oxford Cryosystems 700/800 series nitrogen flow gas systems.
2.8.2 Crystal Structure Determinations and Refinements
X-ray data for all crystals of the compounds were processed and reduced using CrysAlisPro suite of programs. Absorption corrections were performed using empirical methods based upon symmetry-equivalent reflections combined with measurements at different azimuthal angles using SCALE3 ABSPACK.
The crystal structures for 1.nPr2NH2, 1.nPrNH3, 1.Cs, 2.nPr2NH2 and 2.Cs compounds were solved and refined against all F2 values using the SHELX-2016 implemented through Olex2 v1.2 [12, 13].
Pivalate ligands were found disordered and modelling over two positions was possible. C–C bond distances were restrained using DFIX, SAME and SADI commands. The atomic displacement parameters (adp) of the disordered pivalate ligands and solvent molecules have been restrained using RIGU, ISOR EADP and SIMU commands.
Heteroatoms refinement: In all cases, it was noted that there was a variation in Ueq for the metal sites. Therefore, these were refined as mixed metal sites with the relative occupancies of Fe and Cr refined against individual free variables for each site, such that the resulting summed occupancies equalled Ti7M. The positions and atomic displacement parameters were constrained using EADP and EXYZ commands.
There were no overall trends for relative occupancy observed between structures. However, exceptions should be noted. In the case of 1nPr2NH2 and 2nPr2NH2, the heterometal showed the greatest occupancy at the disordered metal edge.
For 1nPr2NH2, the M8 metal site was split due to the apparent disorder in the electron density map. However, the Ti8A metal site occupancy has been fixed to 0.5 and the Ti8/Fe8 site has been refined against a free variable in combination with the other mixed metal site such that the overall formula equal Ti7Fe. The choice of defining a fixed occupancy for the Ti8A site and refining the relative occupancies on the second site was due to the limitations of refining two split sites against a free variable, with the total occupancy of that free variable also refining against a free variable. Initial attempts to refine these sites involved simply removing the EADP and EXYZ commands are refining the respective disordered parts against their free variables. However, it became apparent that the disordered ligands did not share the same occupancy as the Ti8/Fe8 site would have done if it was purely Fe. During the course of the refinement, it became apparent that the relative occupancy of the two disordered parts was 50:50. In addition, attempts to refine the Ti8A site as 50% total occupancy and as mixed Ti/Fe always resulted in 100% occupancy for Ti8A. Ti8A was, therefore, fixed at 50% occupancy. The remaining site was then incorporated into the SUMP command with the other mixed metal sites.
For 2nPr2NH2 the EADP and EXYZ commands were simply removed and the metal sites allowed to shift away from each other, with the relative ocuupancies of the disordered ligands matching the corresponding occupancies of the Ti8 and Cr8A.
2.8.3 EPR Spectroscopy
Continuous wave EPR spectra were recorded at several microwave frequencies using different spectrometers and microwave bridges at the EPSRC EPR National Research Facility at the University of Manchester, and at the University of Stuttgart. A Bruker EMX EPR spectrometer was equipped with a 1.8 T magnet and either a Super-X microwave bridge and a ER4119HS (X-band, ca. 9.4 GHz), or a Bruker ER051 QG microwave bridge with an ER 5106 QT (Q-band, ca. 34 GHz) resonator. K-band (ca. 24 GHz) EPR spectra were recorded with a Bruker ELEXSYS E580 EPR spectrometer with a 1.6 T magnet and equipped with a Super-K bridge and an ER 6706KT K-band resonator. For the low temperature spectra, a liquid He Oxford Cryostat CF935 (cooling range 2.4–300 K) was used. The spectra were collected on polycrystalline powders ground with eicosane. High-frequency EPR spectra (95–190 GHz) were recorded in Stuttgart on a home-built spectrometer [14]. Its radiation source is a 0–20 GHz signal generator (Anritsu) in combination with an amplifier–multiplier chain (VDI) to obtain the required frequencies. It features a quasi-optical bridge (Thomas Keating) and induction mode detection. The detector is a QMC magnetically tuned InSb hot electron bolometer. The sample is located in an Oxford Instruments 15/17 T cryomagnet equipped with a variable temperature insert (1.5–300 K). The spectra were collected on a pressed pellet of polycrystalline powder ground with eicosane.
