
Abstract
Graphene-derived materials attract a great deal of attention because of the peculiar properties that make them suitable for a wide range of applications. Among such materials, nano-sized systems show very interesting behaviour and high reactivity. Often such materials have unpaired electrons that make them suitable for electron paramagnetic resonance (EPR) spectroscopy. In this work we study by continuous wave and pulse EPR spectroscopy undoped and nitrogen-doped graphene quantum dots (GQD) with a size of about 2 nm. The analysis of the spectra allows identifying different types of paramagnetic centers related to electrons localized on large graphenic flakes and molecular-like radicals. By hyperfine spectroscopies on nitrogen-doped samples, we determine the hyperfine coupling constant of paramagnetic centers (limited-size π-delocalized unpaired electrons) with dopant nitrogen atoms. The comparison of the experimental data with models obtained by density functional theory (DFT) calculations supports the interpretation of doping as due to the insertion of nitrogen atoms in the graphene lattice. The dimension of the delocalized regions in the flakes observed by pulse EPR is of about 20–25 carbon atoms; the nitrogen dopant can be classified as pyridinic or graphitic.
Similar content being viewed by others

1 Introduction
Graphene [1] is a flat and regular structure that is arousing technological interest for its peculiar properties [2, 3]. Graphene-derived materials include a large variety of materials, with quite different characteristics that is possible to tune by tuning the synthesis parameters and the presence of heteroatoms in the atomic lattice. Beside the large interest on the material itself, we note that only few techniques are available for the characterization of graphene-like materials [4]; in this respect, electron paramagnetic resonance (EPR) spectroscopy is an appropriate method to study them because such materials exhibit paramagnetic centers [5, 6].
Paradoxically, most of the interest in graphene-like materials derives from the possibility of profoundly affecting their properties by modifying their structure; this possibility stimulated the research in this field both for chemists and physicists. For instance, by reducing graphene down to small flakes (graphene quantum dots, GQDs) one obtains properties that are typical of nano-systems [7]. Besides, pure graphene is known to have a low reactivity; therefore, chemists are considering the substitution of atoms (i.e., doping) as one feasible way to promote it. Quite a number of studies on functionalized or doped graphene and on composite materials appeared in the literature (see e.g. refs [1,2,3, 8,9,10,11,12]), which show the possibility of tuning many of graphene chemical and physical properties and, above all, its reactivity.
Nitrogen-doped GQDs (N-GQDs) are a remarkable example of a very active catalyst in many reactions, including oxygen reduction reaction [13, 14]. N-GQDs can be obtained in many different ways, for example by electrochemical reaction with ammonia [15], exposure to nitrogen plasma [16], chemical vapor deposition on metals using a mixture of hydrocarbons, hydrogen, and nitrogen-containing molecules [14, 17, 18], pyrolysis of polymers [19], solvothermal synthesis [20], and chemical reaction of graphene oxide [21]. A further aspect of interest to physicists, is that doping of graphene with selected nuclei influences the number of electrons either in the conduction or in the valence band.
In this contribution we focus on N-GQDs. In this material each nitrogen substituting a carbon increases the number of π-electrons in conjugated regions. These extra electrons make the material particularly suited to EPR spectroscopy characterization. The EPR-active centers identified so far in graphene-derived materials can be ascribed to a restricted number of typologies [5]. Unfortunately, they all have quite similar features, and often there are interactions among them [22, 23]. We are confident that further investigation is required in this field with respect to the presence of defects that might have specific structure. In a way, doping can help identifying the presence of atomic substitution [24]. The materials studied in this work (GQDs and N-GQDs) were synthetized electrochemically starting from graphene oxide produced by the Hummers’ method [25, 26]. The percentage of doping for the N-GQDs sample, determined by XPS, is 5.1%. GQDs flakes have circular shape, whereas the N-doped flakes have a triangular shape (determined by scanning tunneling microscopy, STM). Flakes have an average size of about 2 nm, but with a rather broad distribution, and a thickness between 0.4 and 0.6 nm, typical of a single graphene layer [25, 27]. Transmission electron microscopy (TEM) investigation suggests that they are made up by a central core of sp2 carbons, whereas dopants and functional groups such as carbonyls, carboxylates or alcohols are mainly localized at the edges. We use continuous wave (cw) and pulse techniques to get structural information on the material, and, at the same time, to characterize the EPR signals related to this type of defects.
