
Abstract
Following periods of haematopoietic cell stress, such as after chemotherapy, radiotherapy, infection and transplantation, patient outcomes are linked to the degree of immune reconstitution, specifically of T cells. Delayed or defective recovery of the T cell pool has significant clinical consequences, including prolonged immunosuppression, poor vaccine responses and increased risks of infections and malignancies. Thus, strategies that restore thymic function and enhance T cell reconstitution can provide considerable benefit to individuals whose immune system has been decimated in various settings. In this Review, we focus on the causes and consequences of impaired adaptive immunity and discuss therapeutic strategies that can recover immune function, with a particular emphasis on approaches that can promote a diverse repertoire of T cells through de novo T cell formation.
Similar content being viewed by others

Introduction
Recovery of immunocompetence after periods of haematopoietic stress or injury is crucial not only for efficient responses against pathogens and tumour antigens but also for optimal responses to immunotherapy for cancer. In contrast to the early recovery of innate cells, including neutrophils, natural killer (NK) cells and monocytes, adaptive immune cells, in particular T cells, recover at a much slower pace and are particularly sensitive to negative insults caused by infections or cytoreductive chemotherapy and radiotherapy. Constrictions in the diversity of the T cell pool have been associated with impaired immune responses to several antigens1,2,3 and adverse clinical outcomes in patients receiving haematopoietic cell transplantation (HCT)4,5.
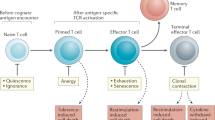
The capacity of T cells to mount and maintain effective responses to a wide variety of antigens depends on a large repertoire of unique T cell receptors (TCRs) generated in the thymus during the process of T cell development. This process is dependent on crosstalk between bone marrow (BM)-derived T cell progenitors and the supportive thymic stromal microenvironment, which primarily consists of thymic epithelial cells (TECs), endothelial cells, mesenchymal stromal cells, dendritic cells and macrophages6. Although, for example, T cell proliferation, driven by interleukin-7 (IL-7) and IL-15, in response to lymphopenic conditions can contribute to numerical reconstitution of T cells, complete long-term recovery of a diverse and functional T cell pool requires reactivation of thymic function and de novo T cell generation (Fig. 1). However, the thymus is sensitive to various injuries, such as those caused by cytoreductive treatments, infection, septic shock and graft-versus-host disease (GVHD). Furthermore, progressive involution of thymic tissue during ageing leads to a decline in T cell output and T cell senescence with restricted TCR repertoire diversity and impaired immune responses.

In the first period following haematopoietic cell transplantation (HCT), immune cells follow a predictable course of reconstitution. In contrast to the relatively early recovery of innate immune cells, recipients of HCT experience prolonged deficiencies in T cells and B cells, which can take more than 2 years to fully recover. This is particularly evident in adult patients, whose thymic function is lessened owing to age-related thymic involution. The ‘first wave’ of T cells after HCT comprises donor T cells that undergo lymphopenia-induced homeostatic proliferation and alloactivation. This results in polyclonal T cells with a restricted T cell receptor (TCR) repertoire and limited antigen specificity, or with alloreactivity causing graft-versus-host disease (GVHD). Overall, the incomplete recovery of the T cell pool has been directly linked to increased risks of infection, malignancy relapse and adverse clinical outcomes. Optimal and complete T cell reconstitution requires the regeneration of thymic function and the reactivation of endogenous T cell development. This allows the generation of a new T cell pool with broad TCR diversity. Multiple pretransplant and post-transplant factors influence the overall process of T cell reconstitution. HSC, haematopoietic stem cell; IL, interleukin; NK, natural killer.
Thymic damage and impaired T cell reconstitution are particularly detrimental in HCT recipients7. Defective quantitative and functional recovery of T cells, in particular of CD4+ T cells8,9,10,11, has been directly linked to increased risks of opportunistic infections9,12, malignant relapse13 and overall adverse clinical outcomes14,15. Defective T cell responses are a clinical hurdle not only for patients receiving HCT but also for patients receiving other modalities of cancer immunotherapy, including immune checkpoint inhibitors, that exert their antitumour effects primarily through the activation of T cell effector function. Although the prognostic significance of this association has still to be further characterized in larger studies, a highly diverse pool of T cells before therapy correlates with improved outcome after immune checkpoint blockade therapy16,17,18,19. Thus, there is considerable interest in developing approaches to evaluate the quantity and quality of T cells before and during different forms of immunotherapy to guide treatment directions, monitor immune responses and ultimately identify functional biomarkers to predict clinical outcomes20.
In this Review, we highlight the primary causes of impaired immune function, with special emphasis on HCT recipients, and discuss regenerative approaches that have been clinically translated to facilitate the recovery of adaptive immune function. We also provide an update on emerging new immune-boosting approaches that have demonstrated promising regenerative properties in preclinical models. We focus on approaches that can broaden the diversity of the T cell pool through the restoration of de novo T cell formation in the thymus and discuss the implications for other cancer immunotherapies. While this Review primarily concentrates on T cell immunity, a brief summary of the B cell defects associated with immunological insults is provided in Box 1.
Conditions leading to immune dysfunction
Infection
In the healthy state, homeostasis of the immune system relies on a fine balance between cell production and cell death. During an infection, this dynamic equilibrium is altered to ensure pathogen clearance without unrestrained immune responses. Haematopoietic stem and progenitor cells (HSPCs) replenish immune cells by responding to infections either indirectly through sensing a depletion of downstream cells (a process termed ‘emergency haematopoiesis’) or directly through sensing pathogen-specific systemic inflammatory signals (such as cytokines or Toll-like receptor ligands)21. During acute inflammation, lineage commitment of HSPCs favours granulopoiesis over lymphopoiesis22. During sepsis, migration of thymic precursors from the BM to the thymus is decreased, leading to a depletion of early thymic progenitors and contributing to lymphopenia23.
In addition, the thymus itself is a target organ of various pathogens, leading to thymic atrophy and lymphocyte depletion, which are both common features of infectious diseases24. Thymic haematopoietic and stromal compartments can both be directly targeted in viral and parasitic infections. CD4+CD8+ double-positive (DP) thymocytes and their immediate precursors CD24hiCD3lowCD8+ single-positive thymocytes are particularly vulnerable, whereas mature CD24mid/lowCD8+ SP cells are the most resistant thymic subsets during infection25,26. Although the precise mechanisms for infection-induced acute thymic involution remain to be further elucidated, stress-responsive hormones (such as glucocorticoids), pro-inflammatory mediators (such as interferon-γ (IFNγ) and tumour necrosis factor (TNF)) and apoptosis pathways (such as those mediated by BAX, BCL-2, JUN amino-terminal kinase, p53, caspase 8 and caspase 9) have all been implicated27. DP thymocytes are particularly sensitive to the increased level of glucocorticoids occurring in infection, providing a potential mechanism by which infections induce depletion of this particular cell subset28.
Functional changes in the thymic microenvironment are also observed following infection24. The thymic epithelium undergoes substantial phenotypic and functional changes after infection with the parasite Trypanosoma cruzi29 and viruses such as HIV30, measles virus31 and Zika virus32. For instance, localization of the TR5 and CK18 antigens restricted to medullary TECs (mTECs) and cortical TECs (cTECs), respectively, was altered in T. cruzi-infected mice along with increased extracellular matrix production by in vitro cultured TECs33,34. Other functional changes in the thymic epithelium, including cell cycle arrest, terminal cell differentiation and abnormal expression of cell adhesion and migration genes, were observed in experimental models of measles virus31 and Zika virus32.
The increases in the levels of pro-inflammatory cytokines, including IL-6, TNF and IFNγ35 (which was previously shown to cause BM and thymic aplasia36,37,38), together with other putative mechanisms, including direct damage of lymphoid tissues and apoptosis of lymphocytes, are presumably the primary causes of the profound lymphopenia observed in patients infected with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)39,40.
Thymic atrophy during acute infection is usually a rapid yet transient response, with thymic rebound occurring within 2 weeks after infection41,42. Nonetheless, delayed or incomplete thymic recovery has been reported and is particularly associated with pathogens that cause chronic infections such as HIV43,44. Sustained immune activation and alteration in T cell homeostasis during chronic infections have been associated with progressive and possibly irreversible loss of thymic function as seen in advanced age. Thymopoiesis, as reflected by the levels of TCR excision circles (TRECs) and naive CD4+ T cells in the periphery (Box 2), is significantly impaired in HIV-infected patients43,44. In addition, the reductions in the ratios of CD4+ T cells to CD8+ T cells and naive T cells to memory T cells, the expansion in CD57+CD8+ and CD28–CD8+ senescent T cell populations, and the overall reduction in vaccine responsiveness have all been correlated with HIV-related disease progression and ageing45. This suggests that HIV-associated immunological abnormalities can induce early onset of immunosenescence and correlate with the higher risk of age-associated diseases typically observed in these patients46.
Cytoreductive therapy
Cytoablative treatments, such as radiotherapy, cell-depleting antibodies and chemotherapy, are designed to target malignant cells as well as the host haematopoietic system to allow successful HCT and markedly deplete all haematopoietic lineages, especially T and B cells47. Whereas many cytoablative therapies target highly proliferative cells while sparing the quiescent haematopoietic stem cells (HSCs) numerically, the resultant immunodepletion impairs haematopoietic cell function by selectively inducing premature senescence of HSCs through upregulation of the cyclin-dependent kinase inhibitors p19ARF and p16INK4a (refs48,49). Recent studies demonstrate that chemotherapy induces fundamental changes in the expression of prohaematopoietic factors by the BM stromal niche, including downregulation of the Notch ligands Delta-like 4 (DLL4) and DLL1 by the vascular endothelium50. Consistent with the key role of DLL4 in sustaining common lymphoid progenitors and providing T-lineage specification before thymic entry51, reduced Notch signalling induces a premature myeloid skewing of HSPCs and can contribute to delayed recovery of lymphoid cells following chemotherapy.
In addition to depleting haematopoietic and lymphoid precursors, cytoablative therapies induce prolonged deficiency in adaptive immunity by damaging the normal process of T cell development in the thymus and impairing B cell lymphopoiesis in the BM (Box 1). The reduction in BM progenitors and T cell development can lead to long-term suppression of thymic function even after a single sublethal irradiation dose and can accelerate age-associated thymic involution52,53. In one study, chemotherapy induced transient thymic atrophy in as many as 90% of patients54. Both alkylating agents, such as cyclophosphamide, and irradiation reduce the numbers of all thymocyte subsets in mice, with DP thymocytes and their immediate progenitors CD4–CD8– double-negative thymocytes being particularly sensitive to such insults55,56.
Further contributing to impaired thymopoiesis, cytoablative agents damage TECs57. Irradiation and cyclophosphamide both deplete cTECs and mTECs which are important for, respectively, positive and negative selection of developing T cells, and this interferes with the generation of a broadly reactive TCR repertoire58,59. Activation of signal transducer and activator of transcription 3 (STAT3) signalling has been linked to the induction of apoptosis in TECs after irradiation60. However, more recent studies show that STAT3-mediated signalling is essential for growth and architectural organization of mTECs but is dispensable for the biology of cTECs61,62. Coinciding with their higher rate of proliferation57, the greatest numerical reduction occurs in the subset of TECs expressing the highest levels of MHC class II, particularly the tolerance-inducing AIRE+MHC-IIhi mTECs63. This imbalance in TEC subsets persists even after thymic cellularity is restored and may promote the development of autoimmunity58. Importantly, certain stromal subsets, such as innate lymphoid cells (ILCs) and endothelial cells, are relatively resistant to cytoreductive therapies and play an important role in thymic regeneration by secreting factors, such as IL-22 and bone morphogenetic protein 4 (BMP4), which promote the survival, proliferation and maintenance of TECs64,65. Full, albeit delayed, recovery of thymic function is possible up until middle age, but the peripheral naive repertoire is never fully restored in older patients66.
Peripheral T cell depletion, especially of CD4+ T cells, is the main cause of clinical immunodeficiency observed in patients with cancer67. T cell recovery following cytoablative therapy is predominantly achieved through two mechanisms: de novo production in the thymus, particularly for CD4+ T cells, or homeostatic expansion of peripheral T cells, preferentially for CD8+ T cells68,69 (Fig. 1). The relatively rapid CD8+ T cell recovery through extrathymic clonal expansion70,71 is typically associated with limited breadth of the TCR repertoire and diminished immune responses72.
Graft-versus-host disease
Although allogeneic HCT offers a potential cure for various malignant and non-malignant disorders, its wider application is limited by the significant morbidity and mortality associated with GVHD and prolonged post-transplantation immunodeficiency73. In GVHD, donor alloreactive T cells in addition to targeting the host gastrointestinal tract, liver and skin also directly destroy primary lymphoid organs (BM and thymus) and delay immune reconstitution after allogeneic HCT. Both mouse models and clinical studies have demonstrated that GVHD targets the BM74, and the resultant medullary aplasia delays donor-derived haematopoiesis, especially of the lymphoid lineage75. IFNγ and TNF disrupt donor haematopoiesis both directly and indirectly through their effects on host BM stromal cells, including osteoblasts and mesenchymal stem cells. As a result, acute GVHD is associated with a significant reduction in the numbers of residual and de novo B cell precursors, in the numbers of mature B cells and in antibody production76,77 (Box 1).