Spectral simulations were performed using the EasySpin 5.0.2 simulation software [15].
3 Synthesis and Structural Refinement
For the {Ti7Fe} cages we used the oxo-centred iron(III) triangle as the starting material, which was reacted with pivalic acid and pivalic anhydride in the presence of the cation prior to addition of [Ti(OiPr)4]. Three new compounds were formed: [Cation][TiIV7FeIIIO8(O2CtBu)16] (for M = FeIII, 1.cation with cation = nPrNH3+, Me2NH2+ or Cs+); the compound where the cation = nPr2NH2 has been reported previously, but the crystallography has been revised. For the {Ti7Cr} ring we started with hydrated chromium(III) nitrate, but in the presence of pivalic acid and pivalic anhydride this probably forms the oxo-centred triangle in situ. The new compound [Cs][TiIV7CrIIIO8(O2CtBu)16] 2.Cs results; we have previously reported 2.Pr2NH2, but the crystallography has been revised.
The crystal structure of sample 1.Me2NH2 was found to be incommensurately modulated. The basic structure unit cell is tetragonal I, a = 19.7135(9) Å, c = 16.3468(9) Å, which is similar to other {[Me2NH2][MIII7MIIF8(O2CR)]} analogues (where MIII = Cr, Ga. MII = Ni, Co, Zn).5–7 However, there are strong, sharp satellite reflections present in the reciprocal lattice in between the main reciprocal lattice points along the c* axis. Initially, it was thought that these could be indexed by tripling the c-axis of the unit cell, to give a unit cell of tetragonal I, a = 19.7135(9) Å, c = 49.0085(17) Å. However, it became apparent that these cannot be indexed to a rational periodicity of the basic structure unit cell, but can be indexed as first order modulation with a modulation vector of 0.370 along the c* axis (Fig. 2). While the data can be processed taking into account the modulation vector, giving an hklfm output, attempts to solve the structure while taking into account the modulation vector, have proved fruitless. This is mainly due to obtaining a suitable structure solution from which to refine, considering the inherent disorder of the t-butyl groups of the pivalate moieties that are prevalent in these systems, in combination with the sheer size and complexity of the system to be treated with a modulation function.

Voxel projection of the reciprocal lattice extracted from the data for 1.Me2NH2. Strong reflections corresponding to the basic structure unit cell appear brighter in the projection, with the satellite reflections appearing dimmer. Note that the spacing between the strong basic unit cell reflections and the satellite reflections are different than the spacing between the pairs of satellite reflections
Elemental analysis and mass spectrometry confirm 1.Me2NH2 has formed, but no metric parameters can be obtained from the X-ray diffraction study.
All other structures crystallise in P21/c, with one molecule in the asymmetric unit. The structures show an octagon of metal sites with each metal…metal edge bridged internally by an oxide and externally by two pivalate ligands (Fig. 1a). For 1.Cs and 2.Cs the cation lies at the middle of the ring and interacts with the bridging oxides. For other structures the secondary ammonium cation resides within the central cavity of the ring such that the ammonium site forms hydrogen bonding interactions with the bridging fluorides of the anionic ring. The alkyl chains project approximately along the line normal to the mean plane of the metal sites. In all the structures, the MIII metal is disordered around each mixed metal site. Each mixed metal site is six-coordinate, bound to four oxygen donors from pivalate ligands and two µ-oxides. The coordination geometry at the metal sites is distorted from octahedral.