2 Experimental Section
2.1 EPR Measurements
The cw and pulse EPR measurements were obtained with an X-band Bruker ELEXSYS spectrometer, equipped with a dielectric resonator and a nitrogen/helium gas-flow cryostat for low temperature measurements. The powder samples were placed inside 2 mm ID quartz tubes and sealed under vacuum after the removal of the adsorbed gases. The EPR signals were recorded as function of temperature, from room temperature down to about 40 K. The magnetic field was calibrated using a standard with a known g-factor (7,7,8,8-tetracyanoquinodimethane lithium salt, LiTCNQ).
The pulse experiments were performed using the standard pulse sequences: π/2-τ-π-τ-echo for the electron spin echo decay measurements (Hahn decay, HD) and π-τ-π/2-τ-π-τ-echo for the echo-detected inversion recovery (IR). The resonance field was set at the maximum of the EPR intensity. The echo-detected EPR (ED-EPR) spectra were obtained by recording the echo intensity as a function of the magnetic field at a delay of 200 ns. The two-pulse electron spin echo envelope modulation (2p-ESEEM) spectra were obtained by Fourier transforming the modulation of the HD, after a proper reconstruction of the signal during the instrumental dead time. The pulse electron nuclear double resonance (ENDOR) spectra were collected using an ENDOR dielectric resonator and an ENI A300RF power amplifier for Davies ENDOR experiments. The experiments were performed using the Davies sequence, π-T-π/2-τ-π-τ-echo with a RF π pulse applied during the delay time T [28]. We used a microwave π/2 pulse of 100 ns, a RF π pulse of 10,000 ns, τ = 500 ns and T = 20,000 ns.
2.2 Simulation of EPR Spectra
The simulations of cw and ED-EPR spectra were obtained by a homemade Matlab code that gives as output the relevant EPR parameters for a variable number of species as described in [29]. To obtain the powder spectra I(B), the program calculates, for each spin packet oriented with an angle Ω (direction of the external field B in the g-frame) and at a given external magnetic field B, the contribution to resonance fi[B—B0(Ω)], where B0 is the resonance field; the calculation of the resonance field is derived after the diagonalization of the Zeeman Hamiltonian. The fi function can be either Lorentzian or Gaussian. The powder spectrum is obtained from the integration over all possible orientations of the magnetic field in the g-tensor reference frame; the presence of more than one species is allowed. Therefore
For ED-EPR, we considered no orientation-dependence of spin relaxation, hence, no lineshape distortion was considered; therefore, the same expression as for cw-EPR spectra was used.
2p-ESEEM and ENDOR spectra were simulated using a home-made program described in ref.s [30] and [31]. Briefly, for every orientation ûB = (l, m, n) of the magnetic field in the molecular frame (or of the hyperfine tensor T), the ENDOR transitions ν+ and ν− are calculated as
with A = l2Txx + m2Tyy + 2lmTxy and B2 = (lTxz + mTyz)2 + [lm(Txx − Tyy) − (l2 − m2)Txy]2. For the 2p-ESEEM, at first, the time-dependent profile was calculated, as function of τ; the modulation fuction depends of the ENDOR transitions as
where the modulation depth of the effect depends on \(k={\left({\nu }_{N}B\right)}^{2}/\left({{\nu }_{+}}^{2}{{\nu }_{-}}^{2}\right)\). Finally, the Fourier transform of the time evolution of the modulation provided the 2p-ESEEM simulation.
2.3 DFT Calculations
The density functional theory calculations reported here have been carried out on isolated molecules. Specifically, we adopted the B3LYP hybrid functional, which is known to provide reliable equilibrium molecular structures for radicals [32]. All the equilibrium molecular geometries and the hyperfine coupling constants (hfcc’s) reported here have been obtained with the B3LYP/6-311G** method using the Gaussian09 package [33].
3 Results
3.1 cw-EPR
We recorded the cw-EPR spectra of sample GQDs and N-GQDs in the range 80–290 K. The spectra recorded at the highest and lowest temperatures are reported in Fig. 1, after subtraction of the broad Mn(II) signal, which is ubiquitous for samples produced by Hummers' method [34] and remained in traces (< 0.1%), even after the purification steps. We determined the total spin concentration of the samples at 290 K by comparing the total area of the spectrum with the spectrum of a standard sample of Mn(II). We obtained 4.0 × 1014 spin/g for GQDs and 3.7 × 1014 spin/g for N-GQDs.

