
Abstract
Protein glycosylation, the enzymatic modification of amino acid sidechains with sugar moieties, plays critical roles in cellular function, human health, and biotechnology. However, studying and producing defined glycoproteins remains challenging. Cell-free glycoprotein synthesis systems, in which protein synthesis and glycosylation are performed in crude cell extracts, offer new approaches to address these challenges. Here, we review versatile, state-of-the-art systems for biomanufacturing glycoproteins in prokaryotic and eukaryotic cell-free systems with natural and synthetic N-linked glycosylation pathways. We discuss existing challenges and future opportunities in the use of cell-free systems for the design, manufacture, and study of glycoprotein biomedicines.
Similar content being viewed by others

Introduction
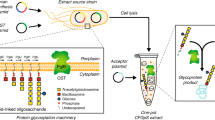
Cell-free protein synthesis (CFPS) systems have emerged as promising platforms to accelerate protein design, biomanufacturing, and testing [14, 30, 50, 93, 115, 125, 143, 151]. CFPS relies on the activation of transcriptional and translational machinery from crude cell extracts to produce proteins without intact cells (Fig. 1). As CFPS technologies have matured, the cost and time required to prepare reactions have decreased, while protein yields have increased, in some cases, to grams of protein produced per liter of reaction volume [16, 52, 84, 125, 129]. Current cell-free systems provide some distinct advantages over cellular expression for high-throughput experimentation. For example, in screening campaigns, CFPS reactions provide excellent speed and flexibility because they can produce protein from linear DNA templates in a matter of hours, avoiding rate-limiting transformation or transfection procedures [119]. Additionally, assembly of CFPS reactions can be automated and tuned using liquid-handling systems, increasing throughput for protein expression, optimization, and characterization [7, 15, 43, 71, 107, 138].

Cell-free protein synthesis schematic. Cell-free protein synthesis is the activation of transcription and translation using crude cellular extracts instead of intact cells. Extracts are supplemented with exogenous resources, including amino acids, nucleotides, a secondary energy substrate, salts, and other necessary factors for protein synthesis. CFPS systems are modular with respect to the protein produced, requiring only changes in DNA or mRNA templates to produce different proteins
Recently, CFPS systems that are tailored to produce proteins with post-translational modifications have been developed, opening the door to cell-free biomanufacturing of therapeutically relevant proteins. A key feature of these efforts has been developing strategies to leverage the open nature of CFPS, which affords rigorous control over the molecular environment of protein expression. This control allows users to study and optimize site-specific protein modifications that are often critical for proper folding and bioactivity of therapeutics and vaccines [38, 39, 51, 105, 116, 125, 151]. For example, CFPS offers flexibility to tune enzymatic protein modifications by varying the enzyme identity, concentration, and available substrates, thereby allowing control over parameters that can be confounding in living organisms [20, 31, 41, 62, 63, 70, 78, 79, 120]. Additionally, CFPS extracts can be prepared from an array of different culturable cell lines, allowing users to leverage strain-specific endogenous (or heterologous) biological machinery. As a result, CFPS systems are now capable of producing products such as antibodies, antibody fragments, multi-subunit enzymes, and conjugate vaccines that may require disulfide bonds and glycosylation for activity. CFPS also offers unique advantages for biomanufacturing, including simplified scalability from microliter to 100 liter reactions [150], and the ability to freeze-dry reactions that are in turn shelf-stable until rehydration at the point of use [4, 53, 107, 127, 131, 132]. The latter point could be transformative for emerging distributed biomanufacturing efforts.
The ability to produce and study glycoproteins is of great importance for engineering therapeutics and vaccines. Protein glycosylation, the covalent conjugation of sugars to amino acid sidechains, is one of the most prevalent and important protein modifications, occurring in all domains of life [27, 32, 66, 90, 118]. Glycosylation occurs on ~ 50% of eukaryotic proteins [68] and on the majority of preclinical and FDA-approved biologics [122], profoundly impacting protein folding [108], stability [47, 154], and immunogenicity [3, 76]. Unfortunately, building and testing defined glycoproteins in cells remains challenging for several reasons. These include glycoprotein heterogeneity [49, 60, 121], gaps in the methods and basic knowledge required to build defined glycoforms with desired pharmacological activities [72, 153], and the high costs and lengthy durations required to generate stable cell lines in mammalian cell culture. To address these issues, diverse CFPS platforms that allow user-defined glycosylation have been developed [26, 72].
Outline and scope
Here, we review CFPS systems for producing defined glycoproteins with asparagine-linked (N-linked) glycosylation (Fig. 2). A wide array of systems that interface glycosylation with cell-free protein synthesis, which we refer to as cell-free glycoprotein synthesis (CFGpS), have been developed to date. While there are many other platforms that utilize chemical techniques [36, 69, 92, 146] or enzymatic reactions with purified components to obtain defined glycoproteins [82, 149], here we restrict our scope to systems where (i) enzymatic glycosylation is used, and (ii) target glycoproteins are synthesized via CFPS in crude extracts. Studies using purified translation components, such as the protein synthesis using recombinant elements (PURE [124]) system are excluded, and systems using purified glycosylation components are excluded unless otherwise noted. More exhaustive reviews of CFPS [10, 14, 64, 115, 125, 143, 151], and glycoengineering can be found elsewhere [6, 22, 26, 29, 46, 72, 94, 102].

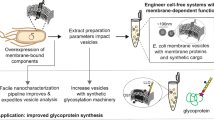
OST-dependent and OST-independent cell-free glycoprotein synthesis systems. Membrane-bound OST-dependent glycosylation systems using endogenous (a) and heterologous (b) glycosylation machinery. a Cell-free glycoprotein synthesis systems derived from eukaryotic cell extracts enriched or supplemented with endoplasmic reticulum (ER) microsomes containing glycosylation components. b Cell-free glycoprotein synthesis systems derived from glycoengineered E. coli cells harboring heterologous, membrane-bound glycosylation machinery. c OST-independent glycosylation enzymes are combined with sugar donors in a mix-and-match fashion to make soluble, synthetic glycosylation pathways
CFGpS platforms that use extracts from a variety of host organisms to install a diverse array of glycoforms have been developed. In this review, we classify CFGpS systems by topology and the origin of the glycosylation machinery (Fig. 2), which are key characteristics when determining their best application areas. The topology of the glycosylation machinery is either membrane-bound and oligosaccharyltransferase (OST)-dependent, or soluble and OST-independent. Glycosylation machinery can be derived from endogenous expression in cells, heterologous expression in cells, or in vitro expression in CFPS. We order the sections from the most “natural systems” (containing endogenous enzymes naturally found in the host strain) to the most synthetic (containing heterologous and novel enzyme combinations, generating glycans that are not found in nature). This distinction illustrates how the use of synthetic biology principles in glycosylation systems has expanded their scope and provided access to new glycoforms [72].
