Abundance Tracking by Long-Read Nanopore Sequencing of Complex Microbial Communities in Samples from 20 Different Biogas/Wastewater Plants


Abstract
:1. Introduction
2. Materials and Methods
2.1. DNA Extraction
2.2. Library Preparation
2.3. Sequencing
2.4. Bioinformatic Tools
3. Results
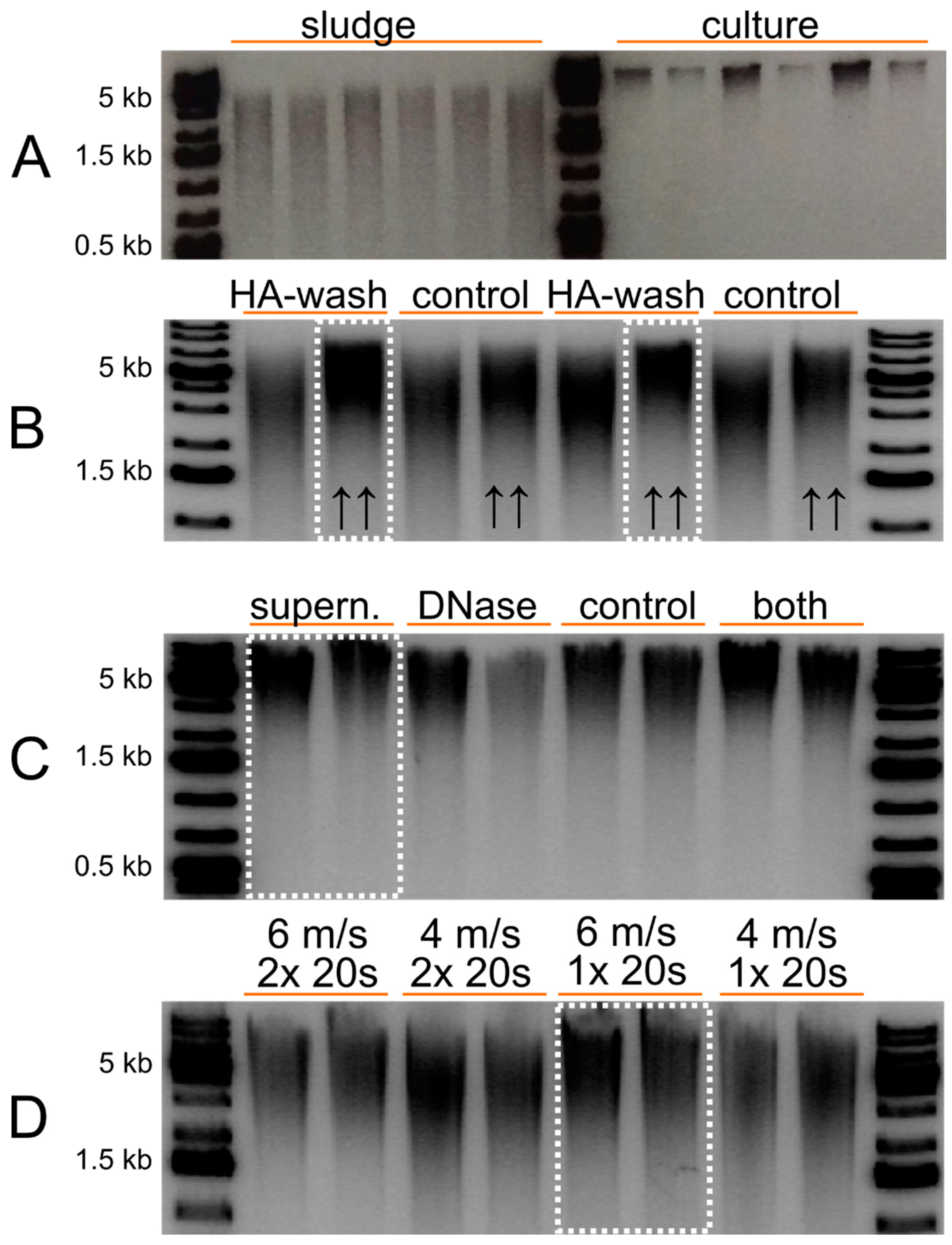
3.1. Digestate Samples and DNA Isolation
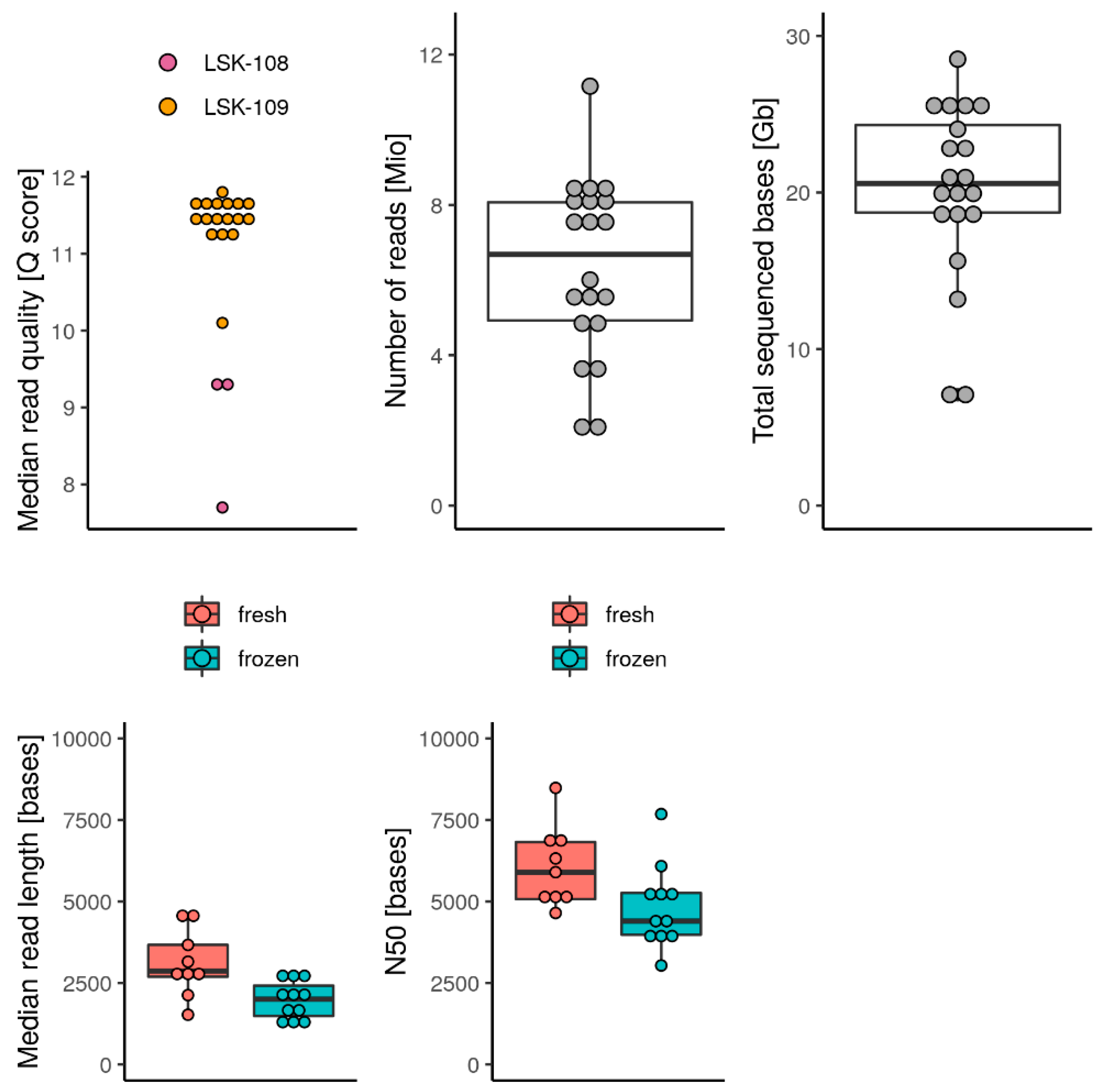
3.2. Sequencing Yield and Quality of Metagenomic DNA
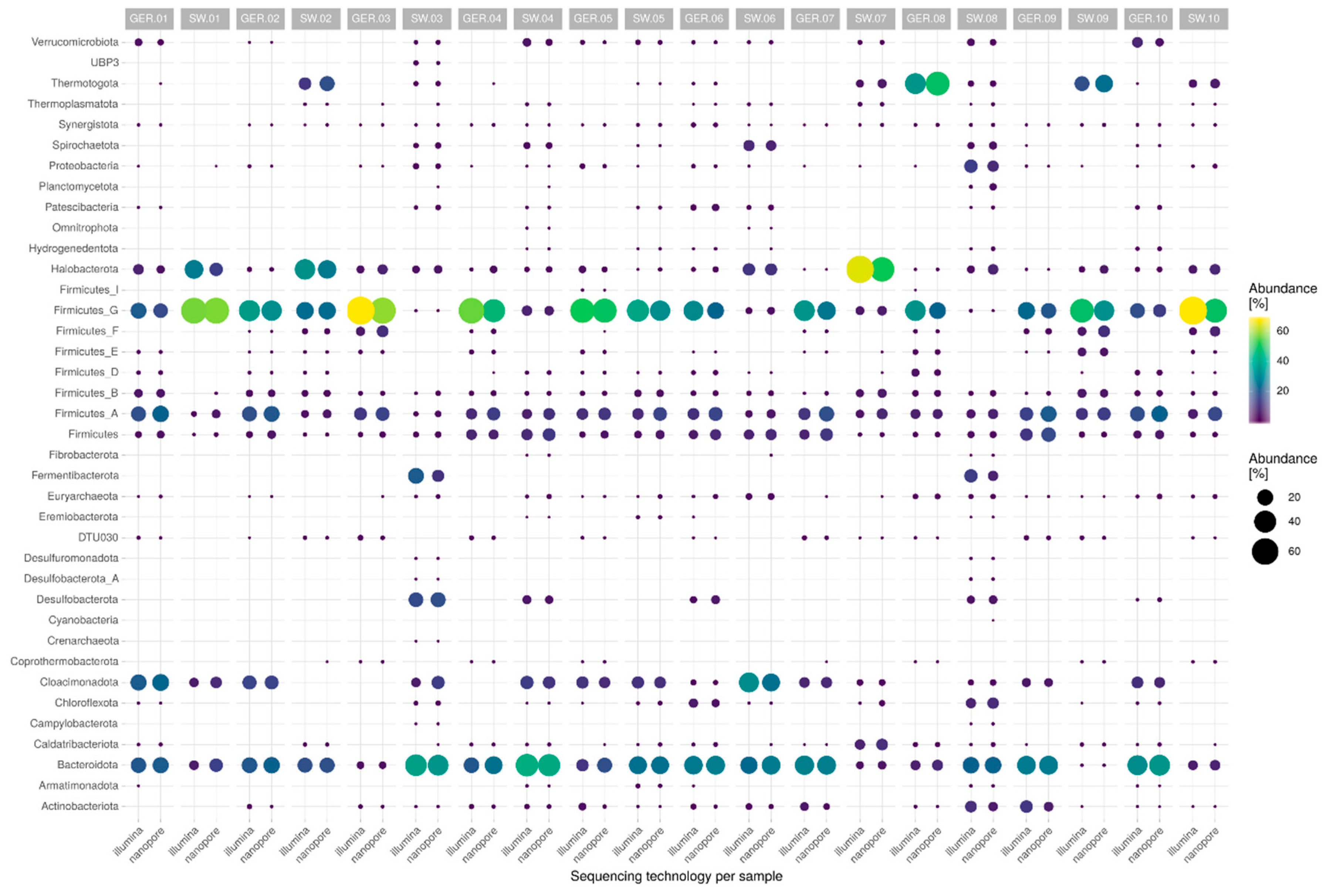
3.3. Abundance Estimation and Taxonomic Read Classification
4. Discussion
4.1. DNA Isolation and Sequencing
4.2. Database Choice and Abundance Estimation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
References
- Campanaro, S.; Treu, L.; Kougias, P.G.; Luo, G.; Angelidaki, I. Metagenomic binning reveals the functional roles of core abundant microorganisms in twelve full-scale biogas plants. Water Res. 2018, 140, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Ziganshin, A.M.; Ziganshina, E.E.; Kleinsteuber, S.; Nikolausz, M. Comparative Analysis of Methanogenic Communities in Different Laboratory-Scale Anaerobic Digesters. Archaea 2016, 2016, 3401272. [Google Scholar] [CrossRef] [PubMed]
- Campanaro, S.; Treu, L.; Rodriguez-R, L.M.; Kovalovszki, A.; Ziels, R.M.; Maus, I.; Zhu, X.; Kougias, P.G.; Basile, A.; Luo, G.; et al. The anaerobic digestion microbiome: A collection of 1600 metagenome-assembled genomes shows high species diversity related to methane production. bioRxiv 2019, 680553. [Google Scholar] [CrossRef]
- Krause, L.; Diaz, N.N.; Edwards, R.A.; Gartemann, K.-H.; Krömeke, H.; Neuweger, H.; Pühler, A.; Runte, K.J.; Schlüter, A.; Stoye, J.; et al. Taxonomic composition and gene content of a methane-producing microbial community isolated from a biogas reactor. J. Biotechnol. 2008, 136, 91–101. [Google Scholar] [CrossRef]