Normalized cw-EPR spectra of samples GQDs and N-GQDs at 290 and 80 K together with their simulations. Simulations were obtained considering single Lorentzian lineshape (except for sample N-GQDs at 80 K for which two components were necessary). Simulations obtained considering Gaussian lineshape are reported in the insets for samples at 290 K to show that a single Gaussian line provides a worse fitting of the spectra
A satisfactory simulation of the spectra at high temperatures was generally obtained using a single species with broad linewidth (see Table 1 for simulation parameters). The g-anisotropy, if barely observed at high temperature, is completely lost at lower temperature because of the broader linewidth. A second component in the cw-EPR spectra needs to be introduced in the simulations only for the N-GQDs sample at low temperature; this second species has a linewidth of about 1.5 G, an isotropic g-tensor (see Fig. 1), and is less saturable than the other. All components display a Curie–Weiss-like behavior [35], with an increase of the EPR intensity upon decreasing of the temperature.
Attempts of fitting were also conducted considering Gaussian lineshape (see insets in Fig. 1) but it was not possible to reproduce satisfactorily the experimental spectra.
3.2 Pulse EPR
All pulse experiments presented were performed at 80 K or below, because they provide a better S/N. For both samples we collected ED-EPR spectra at 80 K (Fig. 2), relative to the paramagnetic centers in GQDs (no metal impurities could be detected). Despite the relatively low-intensity cw-EPR spectrum, the echo intensity was well visible, therefore we expect the inhomogenously-broadened components to be a consistent fraction of the cw-EPR signal.

Normalized ED-EPR spectra of samples GQDs and N-GQDs recorded at 80 K together with their simulations
A satisfactory fit of the spectra was obtained by assuming the presence of two components with Gaussian lineshape. All the parameters obtained from the simulations of the ED-EPR spectra are collected in Table 2.
At the temperature of 80 K we also recorded the HD and IR traces of samples GQDs and N-GQDs (Fig. 3), selecting the magnetic field corresponding to the maximum intensity in the ED-EPR spectra in Fig. 2.

a HD trace of sample GQDs; b HD trace of sample N-GQDs; c IR trace of sample GQDs; d IR trace of sample N-GQDs. All traces were recorded at 80 K selecting the magnetic field corresponding to the maximum intensity in the ED-EPR spectra (Fig. 2). The traces were fitted using a bi-exponential decay function. Tests with mono-exponential functions were done but they were unable to reproduce the fast decay component of the traces
By fitting all the traces in Fig. 3 with bi-exponential decay functions we were able to determine the spin–lattice relaxation time (T1) and the phase memory time (TM). The parameters are collected in Table 3. Attempts to fit the traces with mono-exponential functions resulted in non-acceptable reproduction of the traces, especially of the fast-decaying component.
The weak but evident modulation observed in the HD experiments allowed obtaining the 2p-ESEEM spectra (see Fig. 4a and b). The spectra evidence the weak interaction of unpaired electrons with 1H (with small intensity for sample GQDs; the high-frequency modulation is visible also in the HD trace, Fig. 3b) at 14.9 MHz. From the analysis of the signal, we obtained an estimate of the protons hyperfine coupling constant. For the GQDs we found an average value of about 0.7 G, while for N-GQDs of about 1.9 G. From this experimental information, we derive that the paramagnetic centers are localized in slightly different environment, with GQDs having a less proton-rich environment. Both spectra also show bands at lower frequency. For the GQDs sample such bands, centered at 3.9 MHz, are relative to the coupling with 13C atoms dominated by an isotropic interaction of about 1.2 MHz. For the N-GQDs sample, even if a similar spectrum is obtained, a more careful inspection of the spectrum in the range 1–8 MHz shows that the bands are broader than those of GQDs and slightly shifted to indicate that new bands, relative to the coupling with 14 N, are emerging in this sample. To disentangle such two contributions, we acquired pulse ENDOR spectrum for sample N-GQDs at 80 K. The spectrum (not shown) displayed no signal in the frequency range of interest, likely because both the sequence was unsuited to detect low interacting 13C, and because of fast nuclear relaxation processes of 14 N. Therefore, we cooled the system below 80 K to slowdown such relaxation processes. The pulse ENDOR spectrum at 25 K for sample N-GQDs is reported in Fig. 4c) and clearly shows bands relative to the interaction with nitrogen. The attribution of the interaction to nitrogen is given on the basis of the appearance of such bands at low temperature (expected for 14 N bands), and on the basis of the much better quality of the fit obtained for a coupling to 14 N than that obtained for a coupling to 13C.