During acute GVHD, the thymus is exquisitely sensitive to damage by alloreactive T cells, and its acute atrophy reflects the precipitous contractions of the lymphocytic and stromal compartments78,79. Mouse models of acute GVHD have demonstrated that, in addition to the reduced number of early thymic progenitors migrating from the BM to the thymus, reduction in thymic cellularity is primarily due to loss of the large DP thymocyte population via glucocorticoid-independent apoptotic cell death78.
Within the stromal cell subset, TECs are targets of GVHD-mediated apoptosis. TECs also act as antigen-presenting cells and prime alloreactive T cells directly through their intrinsic expression of MHC class I and class II molecules, such that depletion of host-derived professional antigen-presenting cells does not prevent activation of alloreactive donor T cells and thymic injury79. This raises the possibility that GVHD is restricted to the thymus and remains subclinical despite detrimental consequences for T cell reconstitution. In addition, alloreactive T cells eliminate IL-22-producing thymic group 3 ILCs (ILC3s), resulting in impaired thymic recovery80. Moreover, donor alloreactive CD8+ T cells preferentially target tolerance-inducing mature mTECs, thereby impairing negative selection and allowing de novo generation of autoreactive CD4+ T cells, linking alloimmunity during acute GVHD to the development of autoimmunity during chronic GVHD81,82. These studies underline the importance of functional preservation of TECs as well as numerical restoration of lymphocytes as the goals for immune-regenerative strategies following allogeneic HCT.
Clinically, GVHD-induced immunosuppression has been linked to decreased thymic output of naive T cells, as measured by TRECs, and a distorted TCR repertoire83,84,85,86. Paradoxically, pharmacological immunosuppressants, including corticosteroids and cyclosporine A, used in allogeneic HCT to prevent allograft rejection, as well as GVHD prophylaxis and/or treatment, can induce thymic involution, deplete thymocytes and ablate AIRE+MHC-IIhi mTEC subsets, further contributing to thymic damage58,87. These findings highlight the multifactorial nature of post-transplantation immune disorders and represent the clinical challenges in preventing and treating GVHD after allogeneic HCT.
Immunosenescence
The functionality of the immune system progressively declines with age88, contributing to increased incidence of infections, inadequate vaccine responses and decreased immunosurveillance of malignant cells observed in the elderly population89,90. It is estimated that only ~30–40% of elderly people are capable of mounting sufficient immune responses to the influenza vaccine91,92.
Ageing impairs the normal process of thymic and BM lymphopoiesis at multiple levels. With age, there is an accumulation of HSCs with reduced homing and engraftment capacity93,94,95. In patients receiving allogeneic HCT, younger donor age has been associated with faster immune reconstitution96. Although several intrinsic changes in HSCs and lymphoid progenitors have been identified (including myeloid skewing97,98, defects in DNA repair99,100, epigenetic alterations101 and loss of cell polarity102), substantial changes in the BM stromal microenvironment also contribute to defective lymphopoiesis103. Transplantation of old HSCs into a young microenvironment is sufficient to partially reverse myeloid skewing104,105.
As T cell development depends on the constant output of T cell progenitors from the BM, defective lymphopoiesis, along with impaired developmental potential of intrathymic progenitors106, results in a significant reduction in the number of intrathymic lymphoid progenitors contributing to the reduced T cell output observed in older individuals97,106.
The progressive decline of thymus function during ageing represents one of the most important causes of immune degeneration in the elderly population107. However, the precise kinetics of this chronic physiological process is still under debate. Compared with the mouse thymus, which undergoes a progressive reduction in its volume with age, the human thymus remains almost unchanged in size under normal circumstances and, instead, it is characterized by profound perturbation of the thymic stromal cell microenvironment, including loss of epithelial cells, increased volume of the perivascular space and progressive replacement of healthy tissue with adipose tissue108.
The thymic stroma is one of the main targets of the effects of ageing, as demonstrated by HCT and parabiosis experiments in mice with different age-mismatched donors109,110. For example, intrathymic injection of early thymic progenitors derived from young mice into old mice is not sufficient to restore normal thymic lymphopoiesis111. At a molecular level, several studies have revealed extensive transcriptional changes in mouse thymic stromal cells as early as 3 months of age, particularly in genes associated with cell cycling and inflammatory responses112,113,114. Although ageing significantly erodes its functionality, the thymus maintains a proportion of functional cortical and medullary regions and active thymopoiesis. Indeed, T cell output, as measured by TREC level in the peripheral blood, is maintained in older individuals, albeit substantially reduced115,116. In addition, recent studies demonstrate that the involution of thymic tissue is not as dramatic as previously reported and that residual thymic tissue can be detected by computed tomography scanning in individuals up to 70 years of age117.
As the thymus involutes, the production of T cells is progressively impaired, and with age the naive T cell pool is increasingly maintained by homeostatic expansion3,118,119. Mathematical modelling suggested that thymic T cell export declines exponentially over time with a half-life of 15.7 years; therefore, by the age of 55 years only 5% of naive T cell production is derived from thymic export120. More recently, the average TREC content of the naive T cell population was used to estimate thymic T cell output. This study demonstrated that up to 90% of naive T cells in healthy human adults are generated through proliferation118. By measuring the average TREC content in naive T cells as well as the turnover of T cells, this work also showed that, unlike in humans, the naive T cell pool in mice is almost exclusively sustained throughout life by thymus output118.
The decline in naive T cell production results in a shift towards an oligoclonal pool of memory T cells and an almost linear decrease of T cell diversity with age119,121. These effects are particularly pronounced after the age of 60 years3,122. Some long-lived individuals (average age of 82 years) display a significantly higher percentage of naive CD4+ T cells, decreased abundance of expanded clones and increased TCR diversity compared with a younger cohort, suggesting that these key immune parameters may represent hallmarks of longevity122. Mathematical modelling studies demonstrate a significant inverse correlation between thymic export and the incidence of both infectious diseases and cancer123. During the coronavirus disease 2019 (COVID-19) pandemic, being older than 65 years was identified as a risk factor for morbidity and death from COVID-19 (ref.124).
The age-associated decline of thymic function significantly impairs and delays the endogenous process of thymic repair after immunological insults, resulting in a prolonged recovery time following common cancer cytoreductive therapies12,15. This is particularly problematic in the HCT setting, which is increasingly used in older patients. An inverse relationship exists between the transplant recipient’s age and T cell recovery after transplantation9,125. Insufficient recovery of thymopoiesis has been directly correlated with an increased risk of opportunistic infections, leukaemia relapse and adverse clinical outcome126,127.
Strategies to enhance immune recovery
Over the past few years, several approaches have been proposed to enhance immune function through recovery of the T cell pool (Fig. 2; Table 1). These strategies include the stimulation of T cell development and expansion using cytokines, such as IL-7, IL-12 and IL-21; the administration of cytokines and growth factors, such as stem cell factor (SCF; also known as KITLG), keratinocyte growth factor (KGF; also known as FGF7), IL-22 and FMS-like tyrosine kinase 3 ligand (FLT3LG); the modulation of hormone levels by suppression of sex steroids or by administration of thymosin-α1, growth hormone (GH; also known as somatotropin), insulin-like growth factor 1 (IGF1) and ghrelin; the adoptive transfer of lymphoid progenitors such as precursor T cells and ex vivo expanded thymus-derived endothelial cells; and the use of artificial BM or thymus-like grafts.

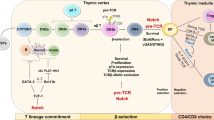
T cell development begins when T cell progenitors, originating from common lymphoid progenitors (CLPs) in the bone marrow, migrate into the thymus and progress through a series of well-characterized developmental steps. Thymocytes go through the double-negative (DN; CD4–CD8–) and double-positive (DP; CD4+CD8+) stages to form single-positive (CD4+CD8– or CD4–CD8+) T cells. During this process, approximately 95% of developing thymocytes produced daily are deleted through β-selection, positive selection and negative selection, resulting in the formation of self-restricted and self-tolerant naive CD4+ and CD8+ single-positive cells that can exit the thymus and migrate to peripheral lymphoid organs. Approaches to enhance T cell recovery act at multiple levels. Factors and approaches such as interleukin-7 (IL-7), IL-12, IL-15, IL-21, FMS-like tyrosine kinase 3 ligand (FLT3LG), growth hormone (GH), insulin-like growth factor 1 (IGF1), sex steroid ablation (SSA), thymosin-α1, stem cell factor (SCF), administration of precursor T cells (pre-T cells) and delivery of ex vivo-generated thymic epithelial cells (exTECs) primarily promote recovery of the haematopoietic compartment. By contrast, keratinocyte growth factor (KGF), IL-22, receptor activator of nuclear factor-κB ligand (RANKL), IGF1, lymphotoxin-α (LTα) and bone morphogenetic protein 4 (BMP4) produced by ex vivo-generated endothelial cells (exECs) enhance reconstitution of the thymic stromal compartment. Question marks denote approaches where the effects on specific targets are not fully understood. ETP, early thymic progenitor; CMP, common myeloid progenitor; cTEC, cortical thymic epithelial cell; DC, dendritic cell: ILC, innate lymphoid cell; HSC, haematopoietic stem cell; MPP, multipotent progenitor; MSC, mesenchymal stromal cell; mTEC, medullary thymic epithelial cell; RTE, recent thymic emigrant.
Here, we focus on immune-boosting strategies that have been translated into clinical studies and provide a brief update on novel approaches that have shown regenerative potential in preclinical models. We put particular emphasis on approaches that can promote de novo T cell formation through the regeneration of thymic function, which is the primary mechanism to generate a pool of naive T cells with a diverse TCR repertoire. We also provide a brief summary of the different methods used to estimate thymic function and the dynamics of T cell reconstitution in Box 2.
Interleukin-7
IL-7 is classified as a type 1 short-chain cytokine crucial for the development of innate and adaptive immune cells128. It is secreted mainly by non-haematopoietic cells, including epithelial cells and fibroblasts in the thymus, BM stromal cells, lymphatic endothelial cells, fibroblastic reticular cells and enterocytes129,130,131. IL-7 is particularly important for the differentiation of T and B cells from common lymphoid progenitors and for the maintenance and survival of mature T cells. IL-7 receptor (IL-7R) is a heterodimer of two chains: IL-7Rα (also known as CD127) and cytokine receptor common subunit-γ (also known as CD132 or IL-2RG). The γ-chain is expressed by all haematopoietic cell types, whereas IL-7Rα is expressed mainly by developing B and T cells, naive and memory T cells, NKT cells, ILC2s and ILC3s.
The crucial role of IL-7 in lymphopoiesis is demonstrated by the development of severe combined immunodeficiency disease in patients carrying mutations affecting the α-chain132,133 or the γ-chain of IL-7R134. While studies in Il7–/– mice showed that IL-7 is a non-redundant cytokine for both T and B cell lymphopoiesis, B cell development in humans does not appear to require IL-7, as B cells are maintained in patients with severe combined immunodeficiency disease with mutations in the gene encoding IL-7Rα132.
The important role of IL-7 in T cell biology is also supported by the inverse correlation between circulating IL-7 levels and peripheral T cells observed in patients with lymphopenia135. The degree of available IL-7 controls the size of the peripheral T cell pool and plays an important role in regulating overall T cell homeostasis.
The effects of exogenous administration of IL-7 on immune reconstitution have been widely investigated128. Several preclinical mouse models have demonstrated the beneficial effects of exogenous IL-7 in promoting immune reconstitution through thymus-dependent and thymus-independent mechanisms. In the setting of HCT, exogenous IL-7 accelerates the reconstitution of donor-derived thymocytes and the peripheral T cell pool, leading to enhanced T cell recovery after both syngeneic and allogeneic HCT135,136,137,138,139. A phase I/IIa dose-escalation study (NCT00477321) of repeated administration of a glycosylated recombinant human IL-7 (rhIL-7; CYT 107) in HIV-1-infected patients demonstrated that rhIL-7 treatment was safe, well tolerated and transiently promoted the expansion of naive and memory CD4+ and CD8+ T cells, and decreased the proportion of exhausted PD1+ T cells140. Importantly, rhIL-7 therapy also increased the numbers of CD4+ recent thymic emigrants (RTEs), the signal joint to β TREC ratio and TCR repertoire diversity in some participants, effects that imply enhanced thymic activity (Box 2). Subsequently, two other clinical trials (NCT01190111 and NCT01241643) demonstrated that repeated doses of rhIL-7 were well tolerated and resulted in sustained CD4+ T cell numbers in the majority of HIV-infected participants140,141,142,143. Similarly, in a phase I/IIa dose-escalation trial (NCT00839436) in patients with idiopathic CD4+ lymphopenia at risk of disease progression, rhIL-7 led to an increase in the number of circulating CD4+ and CD8+ T cells and tissue-resident CD3+ T cells in the gut mucosa and BM. Importantly, enhanced thymopoiesis, measured by TRECs, was observed only in the youngest patients of the cohort (aged 23 and 34 years)144.