- Stolze, Y.; Bremges, A.; Rumming, M.; Henke, C.; Maus, I.; Pühler, A.; Sczyrba, A.; Schlüter, A. Identification and genome reconstruction of abundant distinct taxa in microbiomes from one thermophilic and three mesophilic production-scale biogas plants. Biotechnol. Biofuels 2016, 9, 156. [Google Scholar] [CrossRef] [Green Version]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef]
- Suzuki, M.T.; Giovannoni, S.J. Bias caused by template annealing in the amplification of mixtures of 16S rRNA genes by PCR. Appl. Environ. Microbiol. 1996, 62, 625–630. [Google Scholar] [CrossRef] [Green Version]
- Acinas, S.G.; Sarma-Rupavtarm, R.; Klepac-Ceraj, V.; Polz, M.F. PCR-induced sequence artifacts and bias: Insights from comparison of two 16S rRNA clone libraries constructed from the same sample. Appl. Environ. Microbiol. 2005, 71, 8966–8969. [Google Scholar] [CrossRef] [Green Version]
- Teng, F.; Darveekaran Nair, S.S.; Zhu, P.; Li, S.; Huang, S.; Li, X.; Xu, J.; Yang, F. Impact of DNA extraction method and targeted 16S-rRNA hypervariable region on oral microbiota profiling. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef]
- Louca, S.; Doebeli, M.; Parfrey, L.W. Correcting for 16S rRNA gene copy numbers in microbiome surveys remains an unsolved problem. Microbiome 2018, 6. [Google Scholar] [CrossRef]
- Krehenwinkel, H.; Wolf, M.; Lim, J.Y.; Rominger, A.J.; Simison, W.B.; Gillespie, R.G. Estimating and mitigating amplification bias in qualitative and quantitative arthropod metabarcoding. Sci. Rep. 2017, 7, 17668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quince, C.; Walker, A.W.; Simpson, J.T.; Loman, N.J.; Segata, N. Shotgun metagenomics, from sampling to analysis. Nat. Biotechnol. 2017, 35, 833–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearman, W.S.; Freed, N.E.; Silander, O.K. The advantages and disadvantages of short- and long-read metagenomics to infer bacterial and eukaryotic community composition. bioRxiv 2019, 650788. [Google Scholar] [CrossRef] [Green Version]