a 2p-ESEEM spectrum of sample GQDs at 80 K in which coupling with 13C and 1H nuclei are evident; the pulse sequence is 16–200-32–200-echo; b 2p-ESEEM spectrum of sample N-GQDs taken with the same conditions of spectrum a, c Davies ENDOR spectrum of sample N-GQDs at 25 K; the pulse sequence is 200-20,000-100-500-200-500 with a RF π pulse of length 10,000 ns applied during the delay time of 20,000 ns. The dashed lines are simulated spectra
To extract the hyperfine coupling tensor, we simulated the pulse ENDOR spectrum using a home-made program, as described in the Experimental Section. The hyperfine A tensor of the nitrogen atom with the unpaired electron obtained by the fit is: A = [6.1, 6.1, 11.5] MHz (aiso = 7.9 MHz). Finally, we verified if these obtained parameters are reasonable by superimposing a calculated ESEEM to the experimental one in Fig. 4b. The simulation reproduces the shape and the position of the nitrogen signals (see Fig. 4). Note that this simulation (without fitting) provided a general better reproduction of the low frequency bands than any simulation based on 13C interaction.
4 Discussion
4.1 Analysis of cw and Pulse EPR Spectra
The results obtained in this work show that GQDs and N-GQDs provide EPR spectra that are superposition of contributions attributed to different paramagnetic centers, with different intensities and characteristics. Likely, due the presence of Mn(II), an effective relaxant, some signals are unobservable, because broadened out by the interaction with the metal ion [36]. Those observed are due to unpaired electrons non-interacting with Mn(II) ions.
Based on previous works (see for example refs. [37,38,39,40,41]), it is known that different cw-EPR spectra can be obtained from graphene-like materials depending on the dimension/shape of the flakes, and on the presence and type of defects. So far, the EPR signals can be interpreted by assuming the presence of three types of electrons: mobile electrons, electrons localized at the edge of the flakes or in proximity of defects (whose energy level is at the contact point between the valence and conduction bands), and unpaired electrons in molecular-like radicals [4, 29, 42, 43]; the picture is slightly complicated by the presence of magnetic interactions among them [22]. Therefore, we interpret our results within this framework.
The different contributions are revealed using different approaches, namely the use of both cw and pulse techniques. For the two samples, GQDs and N-GQDs, the cw-EPR spectra at room temperature are quite similar; both are characterized by the presence of an EPR band with a g-factor of ca. 2, typical of organic aromatic systems with low spin–orbit coupling. The concentration of paramagnetic centers is of the same order of magnitude as that found for GQD produced by other methods [44], and confirms that only a small fraction of GQD has one unpaired electron: in most cases GQD are diamagnetic. This is a bit surprising if compared with large-dimension flakes. For example, reduced GO and the N-doped structures, have spin densities (determined by EPR) one order of magnitude larger [45]. Two reasons can be invoked to explain this evidence: in finite extended structures, the interruption of the π-system create edge states at the Fermi level [42, 46], connected to the topology of the edge. In small-dimension systems, the topology of the molecular structure can also result in paramagnetic states [47] but these structures possess rather localized electrons, and, likely, they are more reactive, therefore they can undergo coupling reactions. The second reason is the broadening out of the signals induced by the interaction with extrinsic paramagnetic impurities (Mn(II)) which reduce the EPR-measured spin concentration.
As stated earlier, in heterogeneous samples, the cw-EPR profile is the sum of all the contributions. While inhomogeneous contributions can be revealed by pulse EPR, the isolation of the signals attributed to homogeneous contributions is more difficult to achieve. The cw-EPR spectra of both samples were simulated by a single Lorentzian line, but in parent materials (GO) a Lorentzian-like lineshape was fitted as sum of Gaussians [36]. The response to saturation is a way to determine if the line is homogeneously, or inhomogeneously broadened, but we anticipate that the presence of the two generates a complex saturation profile.