In a phase I clinical trial (NCT00684008) that evaluated the immune-regenerative properties of rhIL-7 in patients receiving T cell-depleted allogeneic HCT138, rhIL-7 induced a rapid increase in peripheral CD4+ and CD8+ T cell numbers. While the estimated half-life of rhIL-7 in this study was 9–35 hours, the biological effects on T cell numbers persisted for several weeks after the circulating levels of IL-7 returned to the baseline. In addition, although rhIL-7 administration resulted in increased numbers of RTEs only in some young patients, most participants had enhanced TCR repertoire diversity that persisted several weeks after the end of rhIL-7 therapy138. The limited effects on thymic output in this study, as represented by minimal changes not only in the numbers of RTEs but also in the levels of TRECs, could be explained by the age and lymphopenic condition of the patients at the time points analysed. It is also possible that extended duration of rhIL-7 administration is necessary to have a greater effect on thymic function. However, compared with mice, in which IL-7 has both thymic-dependent and thymic-independent regenerative effects135, it is still unclear what the direct impact of exogenously administered IL-7 is on the thymus in humans and non-human primates. On the basis of clinical and preclinical observations, it appears that most of the effects on TCR diversity following rhIL-7 treatment are primarily driven by extrathymic sources, including the expansion of less frequent but highly diverse RTEs and naive T cells, which preferentially respond to IL-7 stimulation145,146, as well as their recirculation from lymphoid organs141,147,148.
Lymphopenia, particularly with regard to CD4+ and CD8+ T cell subsets, in patients with COVID-19 on admission to hospital is emerging as one of the key clinical signs of COVID-19 and is closely associated with disease progression39. Enhancing T cell immunity could be a worthwhile strategy for treating these patients. Thus, a clinical trial has recently begun to investigate the possibility that IL-7 can restore T cell immunity in patients with COVID-19.
Keratinocyte growth factor
KGF is a potent growth factor for TECs and is expressed under physiological conditions in the thymus primarily by mesenchymal cells149. KGF binds to its receptor, fibroblast growth factor 2 variant IIIb (FGFR2IIIb), on TECs and induces TEC proliferation through activation of the PI3K–AKT–nuclear factor-κB and p53 pathways150,151,152.
Studies using knockout animals found that although KGF is redundant for thymopoiesis in steady-state conditions, it is crucial for thymic regeneration and peripheral T cell reconstitution after injury such as that caused by total body irradiation and syngeneic or allogeneic HCT153. The impact of exogenous administration of KGF on TEC function and thymic regrowth has been extensively evaluated in several mouse studies. Administration of recombinant KGF transiently accelerated thymic recovery after immune insults such as irradiation, cyclophosphamide therapy and dexamethasone therapy, and enhanced recovery of thymic and peripheral T cell numbers after HCT150,152,154,155. KGF reversed thymic involution and restored thymopoiesis in aged mice for up to 2 months after treatment156. A study performed in rhesus macaques showed that KGF-treated animals displayed accelerated haematological recovery, improved thymopoiesis and enhanced naive T cell recovery following HCT157. Although the effects on thymic function were modest, as measured by minimal changes in thymus mass, KGF-treated animals showed increased numbers of TREC-positive T cells up to 3 months following KGF treatment.
Human recombinant KGF (palifermin; trade name Kepivance, marketed by Biovitrum) is approved by the US Food and Drug Administration for the prevention of mucositis in patients receiving high-dose chemotherapy. Several trials (NCT01233921, NCT03042585, NCT02356159 and NCT00593554) are exploring its effects on T cell reconstitution, but no results have been reported yet. As demonstrated in previous preclinical work158, the benefits of palifermin on immune reconstitution in transplant recipients may derive from its synergistic effects with other immune-boosting therapies rather than as a sole therapeutic agent. However, a recent study of the use of KGF to promote immune reconstitution in patients with relapsing–remitting multiple sclerosis treated with the anti-CD52 lymphocyte-depleting agent alemtuzumab showed reduced thymic output in KGF-treated patients as measured by evaluation of naive CD4+ T cells, RTEs and TRECs (NCT01712945)159. Given that human cTECs express CD52, one possible explanation for these clinical data is that palifermin exacerbates the negative effects of alemtuzumab on thymic function, perhaps through upregulation of CD52 expression on cTECs, rendering these cells more susceptible to antibody-mediated elimination. Thus, the combination of KGF with other drugs should be assessed cautiously as synergistic or deleterious effects on immune regeneration may occur depending on dosing, timing and mechanisms.
Thymic hormones
Thymosins are a group of low molecular weight peptides originally isolated from bovine thymus160. Thymosin-α1, derived from prothymosin-α, is produced by TECs161 and can increase lymphocyte maturation, boost T cell function and promote recovery following immune insults, although its mechanism of action is not completely understood. The receptor for thymosin-α1 is expressed by developing thymocytes, in which it regulates their survival and proliferation. Thymosin-α1 can antagonize dexamethasone-induced apoptosis of DP thymocytes in vitro, as well as the hydrocortisone-induced decrease in thymus and spleen mass162. Thymosin-α1 can also enhance the production of IL-7 by TECs163. Given promising preclinical results, multiple clinical trials have been initiated to evaluate the immunomodulatory effects of thymosin-α1 in the treatment of patients experiencing viral infections, immunodeficiency or haematological malignancies. Safety and efficacy of thymosin-α1 administration were evaluated in recipients of allogeneic HCT in a phase I/II clinical study (NCT00580450). Treatment with thymosin-α1 increased T cell numbers and resulted in earlier appearance of pathogen-specific T cell responses against pathogens such as cytomegalovirus and Aspergillus species. Importantly, thymosin-α1 did not exacerbate acute or chronic GVHD and was associated with significant improvement in phagocytosis and dendritic cell function164.
Recently, thymosin-α1 was given to patients with COVID-19 showing severe lymphopenia to enhance immunity. Thymosin-α1 treatment increased T cell numbers and recovery of thymic function, measured by TREC analysis165. Importantly, thymosin-α1 administration was also associated with increased survival of patients with severe COVID-19 (ref.165).
Sex steroid ablation
In addition to their fundamental role in regulating sex dimorphism, sex hormones can impact haematopoiesis at multiple levels. One of the first observations regarding a relationship between T cell development and sex hormones dates back to 1898, when it was reported that the thymus enlarged after castration of male rabbits166. Several studies confirmed the enlargement of thymic tissue after gonadectomy in both sexes in different experimental animal models. Conversely, androgens and oestrogens induce atrophy of the thymus167,168.
The increase in the levels of sex steroids, and in particular of androgens, during puberty has been directly linked to the age-associated deterioration of immune function and to the process of thymic involution. Although the connection between the increase in the levels of sex steroids after puberty and the initiation of thymic involution is still debated, the regenerative impact of the removal of sex steroids on both thymic and BM lymphopoiesis has been extensively characterized. Indeed, through the use of clinically relevant mouse models of immune reconstitution after haematopoietic injuries, such as chemotherapy and radiotherapy, it has been demonstrated that sex steroid ablation enhances HSC self-renewal and lymphoid differentiation capacity and increases the number of common lymphoid progenitors in the BM169,170,171. Sex steroid ablation also has a direct effect on the BM microenvironment, restoring expression of key haematopoietic factors that are downregulated with age, such as FOXO1 (ref.169). Considerable rejuvenation effects in the thymus have been extensively characterized, demonstrating that sex steroid ablation reverses thymic atrophy, accelerates the recovery of all thymocyte subsets and elicits potent regenerative signals to the thymic stromal microenvironment55,172,173,174. At a molecular level, sex steroid ablation promotes the upregulation of the key thymopoietic factors CC-chemokine ligand 25 (CCL25)175 and DLL4 (ref.167) in mTECs and cTECs, respectively.
Several drugs have been developed to transiently and reversibly block sex steroids for the treatment of precocious puberty, endometriosis, hormone-sensitive prostate cancer and breast cancer. Some of these sex steroid blockers have been tested clinically to boost immune reconstitution after HCT. A non-randomized pilot study demonstrated that administration of the luteinizing hormone-releasing hormone (LHRH) agonist goserelin (Zoladex) before HCT significantly increased neutrophil engraftment, as well as total lymphocyte numbers, particularly those of naive CD4+ T cells, and levels of TRECs and improved recovery of TCR repertoire diversity176. Importantly, an increase in disease-free survival was observed in autologous HCT recipients treated with goserelin. Two trials (NCT01746849 and NCT01338987) are ongoing to evaluate the effects of the LHRH agonist leuprolide (Leuprorelin) and the LHRH antagonist degarelix (Firmagon) to promote immune reconstitution following allogeneic HCT. Notably, the latest androgen receptor inhibitors and LHRH antagonists have the advantage of immediately blocking sex steroids without an initial surge of sex steroids as seen with LHRH agonists167. These novel approaches may provide better therapeutic tools to suppress sex steroids and mediate immune reconstitution.
The regenerative effects of sex steroid ablation on T cell development might continue only as long as the levels of sex steroids are suppressed. However, the duration of such effect, particularly in the setting of surgical castration, remains a subject of debate in the field. After the initial regrowth following castration, the thymus of aged animals has been reported to decline and return approximately to its pretherapy condition 1 month after sex steroid ablation therapy177. While these results support a model in which the regenerative effects induced after surgical sex steroid ablation are transitory and dynamic, additional studies should be done to better characterize the nature of these ‘transient’ effects and the precise kinetics of thymic regeneration, in particular, at later time points. For example, it would be interesting to evaluate whether removal of the gonads, in the long term, can induce additional hormonal changes that negatively impact the process of lymphopoiesis.
Growth hormone
GH is a small peptide hormone secreted primarily in the bloodstream by somatotropic cells in the anterior pituitary gland178. Apart from its anabolic effects and impact on height, GH is also implicated in the regulation of haematopoietic function. Expression of GH receptor (GHR), by which GH mediates most of its effects, has been found on T cells, B cells, NK cells, monocytes, thymocytes and HSCs in humans and in other species179. While the impact of its signalling seems to be dispensable for HSC function, as suggested by the lack of phenotypic defects in HSCs of GHR-deficient mice, GH has important effects on immune function in mice and humans, either directly or through its principal mediator, IGF1. In vivo administration of GH can reverse thymic involution180,181,182. Transgenic mice that overexpress GH have an enlarged thymus. Similarly, the administration of a recombinant form of GH or IGF1 promotes thymic regeneration, increases TCR diversity and enhances recovery of haematopoietic compartments in immunocompromised and aged animals183.
GH administration to immunocompromised patients has been studied in several clinical trials. Daily administration of human recombinant GH (somatropin) enhances thymic function and peripheral immune function in HIV-infected patients (NCT00071240)180,181. The effects on thymic output appear to be transient, as discontinuation of GH treatment is associated with the recurrence of thymic atrophy181. Results from the recently completed studies (NCT00287677, NCT00119769 and NCT00050921) are still pending. The use of GH to reverse chronological ageing of the immune system was assessed in a recent pre‐phase-I study performed in volunteers aged between 51 and 65 years. Magnetic resonance imaging of thymic density showed that the accumulated fat tissue in the thymus was replaced with regenerated tissue in seven of the nine participants receiving the treatment184. In the periphery, GH treatment was associated with a significant increase in both naive CD4+ T cells and naive CD8+ T cells, with a concomitant decrease in PD1+CD8+ T cells184. However, there are several concerns about the side effects associated with GH treatment, such as increased risk of heart disease, diabetes, elevated cholesterol levels and, more importantly, tumour progression. Thus, additional studies are needed to evaluate its potential use in humans.
Emerging approaches for T cell regeneration
Over the past decade, intensive work has been done to further optimize the efficacy of already identified approaches and to identify alternative regenerative mechanisms (Fig. 2). In addition to IL-7, other cytokines have demonstrated efficacy in preclinical mouse models to restore thymic function and/or expand immune cells in the periphery following immune insults. Administration of IL-12 not only induces thymocyte proliferation through increased IL-7 and IL-2 signalling but also enhances engraftment and haematopoietic reconstitution after transplantation185,186. IL-15 can also boost immunity primarily by promoting NK cell, NKT cell and CD8+ T cell proliferation and function. Overall, these effects enhance reconstitution of these cell subsets and graft-versus-tumour responses in mouse models of allogeneic HCT187,188,189.
IL-21 can enhance thymic function in young and aged mice. These effects are primarily mediated by the impact of IL-21 on DP thymocytes, which express high levels of IL-21 receptor after glucocorticoid-induced thymic atrophy, and by activation of the IL-21 downstream target BCL-6 (ref.190). Administration of recombinant IL-21 improved thymic regeneration and reconstitution of the peripheral naive T cell compartment in different models of immune damage, including glucocorticoid-induced thymic atrophy, ageing and allogeneic HCT190,191,192.
IL-22 can also mediate thymic regeneration64,193. Following thymic damage, the loss of DP thymocytes can trigger the production of IL-22 by thymic ILCs in an IL-23-dependent manner (Fig. 2). IL-22 then acts on TECs to promote their survival and proliferation through activation of STAT3 and STAT5 and expression of the downstream antiapoptotic molecule MCL1 (refs64,193). Administration of recombinant IL-22 to sublethally irradiated mice or allogeneic HCT recipients can promote the recovery of thymic function and the development of new thymic-derived peripheral T cells64,80,193.