In each case, there is a degree of disorder to each structure. A look at the structures shows that there is a greater amount of structural averaging in the alkylammonium cation rings than in the Cs+ rings, which implies a greater flexibility to the structure in the alkyl ammonium cation cases. There are two places, where this is seen. First, by larger than expected anisotropic displacement parameters (adps) for the metal sites and the ligands in 1.nPrNH3, 1.nPr2NH2 and 2.nPrNH2, when compared to the Cs+ cation rings. These larger adps are more readily explained, not by increased thermal motion, but by subtle variation in the local ligand geometries at the metal sites around the ring that cannot be accounted for in the model. The slight variances in the metal geometries between the heterometals and the TiIV and the statistical low occupancy distribution of the heterometal precludes clear distinction between the different geometries as the packing is energetically similar for the different conformers. Secondly, there is a distinct asymmetry to the M-Ooxide bonds, which is well ordered in the Cs+ rings and not ordered in the alkyl ammonium cation cases, despite both cases having the similar statistical disorder of the heterometal site. This asymmetry is probably due to the tendency for TiIV to try to form Ti = O Table 1.
This would imply there is a greater structural rigidity in the Cs+ rings, perhaps forcing a greater distortion of the heterometal away from idealized octahedral geometry. The internal O(oxide)-M-O(oxide) angles are most obtuse, ranging from 96.51(17)° to 98.58(16)° in the structures. The O(oxide)-M-O(pivalate) angles range from 89.40(16)° to 95.18(17)°, while all the O(pivalate)-M-O(pivalate) angles range from 80.39(17)° to 88.3(2)°.
4 EPR Spectroscopy
Electron paramagnetic resonance (EPR) spectroscopy is a sensitive probe of the metal site geometry in such circumstances. Compared to magnetometry techniques it provides more accurate information on the ZFS of paramagnetic metal ions. Where more than one metal site is present then EPR can show all sites in contrast to the average thermodynamic response in magnetometry.
The X-band (microwave frequency, ν, ca. 9.80 GHz) EPR spectrum for 1.nPr2NH2 has an intense and relatively well-resolved feature at geff ≈ 4.3 (Figure S1a). This is characteristic of distorted six-coordinate HS FeIII (s = 5/2) ions with a significant (cf. to the microwave energy hν) axial ZFS parameter |D| and high rhombicity (|E/D|; E is the rhombic ZFS parameter, where E ≤|D|/3) [16]. Such spectra are sensitive to the |E/D| ratio, but not to the magnitude of |D| above a threshold value [16].
Determining |D| requires measurement at higher frequencies. We have reported the Q-band (ca. 34 GHz) spectrum for 1.nPr2NH2 previously [9], and spectra for 1.nPrNH3 are very similar (Fig. 3a). Spectra for 1.Cs and 1.Me2NH2 have very similar profiles to each other (Fig. 3b, c), but are subtly different to those of 1.nPr2NH2 and 1.nPrNH3 (see the features at ca. 900 and above 1400 mT, Fig. 3). The spectrum of 1.Me2NH2 is noticeably better resolved than the others (Fig. 3c), but the spread of the features fits under the linewidth of 1.Cs (Fig. 3b). The better resolution for 1.Me2NH2 reveals that the features around 800, 1100 and 1550 mT are split into two or more components. This is also evident in K-band (ca. 24 GHz) spectra (splitting of the features at ca. 750 and 1200 mT; Fig. 4). This could either be due subtle differences in the ZFS parameters (hence microscopic structure) in different sites in the Ti7Fe ring. This is likely also the cause of the asymmetric lineshapes in the more poorly resolved spectra of 1.Cs which otherwise has a very similar profile to 1.Me2NH2.

Experimental (black) and simulated (red) room temperature Q-band (ca. 34 GHz) EPR spectra for powder samples immobilised in eicosane of (a) 1.nPrNH3, (b) 1.Cs and (c) 1.Me2NH2. Simulations with an isotropic Gaussian linewidth of 26 mT for (a) and (b) and 14 mT for (c) (color figure online)

Experimental (black) and simulations (red) room temperature K-band (ca. 24 GHz) EPR spectra for a powder sample of 1.Me2NH2 immobilised in eicosane. Simulation with an isotropic Gaussian linewidth of 16 mT (color figure online)
The spectra were simulated using the spin Hamiltonian:
where the first term corresponds to the electronic Zeeman term. To reduce the number of parameters, we have fixed the g-values as isotropic with g = 2.01 consistent with HS FeIII. For 1.Me2NH2 we have allowed for two equal occupancy sites, modeling to both the Q- and K-band spectra. There are almost certainly more than two independent sites, but we do not wish to over-parameterise the model. It is likely that, given the structural disorder observed by X-ray diffraction, the ZFS parameters are subject to strain effects (i.e., there are statistical distributions of D and E). These are the most likely cause for discrepancies in some relative intensities between calculated and experimental spectra.