As we know that in our samples we have also large dimension fragments, either mobile electrons or edge electrons can provide Lorentzian lineshapes [29]. Conduction electrons can be distinguished from those localized by considering the variation of the EPR intensity with the temperature: the EPR signal due to mobile electrons decreases with the lowering of the temperature, because electrons are depleted from the conduction band [29, 38], whereas edge and defect states have a regular Curie or Curie–Weiss behavior. As for both our samples we observe an increase of the spectrum intensity by lowering the temperature, we conclude that the signals at high temperature are due to localized electrons. The structures of these centers, are likely constituted by unpaired electrons in the proximity of flakes of dimensions large enough to have conduction electrons available. GQDs with larger dimensions of about 30 nm [44] show analogous properties in terms of lineshape and concentration. In ref [44] pulse techniques have not been used to determine the slow-relaxing components.
The narrow signal that becomes evident for the N-GQDs sample at low temperature and at high microwave power has the same lineshape and temperature dependence of the other component, so it can be attributed to edge states as well. These last are characterized by a different exchange interaction strength, which influences both the linewidth and the intensity temperature variation, mostly at low temperatures. Because this component is absent in the GQDs sample, we attribute this band to the presence of extra electrons due to nitrogen inclusion; unfortunately, because of the homogeneous lineshape, we cannot deduce information on the interaction with nitrogen nuclei, and we have not investigated further.
Spin-echo pulse techniques, instead, allow the cancelation of these fast-relaxing contributions, whose spin-echo intensities decay to almost zero within the instrumental dead time. At the same time, spin-echo pulse techniques enable the separation and the identification of the species with slow relaxing spin; previously, the method has been used to identify slow-relaxing species in GO and reduced GO (RGO) [36, 48, 49]. Beside the HD experiments to measure the phase memory time, ED-EPR spectra can reveal the spectral components; they are characterized by inhomogeneously broadened components (Gaussian lineshape) [50, 51], and hyperfine spectroscopies (mostly ESEEM and ENDOR) can be used to extract hyperfine interactions. The absence of Heisenberg exchange interactions in these components is an indication that they are relative to localized electrons in (magnetically isolated) small flakes, not in the proximity of conduction electrons (promoting exchange interactions). Such flakes contain substitutional nitrogen nuclei.
To get a clearer idea on the structure responsible of these signals, we calculated the hyperfine interaction by DFT and compared them with the experimental values obtained form 2p-ESEEM and ENDOR. To this aim, we started from the experimental evidence, generated a guess structure, and introduced small structural variations to approach the experimental values. Such variations are the change of the dimensions of the flakes, and the placement of the substitutional nitrogen. Three types of placements are possible, generating three types of atoms with different characteristics: pyrrolic, pyridinic or graphitic [13]. Pyridinic and pyrrolic nitrogens are along the edges of the quantum dots, in hexagonal or pentagonal rings, and contribute to the π-conjugated system with, respectively, one and two p-electrons. When a nitrogen atom substitutes a carbon atom in the graphene honeycomb layer the nitrogen is called graphitic nitrogen; it contributes to the π-system with two p-electron, one more than carbon. These different types of atoms have been identified also by XPS measurements [25]. In the next section we present and discuss the structures that gave the best results.
4.2 Calculation of the Hyperfine Interactions
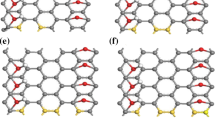
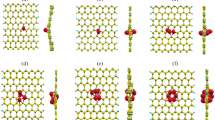
To get information about the nitrogen doping we have carried out DFT calculations on a series of models that are supposed to be similar to the N-GQDs. For such molecules we optimized the geometries and calculated the hfcc’s. From previous STM measurements we know that our N-GQDs have a broad size distribution peaked at 2–3 nm and possess on average about 60 carbon atoms [25]. For the sake of computational speed, we started with smaller models, thereafter we gradually increased their size (Fig. 5). All models were selected as neutral systems with an odd number of electrons (i.e., in doublet states). The only exception is model N07 which has 46 π electrons and can be either in a singlet or a triplet state. The geometry and the EPR calculations of N07 were done assuming a triplet state. We modified the dimension, the shape, the type of edges and the position of the nitrogen dopant inside the molecular models. Most of them have the nitrogen atom on the external edge, which is either zig-zag (N05, N06, N07, N08, N10) or armchair (N09). Two models have the dopant inside (N04, N11). Molecules N02 and N12 have the nitrogen atom in a five-membered ring, whereas all the other models have the typical graphenic structure with all six-membered rings. Molecules N01, N02, N06 and N07 have pyridinic nitrogen, molecule N12 has pyrrolic nitrogen and all the other have graphenic nitrogen.