In Sections “OST-dependent glycosylation with endogenous eukaryotic glycosylation machinery” and “OST-dependent glycosylation in glycoengineered E. coli extracts”, we review CFGpS systems that use OST-dependent N-linked glycosylation systems. These systems rely on membrane-bound OSTs to transfer complex, prebuilt glycans from lipid-linked oligosaccharide (LLO) donors to target proteins but vary in application depending on the glycan transferred. Section “OST-dependent glycosylation with endogenous eukaryotic glycosylation machinery” describes systems using endogenous glycosylation machinery from eukaryotic cells (Fig. 2a). Section “OST-dependent glycosylation in glycoengineered E. coli extracts” describes Escherichia coli-based systems that contain heterologous glycosylation machinery (Fig. 2b). Thus far, the ability of eukaryotic systems to install human-like glycans points to greater utility in expression of functional therapeutics such as antibodies [88, 134]. E. coli-based systems have shown greater promise for expressing antimicrobial glycoconjugate vaccines that can be biomanufactured with low cost and in an on-demand format [133]. In Section “E. coli cell-free systems for OST-independent glycoprotein synthesis”, we discuss platforms that utilize CFPS to characterize, assemble, and prototype OST-independent pathways composed of soluble glycosyltransferase (GT) enzymes [70, 71]. In these systems, cell-free derived GTs sequentially decorate glycoproteins with minimal, synthetic glycans (Fig. 2c) [70, 81]. We anticipate that minimal glycosylation could enable fundamental understanding of the properties of isolated glycan motifs and open doors for production of new proteins with optimized activities.
Main text
OST-dependent glycosylation systems
OST-dependent N-linked glycosylation consists of three key steps that are conserved across the domains of life, all taking place on or across lipid membranes (the periplasmic membrane in prokaryotes, and the endoplasmic reticulum (ER) in eukaryotes) [27, 121]. First, the LLO donor is assembled in a stepwise fashion as biosynthetic enzymes incorporate monosaccharides into a growing polysaccharide chain. Second, the LLO is flipped across the lipid membrane into the periplasm or ER. Finally, the oligosaccharide from the LLO donor is transferred to asparagine residues within an acceptor polypeptide by a membrane-bound OST. OSTs mediate glycosylation by their substrate specificity, glycosylating only when specific polypeptide acceptor sequences and LLO donor structures are recognized. While the general acceptor peptide motif (or “sequon”) N-X-S/T is conserved, specific sequons and LLOs recognized by OSTs vary across organisms [72, 121]. In eukaryotes, OSTs are multi-subunit complexes assembled around a core catalytic subunit called STT3 [66, 85]. In prokaryotes, OSTs called PglBs are single-subunit enzymes that bear homology to STT3, but are more tractable for heterologous expression [56]. Even the simplest OSTs, however, are large enzymes with ~ 13 transmembrane passes and require proper membrane embedding for activity [97]. Due to the complexity of OST-dependent glycosylation, the use of cellular extracts enriched with, or supplemented with, native cellular lipids and membrane-bound machinery has been the main strategy for obtaining active CFGpS systems [58].
OST-dependent glycosylation with endogenous eukaryotic glycosylation machinery
The coactivation of CFPS and OST-dependent protein glycosylation was first observed in eukaryotic cell extracts supplemented with ER microsomes. CFGpS was reported in extracts from various origins (wheat germ cells [9], rabbit reticulocytes [5, 18, 25, 83, 145], and other higher eukaryotes [87]) supplemented with mammalian-derived microsomes. Additionally, homologous systems were developed from yeast [113, 114, 147] and other fungal cells [28] by supplementing microsomes derived from the same strain as the extract. Taken together, these studies demonstrated that proteins fused to a proper microsome-targeting leader sequence could be produced and translocated into ER microsomes in vitro, and that N-linked glycosylation could occur on these microsome-targeted proteins.
While the original eukaryotic CFGpS systems were intended to study protein secretion and processing, the realization that glycosylation could be combined with the benefits of CFPS prompted the development of more robust, biomanufacturing-oriented systems. Other useful features of eukaryotic CFPS systems for manufacturing therapeutics include the presence of endogenous folding chaperones (e.g., protein disulfide isomerase) and a lack of endotoxins.
Recent advances in eukaryotic CFPS methods have enabled increases in protein titers, throughput, and glycosylation efficiency. Protein expression titers in commonly used eukaryotic strains have now reached hundreds of μg/mL of model and non-model proteins by employing semi-continuous reaction conditions, in which the CFPS reaction can exchange small-molecule byproducts and nutrients through a dialysis membrane [44, 112]. Additionally, the implementation of internal ribosome-mediated entry site (IRES)-mediated translation has enabled programming of eukaryotic CFPS reactions with DNA templates instead of mRNA templates, increasing throughput by obviating the need to prepare mRNA templates [12, 112]. Processing methods have also been established for enriching extracts with intact microsomes, circumventing the need for tedious microsome purification/supplementation protocols. Using optimized lysis and extract preparation techniques, the ER is rearranged into well-defined microsomes that remain in the final extract and are active for glycosylation (Fig. 2a). Microsome-enriched extracts can now be made without the need for specialized cell disruption equipment or chromatography steps, simplifying extract preparation [65]. Moving forward, further improvement of batch-mode eukaryotic CFPS reactions to match the comparatively low costs and high CFPS titers of their E. coli-based counterparts could make the technology more accessible [13, 125, 151].
Importantly, for the production of defined glycoproteins, the cell lines—and thus, the diversity of glycosylation systems—available for preparing microsome-enriched extracts has expanded to include tobacco BY-2 [13], hybridoma [89], human [12, 89], insect [12, 65, 136, 137, 152], and Chinese hamster ovary (CHO) [11, 12, 44, 134, 140, 141] cells. These systems have been used to produce a variety of complex, active glycoprotein targets. Table 1 shows representative yields of proteins and glycoproteins synthesized in selected CFGpS systems. Notably, expression of active, multi-subunit glycoproteins stabilized by disulfide bonds, such as antibodies [134] and glucose oxidase [13], is achievable in eukaryotic systems.
Toward advanced biomanufacturing applications, the well-developed CHO and insect platforms have been interfaced with non-canonical amino acid (ncAA) incorporation [45, 110, 130], enabling glycosylation and incorporation of site-specific ncAAs [134, 152]. These advances enable, for the first time, high-throughput screening of valuable, chemically-defined glycoprotein therapeutics [152] and antibody–drug conjugates [134].