For all the 1.Cation compounds we find |D|= 0.17–0.19 cm−1 with |E/D|= 0.2–0.3 (Table 2): these are comparable with the values found for other {FeO6} coordination spheres [17,18,19,20]. We have confirmed the parameters for 1.nPr2NH2 by high-frequency EPR (HFEPR; Fig. 5), which allows access to the high-field regime (gμBB > >|D|) thus hugely simplifying spectral analysis. Modeling the HFEPR of 1.nPr2NH2 at low temperature also gives the sign of D as negative. Gatteschi and co-workers were pioneers in the application of HFEPR to large ZFS transition ion materials [21]. Although the variations are small, 1.Cs has marginally larger |D| (0.19 cm−1) than the ammonium salts (0.17–0.18 cm−1; Table 2). 1.nPr2NH2 and 1.nPrNH3 have |E/D| ca. 0.22, while that 1.Cs and 1.Me2NH2 have slightly larger |E/D| of ca. 0.3. This appears to be due to a larger E more than any change in D.

a HFEPR spectrum at 95 GHz for 1.nPr2NH2 at 10 K experimental (black) and simulated (red). b Spectra for 1.nPr2NH2 in higher frequencies at 10 K. The asterisks mark the transitions resulting from the third harmonics of the fundamental microwave frequencies used in the experiments (color figure online)
X-band spectra for the {Ti7Cr} compounds 2.nPr2NH2 and 2.Cs (Figure S1) are dominated by features at geff ≈ 4 and 2, which is characteristic of large |D| (cf. the microwave energy hν) for CrIII (s = 3/2) but with near axial symmetry (|E/D| ≈ 0). Q-band spectra of 2.nPr2NH2 and 2.Cs are similar in profile (Fig. 6) but for the former, (i) there is noticeably greater spread in magnetic field range in the transitions (see the feature close to zero-field), and (ii) the linewidth is significantly narrower revealing multiple sites (e.g., see the feature at ca. 700 mT). The band-shapes in the spectrum of 2.Cs cover the spread of the features in the spectrum of 2.nPr2NH2. Simulation with the same spin Hamiltonian (with fixed isotropic g = 1.98) gives the parameters in Table 2. We previously reported a model of the Q-band spectrum of 2.nPr2NH2 with three distinct components with similar ZFS parameters (Table 2) [9].

Experimental (black) and simulations (red) room temperature Q-band (ca. 34 GHz) EPR spectra for powder samples immobilised in eicosane of (a) 2.Pr2NH3, (b) 2.Cs. Simulations with an isotropic Gaussian linewidth of 16 mT for (a) and 25 mT for (b). The broad peak at g ~ 1.98, labeled *, is attributed to a coupled CrIII impurity (most likely a {Cr3O} triangle) [22] (color figure online)
We find |D|= 0.48–0.53 cm−1 with |E/D|= 0.04–0.07 (Table 2). The values reported in Table 2 are within the range typical for octahedral CrIII{O6} complexes [23, 24]. The |D| value is slightly smaller for 2.Cs than for 2.nPr2NH2 (or at least at the lower end of the range of the |D| values observed for the different sites in the latter), but we are cautious to compare the rhombicities given the small values and uncertainties in E. Q-band spectra of 2.Pr2NH2, broaden noticeably on lowering the temperature to 5 K (Figure S2). At the lowest temperatures, the high-field xy features of the spectrum are marginally enhanced in intensity relative to the low-field features, which is consistent with negative D. This is also consistent with other CrIII{O6} species [23, 24].