ILCs play a fundamental role in thymic reconstitution not only via IL-22 production but also through production of receptor activator of nuclear factor-κΒ ligand (RANKL), a potent factor known for its fundamental role in TEC maintenance and maturation194,195,196. Recent studies have characterized the role of RANKL in the regeneration of the thymic microenvironment and T cell recovery in mouse models of allogeneic HCT. RANKL is expressed early after thymic damage by CD4+ thymocytes and, to a great extent, by lymphoid tissue inducer (LTi) cells64,197. RANKL acts on its cognate receptor, RANK, expressed on LTi cells, and induces upregulation of lymphotoxin-α (LTα). Following thymic damage, LTα can bind to LTβ receptor on thymic epithelial progenitor cells and TECs and promote their regeneration. Exogenous administration of recombinant RANKL boosts regeneration of thymic epithelial progenitor cells and TECs and improves T cell progenitor homing and de novo thymopoiesis. Overall, these effects lead to enhanced peripheral T cell reconstitution197. LTα can activate LTβ receptor on intestinal dendritic cells to induce IL-23 production, which in turn acts on intestinal ILCs to promote IL-22 production198.
As cells producing RANKL, IL-22, LTα or LTβ are responsive to IL-7R signalling, which can promote their expansion and function199,200, it is possible that these molecules contribute to the regenerative effects mediated by IL-7 in the thymus. Although IL-7 has not been shown to directly regulate IL-22, it can regulate LTα1β2 expression by LTi cells201, thus providing a mechanism by which IL-7R signalling integrates regenerative pathways. In addition, IL-7 can directly induce RANKL expression by T cells and aid in thymic regeneration202.
In addition to its well-described role during thymus organogenesis203,204, BMP4 can promote thymic regeneration after thymic damage. Indeed, BMP4 produced by thymic endothelial cells drives thymic regeneration by binding to its receptor expressed on TECs and stimulating the upregulation of FOXN1 and its target genes65,205. Importantly, adoptive transfer of ex vivo expanded thymic endothelial cells improves thymic reconstitution after a sublethal dose of total body irradiation through the delivery of BMP4.
Concluding remarks
At present, there are no approved therapies to enhance T cell function in patients with lymphopenia. The development of such approaches would not only benefit patients whose immune system has been decimated by multiple cycles of chemotherapy and radiotherapy or by viral infections but could also improve T cell responses in other clinical settings.
Cancer immunotherapy with immune checkpoint inhibitors is emerging as one of the most promising new treatments for a variety of solid and liquid malignancies. To be effective, these treatments rely on the presence of an adequate pool of T cells capable of recognizing specific tumour antigens206. Previous studies demonstrated that the limited response rate to checkpoint inhibitor therapy may be linked to a restricted TCR repertoire observed in the periphery of patients with cancer before therapy. Non-synonymous mutations and neoantigens are associated with clinical efficacy of immune checkpoint blockade207,208,209. Thus, treatments capable of improving immune functions and enhancing TCR repertoire diversity may have the potential to significantly extend the clinical benefit of immune checkpoint blockade.
Therapeutic approaches that can rejuvenate the peripheral T cell pool will also be relevant for the treatment of elderly patients not only to enhance responses to pathogens but also to increase the efficacy of vaccines. The progressive expansion of peripheral TCR clonality and loss of specific T cell clones observed during ageing contribute to the moderate success rate of vaccination in elderly patients.
However, important aspects should be taken in consideration when one is designing approaches to restore immunocompetence in aged individuals. Recent work performed in old mice and non-human primates demonstrated that additional barriers limit the impact of immune regeneration in the periphery even when thymic regeneration is achieved210. Although the thymuses of old mice treated with sex steroid ablation or KGF can be rejuvenated, this did not translate into increased frequencies of naive CD8+ T cells and naive CD4+ T cells in peripheral blood. Age-related intrinsic defects of RTEs210,211 and a defective thymic stromal microenvironment113,114,177, together with reduced responses to homeostatic cytokines212, could explain the defective maintenance and function of naive T cells in old recipients.
One promising, recently identified strategy to expedite immune reconstitution following HCT is the use of non-genotoxic conditioning approaches. The use of depleting antibodies, such as anti-CD117 and anti-CD45, which can recognize and eliminate HSCs and other haematopoietic cells in a targeted manner, allows remarkably efficient HSC engraftment while sparing non-haematopoietic cells, with minimal off-target toxicity213,214,215. This novel approach has the potential to reduce HCT-related toxicity, promote faster immune recovery and significantly improve patient clinical outcome.
References
Nikolich-Žugich, J., Slifka, M. K. & Messaoudi, I. The many important facets of T-cell repertoire diversity. Nat. Rev. Immunol. 4, 123–132 (2004). This review provides an overview of the diversity of TCRαβ T cells in relation to the maintenance of an optimal immune response.
Čičin-Šain, L. et al. Loss of naive T cells and repertoire constriction predict poor response to vaccination in old primates. J. Immunol. 184, 6739–6745 (2010).
Yager, E. J. et al. Age-associated decline in T cell repertoire diversity leads to holes in the repertoire and impaired immunity to influenza virus. J. Exp. Med. 205, 711–723 (2008).
Yew, P. Y. et al. Quantitative characterization of T-cell repertoire in allogeneic hematopoietic stem cell transplant recipients. Bone Marrow Transpl. 50, 1227–1234 (2015).
Van Heijst, J. W. J. et al. Quantitative assessment of T cell repertoire recovery after hematopoietic stem cell transplantation. Nat. Med. 19, 372–377 (2013). This study analyses changes in the TCRβ chain diversity following HSCT and their association with infections, GVHD and age.
Takahama, Y. Journey through the thymus: stromal guides for T-cell development and selection. Nat. Rev. Immunol. 6, 127–135 (2006).
Toubert, A., Glauzy, S., Douay, C. & Clave, E. Thymus and immune reconstitution after allogeneic hematopoietic stem cell transplantation in humans: never say never again. Tissue Antigens 79, 83–89 (2012). This is a detailed review of the role of the thymus in allogeneic HSCT recipients and the use of TRECs to quantify its function.
Admiraal, R. et al. Leukemia-free survival in myeloid leukemia, but not in lymphoid leukemia, is predicted by early CD4+ reconstitution following unrelated cord blood transplantation in children: a multicenter retrospective cohort analysis. Bone Marrow Transpl. 51, 1376–1378 (2016).
Small, T. N. et al. Comparison of immune reconstitution after unrelated and related T-cell-depleted bone marrow transplantation: effect of patient age and donor leukocyte infusions. Blood 93, 467–480 (1999).
Goldberg, J. D. et al. Early recovery of T-cell function predicts improved survival after T-cell depleted allogeneic transplant. Leuk. Lymphoma 58, 1859–1871 (2017).
Fedele, R. et al. The impact of early CD4+ lymphocyte recovery on the outcome of patients who undergo allogeneic bone marrow or peripheral blood stem cell transplantation. Blood Transfus. 10, 174–180 (2012).
Storek, J., Gooley, T., Witherspoon, R. P., Sullivan, K. M. & Storb, R. Infectious morbidity in long-term survivors of allogeneic marrow transplantation is associated with low CD4 T cell counts. Am. J. Hematol. 54, 131–138 (1997).
Powles, R. et al. Identification of patients who may benefit from prophylactic immunotherapy after bone marrow transplantation for acute myeloid leukemia on the basis of lymphocyte recovery early after transplantation. Blood 91, 3481–3486 (1998).
Le Blanc, K. et al. Lymphocyte recovery is a major determinant of outcome after matched unrelated myeloablative transplantation for myelogenous malignancies. Biol. Blood Marrow Transplant. 15, 1108–1115 (2009).
Maury, S. et al. Prolonged immune deficiency following allogeneic stem cell transplantation: risk factors and complications in adult patients. Br. J. Haematol. 115, 630–641 (2001).
Hopkins, A. C. et al. T cell receptor repertoire features associated with survival in immunotherapy-treated pancreatic ductal adenocarcinoma. JCI Insight 3, e122092 (2018).
Snyder, A. et al. Contribution of systemic and somatic factors to clinical response and resistance to PD-L1 blockade in urothelial cancer: an exploratory multi-omic analysis. PLoS Med. 14, e1002309 (2017).
Arakawa, A. et al. Clonality of CD4+ blood T cells predicts longer survival with CTLA4 or PD-1 checkpoint inhibition in advanced melanoma. Front. Immunol. 10, 1336 (2019).
Postow, M. A. et al. Peripheral T cell receptor diversity is associated with clinical outcomes following ipilimumab treatment in metastatic melanoma. J. Immunother. Cancer 3, 23 (2015).
Nixon, A. B. et al. Peripheral immune-based biomarkers in cancer immunotherapy: can we realize their predictive potential? J. Immunother. Cancer 7, 325 (2019).
Baldridge, M. T., King, K. Y. & Goodell, M. A. Inflammatory signals regulate hematopoietic stem cells. Trends Immunol. 32, 57–65 (2011).
Ueda, Y., Kondo, M. & Kelsoe, G. Inflammation and the reciprocal production of granulocytes and lymphocytes in bone marrow. J. Exp. Med. 201, 1771–1780 (2005).
Kong, Y. et al. Sepsis-induced thymic atrophy is associated with defects in early lymphopoiesis. Stem Cell 34, 2902–2915 (2016).
Savino, W. The thymus is a common target organ in infectious diseases. PLoS Pathog. 2, e62 (2006).
Chen, W. et al. Low dose aerosol infection of mice with virulent type A Francisella tularensis induces severe thymus atrophy and CD4+CD8+ thymocyte depletion. Microb. Pathog. 39, 189–196 (2005).
Majumdar, S. et al. Differential susceptibility and maturation of thymocyte subsets during Salmonella Typhimurium infection: insights on the roles of glucocorticoids and Interferon-gamma. Sci. Rep. 7, 40793 (2017).
Majumdar, S. & Nandi, D. Thymic atrophy: experimental studies and therapeutic interventions. Scand. J. Immunol. 87, 4–14 (2018). This review provides an overview of the numerous factors linked to the atrophy of the thymus.
Berki, T., Palinkas, L., Boldizsar, F. & Nemeth, P. Glucocorticoid (GC) sensitivity and GC receptor expression differ in thymocyte subpopulations. Int. Immunol. 14, 463–469 (2002).
Linhares-Lacerda, L. et al. Differential expression of microRNAs in thymic epithelial cells from Trypanosoma cruzi acutely infected mice: putative role in thymic atrophy. Front. Immunol. 6, 428 (2015).
Autran, B. et al. Thymocyte and thymic microenvironment alterations during a systemic HIV infection in a severe combined immunodeficient mouse model. AIDS 10, 717–727 (1996).
Valentin, H. et al. Measles virus infection induces terminal differentiation of human thymic epithelial cells. J. Virol. 73, 2212–2221 (1999).
Messias, C. V. et al. Zika virus targets the human thymic epithelium. Sci. Rep. 10, 1378 (2020).
Savino, W., Leite-de-Moraes, M. C., Hontebeyrie-Joskowicz, M. & Dardenne, M. Studies on the thymus in Chagas’ disease. I. Changes in the thymic microenvironment in mice acutely infected with Trypanosoma cruzi. Eur. J. Immunol. 19, 1727–1733 (1989).
Cotta-de-Almeida, V., Bertho, A. L., Villa-Verde, D. M. & Savino, W. Phenotypic and functional alterations of thymic nurse cells following acute Trypanosoma cruzi infection. Clin. Immunol. Immunopathol. 82, 125–132 (1997).
Huang, C. et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395, 497–506 (2020).
Sempowski, G. D. et al. Leukemia inhibitory factor, oncostatin M, IL-6, and stem cell factor mRNA expression in human thymus increases with age and is associated with thymic atrophy. J. Immunol. 164, 2180–2187 (2000).
Carbajosa, S. et al. Altered bone marrow lymphopoiesis and interleukin-6-dependent inhibition of thymocyte differentiation contribute to thymic atrophy during Trypanosoma cruzi infection. Oncotarget 8, 17551–17561 (2017).
Liepinsh, D. J. et al. Accelerated thymic atrophy as a result of elevated homeostatic expression of the genes encoded by the TNF/lymphotoxin cytokine locus. Eur. J. Immunol. 39, 2906–2915 (2009).
Zhang, X. et al. Viral and host factors related to the clinical outcome of COVID-19. Nature 583, 437–440 (2020).
Tay, M. Z., Poh, C. M., Renia, L., MacAry, P. A. & Ng, L. F. P. The trinity of COVID-19: immunity, inflammation and intervention. Nat. Rev. Immunol. 20, 363–374 (2020).
Ross, E. A. et al. Thymic function is maintained during Salmonella-induced atrophy and recovery. J. Immunol. 189, 4266–4274 (2012).
Anz, D. et al. Activation of melanoma differentiation-associated gene 5 causes rapid involution of the thymus. J. Immunol. 182, 6044–6050 (2009).
Douek, D. C. et al. Changes in thymic function with age and during the treatment of HIV infection. Nature 396, 690–695 (1998).
Dion, M. L. et al. HIV infection rapidly induces and maintains a substantial suppression of thymocyte proliferation. Immunity 21, 757–768 (2004).