A compelling application of eukaryotic CFGpS systems is producing and screening therapeutic proteins, whose activity and immunogenicity can be strongly affected by differences in glycan structures [59, 92, 153, 154]. Therefore, developing methods to produce homogeneous glycoproteins with multiple, distinct, human-like glycans present on therapeutic proteins is critical. Figure 3a summarizes the information inferred from glycan analysis in a variety of eukaryotic, OST-dependent CFGpS platforms. Glycosylation in eukaryotic CFGpS is typically confirmed using enzymatic deglycosylation with PNGase F (specific for all eukaryotic glycans with a Man3GlcNAc2 core without α1-3 fucosylation) and/or Endo H (specific for Man5GlcNAc2 hybrid and high mannose glycans). Knowledge of these minimum recognition motifs, and of the LLO specificity of eukaryotic OSTs, indicates that proteins produced in eukaryotic CFGpS systems have been modified by ER-dependent N-linked glycosylation systems with glycans resembling (Glc3Man9GlcNAc2) [123]. The extent to which these glycans are trimmed and elaborated—as they would be in the ER and Golgi apparatus in living cells—still requires further characterization. The implementation of higher-resolution assays, such as those recently performed on insect and CHO systems (Fig. 3a), is helping to clarify the diversity of glycans that can be produced by CFGpS [44, 152]. For example, a mass spectrometry (MS)-based analysis of erythropoietin (EPO) derived from insect CFGpS revealed that glycans were trimmed down to structures as minimal as Man5GlcNAc2 [152]. Lectin-based analysis of EPO derived from CHO CFGpS supplemented with ER and Golgi vesicles showed the presence of high mannose, fucosylated, and galactose-terminated structures, indicating that enzymes present in the Golgi of the host strain can remain active in CFGpS reactions when microsomes are prepared appropriately [44].

Glycosylation in cell-free glycoprotein synthesis systems. a OST-dependent glycosylation in eukaryotic and E. coli extracts. b Bottom-up synthesis of glycoproteins in an OST-independent manner. When structural characterization was performed by deglycosylation studies, minimal recognition motifs (shown as structures with elaboration arrows) were inferred based on known glycosidase and OST specificities. References (‘Ref.’) for each structure are listed in the top right-hand corner of each section
Further characterization of CFGpS products using high-resolution techniques such as MS and nuclear magnetic resonance spectroscopy are needed to define glycoforms in the future. These methods will be critical to develop strategies to control and remodel glycan structures. Promising avenues to achieve defined glycoforms include the use of glyco-engineered cell lines containing edited glycosylation pathways [17] and the supplementation of reactions with glycosyltransferases and/or glycosidases [139].
OST-dependent glycosylation in glycoengineered E. coli extracts
E. coli-based platforms are the most robust and cost-effective CFPS systems currently available. With protein yields of thousands of μg/mL and costs at less than $5/mL reaction volume [125], E. coli systems surpass eukaryotic systems in batch yields by an order of magnitude and are two orders of magnitude less expensive than commonly used CHO and insect systems [13, 125, 151]. Additionally, E. coli is faster and easier to grow than eukaryotic strains, enabling cell growth, harvest, and extract preparation to be completed in less than one day [77, 86, 126]. Furthermore, extracts from laboratory strains of E. coli provide a ‘blank slate’ for N-linked glycosylation because they contain no endogenous N-linked glycosylation machinery. Despite these advantages, laboratory E. coli strains could not be used to produce glycoproteins until the recent discovery of bacterial N-linked glycosylation systems [135, 144]. Efforts to harness these systems by transferring them into E. coli expression systems for engineering functional therapeutics, vaccines, and materials has spawned the new discipline of bacterial glycoengineering [6, 100, 101].
Of emerging interest for glycoengineering are single-subunit bacterial OSTs, which, despite having stringent sequon specificity [19, 75, 103], can be used as a tool to transfer diverse glycans to acceptor proteins engineered with proper sequons [6]. Since the functional transfer of the model N-linked glycosylation pathway from Campylobacter jejuni into E. coli [144], E. coli has been engineered with myriad OST-dependent glycosylation pathways. Glycan structures including the eukaryotic Man3GlcNAc2 core for mimicking eukaryotic glycosylation [142], microbial O-antigens for glycoconjugate vaccine development [24, 35, 106], Lewis structures for therapeutic development [54], and exotic bacterial glycans [57] have all been ported into living E. coli. In practice, recapitulating eukaryotic glycosylation in E. coli systems remains challenging, but the expression of bacterial glycosylation systems is relatively straightforward, enabling prototyping and biomanufacturing of antimicrobial conjugate vaccines [46].
The first E. coli CFGpS system was developed via supplementation of purified C. jejuni OST (CjOST) and LLO (CjLLO) into CFPS reactions where the nascent acceptor protein was glycosylated via purified components [42]. This system provided a proof-of-principle that bacterial N-linked glycosylation is possible in the absence of intact cellular membranes. Toward a lower cost, simpler system, the pathway from C. jejuni was recapitulated in vitro using crude E. coli extracts prepared from strains overexpressing CjOST and CjLLO (Fig. 2b) [57]. The key idea is that OST and LLOs are overexpressed in the E. coli cells, enriching the crude extract with these components and obviating the need for exogeneous addition of purified components. Glycosylation components are present in E. coli extracts in nanoscale [48] membrane vesicles which serve the dual purpose of enabling (i) the activation of ATP regeneration through oxidative phosphorylation [61] and (ii) harboring active membrane-bound LLOs and OSTs [57]. Glycoengineered extracts of E. coli have been used to synthesize a variety of glycoproteins, such as EPO, with diverse glycan structures (Table 1).
Recently, the E. coli CFGpS platform has been combined with efforts in decentralized biomanufacturing to enable in vitro bioconjugate vaccine expression (iVAX) from freeze-dried, shelf-stable reactions. iVAX is modular, allowing the transfer of diverse microbial O-antigens to protein targets (Fig. 3a) and the expression of conjugate vaccine carriers including detoxified Corynebacterium diphtheriae toxin (known as CRM197) and the Clostridium tetani toxin [133]. Importantly, iVAX-derived vaccines against the pathogen Francisella tularensis have proven to be efficacious in vivo, protecting vaccinated mice from a lethal pathogen challenge [133]. iVAX glycoconjugate titers (Table 1) enable individual vaccine doses of 10 μg to be produced in one hour for ~ $6 [133]. In efforts to further decrease biomanufacturing costs, E. coli CFGpS was recently optimized to synthesize glycoprotein titers of > 100 μg/mL in batch by increasing the concentrations of the LLO- and OST-harboring vesicles during extract preparation [48].