The axial ZFS in the CrIII complexes 2.Cation are greater than in the FeIII complexes 1.Cation. This is because CrIII (4A ground state) has low-lying excited states of the same spin multiplicity as the ground state, whereas HS FeIII (6A) does not. Mixing with excited states via spin–orbit coupling is a dominant mechanism for |D| for transition ions. The spread of D values as a percentage of |D| is similar in the 2.Cation and 1.Cation series, being ca. 10%. However, we find that for 1 the Cs+ salt has a marginally larger |D| than the alkylammonium salts, while for 2, the reverse is the case. While the link between geometric structure and electronic structure is not straightforward, it is clear that the H-bonding to the bridging oxides has a subtle effect on the ZFS of the hetero-ion.
The slightly larger |D| values found for the 1.Cs compounds could be related to the slightly greater distortion in the M–O(oxide) bonds in these compounds (see Table 1). However, that greater distortion is also present in 2.Cs which has a smaller |D| value. Also, for both the crystallography and the EPR spectroscopy the difference between the parameters measured are barely statistically significant. This is unfortunate. The crystallography is, in all cases, disordered and in the case of 1.Me2NH2 we cannot even solve a structure. This mitigates against being able to explain why we see multi-site resolution in 1.Me2NH2 and 2.Pr2NH2. Unfortunately, we have to conclude that while these rings crystallise beautifully the analytical challenge they present in drawing information about localised geometries from a bulk averaging technique prevent us drawing simple magneto-structural correlations. The EPR data are extremely beautiful.
5 Conclusions
We have shown that heterometallic, eight-membered rings of the formula [CAT][TiIV7MIIIO8(O2CtBu)16] can encapsulate different templating cations, which have short contacts with the first coordination sphere of the metal ions in the ring. The availability of such a family of compounds, where there is control in the geometrical variance while having a structural rigidity has enabled an in-depth investigation into the effects of different counter-cations on the first coordination sphere by monitoring the anisotropy of FeIII and CrIII via EPR spectroscopy. With the use of appropriate EPR frequency compared to the magnitude of D, it is possible to extract the ZFS parameters of the trivalent metal ion and the number of unique metal sites. Within the series of FeIII-containing compounds, stronger binding of the cation to the bridging oxide decreases the magnitude of D and the rhombicity, while for CrIII, an opposite effect is observed. In the 1.cation family of compounds up to two unique metal centres are resolved in the EPR spectra and up to three for the 2.cation series, owning to the higher anisotropy of CrIII compared to FeIII.
This approach for the investigation of the magnetic anisotropy should be applicable to a diverse range of compounds without the need to study the sample as a single crystal. The only real requirement is simply that the crystal or the compound provide a framework symmetry to quantify the structural effect and the number of unique centres.
6 Notes and References
‡ CCDC 2012978, 2012979, 2012980, 2012981, and 2012982 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/conts/retrieving.html (or from the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB21EZ, UK; fax: (+ 44)1223-336-033; or deposit@ccdc.cam.ac.uk).