Kalayjian, R. C. et al. Age-related immune dysfunction in health and in human immunodeficiency virus (HIV) disease: association of age and HIV infection with naive CD8+ cell depletion, reduced expression of CD28 on CD8+ cells, and reduced thymic volumes. J. Infect. Dis. 187, 1924–1933 (2003).
Deeks, S. G. HIV infection, inflammation, immunosenescence, and aging. Annu. Rev. Med. 62, 141–155 (2011).
Mackall, C. L. et al. Lymphocyte depletion during treatment with intensive chemotherapy for cancer. Blood 84, 2221–2228 (1994).
Meng, A., Wang, Y., Van Zant, G. & Zhou, D. Ionizing radiation and busulfan induce premature senescence in murine bone marrow hematopoietic cells. Cancer Res. 63, 5414–5419 (2003).
Wang, Y., Schulte, B. A., LaRue, A. C., Ogawa, M. & Zhou, D. Total body irradiation selectively induces murine hematopoietic stem cell senescence. Blood 107, 358–366 (2006).
Tikhonova, A. N. et al. The bone marrow microenvironment at single-cell resolution. Nature 569, 222–228 (2019).
Yu, V. W. et al. Specific bone cells produce DLL4 to generate thymus-seeding progenitors from bone marrow. J. Exp. Med. 212, 759–774 (2015).
Xiao, S. et al. Sublethal total body irradiation causes long-term deficits in thymus function by reducing lymphoid progenitors. J. Immunol. 199, 2701–2712 (2017).
Ito, R. et al. Late effects of exposure to ionizing radiation and age on human thymus morphology and function. Radiat. Res. 187, 589–598 (2017).
Choyke, P. L. et al. Thymic atrophy and regrowth in response to chemotherapy: CT evaluation. Am. J. Roentgenol. 149, 269–272 (1987).
Heng, T. S. et al. Effects of castration on thymocyte development in two different models of thymic involution. J. Immunol. 175, 2982–2993 (2005).
Gentil Dit Maurin, A. et al. Developmental regulation of p53-dependent radiation-induced thymocyte apoptosis in mice. Clin. Exp. Immunol. 179, 30–38 (2015).
Gray, D. H. et al. Developmental kinetics, turnover, and stimulatory capacity of thymic epithelial cells. Blood 108, 3777–3785 (2006).
Fletcher, A. L. et al. Ablation and regeneration of tolerance-inducing medullary thymic epithelial cells after cyclosporine, cyclophosphamide, and dexamethasone treatment. J. Immunol. 183, 823–831 (2009).
Williams, K. M. et al. Single cell analysis of complex thymus stromal cell populations: rapid thymic epithelia preparation characterizes radiation injury. Clin. Transl. Sci. 2, 279–285 (2009).
Sano, S. et al. Stat3 in thymic epithelial cells is essential for postnatal maintenance of thymic architecture and thymocyte survival. Immunity 15, 261–273 (2001).
Lomada, D. et al. Stat3 signaling promotes survival and maintenance of medullary thymic epithelial cells. PLoS Genet. 12, e1005777 (2016).
Satoh, R. et al. Requirement of Stat3 signaling in the postnatal development of thymic medullary epithelial cells. PLoS Genet. 12, e1005776 (2016).
Gray, D., Abramson, J., Benoist, C. & Mathis, D. Proliferative arrest and rapid turnover of thymic epithelial cells expressing Aire. J. Exp. Med. 204, 2521–2528 (2007).
Dudakov, J. A. et al. Interleukin-22 drives endogenous thymic regeneration in mice. Science 336, 91–95 (2012).
Wertheimer, T. et al. Production of BMP4 by endothelial cells is crucial for endogenous thymic regeneration. Sci. Immunol. 3, eaal2736 (2018).
Sfikakis, P. P. et al. Age-related thymic activity in adults following chemotherapy-induced lymphopenia. Eur. J. Clin. Invest. 35, 380–387 (2005).
Mackall, C. L. T-cell immunodeficiency following cytotoxic antineoplastic therapy: a review. Stem Cell 18, 10–18 (2000).
Mackall, C. L. et al. Age, thymopoiesis, and CD4+ T-lymphocyte regeneration after intensive chemotherapy. N. Engl. J. Med. 332, 143–149 (1995).
Hakim, F. T. et al. Age-dependent incidence, time course, and consequences of thymic renewal in adults. J. Clin. Invest. 115, 930–939 (2005).
Mackall, C. L. et al. Distinctions between CD8+ and CD4+ T-cell regenerative pathways result in prolonged T-cell subset imbalance after intensive chemotherapy. Blood 89, 3700–3707 (1997).
Fagnoni, F. F. et al. T-cell dynamics after high-dose chemotherapy in adults: elucidation of the elusive CD8+ subset reveals multiple homeostatic T-cell compartments with distinct implications for immune competence. Immunology 106, 27–37 (2002).
Lin, S. J., Chen, A. T. & Welsh, R. M. Immune system derived from homeostatic proliferation generates normal CD8 T-cell memory but altered repertoires and diminished heterologous immune responses. Blood 112, 680–689 (2008).
Zeiser, R. Introduction to a review series on pathophysiology and treatment of acute GVHD. Blood 136, 375–376 (2020).
Arnold, R. et al. Hemopoietic reconstitution after bone marrow transplantation. Exp. Hematol. 14, 271–277 (1986).
Krenger, W. & Hollander, G. A. The immunopathology of thymic GVHD. Semin. Immunopathol. 30, 439–456 (2008).
Shono, Y. et al. Bone marrow graft-versus-host disease: early destruction of hematopoietic niche after MHC-mismatched hematopoietic stem cell transplantation. Blood 115, 5401–5411 (2010).
Mensen, A. et al. Bone marrow T-cell infiltration during acute GVHD is associated with delayed B-cell recovery and function after HSCT. Blood 124, 963–972 (2014).
Krenger, W., Rossi, S. & Hollander, G. A. Apoptosis of thymocytes during acute graft-versus-host disease is independent of glucocorticoids. Transplantation 69, 2190–2193 (2000).
Hauri-Hohl, M. M. et al. Donor T-cell alloreactivity against host thymic epithelium limits T-cell development after bone marrow transplantation. Blood 109, 4080–4088 (2007). This study investigates the molecular and cellular mechanisms underlying the impact of GVHD on TEC function.
Dudakov, J. A. et al. Loss of thymic innate lymphoid cells leads to impaired thymopoiesis in experimental graft-versus-host disease. Blood 130, 933–942 (2017).
Wu, T. et al. Thymic damage, impaired negative selection, and development of chronic graft-versus-host disease caused by donor CD4+ and CD8+ T cells. J. Immunol. 191, 488–499 (2013).
Dertschnig, S., Hauri-Hohl, M. M., Vollmer, M., Hollander, G. A. & Krenger, W. Impaired thymic expression of tissue-restricted antigens licenses the de novo generation of autoreactive CD4+ T cells in acute GVHD. Blood 125, 2720–2723 (2015).
Przybylski, G. K., Kreuzer, K. A., Siegert, W. & Schmidt, C. A. No recovery of T-cell receptor excision circles (TRECs) after non-myeloablative allogeneic hematopoietic stem cell transplantation is correlated with the onset of GvHD. J. Appl. Genet. 48, 397–404 (2007).
Link-Rachner, C. S. et al. T-cell receptor-alpha repertoire of CD8+ T cells following allogeneic stem cell transplantation using next-generation sequencing. Haematologica 104, 622–631 (2019).
Meyer, E. H. et al. A distinct evolution of the T-cell repertoire categorizes treatment refractory gastrointestinal acute graft-versus-host disease. Blood 121, 4955–4962 (2013).
Clave, E. et al. Acute graft-versus-host disease transiently impairs thymic output in young patients after allogeneic hematopoietic stem cell transplantation. Blood 113, 6477–6484 (2009).
Blazar, B. R., Murphy, W. J. & Abedi, M. Advances in graft-versus-host disease biology and therapy. Nat. Rev. Immunol. 12, 443–458 (2012).
Nikolich-Zugich, J. The twilight of immunity: emerging concepts in aging of the immune system. Nat. Immunol. 19, 10–19 (2018).
Weinberger, B., Herndler-Brandstetter, D., Schwanninger, A., Weiskopf, D. & Grubeck-Loebenstein, B. Biology of immune responses to vaccines in elderly persons. Clin. Infect. Dis. 46, 1078–1084 (2008).
McElhaney, J. E. & Effros, R. B. Immunosenescence: what does it mean to health outcomes in older adults? Curr. Opin. Immunol. 21, 418–424 (2009).
Thompson, W. W. et al. Mortality associated with influenza and respiratory syncytial virus in the United States. JAMA 289, 179–186 (2003).
Tsutsumi, Y. et al. Monitoring of T-cell repertoire was useful for predicting graft-versus-host disease prognosis in a patient with chronic myelogeneous leukemia after allogeneic bone marrow transplantation. Transplant. Proc. 36, 3200–3202 (2004).
Kamminga, L. M. et al. Impaired hematopoietic stem cell functioning after serial transplantation and during normal aging. Stem Cell 23, 82–92 (2005).
Zediak, V. P., Maillard, I. & Bhandoola, A. Multiple prethymic defects underlie age-related loss of T progenitor competence. Blood 110, 1161–1167 (2007).
Beerman, I., Maloney, W. J., Weissmann, I. L. & Rossi, D. J. Stem cells and the aging hematopoietic system. Curr. Opin. Immunol. 22, 500–506 (2010).
Gonzalez-Vicent, M. et al. Donor age matters in T-cell depleted haploidentical hematopoietic stem cell transplantation in pediatric patients: Faster immune reconstitution using younger donors. Leuk. Res. 57, 60–64 (2017).
Montecino-Rodriguez, E. et al. Lymphoid-biased hematopoietic stem cells are maintained with age and efficiently generate lymphoid progeny. Stem Cell Rep. 12, 584–596 (2019). This study shows that the lymphoid-biased HSCs do not decline in number with age but exhibit changes in gene expression and acquire a myeloid-biased genetic profile.
Rossi, D. J. et al. Cell intrinsic alterations underlie hematopoietic stem cell aging. Proc. Natl Acad. Sci. USA 102, 9194–9199 (2005).
Nijnik, A. et al. DNA repair is limiting for haematopoietic stem cells during ageing. Nature 447, 686–690 (2007).
Rossi, D. J. et al. Deficiencies in DNA damage repair limit the function of haematopoietic stem cells with age. Nature 447, 725–729 (2007).
Fraga, M. F. Genetic and epigenetic regulation of aging. Curr. Opin. Immunol. 21, 446–453 (2009).
Florian, M. C. et al. Aging alters the epigenetic asymmetry of HSC division. PLoS Biol. 16, e2003389 (2018).
Pinho, S. & Frenette, P. S. Haematopoietic stem cell activity and interactions with the niche. Nat. Rev. Mol. Cell. Biol. 20, 303–320 (2019).
Donnini, A., Re, F., Orlando, F. & Provinciali, M. Intrinsic and microenvironmental defects are involved in the age-related changes of Lin- c-kit+ hematopoietic progenitor cells. Rejuvenation Res. 10, 459–472 (2007).
Ergen, A. V., Boles, N. C. & Goodell, M. A. Rantes/Ccl5 influences hematopoietic stem cell subtypes and causes myeloid skewing. Blood 119, 2500–2509 (2012).
Min, H., Montecino-Rodriguez, E. & Dorshkind, K. Reduction in the developmental potential of intrathymic T cell progenitors with age. J. Immunol. 173, 245–250 (2004).
Hakim, F. T. & Gress, R. E. Immunosenescence: deficits in adaptive immunity in the elderly. Tissue Antigens 70, 179–189 (2007).
Flores, K. G., Li, J., Sempowski, G. D., Haynes, B. F. & Hale, L. P. Analysis of the human thymic perivascular space during aging. J. Clin. Invest. 104, 1031–1039 (1999).
Mackall, C. L., Punt, J. A., Morgan, P., Farr, A. G. & Gress, R. E. Thymic function in young/old chimeras: substantial thymic T cell regenerative capacity despite irreversible age-associated thymic involution. Eur. J. Immunol. 28, 1886–1893 (1998).
Kim, M. J., Miller, C. M., Shadrach, J. L., Wagers, A. J. & Serwold, T. Young, proliferative thymic epithelial cells engraft and function in aging thymuses. J. Immunol. 194, 4784–4795 (2015).
Zhu, X. et al. Lymphohematopoietic progenitors do not have a synchronized defect with age-related thymic involution. Aging Cell 6, 663–672 (2007).
Wu, H., Qin, X., Dai, H. & Zhang, Y. Time-course transcriptome analysis of medullary thymic epithelial cells in the early phase of thymic involution. Mol. Immunol. 99, 87–94 (2018).
Ki, S. et al. Global transcriptional profiling reveals distinct functions of thymic stromal subsets and age-related changes during thymic involution. Cell Rep. 9, 402–415 (2014). This work analyses age-associated transcriptional changes of thymic stromal cells and provides an intuitive online tool to visualize the data.
Ferrando-Martinez, S. et al. WNT signaling suppression in the senescent human thymus. J. Gerontol. A. Biol. Sci. Med. Sci. 70, 273–281 (2015).