E. coli systems are a promising venue for producing therapeutic glycoproteins and on-demand vaccines. From a protein biomanufacturing perspective, concerns over producing disulfide bond proteins and the presence of endotoxin have recently been overcome [38, 148]. From the perspective of glycosylation, a major advantage of the E. coli system is the possible breadth of glycans that can be installed with OSTs, which will continue to expand as new glycosylation pathways are engineered and characterized. Toward this goal, a class of bacterial OSTs termed O-OSTs (which carry out O-linked glycosylation on serine and threonine residues) that are structurally similar to bacterial N-OSTs, but with relaxed LLO specificities, have recently been characterized [33, 34]. O-OSTs, which further expand the palette of potential glycoconjugate vaccines available for manufacture in E. coli, [67, 132] have recently been shown to be active in E. coli CFGpS [48, 98].
A major challenge that remains for synthesizing therapeutic proteins is the transfer of eukaryotic-type N-linked glycosylation in a high-yielding E. coli system. While advances have been made toward increasing eukaryotic LLO production in E. coli [37], and Man3GlcNAc2 has been installed both with purified E.coli-derived components in vitro [57] and in living E. coli cells [142], efforts have been limited by low transfer efficiency of Man3GlcNAc2 (Fig. 3a) by bacterial OSTs. Additionally, because the polypeptide substrates of bacterial OSTs differ from eukaryotic OSTs, naturally occurring sequons in therapeutic proteins must be replaced with synthetic sequons, changing the primary protein sequence [57, 103, 142]. To address these issues, characterization of natural [103] and engineered [55, 104] bacterial OSTs to improve conjugation efficiency of diverse glycans is of key importance for advancing E. coli CFGpS. To this end, the structure of a widely-used bacterial OST in complex with an LLO and an acceptor sequon was recently solved, providing high-resolution information for rational engineering of OSTs and providing new opportunities for glycoprotein biomanufacturing [55, 104].
OST-independent glycosylation systems
E. coli cell-free systems for OST-independent glycoprotein synthesis
In addition to biomanufacturing, cell-free systems provide a flexible environment to construct and optimize new biosynthetic glycosylation pathways, and to interface glycosylation with high-throughput experimental workflows. A key determinant of the throughput accessible to study and engineer glycosylation systems in cell-free platforms is the ability to utilize or synthesize glycosylation components outside of living cells, where they can be more easily varied, sampled, and controlled. The innovations described in Sections “OST-dependent glycosylation with endogenous eukaryotic glycosylation machinery” and “OST-dependent glycosylation in glycoengineered E. coli extracts” provide methodologies to utilize OST-dependent glycosylation components generated inside of living cells in a cell-free environment. However, taking full advantage of the cell-free paradigm for glycosyltransferase (GT) characterization, engineering, and biosynthetic pathway prototyping, requires the synthesis of glycosylation components outside of living cells.
A key challenge with OST-dependent N-linked glycosylation is that LLOs and OSTs are membrane-associated and are, therefore, more difficult to synthesize than soluble proteins. This challenge was partially overcome by supplementing E. coli CFPS reactions with lipid–protein nanodiscs to enable the synthesis of active OSTs at high titers in vitro [120], opening the door to high-throughput OST characterization. However, the in vitro, bottom-up synthesis of LLOs remains challenging [146] and the co-activation of LLO biosynthesis and CFGpS has not been demonstrated. Additionally, cell-free synthesis of eukaryotic OST complexes (e.g., STT3) has not yet been reported to our knowledge, limiting the diversity of OSTs that can be synthesized using current in vitro systems. Given the challenges associated with synthesizing OSTs and LLOs in vitro, recent efforts have sought to study and engineer OST-independent glycosylation pathways in a cell-free environment [70, 71, 81, 138]. The absence of membrane-associated enzymes or substrates in OST-independent glycosylation pathways make them easier to implement in cell-free systems and permits the direct transfer of developed pathways into the bacterial cytoplasm. Furthermore, OST-independent systems may be more modular, as they circumvent the specificities of OSTs for LLOs by sequentially installing monosaccharides onto proteins.
Thus far, OST-independent cell-free glycoengineering efforts have focused on a recently discovered class of cytoplasmic enzymes known as N-glycosyltransferases (NGTs). NGTs transfer a single glucose residue from a nucleotide-activated sugar (UDP-Glc) onto an asparagine residue within an acceptor sequon. Sequons recognized by NGTs resemble the eukaryotic N-X-S/T glycosylation motif [21, 40, 95, 96]. Because the acceptor sequence specificity of NGTs had not been rigorously characterized, and this information is required for site-specific modification of glycoproteins, initial efforts in this area used CFPS with a high-throughput experimentation platform for glycosylation sequence characterization and optimization by rapid expression and screening (GlycoSCORES) [71]. GlycoSCORES uses CFPS to produce a polypeptide-modifying glycosyltransferase of interest and self-assembled monolayers for matrix-assisted laser desorption/ionization mass spectrometry (SAMDI-MS) to determine its specificity [91]. This method was applied to determine the sugar donor and peptide acceptor sequence specificities of both N- and O-linked polypeptide modifying GTs from bacteria and humans using 3,480 unique peptides and 13,903 unique reaction conditions. This information was then used to redesign glycosylation sites within heterologous proteins (including the Fc region of human IgG) to increase their glycosylation efficiency by up to fivefold in living E. coli and in a cell-free environment [71]. This method was later adapted to intact proteins, enabling the analysis of an 87-member protein library containing a single glycosylation site at all positions along the protein backbone. This assay provided insight into how the position of the acceptor sequon within a target protein can affect glycosylation [138], an approach called shotgun glycomutagenesis [80]. Another effort used the GlycoSCORES method to produce 41 putative NGT homologs in CFPS and rigorously characterize their acceptor sequence specificities. This campaign discovered four NGT variants with conditionally orthogonal peptide acceptor specificities that were used to develop new workflows for sequential and site-specific glycosylation at up to four distinct locations within a single protein [81]. These works show how cell-free systems have been interfaced with OST-dependent glycosylation to accelerate glycoprotein design and testing.
Besides controlling the efficiency and position of glycan modifications, OST-independent cell-free systems have also enabled the bottom-up construction of multi-enzyme synthetic glycosylation pathways in vitro to generate proteins modified with a wide variety of glycan structures. A recent study reported the development and application of a modular, cell-free platform for glycosylation pathway assembly by rapid in vitro mixing and expression (GlycoPRIME) in which a target protein and GTs were synthesized in separate CFPS reactions and then combined to generate unique protein glycosylation pathways (Fig. 2c) [70]. The key idea is that cell-free biosynthesis “units” are made from crude cell lysates that are selectively enriched with pathway enzymes produced directly in lysates by cell-free protein synthesis. Then, these units are assembled modularly, in a mix-and-match fashion, to build and study biosynthetic pathways. Biosynthetic pathways yielding 23 unique glycosylation motifs were developed using this method (Fig. 3b). Once discovered in vitro, the pathways developed using GlycoPRIME were successfully transferred to living E. coli, enabling the cytoplasmic production of glycoproteins. These pathways were also shown to be functional in a one-pot format in which all plasmids for the target protein and GTs are combined in the CFPS reaction supplemented with activated sugar donors to generate glycoprotein in 24 h. The use of OST-independent glycosylation systems has greatly expanded the diversity of glycosylation structures available for production in cell-free [70, 71, 81, 138] and cellular [23, 67, 71, 109] systems. Additionally, OST-independent glycosylation systems hold promise for applications including adjuvants and antigens for vaccines [1, 2, 23, 109, 111], glycoprotein antitoxins [73, 99], biomaterials that promote cell growth or differentiation by interfacing with cellular lectins [8, 117], and stabilized therapeutics [128].