References
C. Benelli, D. Gatteschi, Introduction to Molecular Magnetism (Wiley-VCH, Weinheim, 2015)
M. Ardini, B.D. Howes, A. Fiorillo, E. Falvo, S. Sottini, D. Rovai, M. Lantieri, A. Ilari, D. Gatteschi, G. Spina, E. Chiancone, S. Stefanini, M. Fittipaldi, J. Inorg. Biochem. 182, 103 (2018)
B. Cahier, M. Perfetti, G. Zakhai, D. Naoufal, F. El-Khatib, R. Guillot, E. Rivière, R. Sessoli, A.-L. Barra, N. Guihéry, T. Mallah, Chem. Eur. J. 23, 3648 (2017)
A. Bencini, D. Gatteschi, Electron Paramagnetic Resonance of Exchange Coupled Systems (Springer-Verlag, Berlin Heidelberg, 1990)
J. van Slageren, R. Sessoli, D. Gatteschi, A.A. Smith, M. Helliwell, R.E.P. Winpenny, A. Cornia, A.-L. Barra, A.G.M. Jansen, E. Rentschler, G.A. Timco, Chem. Eur. J. 8, 277 (2002)
M.L. Baker, T. Guidi, S. Carretta, J. Ollivier, H. Mutka, H.U. Gudel, G.A. Timco, E.J.L. McInnes, G. Amoretti, R.E.P. Winpenny, P. Santini, Nature Phys. 8, 906 (2002)
E.J.L. McInnes, G.A. Timco, G.F.S. Whitehead, R.E.P. Winpenny, Angew. Chem. Int. Ed. Engl. 54, 14244 (2015)
S. Piligkos, H. Weihe, E. Bill, F. Neese, H. El Mkami, G.M. Smith, D. Collison, G. Rajaraman, G.A. Timco, R.E.P. Winpenny, E.J.L. McInnes, Chem. Eur. J. 15, 3152 (2009)
G.A. Timco, A. Fernandez, A.K. Kostopoulos, C.A. Muryn, R.G. Pritchard, I. Strashnov, I.J. Vitorica-Yrezebal, G.F.S. Whitehead, R.E.P. Winpenny, Angew. Chem. Int. Ed. 56, 13629 (2017)
K. Abdulwahab, M.A. Malik, P. O’Brien, K. Govender, C.A. Muryn, G.A. Timco, F. Tuna, R.E.P. Winpenny, Dalton Trans. 42, 196 (2013)
T.B. Faust, P.G. Heath, C.A. Muryn, G.A. Timco, R.E.P. Winpenny, Chem. Commun. 46, 6258 (2010)
G.M. Sheldrick, Acta Crystallogr. C Struct. Chem. 71, 3 (2015)
O.V. Dolomanov, L.J. Bourhis, R.J. Gildea, J.A.K. Howard, H. Puschmann, J. Appl. Cryst. 42, 339 (2009)
P. Neugebauer, D. Bloos, R. Marx, P. Lutz, M. Kern, D. Aguilà, J. Vaverka, O. Laguta, C. Dietrich, R. Clérac, J. van Slageren, Phys. Chem. Chem. Phys. 20, 15528 (2018)
S. Stoll, A. Schweiger, J. Magn. Reson. 178, 42 (2006)
F.E. Mabbs, D. Collison, Electron Paramagnetic Resonance of d Transition Metal Compounds, Studies in Inorganic Chemistry 16 (Elsevier, Amsterdam, 1992)
D. Collison, A.K. Powell, Inorg. Chem. 29, 4735 (1990)
A.L. Barra, A. Caneschi, A. Cornia, F. Fabrizi de Biani, D. Gatteschi, C. Sangregorio, R. Sessoli, L. Sorace, J. Am. Chem. Soc. 121, 5302 (1999)
J.M. Zadrozny, D.E. Freedman, Inorg. Chem. 54, 12027 (2015)
J.M. Zadrozny, M.J. Graham, M.D. Krzyaniak, M.R. Wasielewski, D.E. Freedman, Chem. Commun. 52, 10175 (2016)
A.-L. Barra, L.-C. Brunel, D. Gatteschi, L. Pardi, R. Sessoli, Acc. Chem. Res. 31, 460 (1998)
A.K. Boudalis, G. Rogez, P. Turek, Inorg. Chem. 57, 13259 (2018)
G. Elbers, S. Remme, G. Lehmann, Inorg. Chem. 25, 896 (1986)
M.S. Fataftah, J.M. Zadrozny, S.C. Coste, M.J. Graham, D.M. Rogers, D.E. Freedman, J. Am. Chem. Soc. 138, 1344 (2016)
Acknowledgements
This work was supported by the EPSRC (UK), by funding the National EPR Facility (NS/A000055/1) and an X-ray diffractometer (EP/K039547/1). We also thank the COST network in Molecular Spintronics for funding a scientific visit for AK to Stuttgart.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kostopoulos, A.K., Timco, G.A., Tuna, F. et al. Single Ion Anisotropy of CrIII and FeIII in a Series of {Ti7M} Rings. Appl Magn Reson 51, 1251–1265 (2020). https://doi.org/10.1007/s00723-020-01285-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00723-020-01285-z