- Sanderson, N.D.; Street, T.L.; Foster, D.; Swann, J.; Atkins, B.L.; Brent, A.J.; McNally, M.A.; Oakley, S.; Taylor, A.; Peto, T.E.A.; et al. Real-time analysis of nanopore-based metagenomic sequencing from infected orthopaedic devices. BMC Genom. 2018, 19, 714. [Google Scholar] [CrossRef] [Green Version]
- Somerville, V.; Lutz, S.; Schmid, M.; Frei, D.; Moser, A.; Irmler, S.; Frey, J.E.; Ahrens, C.H. Long read-based de novo assembly of low complex metagenome samples results in finished genomes and reveals insights into strain diversity and an active phage system. bioRxiv 2018, 476747. [Google Scholar] [CrossRef] [Green Version]
- Nicholls, S.M.; Quick, J.C.; Tang, S.; Loman, N.J. Ultra-deep, long-read nanopore sequencing of mock microbial community standards. Gigascience 2019, 8. [Google Scholar] [CrossRef]
- De Boer, R.; Peters, R.; Gierveld, S.; Schuurman, T.; Kooistra-Smid, M.; Savelkoul, P. Improved detection of microbial DNA after bead-beating before DNA isolation. J. Microbiol. Methods 2010, 80, 209–211. [Google Scholar] [CrossRef]
- Güllert, S.; Fischer, M.A.; Turaev, D.; Noebauer, B.; Ilmberger, N.; Wemheuer, B.; Alawi, M.; Rattei, T.; Daniel, R.; Schmitz, R.A.; et al. Deep metagenome and metatranscriptome analyses of microbial communities affiliated with an industrial biogas fermenter, a cow rumen, and elephant feces reveal major differences in carbohydrate hydrolysis strategies. Biotechnol. Biofuels 2016, 9, 121. [Google Scholar] [CrossRef] [Green Version]
- Brandt, C. Long-read DNA Preparation for Metagenomic Samples. ProtocolsIo 2019. [Google Scholar] [CrossRef]
- Quick, J. One-pot Ligation Protocol for Oxford Nanopore Libraries. ProtocolsIo 2018. [Google Scholar] [CrossRef]