The ease of implementation of OST-independent CFGpS systems has enabled enzyme characterization and glycoprotein analysis at high throughput and may offer new paradigms for glycoprotein biomanufacturing methods. The glycans installed using these methods, however, are generally smaller than those installed by OSTs (Fig. 3) and do not occur in exactly the same form in nature. Further characterization of the functionality of minimal glycans will be critical for understanding and leveraging OST-independent glycosylation systems in the future. Several hurdles remain before human N-glycosylation can be precisely mimicked using OST-independent machinery. Specifically, NGTs discovered to date are unable to transfer GlcNAc, the reducing end sugar in all eukaryotic N-linked glycans [95]. Therefore, the discovery and engineering of NGTs capable of transferring GlcNAc remains an active area of research [71, 74, 81].
Conclusion and outlook
Cell-free systems hold great promise for expediting expression, testing, and biomanufacturing glycoproteins. Because cell-free glycosylation systems are modular, and can be easily interfaced with automated experimental workflows, the pace of optimizing and discovering glycoprotein variants can be accelerated compared with traditional cell culture approaches. This feature promises to be particularly impactful for the optimization of important glycoprotein targets such as native glycoproteins that rely on eukaryotic glycosylation patterns (such as antibodies), engineered conjugate vaccines that rely on the conjugation of bacterial antigens, and fully synthetic glycoproteins with an array of potential applications.
While cell-free systems are accelerating bio-discovery and bio-design today, large-scale cell-free synthesis of glycoproteins remains on the horizon. Three key challenges to be addressed include: increasing glycoprotein synthesis yields, reducing costs, and creating continuous biomanufacturing systems. In addition, the development of an expanded set of glycoengineered chassis strains for creating cell-free systems that can freely build any glycosylation structure on any glycoprotein of choice is needed. Even though challenges exist, the ability to readily store, distribute, and activate freeze-dried cell-free systems by simply adding water has already opened new opportunities for on-demand, decentralized biomanufacturing.
Looking forward, we anticipate that the modularity and flexibility of cell-free glycosylation systems will continue to increase our understanding of glycosylation, advance applications in on-demand biomanufacturing, and accelerate glycoprotein research and development timelines. In years to come, these advances will enable CFGpS systems to complement more traditional fermentation and cell culture production systems at the commercial-scale. CFGpS systems are likely to be adopted first in applications where batches of glycoprotein are needed quickly, or the required glycosylation structures must be tightly controlled or cannot be easily produced using existing cellular systems (including those from pathogenic bacteria or those containing non-standard sugar monomers or linkages).
Abbreviations
- CFGpS:
-
Cell-free glycoprotein synthesis
- CFPS:
-
Cell-free protein synthesis
- CHO:
-
Chinese hamster ovary
- Cj:
-
Campylobacter jejuni
- DSB:
-
Disulfide bond
- EGFR:
-
Epidermal growth factor receptor
- ELISA:
-
Enzyme-linked immunosorbent assay
- EPO:
-
Erythropoietin
- ER:
-
Endoplasmic reticulum
- Ft:
-
Francisella tularensis
- Gal:
-
Galactose
- GalNAc:
-
N-acetyl galactosamine
- Glc:
-
Glucose
- GlcNAc:
-
N-acetyl glucosamine
- GlycoPRIME:
-
Glycosylation pathway assembly by rapid in vitro mixing and expression
- GlycoSCORES:
-
Glycosylation sequence characterization and optimization by rapid expression and screening
- GOx:
-
Glucose oxidase
- gp120:
-
HIV-1 envelope glycoprotein
- GT:
-
Glycosyltransferase
- IgG:
-
Immunoglobulin G
- IRES:
-
Internal ribosome mediated entry site
- iVAX:
-
In vitro bioconjugate vaccine expression
- LLO:
-
Lipid-linked oligosaccharide
- Luc:
-
Firefly luciferase
- Man:
-
Mannose
- MBP:
-
Maltose binding protein
- MS:
-
Mass spectrometry
- N-linked glycosylation:
-
Asparagine-linked glycosylation
- ncAA:
-
Non-canonical amino acid
- NGT:
-
N-Glycosyltransferase
- O-linked glycosylation:
-
Serine/ threonine-linked glycosylation
- OST:
-
Oligosaccharyltransferase
- PURE:
-
Protein synthesis using recombinant elements
- SAMDI-MS:
-
Self-assembled monolayers for matrix-assisted laser desorption/ionization mass spectrometry
- sfGFP:
-
Superfolder green fluorescent protein
- Sia:
-
Sialic acid
- UDP:
-
Uridine diphosphate
- WB:
-
Western blot
References
Abdel-Motal UM, Guay HM, Wigglesworth K, Welsh RM, Galili U (2007) Immunogenicity of influenza virus vaccine is increased by anti-gal-mediated targeting to antigen-presenting cells. J Virol 81:9131–9141. https://doi.org/10.1128/jvi.00647-07
Abdel-Motal UM, Wang S, Awad A, Lu S, Wigglesworth K, Galili U (2010) Increased immunogenicity of HIV-1 p24 and gp120 following immunization with gp120/p24 fusion protein vaccine expressing α-gal epitopes. Vaccine 28:1758–1765. https://doi.org/10.1016/j.vaccine.2009.12.015
Adamo R, Nilo A, Castagner B, Boutureira O, Berti F, De GJLB (2013) Synthetically defined glycoprotein vaccines: current status and future directions. Chem Sci 4:2995–3008. https://doi.org/10.1039/c3sc50862e
Adiga R, Al-adhami M, Andar A, Borhani S, Brown S, Burgenson D, Cooper MA, Deldari S, Frey DD, Ge X, Guo H, Gurramkonda C, Jensen P, Kostov Y, Lacourse W, Liu Y, Moreira A, Mupparapu K, Peñalber-Johnstone C, Pilli M, Punshon-smith B, Rao A, Rao G, Rauniyar P, Snovida S, Taurani K, Tilahun D, Tolosa L, Tolosa M, Tran K, Vattem K, Veeraraghavan S, Wagner B, Wilhide J, Wood DW, Zuber A (2018) Point-of-care production of therapeutic proteins of good-manufacturing-practice quality. Nat Biomed Eng 2:675–686. https://doi.org/10.1038/s41551-018-0259-1