Lynch, H. E. et al. Thymic involution and immune reconstitution. Trends Immunol. 30, 366–373 (2009).
Jamieson, B. D. et al. Generation of functional thymocytes in the human adult. Immunity 10, 569–575 (1999).
Drabkin, M. J. et al. Age-stratified patterns of thymic involution on multidetector CT. J. Thorac. Imaging 33, 409–416 (2018).
den Braber, I. et al. Maintenance of peripheral naive T cells is sustained by thymus output in mice but not humans. Immunity 36, 288–297 (2012).
Mold, J. E. et al. Cell generation dynamics underlying naive T-cell homeostasis in adult humans. PLoS Biol. 17, e3000383 (2019).
Murray, J. M. et al. Naive T cells are maintained by thymic output in early ages but by proliferation without phenotypic change after age twenty. Immunol. Cell. Biol. 81, 487–495 (2003).
Nikolich-Zugich, J. & Rudd, B. D. Immune memory and aging: an infinite or finite resource? Curr. Opin. Immunol. 22, 535–540 (2010).
Britanova, O. V. et al. Age-related decrease in TCR repertoire diversity measured with deep and normalized sequence profiling. J. Immunol. 192, 2689–2698 (2014).
Palmer, S., Albergante, L., Blackburn, C. C. & Newman, T. J. Thymic involution and rising disease incidence with age. Proc. Natl Acad. Sci. USA 115, 1883–1888 (2018).
Nikolich-Zugich, J. et al. SARS-CoV-2 and COVID-19 in older adults: what we may expect regarding pathogenesis, immune responses, and outcomes. Geroscience 42, 505–514 (2020).
Small, T. N. et al. Immune reconstitution following T-cell depleted bone marrow transplantation: effect of age and posttransplant graft rejection prophylaxis. Biol. Blood Marrow Transpl. 3, 65–75 (1997).
Wils, E. J. et al. Insufficient recovery of thymopoiesis predicts for opportunistic infections in allogeneic hematopoietic stem cell transplant recipients. Haematologica 96, 1846–1854 (2011).
Kim, D. H. et al. Rapid helper T-cell recovery above 200 × 106/l at 3 months correlates to successful transplant outcomes after allogeneic stem cell transplantation. Bone Marrow Transpl. 37, 1119–1128 (2006).
Mackall, C. L., Fry, T. J. & Gress, R. E. Harnessing the biology of IL-7 for therapeutic application. Nat. Rev. Immunol. 11, 330–342 (2011).
Link, A. et al. Fibroblastic reticular cells in lymph nodes regulate the homeostasis of naive T cells. Nat. Immunol. 8, 1255–1265 (2007).
Onder, L. et al. IL-7-producing stromal cells are critical for lymph node remodeling. Blood 120, 4675–4683 (2012).
Laky, K. et al. Enterocyte expression of interleukin 7 induces development of gammadelta T cells and Peyer’s patches. J. Exp. Med. 191, 1569–1580 (2000).
Puel, A., Ziegler, S. F., Buckley, R. H. & Leonard, W. J. Defective IL7R expression in T-B+NK+ severe combined immunodeficiency. Nat. Genet. 20, 394–397 (1998).
Noguchi, M. et al. Interleukin-2 receptor gamma chain mutation results in X-linked severe combined immunodeficiency in humans. Cell 73, 147–157 (1993).
Fischer, A., Notarangelo, L. D., Neven, B., Cavazzana, M. & Puck, J. M. Severe combined immunodeficiencies and related disorders. Nat. Rev. Dis. Prim. 1, 15061 (2015).
Fry, T. J. et al. A potential role for interleukin-7 in T-cell homeostasis. Blood 97, 2983–2990 (2001).
Alpdogan, O. et al. IL-7 enhances peripheral T cell reconstitution after allogeneic hematopoietic stem cell transplantation. J. Clin. Invest. 112, 1095–1107 (2003).
Alpdogan, O. et al. Administration of interleukin-7 after allogeneic bone marrow transplantation improves immune reconstitution without aggravating graft-versus-host disease. Blood 98, 2256–2265 (2001).
Perales, M. A. et al. Recombinant human interleukin-7 (CYT107) promotes T-cell recovery after allogeneic stem cell transplantation. Blood 120, 4882–4891 (2012).
Abdul-Hai, A. et al. Stimulation of immune reconstitution by interleukin-7 after syngeneic bone marrow transplantation in mice. Exp. Hematol. 24, 1416–1422 (1996).
Levy, Y. et al. Effects of recombinant human interleukin 7 on T-cell recovery and thymic output in HIV-infected patients receiving antiretroviral therapy: results of a phase I/IIa randomized, placebo-controlled, multicenter study. Clin. Infect. Dis. 55, 291–300 (2012).
Sportes, C. et al. Administration of rhIL-7 in humans increases in vivo TCR repertoire diversity by preferential expansion of naive T cell subsets. J. Exp. Med. 205, 1701–1714 (2008).
Rosenberg, S. A. et al. IL-7 administration to humans leads to expansion of CD8+ and CD4+ cells but a relative decrease of CD4+ T-regulatory cells. J. Immunother. 29, 313–319 (2006).
Tredan, O. et al. ELYPSE-7: a randomized placebo-controlled phase IIa trial with CYT107 exploring the restoration of CD4+ lymphocyte count in lymphopenic metastatic breast cancer patients. Ann. Oncol. 26, 1353–1362 (2015).
Sheikh, V. et al. Administration of interleukin-7 increases CD4 T cells in idiopathic CD4 lymphocytopenia. Blood 127, 977–988 (2016).
Kim, H. K. et al. Distinct IL-7 signaling in recent thymic emigrants versus mature naive T cells controls T-cell homeostasis. Eur. J. Immunol. 46, 1669–1680 (2016).
Azevedo, R. I. et al. IL-7 sustains CD31 expression in human naive CD4+ T cells and preferentially expands the CD31+ subset in a PI3K-dependent manner. Blood 113, 2999–3007 (2009).
Chu, Y. W. et al. Exogenous IL-7 increases recent thymic emigrants in peripheral lymphoid tissue without enhanced thymic function. Blood 104, 1110–1119 (2004).
Okoye, A. A. et al. Effect of IL-7 therapy on naive and memory T cell homeostasis in aged rhesus macaques. J. Immunol. 195, 4292–4305 (2015).
Finch, P. W. & Rubin, J. S. Keratinocyte growth factor/fibroblast growth factor 7, a homeostatic factor with therapeutic potential for epithelial protection and repair. Adv. Cancer. Res. 91, 69–136 (2004).
Rossi, S. W. et al. Keratinocyte growth factor (KGF) enhances postnatal T-cell development via enhancements in proliferation and function of thymic epithelial cells. Blood 109, 3803–3811 (2007).
Erickson, M. et al. Regulation of thymic epithelium by keratinocyte growth factor. Blood 100, 3269–3278 (2002).
Rossi, S. et al. Keratinocyte growth factor preserves normal thymopoiesis and thymic microenvironment during experimental graft-versus-host disease. Blood 100, 682–691 (2002).
Alpdogan, O. et al. Keratinocyte growth factor (KGF) is required for postnatal thymic regeneration. Blood 107, 2453–2460 (2006).
Min, D. et al. Protection from thymic epithelial cell injury by keratinocyte growth factor: a new approach to improve thymic and peripheral T-cell reconstitution after bone marrow transplantation. Blood 99, 4592–4600 (2002).
Wang, Y. et al. Keratinocyte growth factor enhanced immune reconstitution in murine allogeneic umbilical cord blood cell transplant. Leuk. Lymphoma 52, 1556–1566 (2011).
Min, D. et al. Sustained thymopoiesis and improvement in functional immunity induced by exogenous KGF administration in murine models of aging. Blood 109, 2529–2537 (2007).
Wils, E. J. et al. Keratinocyte growth factor and stem cell factor to improve thymopoiesis after autologous CD34+ cell transplantation in rhesus macaques. Biol. Blood Marrow Transpl. 18, 55–65 (2012).
Kelly, R. M. et al. Keratinocyte growth factor and androgen blockade work in concert to protect against conditioning regimen-induced thymic epithelial damage and enhance T-cell reconstitution after murine bone marrow transplantation. Blood 111, 5734–5744 (2008). This article shows that the combination of sex steroid ablation and KGF results in the preservation of thymic architecture, enhancement of T cell development and improvement of the T cell repertoire.
Coles, A. J. et al. Keratinocyte growth factor impairs human thymic recovery from lymphopenia. JCI Insight 4, e125377 (2019).
Goldstein, A. L., Guha, A., Zatz, M. M., Hardy, M. A. & White, A. Purification and biological activity of thymosin, a hormone of the thymus gland. Proc. Natl Acad. Sci. USA 69, 1800–1803 (1972).
Hirokawa, K., McClure, J. E. & Goldstein, A. L. Age-related changes in localization of thymosin in the human thymus. Thymus 4, 19–29 (1982).
Wang, F., Yu, T., Zheng, H. & Lao, X. Thymosin alpha1-Fc modulates the immune system and down-regulates the progression of melanoma and breast cancer with a prolonged half-life. Sci. Rep. 8, 12351 (2018).
Knutsen, A. P., Freeman, J. J., Mueller, K. R., Roodman, S. T. & Bouhasin, J. D. Thymosin-alpha1 stimulates maturation of CD34+ stem cells into CD3+4+ cells in an in vitro thymic epithelia organ coculture model. Int. J. Immunopharmacol. 21, 15–26 (1999).
Perruccio, K. et al. Thymosin alpha1 to harness immunity to pathogens after haploidentical hematopoietic transplantation. Ann. N. Y. Acad. Sci. 1194, 153–161 (2010).
Liu, Y. et al. Thymosin alpha 1 (Talpha1) reduces the mortality of severe COVID-19 by restoration of lymphocytopenia and reversion of exhausted T cells. Clin. Infect. Dis. https://doi.org/10.1093/cid/ciaa630 (2020).
Calzolari, A. Recherches experimentales sur un rapport probable entre la function du thymus et celle des testicules. Arch. Ital. Biol. 30, 71–77 (1898). This article presents one of the very first observations suggesting a relationship between sex steroids and thymic function.
Velardi, E. et al. Sex steroid blockade enhances thymopoiesis by modulating Notch signaling. J. Exp. Med. 211, 2341–2349 (2014). This study demonstrates that androgens suppress thymic function through the direct negative regulation of DLL4 expression.
Zoller, A. L. & Kersh, G. J. Estrogen induces thymic atrophy by eliminating early thymic progenitors and inhibiting proliferation of beta-selected thymocytes. J. Immunol. 176, 7371–7378 (2006).
Khong, D. M. et al. Enhanced hematopoietic stem cell function mediates immune regeneration following sex steroid blockade. Stem Cell Rep. 4, 445–458 (2015).
Dudakov, J. A. et al. Sex steroid ablation enhances hematopoietic recovery following cytotoxic antineoplastic therapy in aged mice. J. Immunol. 183, 7084–7094 (2009).
Dudakov, J. A., Goldberg, G. L., Reiseger, J. J., Chidgey, A. P. & Boyd, R. L. Withdrawal of sex steroids reverses age- and chemotherapy-related defects in bone marrow lymphopoiesis. J. Immunol. 182, 6247–6260 (2009).
Goldberg, G. L. et al. Luteinizing hormone-releasing hormone enhances T cell recovery following allogeneic bone marrow transplantation. J. Immunol. 182, 5846–5854 (2009).
Sutherland, J. S. et al. Activation of thymic regeneration in mice and humans following androgen blockade. J. Immunol. 175, 2741–2753 (2005).
Windmill, K. F. & Lee, V. W. Effects of castration on the lymphocytes of the thymus, spleen and lymph nodes. Tissue Cell 30, 104–111 (1998).
Williams, K. M. et al. CCL25 increases thymopoiesis after androgen withdrawal. Blood 112, 3255–3263 (2008).
Sutherland, J. S. et al. Enhanced immune system regeneration in humans following allogeneic or autologous hemopoietic stem cell transplantation by temporary sex steroid blockade. Clin. Cancer Res. 14, 1138–1149 (2008). This is the first observation showing that pharmacological ablation of sex steroids enhances immune recovery in HCT recipients.
Griffith, A. V., Fallahi, M., Venables, T. & Petrie, H. T. Persistent degenerative changes in thymic organ function revealed by an inducible model of organ regrowth. Aging Cell 11, 169–177 (2012).
Taub, D. D., Murphy, W. J. & Longo, D. L. Rejuvenation of the aging thymus: growth hormone-mediated and ghrelin-mediated signaling pathways. Curr. Opin. Pharmacol. 10, 408–424 (2010).
Hattori, N. Expression, regulation and biological actions of growth hormone (GH) and ghrelin in the immune system. Growth Horm. IGF Res. 19, 187–197 (2009).
Napolitano, L. A. et al. Growth hormone enhances thymic function in HIV-1-infected adults. J. Clin. Invest. 118, 1085–1098 (2008).
Napolitano, L. A. et al. Increased thymic mass and circulating naive CD4 T cells in HIV-1-infected adults treated with growth hormone. AIDS 16, 1103–1111 (2002).