- Kim, D.; Song, L.; Breitwieser, F.P.; Salzberg, S.L. Centrifuge: Rapid and sensitive classification of metagenomic sequences. Genome Res. 2016, 26, 1721–1729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Méric, G.; Wick, R.R.; Watts, S.C.; Holt, K.E.; Inouye, M. Correcting index databases improves metagenomic studies. bioRxiv 2019, 712166. [Google Scholar] [CrossRef]
- Parks, D.H.; Chuvochina, M.; Chaumeil, P.-A.; Rinke, C.; Mussig, A.J.; Hugenholtz, P. Selection of representative genomes for 24,706 bacterial and archaeal species clusters provide a complete genome-based taxonomy. bioRxiv 2019, 771964. [Google Scholar] [CrossRef] [Green Version]
- Gweon, H.S.; Shaw, L.P.; Swann, J.; De Maio, N.; AbuOun, M.; Niehus, R.; Hubbard, A.T.M.; Bowes, M.J.; Bailey, M.J.; Peto, T.E.A.; et al. The impact of sequencing depth on the inferred taxonomic composition and AMR gene content of metagenomic samples. Environ. Microbiome 2019, 14, 7. [Google Scholar] [CrossRef] [Green Version]
- Westerholm, M.; Schnürer, A. Microbial Responses to Different Operating Practices for Biogas Production Systems. Anaerob. Dig. 2019. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Liu, T.; Müller, B.; Schnürer, A. The microbial community structure in industrial biogas plants influences the degradation rate of straw and cellulose in batch tests. Biotechnol. Biofuels 2016, 9, 128. [Google Scholar] [CrossRef] [Green Version]
- Kazda, M.; Langer, S.; Bengelsdorf, F.R. Fungi open new possibilities for anaerobic fermentation of organic residues. Energy Sustain. Soc. 2014, 4, 6. [Google Scholar] [CrossRef]
- Stalder, T.; Press, M.O.; Sullivan, S.; Liachko, I.; Top, E.M. Linking the resistome and plasmidome to the microbiome. ISME J. 2019, 13, 2437–2446. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | Substrate | Operation | Status | c(dsDNA) (ng/µL) | |||||
|---|---|---|---|---|---|---|---|---|---|
| Organic Household Waste | Slaughter-House Waste | Manure | Sewage Sludge | Green +-Based | |||||