Bailey CA, Gerber L, Howard AD, Udenfriend S (1989) Processing at the carboxyl terminus of nascent placental alkaline phosphatase in a cell-free system: Evidence for specific cleavage of a signal peptide. Proc Natl Acad Sci USA 86:22–26. https://doi.org/10.1073/pnas.86.1.22
Baker JL, Çelik E, DeLisa MP (2013) Expanding the glycoengineering toolbox: the rise of bacterial N-linked protein glycosylation. Trends Biotechnol 31:313–323. https://doi.org/10.1016/j.tibtech.2013.03.003
Beebe ET, Makino S, Nozawa A, Matsubara Y, Frederick RO, Primm JG, Goren MA, Fox BG (2011) Robotic large-scale application of wheat cell-free translation to structural studies including membrane proteins. N Biotechnol 28:239–249. https://doi.org/10.1016/j.nbt.2010.07.003
Beer M, Rech C, Gasteier P, Sauerzapfe B, Salber J, Ewald A, Möller M, Elling L, Groll J (2013) The next step in biomimetic material design: poly-LacNAc-mediated reversible exposure of extra cellular matrix components. Adv Healthc Mater 2:306–311. https://doi.org/10.1002/adhm.201200080
Blobel G, Dobberstein B (1975) Transfer of proteins across membranes. II. Reconstitution of functional rough microsomes from heterologous components. J Cell Biol 852–862
Brödel AK, Kubick S (2014) Developing cell-free protein synthesis systems: a focus on mammalian cells. Pharm Bioprocess 2:339–348
Brödel AK, Sonnabend A, Kubick S (2014) Cell-free protein expression based on extracts from CHO cells. Biotechnol Bioeng 111:25–36. https://doi.org/10.1002/bit.25013
Brödel AK, Sonnabend A, Roberts LO, Stech M, Wüstenhagen DA, Kubick S (2013) IRES-mediated translation of membrane proteins and glycoproteins in eukaryotic cell-free systems. PLoS ONE. https://doi.org/10.1371/journal.pone.0082234
Buntru M, Vogel S, Stoff K, Spiegel H, Schillberg S (2015) A versatile coupled cell-free transcription-translation system based on tobacco BY-2 cell lysates. Biotechnol Bioeng 112:867–878. https://doi.org/10.1002/bit.25502
Carlson ED, Gan R, Hodgman CE, Jewett MC (2012) Cell-free protein synthesis: applications come of age. Biotechnol Adv 30:1185–1194. https://doi.org/10.1016/j.biotechadv.2011.09.016
Caschera F, Karim AS, Gazzola G, d’Aquino AE, Packard NH, Jewett MC (2018) High-throughput optimization cycle of a cell-free ribosome assembly and protein synthesis system. ACS Synth Biol 7:2841–2853. https://doi.org/10.1021/acssynbio.8b00276
Caschera F, Noireaux V (2014) Synthesis of 2.3 mg/ml of protein with an all Escherichia coli cell-free transcription-translation system. Biochimie 99:162–168. https://doi.org/10.1016/j.biochi.2013.11.025
Chang GD, Chen CJ, Lin CY, Chen HC, Chen H (2003) Improvement of glycosylation in insect cells with mammalian glycosyltransferases. J Biotechnol 102:61–71. https://doi.org/10.1016/S0168-1656(02)00364-4
Chao CC, Bird P, Gething MJ, Sambrook J (1987) Posttranslational translocation of influenza virus hemagglutinin across microsomal membranes. Mol Cell Biol 7:3842–3845. https://doi.org/10.1128/mcb.7.10.3842
Chen MM, Glover KJ, Imperiali B (2007) From peptide to protein: Comparative analysis of the substrate specificity of N-linked glycosylation in C. jejuni. Biochemistry 46:5579–5585. https://doi.org/10.1021/bi602633n
Chen Z, Kibler RD, Hunt A, Busch F, Pearl J, Jia M, VanAernum ZL, Wicky BIM, Dods G, Liao H (2020) De novo design of protein logic gates. Science 368:78–84
Choi KJ, Grass S, Paek S, St. Geme JW, Yeo HJ, (2010) The actinobacillus pleuropneumoniae HMW1C-like glycosyltransferase mediates N-Linked glycosylation of the haemophilus influenzae HMW1 adhesin. PLoS ONE. https://doi.org/10.1371/journal.pone.0015888
Clausen H, Wandall H, Steentoft C, Stanley P, Schnaar R (2015) Glycosylation Engineering. In: Varki A, Cummings RD, Esko JD, et al. (eds) Essentials of Glycobiology, 3rd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York, pp 713–728
Cuccui J, Terra VS, Bossé JT, Naegeli A, Abouelhadid S, Li Y, Lin C, Vohra P, Tucker AW, Rycroft AN, Maskell DJ, Aebi M, Langford PR, Wren BW (2017) The N-linking glycosylation system from Actinobacillus pleuropneumoniae is required for adhesion and has potential use in glycoengineering. Open Biol. https://doi.org/10.1098/rsob.160212
Cuccui J, Thomas RM, Moule MG, D’Elia RV, Laws TR, Mills DC, Williamson D, Atkins TP, Prior JL, Wren BW (2013) Exploitation of bacterial N-linked glycosylation to develop a novel recombinant glycoconjugate vaccine against Francisella tularensis. Open Biol 3:130002
Dalley JA, Bulleid NJ (2003) The endoplasmic reticulum (ER) translocon can differentiate between hydrophobic sequences allowing signals for glycosylphosphatidylinositol anchor addition to be fully translocated into the ER lumen. J Biol Chem 278:51749–51757. https://doi.org/10.1074/jbc.M303978200
DeLisa M, Jaroentomeechai T, Taw M, Li M, Aquino A, Agashe N, Chung S (2020) Cell-free synthetic glycobiology: designing and engineering glycomolecules outside of living cells. Front Chem 8:645
Dell A, Galadari A, Sastre F, Hitchen P (2010) Similarities and differences in the glycosylation mechanisms in prokaryotes and eukaryotes. Int J Microbiol. https://doi.org/10.1155/2010/148178
Devchand M, Gwynne D, Buxton FP, Davies RW (1988) An efficient cell-free translation system from Aspergillus nidulans and in vitro translocation of prepro-a-factor across Aspergillus microsomes. Curr Genet 14:561–566. https://doi.org/10.1007/BF00434081
Dicker M, Strasser R (2015) Using glyco-engineering to produce therapeutic proteins. Expert Opin Biol Ther 15:1501–1516
Dudley QM, Karim AS, Jewett MC (2015) Cell-free metabolic engineering: Biomanufacturing beyond the cell. Biotechnol J 10:69–82. https://doi.org/10.1002/biot.201400330
Dudley QM, Karim AS, Nash CJ, Jewett MC (2020) Cell-free prototyping of limonene biosynthesis using cell-free protein synthesis. Metab Eng