Chen, B. J., Cui, X., Sempowski, G. D. & Chao, N. J. Growth hormone accelerates immune recovery following allogeneic T-cell-depleted bone marrow transplantation in mice. Exp. Hematol. 31, 953–958 (2003).
Morrhaye, G. et al. Impact of growth hormone (GH) deficiency and GH replacement upon thymus function in adult patients. PLoS ONE 4, e5668 (2009).
Fahy, G. M. et al. Reversal of epigenetic aging and immunosenescent trends in humans. Aging Cell 18, e13028 (2019).
Chen, T. et al. IL-12 facilitates both the recovery of endogenous hematopoiesis and the engraftment of stem cells after ionizing radiation. Exp. Hematol. 35, 203–213 (2007).
Li, L. et al. IL-12 inhibits thymic involution by enhancing IL-7- and IL-2-induced thymocyte proliferation. J. Immunol. 172, 2909–2916 (2004).
Bailey, C. P. et al. New interleukin-15 superagonist (IL-15SA) significantly enhances graft-versus-tumor activity. Oncotarget 8, 44366–44378 (2017).
Alpdogan, O. et al. Interleukin-15 enhances immune reconstitution after allogeneic bone marrow transplantation. Blood 105, 865–873 (2005).
Sauter, C. T. et al. Interleukin-15 administration increases graft-versus-tumor activity in recipients of haploidentical hematopoietic SCT. Bone Marrow Transpl. 48, 1237–1242 (2013).
Rafei, M., Dumont-Lagace, M., Rouette, A. & Perreault, C. Interleukin-21 accelerates thymic recovery from glucocorticoid-induced atrophy. PLoS ONE 8, e72801 (2013).
Tormo, A. et al. Interleukin-21 promotes thymopoiesis recovery following hematopoietic stem cell transplantation. J. Hematol. Oncol. 10, 120 (2017).
Al-Chami, E. et al. Interleukin-21 administration to aged mice rejuvenates their peripheral T-cell pool by triggering de novo thymopoiesis. Aging Cell 15, 349–360 (2016).
Pan, B. et al. Donor T-cell-derived interleukin-22 promotes thymus regeneration and alleviates chronic graft-versus-host disease in murine allogeneic hematopoietic cell transplant. Int. Immunopharmacol. 67, 194–201 (2019).
Hikosaka, Y. et al. The cytokine RANKL produced by positively selected thymocytes fosters medullary thymic epithelial cells that express autoimmune regulator. Immunity 29, 438–450 (2008).
Roberts, N. A. et al. Rank signaling links the development of invariant γδ T cell progenitors and Aire+ medullary epithelium. Immunity 36, 427–437 (2012).
Rossi, S. W. et al. RANK signals from CD4+3- inducer cells regulate development of Aire-expressing epithelial cells in the thymic medulla. J. Exp. Med. 204, 1267–1272 (2007).
Lopes, N., Vachon, H., Marie, J. & Irla, M. Administration of RANKL boosts thymic regeneration upon bone marrow transplantation. EMBO Mol. Med. 9, 835–851 (2017).
Tumanov, A. V. et al. Lymphotoxin controls the IL-22 protection pathway in gut innate lymphoid cells during mucosal pathogen challenge. Cell Host Microbe 10, 44–53 (2011).
Cella, M., Otero, K. & Colonna, M. Expansion of human NK-22 cells with IL-7, IL-2, and IL-1beta reveals intrinsic functional plasticity. Proc. Natl Acad. Sci. USA 107, 10961–10966 (2010).
Vonarbourg, C. et al. Regulated expression of nuclear receptor RORgammat confers distinct functional fates to NK cell receptor-expressing RORgammat+ innate lymphocytes. Immunity 33, 736–751 (2010).
Yoshida, H. et al. Different cytokines induce surface lymphotoxin-alphabeta on IL-7 receptor-alpha cells that differentially engender lymph nodes and Peyer’s patches. Immunity 17, 823–833 (2002).
Gendron, S., Boisvert, M., Chetoui, N. & Aoudjit, F. Alpha1beta1 integrin and interleukin-7 receptor up-regulate the expression of RANKL in human T cells and enhance their osteoclastogenic function. Immunology 125, 359–369 (2008).
Gordon, J., Patel, S. R., Mishina, Y. & Manley, N. R. Evidence for an early role for BMP4 signaling in thymus and parathyroid morphogenesis. Dev. Biol. 339, 141–154 (2010).
Bleul, C. C. & Boehm, T. BMP signaling is required for normal thymus development. J. Immunol. 175, 5213–5221 (2005).
Tsai, P. T., Lee, R. A. & Wu, H. BMP4 acts upstream of FGF in modulating thymic stroma and regulating thymopoiesis. Blood 102, 3947–3953 (2003).
Havel, J. J., Chowell, D. & Chan, T. A. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat. Rev. Cancer 19, 133–150 (2019).
McGranahan, N. et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 351, 1463–1469 (2016).
Snyder, A. et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 371, 2189–2199 (2014).
Rizvi, N. A. et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 348, 124–128 (2015).
Thompson, H. L. et al. Lymph nodes as barriers to T-cell rejuvenation in aging mice and nonhuman primates. Aging Cell 18, e12865 (2019).
Clise-Dwyer, K., Huston, G. E., Buck, A. L., Duso, D. K. & Swain, S. L. Environmental and intrinsic factors lead to antigen unresponsiveness in CD4+ recent thymic emigrants from aged mice. J. Immunol. 178, 1321–1331 (2007).
Becklund, B. R. et al. The aged lymphoid tissue environment fails to support naive T cell homeostasis. Sci. Rep. 6, 30842 (2016).
Czechowicz, A. et al. Selective hematopoietic stem cell ablation using CD117-antibody-drug-conjugates enables safe and effective transplantation with immunity preservation. Nat. Commun. 10, 617 (2019).
Palchaudhuri, R. et al. Non-genotoxic conditioning for hematopoietic stem cell transplantation using a hematopoietic-cell-specific internalizing immunotoxin. Nat. Biotechnol. 34, 738–745 (2016).
Czechowicz, A., Kraft, D., Weissman, I. L. & Bhattacharya, D. Efficient transplantation via antibody-based clearance of hematopoietic stem cell niches. Science 318, 1296–1299 (2007). Together with the articles by Czechowicz et al. (2019) and Palchaudhuri et al. (2016), this article documents the use of non-genotoxic conditioning approaches to deplete host haematopoietic cells in the BM niche and allow donor HSC engraftment.
Mackall, C. L. et al. IL-7 increases both thymic-dependent and thymic-independent T-cell regeneration after bone marrow transplantation. Blood 97, 1491–1497 (2001).
Fry, T. J. et al. IL-7 therapy dramatically alters peripheral T-cell homeostasis in normal and SIV-infected nonhuman primates. Blood 101, 2294–2299 (2003).
Montero-Herradon, S., Garcia-Ceca, J. & Zapata, A. G. altered maturation of medullary TEC in EphB-deficient thymi is recovered by RANK signaling stimulation. Front. Immunol. 9, 1020 (2018).
Akiyama, T. et al. The tumor necrosis factor family receptors RANK and CD40 cooperatively establish the thymic medullary microenvironment and self-tolerance. Immunity 29, 423–437 (2008).
Rizwan, R. et al. Peritransplant palifermin use and lymphocyte recovery after T-cell replete, matched related allogeneic hematopoietic cell transplantation. Am. J. Hematol. 86, 879–882 (2011).
Seggewiss, R. et al. Keratinocyte growth factor augments immune reconstitution after autologous hematopoietic progenitor cell transplantation in rhesus macaques. Blood 110, 441–449 (2007).
Goldberg, J. D. et al. Palifermin is efficacious in recipients of TBI-based but not chemotherapy-based allogeneic hematopoietic stem cell transplants. Bone Marrow Transpl. 48, 99–104 (2013).
Jacobson, J. M. et al. A randomized controlled trial of palifermin (recombinant human keratinocyte growth factor) for the treatment of inadequate CD4+ T-lymphocyte recovery in patients with HIV-1 infection on antiretroviral therapy. J. Acquir. Immune. Defic. Syndr. 66, 399–406 (2014).
Fry, T. J. et al. Flt3 ligand enhances thymic-dependent and thymic-independent immune reconstitution. Blood 104, 2794–2800 (2004).
Wils, E. J. et al. Flt3 ligand expands lymphoid progenitors prior to recovery of thymopoiesis and accelerates T cell reconstitution after bone marrow transplantation. J. Immunol. 178, 3551–3557 (2007).
Williams, K. M. et al. FLT3 ligand regulates thymic precursor cells and hematopoietic stem cells through interactions with CXCR4 and the marrow niche. Exp. Hematol. 52, 40–49 (2017).
Alpdogan, O. et al. Insulin-like growth factor-I enhances lymphoid and myeloid reconstitution after allogeneic bone marrow transplantation. Transplantation 75, 1977–1983 (2003).
Chu, Y. W. et al. Exogenous insulin-like growth factor 1 enhances thymopoiesis predominantly through thymic epithelial cell expansion. Blood 112, 2836–2846 (2008).
Patchen, M. L., Fischer, R., Schmauder-Chock, E. A. & Williams, D. E. Mast cell growth factor enhances multilineage hematopoietic recovery in vivo following radiation-induced aplasia. Exp. Hematol. 22, 31–39 (1994).
Chung, B. et al. Combined effects of interleukin-7 and stem cell factor administration on lymphopoiesis after murine bone marrow transplantation. Biol. Blood Marrow Transpl. 17, 48–60 (2011).
Wils, E. J. et al. Stem cell factor consistently improves thymopoiesis after experimental transplantation of murine or human hematopoietic stem cells in immunodeficient mice. J. Immunol. 187, 2974–2981 (2011).
Ding, J.-H. et al. The role of Tα1 on the infective patients after hematopoietic stem cell transplantation. Int. J. Hematol. 97, 280–283 (2013).
Sirohi, B. et al. Use of physiological doses of human growth hormone in haematological patients receiving intensive chemotherapy promotes haematopoietic recovery: a double-blind randomized, placebo-controlled study. Bone Marrow Transpl. 39, 115–120 (2007).
Carlo-Stella, C. et al. Age- and irradiation-associated loss of bone marrow hematopoietic function in mice is reversed by recombinant human growth hormone. Exp. Hematol. 32, 171–178 (2004).
Dixit, V. D. et al. Ghrelin promotes thymopoiesis during aging. J. Clin. Invest. 117, 2778–2790 (2007).
Lai, K. P. et al. Targeting thymic epithelia AR enhances T-cell reconstitution and bone marrow transplant grafting efficacy. Mol. Endocrinol. 27, 25–37 (2013).
Heng, T. S. et al. Impact of sex steroid ablation on viral, tumour and vaccine responses in aged mice. PLoS ONE 7, e42677 (2012).
Goldberg, G. L. et al. Sex steroid ablation enhances immune reconstitution following cytotoxic antineoplastic therapy in young mice. J. Immunol. 184, 6014–6024 (2010).
Goldberg, G. L. et al. Enhanced immune reconstitution by sex steroid ablation following allogeneic hemopoietic stem cell transplantation. J. Immunol. 178, 7473–7484 (2007).
Velardi, E. et al. Suppression of luteinizing hormone enhances HSC recovery after hematopoietic injury. Nat. Med. 24, 239–246 (2018).
Zakrzewski, J. L. et al. Tumor immunotherapy across MHC barriers using allogeneic T-cell precursors. Nat. Biotechnol. 26, 453–461 (2008).
Holland, A. M., Zakrzewski, J. L., Goldberg, G. L., Ghosh, A. & van den Brink, M. R. Adoptive precursor cell therapy to enhance immune reconstitution after hematopoietic stem cell transplantation in mouse and man. Semin. Immunopathol. 30, 479–487 (2008).
Zakrzewski, J. L. et al. Adoptive transfer of T-cell precursors enhances T-cell reconstitution after allogeneic hematopoietic stem cell transplantation. Nat. Med. 12, 1039–1047 (2006).
Dallas, M. H., Varnum-Finney, B., Martin, P. J. & Bernstein, I. D. Enhanced T-cell reconstitution by hematopoietic progenitors expanded ex vivo using the Notch ligand Delta1. Blood 109, 3579–3587 (2007).
Awong, G. et al. Human proT-cells generated in vitro facilitate hematopoietic stem cell-derived T-lymphopoiesis in vivo and restore thymic architecture. Blood 122, 4210–4219 (2013).
Smith, M. J. et al. T cell progenitor therapy–facilitated thymopoiesis depends upon thymic input and continued thymic microenvironment interaction. JCI Insight 2, e92056 (2017).
Batorov, E. V. et al. Mesenchymal stromal cells improve early lymphocyte recovery and T cell reconstitution after autologous hematopoietic stem cell transplantation in patients with malignant lymphomas. Cell Immunol. 297, 80–86 (2015).
Choi, D. W. et al. Cotransplantation of tonsil derived mesenchymal stromal cells in bone marrow transplantation promotes thymus regeneration and T cell diversity following cytotoxic conditioning. Int. J. Mol. Med. 46, 1166–1174 (2020).
Su, M. et al. Efficient in vitro generation of functional thymic epithelial progenitors from human embryonic stem cells. Sci. Rep. 5, 9882 (2015).