Structures of the molecular models used to describe the N-GQDs sample by DFT calculations. The N atoms are marked with blue circles. The reported equilibrium geometries have been computed at the B3LYP/6-311G** level. With the exception of N07 (see text) all these models have at least one unpaired π-electron
For all such models we obtained the equilibrium geometries and, starting from them, we computed the hyperfine coupling constants. The values obtained for each molecular model are collected in Table 4. The highest hfcc’s values have been obtained for N02 and N06 (17.14 and 11.53 MHz, respectively). In such models the sp2-hybridized nitrogen atom contributes to the conjugated system only with one electron and has no hydrogen atom directly bound, even if it is located along the edge (the nitrogen lone pair is formally described by one the sp2 hybrids, with the lobe extending away from the edge). Besides N02 and N06, high hfcc’s values (7–9 MHz) have been found for the smaller models with 23 carbon atoms (N01, N03, N04), independently from the position of nitrogen within the molecule. For the larger models, the computed values of the hfcc’s are about 1 MHz or lower. From such calculations we found that the isotropic part of the hyperfine coupling tensor of nitrogen strongly depends on the dimension of the molecule and, instead, it’s almost unaffected by the type of nitrogen.
We note that the molecules can be well separated in three groups on the base of the hyperfine interaction, with aiso relatively large (aiso > 10 MHz), intermediate (aiso = 7–9 MHz ca) or small (aiso < 2 MHz). The AZZ value follows the same trend. The experimental value for the N-GQDs sample (aiso = 7.9 MHz) falls into the intermediate interaction group (Table 4), therefore, we find a close similarity with the smallest models, namely N01, N03 and N04, that are rather distant from those with much smaller aiso values. We note that the experimental dipolar part is smaller than the calculated one for these structures, but we remark that in the model samples, for simplicity, we did not consider side groups that can perturb the electron density distribution (e.g., hydroxyl, carboxyl, and other functional groups that can be introduced in graphene by the Hummers method). Since the mean dimension of N-GQDs (20 nm–see Introduction) is larger than the dimension suggested by DFT calculations (see above), we think that the paramagnetic N-GQDs (a small fraction of the flakes) have a dimension smaller than the average.
5 Conclusions
The analysis of the EPR spectra of GQDs and N-doped GQDs allows identifying different types of paramagnetic centers. Both in undoped and N-doped GQDs the most abundant contribution derived from cw-EPR spectra is assigned to electrons localized on relatively large graphenic flakes, which anyway represent a minority fraction of the investigated colloidal solutions. These electrons, interacting with the conduction ones, are characterized by a Lorentzian lineshape. The absence of any hyperfine interaction prevents any further investigation about the inclusion of nitrogen atoms in the graphene structure.
More interesting is the analysis of the pulse-EPR spectra, which reveals the presence of electrons with slow spin relaxation in flakes with small dimensions. A qualitative analysis makes us suppose that the relative concentration of such paramagnetic centers is relevant in the cw-EPR spectrum. Pulse-EPR experiments allowed identification of the hyperfine interaction of electrons with slow spin relaxation and nitrogen nuclei. From the interpretation of the spectra and the description of N-doped GQDs by selected DFT models, we confirm that doping in N-GQDs is due to the insertion of nitrogen atoms in the graphene lattice. The region of the flakes in which electrons are delocalized, as observed by pulse-EPR, is a fraction of the paramagnetic N-GQD, and it is composed of about 20–25 carbon atoms in an aromatic system. In these flakes, the nitrogen dopant can be classified as pyridinic or graphitic, and the pyrrolic structure in this type of defects is ruled out.
Data Availability
Not applicable.
Code Availability
Not applicable.