Eichler J (2013) Extreme sweetness: protein glycosylation in archaea. Nat Rev Microbiol. https://doi.org/10.1038/nrmicro2957
Faridmoayer A, Fentabil MA, Haurat MF, Yi W, Woodward R, Wang PG, Feldman MF (2008) Extreme substrate promiscuity of the Neisseria oligosaccharyl transferase involved in protein O-glycosylation. J Biol Chem 283:34596–34604. https://doi.org/10.1074/jbc.M807113200
Faridmoayer A, Fentabil MA, Mills DC, Klassen JS, Feldman MF (2007) Functional characterization of bacterial oligosaccharyltransferases involved in O-linked protein glycosylation. J Bacteriol 189:8088–8098. https://doi.org/10.1128/JB.01318-07
Feldman MF, Wacker M, Hernandez M, Hitchen PG, Marolda CL, Kowarik M, Morris HR, Dell A, Valvano MA, Aebi M (2005) Engineering N-linked protein glycosylation with diverse O antigen lipopolysaccharide structures in Escherichia coli. Proc Natl Acad Sci USA 102:3016–3021. https://doi.org/10.1073/pnas.0500044102
Fernández-tejada A, Brailsford J (2015) Total synthesis of glycosylated proteins. Top Curr Chem. https://doi.org/10.1007/128
Glasscock CJ, Yates LE, Jaroentomeechai T, Wilson JD, Merritt JH, Lucks JB, DeLisa MP (2018) A flow cytometric approach to engineering Escherichia coli for improved eukaryotic protein glycosylation. Metab Eng 47:488–495. https://doi.org/10.1016/j.ymben.2018.04.014
Goerke AR, Swartz JR (2008) Development of cell-free protein synthesis platforms for disulfide bonded proteins. Biotechnol Bioeng 99:351–367. https://doi.org/10.1002/bit.21567
Goerke AR, Swartz JR (2009) High-level cell-free synthesis yields of proteins containing site-specific non-natural amino acids. Biotechnol Bioeng 102:400–416. https://doi.org/10.1002/bit.22070
Grass S, Lichti CF, Townsend RR, Gross J, St. Geme JW, (2010) The haemophilus influenzae HMW1c protein is a glycosyltransferase that transfers hexose residues to asparagine sites in the HMW1 adhesin. PLoS Pathog 6:1–9. https://doi.org/10.1371/journal.ppat.1000919
Grubbe WS, Karim AS, Rasor BJ, Krüger A, Jewett MC (2020) Cell-free styrene biosynthesis at high titers. Metab Eng 61:89–95. https://doi.org/10.1016/j.ymben.2020.05.009
Guarino C, Delisa MP (2012) A prokaryote-based cell-free translation system that efficiently synthesizes glycoproteins. Glycobiology 22:596–601. https://doi.org/10.1093/glycob/cwr151
Guo W, Sheng J, Feng X (2017) Mini-review: In vitro Metabolic Engineering for Biomanufacturing of High-value Products. Comput Struct Biotechnol J 15:161–167. https://doi.org/10.1016/j.csbj.2017.01.006
Gurramkonda C, Rao A, Borhani S, Pilli M, Deldari S, Ge X, Pezeshk N, Han TC, Tolosa M, Kostov Y, Tolosa L, Wood DW, Vattem K, Frey DD, Rao G (2018) Improving the recombinant human erythropoietin glycosylation using microsome supplementation in CHO cell-free system. Biotechnol Bioeng 115:1253–1264. https://doi.org/10.1002/bit.26554
Hammerling MJ, Krüger A, Jewett MC (2020) Strategies for in vitro engineering of the translation machinery. Nucleic Acids Res 48:1068–1083
Harding CM, Feldman MF (2019) Glycoengineering bioconjugate vaccines, therapeutics, and diagnostics in E. coli. Glycobiology 29:519–529. https://doi.org/10.1093/glycob/cwz031
Helenius A, Aebi M (2004) Roles of N-linked glycans in the endoplasmic reticulum. Annu Rev Biochem 73:1019–1049. https://doi.org/10.1146/annurev.biochem.73.011303.073752
Hershewe J, Warfel K, Iyer S, Peruzzi J, Roth E, Sullivan C, Kamat N, Jewett M (2020) Improving cell-free glycoprotein synthesis by characterizing and enriching native membrane vesicles. bioRxiv. https://doi.org/10.1101/2020.07.19.211201
Higel F, Seidl A, Sorgel F, Friess W (2016) N-glycosylation heterogeneity and the influence on structure, function and pharmacokinetics of monoclonal antibodies and Fc fusion proteins. Eur J Pharm Biopharm 100:94–100. https://doi.org/10.1016/j.ejpb.2016.01.005
Hodgman CE, Jewett MC (2012) Cell-free synthetic biology: thinking outside the cell. Metab Eng 14:261–269. https://doi.org/10.1016/j.ymben.2011.09.002
Hong SH, Kwon Y-C, Jewett MC (2014) Non-standard amino acid incorporation into proteins using Escherichia coli cell-free protein synthesis. Front Chem. https://doi.org/10.3389/fchem.2014.00034
Hong SH, Kwon Y, Martin RW, Des Soye BJ, de Paz AM, Swonger KN, Ntai I, Kelleher NL, Jewett MC (2015) Improving cell-free protein synthesis through genome engineering of Escherichia coli lacking release factor 1. ChemBioChem 16:844–853. https://doi.org/10.1002/cbic.201402708
Huang A, Nguyen PQ, Stark JC, Takahashi MK, Donghia N, Ferrante T, Dy AJ, Hsu KJ, Dubner RS, Pardee K, Jewett MC, Collins JJ (2018) BiobitsTM explorer: A modular synthetic biology education kit. Sci Adv. https://doi.org/10.1126/sciadv.aat5105
Hug I, Zheng B, Reiz B, Whittal RM, Fentabil MA, Klassen JS, Feldman MF (2011) Exploiting bacterial glycosylation machineries for the synthesis of a Lewis antigen-containing glycoprotein. J Biol Chem 286:37887–37894. https://doi.org/10.1074/jbc.M111.287755
Ihssen J, Kowarik M, Wiesli L, Wacker M, Schwede T, Tho L (2015) Increased efficiency of Campylobacter jejuni N -oligosaccharyltransferase PglB by structure-guided engineering. Open Biol 5
Jaffee MB, Imperiali B (2013) Optimized protocol for expression and purification of membrane-bound PglB, a bacterial oligosaccharyl transferase. Protein Expr Purif 89:241–250. https://doi.org/10.1016/j.pep.2013.04.001
Jaroentomeechai T, Stark JC, Natarajan A, Glasscock CJ, Yates LE, Hsu KJ, Mrksich M, Jewett MC, Delisa MP (2018) Single-pot glycoprotein biosynthesis using a cell-free transcription-translation system enriched with glycosylation machinery. Nat Commun 9:1–11. https://doi.org/10.1038/s41467-018-05110-x
Jaroentomeechai T, Zheng X, Hershewe J, Stark JC, Jewett MC, DeLisa MP (2017) A Pipeline for Studying and Engineering Single-Subunit Oligosaccharyltransferases, first ed. Elsevier Inc.