Bredenkamp, N. et al. An organized and functional thymus generated from FOXN1-reprogrammed fibroblasts. Nat. Cell. Biol. 16, 902–908 (2014). This study demonstrates that enforced expression of FOXN1 is sufficient to reprogramme fibroblasts into functional TECs capable of supporting T cell differentiation when transferred in vivo.
Lai, L. et al. Mouse embryonic stem cell-derived thymic epithelial cell progenitors enhance T-cell reconstitution after allogeneic bone marrow transplantation. Blood 118, 3410–3418 (2011).
Parent, A. V. et al. Generation of functional thymic epithelium from human embryonic stem cells that supports host T cell development. Cell Stem. Cell 13, 219–229 (2013).
Bortolomai, I. et al. Gene modification and three-dimensional scaffolds as novel tools to allow the use of postnatal thymic epithelial cells for thymus regeneration approaches. Stem Cell Transl. Med. 8, 1107–1122 (2019).
Otsuka, R. et al. Efficient generation of thymic epithelium from induced pluripotent stem cells that prolongs allograft survival. Sci. Rep. 10, 224 (2020).
Shah, N. J. et al. An injectable bone marrow–like scaffold enhances T cell immunity after hematopoietic stem cell transplantation. Nat. Biotechnol. 7, 293–302 (2019). This work shows that the injection of an engineered BM-like scaffold enhances T cell neogenesis and diversification of TCRs in mouse models of HCT.
Chung, B. et al. Engineering the human thymic microenvironment to support thymopoiesis in vivo. Stem Cell 32, 2386–2396 (2014).
Fan, Y. et al. Bioengineering thymus organoids to restore thymic function and induce donor-specific immune tolerance to allografts. Mol. Ther. 23, 1262–1277 (2015).
van der Maas, N. G., Berghuis, D., van der Burg, M. & Lankester, A. C. B cell reconstitution and influencing factors after hematopoietic stem cell transplantation in children. Front. Immunol. 10, 782 (2019).
Abdel-Azim, H., Elshoury, A., Mahadeo, K. M., Parkman, R. & Kapoor, N. Humoral immune reconstitution kinetics after allogeneic hematopoietic stem cell transplantation in children: a maturation block of IgM memory B cells may lead to impaired antibody immune reconstitution. Biol. Blood Marrow Transpl. 23, 1437–1446 (2017).
Sarantopoulos, S., Blazar, B. R., Cutler, C. & Ritz, J. B cells in chronic graft-versus-host disease. Biol. Blood Marrow Transpl. 21, 16–23 (2015).
Park, B. G. et al. Reconstitution of lymphocyte subpopulations after hematopoietic stem cell transplantation: comparison of hematologic malignancies and donor types in event-free patients. Leuk. Res. 39, 1334–1341 (2015).
Marie-Cardine, A. et al. Transitional B cells in humans: characterization and insight from B lymphocyte reconstitution after hematopoietic stem cell transplantation. Clin. Immunol. 127, 14–25 (2008).
Buser, A. et al. Impaired B-cell reconstitution in lymphoma patients undergoing allogeneic HSCT: an effect of pretreatment with rituximab? Bone Marrow Transpl. 42, 483–487 (2008).
Scarselli, A. et al. Longitudinal evaluation of immune reconstitution and B-cell function after hematopoietic cell transplantation for primary immunodeficiency. J. Clin. Immunol. 35, 373–383 (2015).
Small, T. N. et al. B-cell differentiation following autologous, conventional, or T-cell depleted bone marrow transplantation: a recapitulation of normal B-cell ontogeny. Blood 76, 1647–1656 (1990).
Pao, M. et al. Response to pneumococcal (PNCRM7) and haemophilus influenzae conjugate vaccines (HIB) in pediatric and adult recipients of an allogeneic hematopoietic cell transplantation (alloHCT). Biol. Blood Marrow Transpl. 14, 1022–1030 (2008).
Omazic, B., Lundkvist, I., Mattsson, J., Permert, J. & Näsman-Björk, I. Memory B lymphocytes determine repertoire oligoclonality early after haematopoietic stem cell transplantation. Clin. Exp. Immunol. 134, 159–166 (2003).
Glas, A. M. et al. B-cell-autonomous somatic mutation deficit following bone marrow transplant. Blood 96, 1064–1069 (2000).
Sethi, M. K. et al. VH1 family immunoglobulin repertoire sequencing after allogeneic hematopoietic stem cell transplantation. PLoS ONE 12, e0168096 (2017).
Lakshmikanth, T. et al. Mass cytometry and topological data analysis reveal immune parameters associated with complications after allogeneic stem cell transplantation. Cell Rep. 20, 2238–2250 (2017).
van den Broek, T., Borghans, J. A. M. & van Wijk, F. The full spectrum of human naive T cells. Nat. Rev. Immunol. 18, 363–373 (2018).
Haines, C. J. et al. Human CD4+ T cell recent thymic emigrants are identified by protein tyrosine kinase 7 and have reduced immune function. J. Exp. Med. 206, 275–285 (2009).
McFarland, R. D., Douek, D. C., Koup, R. A. & Picker, L. J. Identification of a human recent thymic emigrant phenotype. Proc. Natl Acad. Sci. USA 97, 4215–4220 (2000).
Brezinschek, R. I., Oppenheimer-Marks, N. & Lipsky, P. E. Activated T cells acquire endothelial cell surface determinants during transendothelial migration. J. Immunol. 162, 1677–1684 (1999).
Pekalski, M. L. et al. Neonatal and adult recent thymic emigrants produce IL-8 and express complement receptors CR1 and CR2. JCI Insight 2, e93739 (2017).
Kimmig, S. et al. Two subsets of naive T helper cells with distinct T cell receptor excision circle content in human adult peripheral blood. J. Exp. Med. 195, 789–794 (2002).
van den Beemd, R. et al. Flow cytometric analysis of the Vbeta repertoire in healthy controls. Cytometry 40, 336–345 (2000).
Verfuerth, S. et al. Longitudinal monitoring of immune reconstitution by CDR3 size spectratyping after T-cell-depleted allogeneic bone marrow transplant and the effect of donor lymphocyte infusions on T-cell repertoire. Blood 95, 3990–3995 (2000).
Acknowledgements
The authors are grateful for support from the US National Cancer Institute (R01-CA228358, R01-CA228308, R01-HL147584, P30-CA008748 Memorial Sloan Kettering Cancer Center Support Grant/Core Grant and Project 2 of P01-CA023766), the US National Heart, Lung, and Blood Institute (R01-HL125571 and R01-HL123340), the US National Institute on Aging (Project 2 of P01-AG052359), the Tri-Institutional Stem Cell Initiative (2016-013) and the US National Institute of Allergy and Infectious Diseases (U01-AI124275) to M.R.M.v.d.B., the Amy Strelzer Manasevit Research Program, the Italian Association for Cancer Research and the Italian Ministry of Health (Ricerca Corrente programme) to E.V. and a Society of Memorial Sloan Kettering Cancer Center Scholars Award to J.J.T. Additional funding was received from The Lymphoma Foundation, the Susan and Peter Solomon Divisional Genomics Program and the Parker Institute for Cancer Immunotherapy at Memorial Sloan Kettering Cancer Center and Seres Therapeutics.
Author information
Authors and Affiliations
Contributions
The authors contributed equally to all aspects of the article.
Corresponding authors
Ethics declarations
Competing interests
M.R.M.v.d.B. has received research support and stock options from Seres Therapeutics, has received royalties from Wolters Kluwer, has consulted for, received honoraria from or participated in advisory boards for Seres Therapeutics, Jazz Pharmaceuticals, Rheos, Therakos, WindMIL Therapeutics, Amgen, Merck & Co. Inc., Magenta Therapeutics, DKMS Medical Council (board), Forty Seven Inc. (spouse), Pharmacyclics (spouse) and Kite Pharmaceuticals (spouse) and has intellectual property licensing agreements with Seres Therapeutics and Juno Therapeutics. E.V. has acted as a consultant for and received honoraria from Ferring Pharmaceuticals. M.R.M.v.d.B. is an inventor on a patent application (US2015/058095) submitted by Memorial Sloan Kettering Cancer Center. Two provisional patent applications have been filed (US 15/033,178 and US 62/566,897) with E.V. and M.R.M.v.d.B. listed as inventors. J.J.T. declares no competing interests.
Additional information
Peer review information
Nature Reviews Immunology thanks K. Weinberg and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Glossary
- Thymic epithelial cells
-
(TECs). The major component of thymic stroma that supports all stages of thymocyte development. They are further divided into cortical and medullary TECs on the basis of their localization within the thymus and are crucial for the positive and negative selection of thymocytes, respectively.
- Graft-versus-host disease
-
(GVHD). Following allogeneic bone marrow transplantation, donor-derived T cells can be activated by residual host-derived antigen-presenting cells. The resulting T cell reactivity can escalate into the life-threatening condition known as GVHD, which targets mainly the skin, liver and intestines. Acute GVHD is a rapid response against recipient tissues that usually manifests itself within 100 days following haematopoietic cell transplantation, whereas chronic GVHD is reactions that occur after 100 days.
- Sepsis
-
A severe, life-threatening form of infection characterized by systemic inflammatory response with resultant multi-organ failure followed by immunosuppression.
- Glucocorticoids
-
A group of compounds that belong to the corticosteroid family. These compounds can be either naturally produced (hormones) or synthetic. They affect metabolism and have anti-inflammatory and immunosuppressive effects. Many synthetic glucocorticoids (for example, dexamethasone) are used in clinical medicine as anti-inflammatory drugs.
- Conditioning regimens
-
Also known as preparative regimens. A combination of chemotherapy, radiotherapy and/or immunosuppressive medications that is designed not only to destroy residual malignant cells but also to provide space for donor stem cell engraftment and to provide immunosuppression to prevent host rejection of the donor stem cells.
- TCR excision circles
-
(TRECs). Non-replicative DNA episomes that are normally produced as by-products during T cell receptor (TCR) rearrangement in thymocytes. They are therefore expressed only in T cells of thymic origin and provide a useful tool to assess thymic function and recovery of the T cell pool.
- Common lymphoid progenitors
-
Progenitors of lymphoid cell lineages, which include B cells, T cells, natural killer cells and innate lymphoid cells. Bone marrow common lymphoid progenitors are defined by their expression of the interleukin-7 receptor, FMS-related tyrosine kinase 3 (FLT3) and KIT, and the absence of all conventional lineage markers.
- Innate lymphoid cells
-
(ILCs). A group of innate immune cells that are lymphoid in morphology and developmental origin but lack properties of adaptive B cells and T cells such as recombined antigen-specific receptors. They function in the regulation of immunity, tissue homeostasis and inflammation in response to cytokine stimulation.
- Allogeneic HCT
-
Transplantation approach involving transfer of haematopoietic cells from a healthy donor to a patient after conditioning with high-intensity chemotherapy or irradiation. This approach can be used to treat either malignant or non-malignant disorders. Mismatches between the histocompatibility antigens of the donor and the patient can lead to adverse events, such as rejection of the transplanted graft or pathological immune responses to normal tissues in the patient.
- Cytokine receptor common subunit-γ
-
A chain common to type I cytokine receptors. It was first discovered as the γ-chain of the interleukin-2 (IL-2) receptor and was subsequently shown also to be present in the receptors for IL-4, IL-7, IL-9, IL-15 and IL-21. Gene mutations affecting this γ-chain in humans result in absence of T cells and natural killer cells, a condition termed X-linked severe combined immunodeficiency.
- Recent thymic emigrants
-
(RTEs). Semimature T cells that have left the thymus but have yet to undergo the final stages of maturation. Typically a window of around 2 weeks after thymic maturation is used to differentiate between RTEs and fully mature T cells.
- Lymphoid tissue inducer (LTi) cells
-
Cells that are present in developing lymph nodes, Peyer’s patches and nasopharynx-associated lymphoid tissue. They are required for the development of these lymphoid organs and are characterized by expression of the transcription factor RORγt, interleukin-7 receptor-α and lymphotoxin-α1β2.
Rights and permissions
About this article

Cite this article
Velardi, E., Tsai, J.J. & van den Brink, M.R.M. T cell regeneration after immunological injury. Nat Rev Immunol 21, 277–291 (2021). https://doi.org/10.1038/s41577-020-00457-z
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41577-020-00457-z
This article is cited by
-
Oleic acid availability impacts thymocyte preprogramming and subsequent peripheral Treg cell differentiation
Nature Immunology (2024)
-
Cyclosporine A-resistant CAR-T cells mediate antitumour immunity in the presence of allogeneic cells
Nature Communications (2023)
-
Profiling the epigenetic landscape of the antigen receptor repertoire: the missing epi-immunogenomics data
Nature Methods (2023)
-
Donor HLA mismatch promotes full donor T-cell chimerism in the allogeneic stem cell transplant with reduced-intensity conditioning and post-transplant cyclophosphamide GVHD prophylaxis
Annals of Hematology (2023)
-
Real clinical outcomes of nivolumab plus ipilimumab for renal cell carcinoma in patients over 75 years old
International Journal of Clinical Oncology (2023)