| Sweden | 01-SW # | o | Mesophilic | Fresh | 250 | ||||
| 02-SW | o | o | Thermophilic | Fresh | 450 | ||||
| 03-SW | o | Mesophilic | Fresh | 200 | |||||
| 04-SW | o | Mesophilic | Fresh | 366 | |||||
| 05-SW | o | Mesophilic | Frozen | 362 | |||||
| 06-SW | o | Mesophilic | Fresh | 748 | |||||
| 07-SW | o | o | Mesophilic | Fresh | 436 | ||||
| 08-SW | o | Mesophilic | Fresh | 454 | |||||
| 09-SW | o | o | Thermophilic | Fresh | 418 | ||||
| 10-SW | o | Mesophilic | Fresh | 228 | |||||
| Germany | 01-GER | o | Mesophilic | Frozen | 533 | ||||
| 02-GER | o | o | Hyper-mesophilic | Frozen | 282 | ||||
| 03-GER | o | o | Hyper-mesophilic | Frozen | 410 | ||||
| 04-GER | o | o | Thermophilic | Frozen | 594 | ||||
| 05-GER | o | o | Hyper-mesophilic | Frozen | 190 | ||||
| 06-GER | o | o | Mesophilic | Frozen | 242 | ||||
| 07-GER * | o | o | Mesophilic | Frozen | 106 | ||||
| 08-GER | o | o | Thermophilic | Frozen | 64 | ||||
| 09-GER * | o | o | Mesophilic | Frozen | 260 | ||||
| 10-GER | o | o | Mesophilic | Frozen | 320 | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brandt, C.; Bongcam-Rudloff, E.; Müller, B. Abundance Tracking by Long-Read Nanopore Sequencing of Complex Microbial Communities in Samples from 20 Different Biogas/Wastewater Plants. Appl. Sci. 2020, 10, 7518. https://doi.org/10.3390/app10217518
Brandt C, Bongcam-Rudloff E, Müller B. Abundance Tracking by Long-Read Nanopore Sequencing of Complex Microbial Communities in Samples from 20 Different Biogas/Wastewater Plants. Applied Sciences. 2020; 10(21):7518. https://doi.org/10.3390/app10217518
Chicago/Turabian StyleBrandt, Christian, Erik Bongcam-Rudloff, and Bettina Müller. 2020. "Abundance Tracking by Long-Read Nanopore Sequencing of Complex Microbial Communities in Samples from 20 Different Biogas/Wastewater Plants" Applied Sciences 10, no. 21: 7518. https://doi.org/10.3390/app10217518