References
L. Gong, I.A. Kinloch, R.J. Young, I. Riaz, R. Jalil, K.S. Novoselov, Adv. Mater. 22, 2694 (2010)
V. Singh, D. Joung, L. Zhai, S. Das, S.I. Khondaker, S. Seal, Prog. Mater. Sci. 56, 1178 (2011)
S. Stankovich, D.A. Dikin, G.H.B. Dommett, K.M. Kohlhaas, E.J. Zimney, E.A. Stach, R.D. Piner, S.T. Nguyen, R.S. Ruoff, Nature 442, 282 (2006)
A. Barbon, in Specialist Periodical Reports Electron Paramagnetic Resonance, ed. by V. Chechik, D.M. Murphy (Royal Society of Chemistry, London, 2018), pp. 38–65
C.N.R. Rao, A.K. Sood (eds.), Graphene: Synthesis, Properties, and Phenomena (Wiley-VCHVerlag GmbH & Co. KGaA, Weinheim, 2013)
D. Savchenko, A.H. Kassiba (eds.), Frontiers in Magnetic Resonance. Electron Paramagnetic Resonance in Modern Carbon-Based Nanomaterials (Bentham Science Publishers, Sharjah, 2018)
S.N. Baker, G.A. Baker, Angew. Chem. Int. Ed. 49, 6726 (2010)
S. Stankovich, D.A. Dikin, R.D. Piner, K.A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S.T. Nguyen, R.S. Ruoff, Carbon NY 45, 1558 (2007)
S. Agnoli, G. Granozzi, Surf. Sci. 609, 1 (2013)
M.J. McAllister, J.-L. Li, D.H. Adamson, H.C. Schniepp, A.A. Abdala, J. Liu, M. Herrera-Alonso, D.L. Milius, R. Car, R.K. Prudhomme, I.A. Aksay, Chem. Mater. 19, 4396 (2007)
T. Ramanathan, A.A. Abdala, S. Stankovich, D.A. Dikin, M. Herrera-Alonso, R.D. Piner, D.H. Adamson, H.C. Schniepp, X. Chen, R.S. Ruoff, S.T. Nguyen, I.A. Aksay, R.K. PrudHomme, L.C. Brinson, Nat. Nanotechnol. 3, 327 (2008)
H.C. Schniepp, J.-L. Li, M.J. McAllister, H. Sai, M. Herrera-Alonso, D.H. Adamson, R.K. Prudhomme, R. Car, D.A. Saville, I.A. Aksay, J. Phys. Chem. B 110, 8535 (2006)
H. Wang, T. Maiyalagan, X. Wang, ACS Catal. 2, 781 (2012)
D. Usachov, O. Vilkov, A. Grüneis, D. Haberer, A. Fedorov, V.K. Adamchuk, A.B. Preobrajenski, P. Dudin, A. Barinov, M. Oehzelt, C. Laubschat, D.V. Vyalikh, Nano Lett. 11, 5401 (2011)
X. Wang, X. Li, L. Zhang, Y. Yoon, P.K. Weber, H. Wang, J. Guo, H. Dai, Science 324, 768 (2009)
Y. Shao, S. Zhang, M.H. Engelhard, G. Li, G. Shao, Y. Wang, J. Liu, I.A. Aksay, Y. Lin, J. Mater. Chem. 20, 7491 (2010)
L. Qu, Y. Liu, J.-B. Baek, L. Dai, ACS Nano 4, 1321 (2010)
Z. Jin, J. Yao, C. Kittrell, J.M. Tour, ACS Nano 5, 4112 (2011)
Z. Sun, Z. Yan, J. Yao, E. Beitler, Y. Zhu, J.M. Tour, Nature 468, 549 (2010)
D. Deng, X. Pan, L. Yu, Y. Cui, Y. Jiang, J. Qi, W.-X. Li, Q. Fu, X. Ma, Q. Xue, G. Sun, X. Bao, Chem. Mater. 23, 1188 (2011)
Z.-H. Sheng, L. Shao, J.-J. Chen, W.-J. Bao, F.-B. Wang, X.-H. Xia, ACS Nano 5, 4350 (2011)
V.L.J. Joly, K. Takahara, K. Takai, K. Sugihara, T. Enoki, M. Koshino, H. Tanaka, Phys. Rev. B 81, 115408 (2010)
M.A. Augustyniak-Jabłokow, K. Tadyszak, M. Maćkowiak, S. Lijewski, Chem. Phys. Lett. 557, 118 (2013)
W.B. Fowler, Encyclopedia of Condensed Matter Physics (Elsevier, Amsterdam, 2005), pp. 318–323
M. Favaro, L. Ferrighi, G. Fazio, L. Colazzo, C. Di Valentin, C. Durante, F. Sedona, A. Gennaro, S. Agnoli, G. Granozzi, ACS Catal. 5, 129 (2015)
M. Favaro, S. Agnoli, M. Cattelan, A. Moretto, C. Durante, S. Leonardi, J. Kunze-Liebhäuser, O. Schneider, A. Gennaro, G. Granozzi, Carbon NY 77, 405 (2014)
M. Favaro, F. Carraro, M. Cattelan, L. Colazzo, C. Durante, M. Sambi, A. Gennaro, S. Agnoli, G. Granozzi, J. Mater. Chem. A 3, 14334 (2015)
A. Schweiger, G. Jeschke, Principles of Pulse Electron Paramagnetic Resonance (Oxford University Press, New York, 2001)
F. Tampieri, S. Silvestrini, R. Riccò, M. Maggini, A. Barbon, J. Mater. Chem. C 2, 8105 (2014)
M. Brustolon, A.L. Maniero, S. Jovine, U. Segre, Res. Chem. Intermed. 22, 359 (1996)
A. Barbon, M. Brustolon, A.L. Maniero, M. Romanelli, L.-C. Brunel, Phys. Chem. Chem. Phys. 1, 4015 (1999)
M. Kaupp, M. Bühl, V.G. Malkin, Calculation of NMR and EPR Parameters (Wiley, Amsterdam, 2004)
Gaussian 09, Revision D.01, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, D. J. Fox, Gaussian, Inc., Wallingford CT (2016)
W.S. Hummers, R.E. Offeman, J. Am. Chem. Soc. 80, 1339 (1958)
N. Ashcroft, D. Mermin, Solid State Physics (Saunders College, New York, 1976)
M.A. Augustyniak-Jabłokow, R. Fedaruk, R. Strzelczyk, Ł. Majchrzycki, Appl. Magn. Reson. 50, 761 (2019)
G. Wagoner, Phys. Rev. 118, 647 (1960)
L. Ćirić, A. Sienkiewicz, B. Náfrádi, M. Mionić, A. Magrez, L. Forró, Phys. Status Solidi 246, 2558 (2009)
S.S. Rao, A. Stesmans, D.V. Kosynkin, A. Higginbotham, J.M. Tour, New J. Phys. 13, 113004 (2011)
B.S. Paratala, B.D. Jacobson, S. Kanakia, L.D. Francis, B. Sitharaman, PLoS ONE 7, e38185 (2012)
A. Barbon, M. Brustolon, Appl. Magn. Reson. 42, 197 (2012)
M. Tommasini, C. Castiglioni, G. Zerbi, A. Barbon, M. Brustolon, Chem. Phys. Lett. 516, 220 (2011)
S.R. Singamaneni, J. van Tol, R. Ye, J.M. Tour, Appl. Phys. Lett. 107, 212402 (2015)
K. Tadyszak, A. Musiał, A. Ostrowski, J.K. Wychowaniec, Nanomaterials 10, 798 (2020)
B. Wang, V. Likodimos, A.J. Fielding, R.A.W. Dryfe, Carbon NY 160, 236 (2020)
G. Zerbi, A. Barbon, R. Bengalli, A. Lucotti, T. Catelani, F. Tampieri, M. Gualtieri, M. D’Arienzo, F. Morazzoni, M. Camatini, Nanoscale 9, 13640 (2017)
Y. Morita, S. Suzuki, K. Sato, T. Takui, Nat. Chem. 3, 197 (2011)
M.A. Augustyniak-Jabłokow, K. Tadyszak, R. Strzelczyk, R. Fedaruk, R. Carmieli, Carbon NY 152, 98 (2019)
M.A. Augustyniak-Jabłokow, R. Strzelczyk, R. Fedaruk, Carbon NY 168, 665 (2020)
A. Barbon, F. Tampieri, A.I.M.S. Mater, Science 4, 147 (2017)
F. Tampieri, A. Barbon, in Frontiers in Magnetic Resonance, ed. by D. Savchenko, A.H. Kassiba (Bentham Science, Sharjah, 2018), pp. 36–66
Funding
Open access funding provided by Università degli Studi di Padova within the CRUI-CARE Agreement.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of Interest
The Authors declare that there is no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tampieri, F., Tommasini, M., Agnoli, S. et al. N-Doped Graphene Oxide Nanoparticles Studied by EPR. Appl Magn Reson 51, 1481–1495 (2020). https://doi.org/10.1007/s00723-020-01276-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00723-020-01276-0