Jefferis R (2009) Glycosylation as a strategy to improve antibody-based therapeutics. Nat Rev 8:226–234
Jenkins N, Parekh RB, James DC (1996) Getting the glycosylation right: implications for the biotechnology industry. Nat Biotechnol 14:975–981. https://doi.org/10.1038/nbt0896-975
Jewett MC, Calhoun KA, Voloshin A, Wuu JJ, Swartz JR (2008) An integrated cell-free metabolic platform for protein production and synthetic biology. Mol Syst Biol. https://doi.org/10.1038/msb.2008.57
Karim AS, Dudley QM, Juminaga A, Yuan Y, Crowe SA, Heggestad JT, Garg S, Abdalla T, Grubbe WS, Rasor BJ, Coar DN, Torculas M, Krein M, Liew F, Quattlebaum A, Jensen RO, Stuart JA, Simpson SD, Köpke M, Jewett MC (2020) In vitro prototyping and rapid optimization of biosynthetic enzymes for cell design. Nat Chem Biol. https://doi.org/10.1038/s41589-020-0559-0
Karim AS, Heggestad JT, Crowe SA (2018) Controlling cell-free metabolism through physiochemical perturbations. Metab Eng 45:86–94. https://doi.org/10.1016/j.ymben.2017.11.005
Katzen F, Chang G, Kudlicki W (2005) The past, present and future of cell-free protein synthesis. Trends Biotechnol 23:150–156. https://doi.org/10.1016/j.tibtech.2005.01.003
Katzen F, Kudlicki W (2006) Efficient generation of insect-based cell-free translation extracts active in glycosylation and signal sequence processing. J Biotechnol 125:194–197. https://doi.org/10.1016/j.jbiotec.2006.03.002
Kelleher DJ, Gilmore R (2006) An evolving view of the eukaryotic oligosaccharyltransferase. Glycobiology 16:47–62. https://doi.org/10.1093/glycob/cwj066
Keys TG, Wetter M, Hang I, Rutschmann C, Russo S, Mally M, Steffen M, Zuppiger M, Müller F, Schneider J, Faridmoayer A, Linwei Aebi CM (2017) A biosynthetic route for polysialylating proteins in Escherichia coli. Metab Eng 44:293–301. https://doi.org/10.1016/j.ymben.2017.10.012
Khoury GA, Baliban RC, Floudas CA (2011) Proteome-wide post-translational modification statistics: frequency analysis and curation of the swiss-prot database. Sci Rep 1:90. https://doi.org/10.1038/srep00090
Kiessling LL, Splain RA (2010) Chemical approaches to glycobiology. Annu Rev Biochem 79:619–653. https://doi.org/10.1146/annurev.biochem.77.070606.100917
Kightlinger W, Duncker KE, Ramesh A, Thames AH, Natarajan A, Stark JC, Yang A, Lin L, Mrksich M, DeLisa MP, Jewett MC (2019) A cell-free biosynthesis platform for modular construction of protein glycosylation pathways. Nat Commun 101(10):1–13. https://doi.org/10.1038/s41467-019-12024-9
Kightlinger W, Lin L, Rosztoczy M, Li W, Delisa MP, Mrksich M, Jewett MC (2018) Design of glycosylation sites by rapid synthesis and analysis of glycosyltransferases. Nat Chem Biol 14:627–635. https://doi.org/10.1038/s41589-018-0051-2
Kightlinger W, Warfel KF, Delisa MP, Jewett MC (2020) Synthetic glycobiology: parts, systems, and applications. ACS Synth Biol. https://doi.org/10.1021/acssynbio.0c00210
Kitov PI, Sadowska JM, Mulvey G, Armstrong GD, Ling H, Pannu NS, Read RJ, Bundle DR (2000) Shiga-like toxins are neutralized by tailored multivalent carbohydrate ligands. Nature 403:669–672. https://doi.org/10.1038/35001095
Kong Y, Li J, Hu X, Wang Y, Meng Q, Gu G, Wang PG, Chen M (2018) N-Glycosyltransferase from Aggregatibacter aphrophilus synthesizes glycopeptides with relaxed nucleotide-activated sugar donor selectivity. Carbohydr Res 462:7–12. https://doi.org/10.1016/j.carres.2018.03.008
Kowarik M, Young NM, Numao S, Schulz BL, Hug I, Callewaert N, Mills DC, Watson DC, Hernandez M, Kelly JF, Wacker M, Aebi M (2006) Definition of the bacterial N-glycosylation site consensus sequence. EMBO J 25:1957–1966. https://doi.org/10.1038/sj.emboj.7601087
Kuriakose A, Chirmule N, Nair P (2016) Immunogenicity of Biotherapeutics: Causes and Association with Posttranslational Modifications. J Immunol Res
Kwon YC, Jewett MC (2015) High-throughput preparation methods of crude extract for robust cell-free protein synthesis. Sci Rep 5:1–8. https://doi.org/10.1038/srep08663
Lee J, Schwieter KE, Watkins AM, Kim DS, Yu H, Schwarz KJ, Lim J, Coronado J, Byrom M, Anslyn EV, Ellington AD, Moore JS, Jewett MC (2019) Expanding the limits of the second genetic code with ribozymes. Nat Commun 10:5097. https://doi.org/10.1038/s41467-019-12916-w
Lee J, Torres R, Kim DS, Byrom M, Ellington AD, Jewett MC (2020) Ribosomal incorporation of cyclic β-amino acids into peptides using in vitro translation. Chem Commun (Camb) 56:5597–5600. https://doi.org/10.1039/d0cc02121k
Li M, Zheng X, Shanker S, Jaroentomeechai T, Kocer I, Byrne J, Cox E, Labonte J, Gray J, DeLisa M (2020) High-resolution mapping of glycoprotein structure–activity relationships by shotgun scanning glycomutagenesis. bioRxiv. https://doi.org/10.1101/2020.06.28.176198
Lin L, Kightlinger W, Prabhu SK, Hockenberry AJ, Li C, Wang LX, Jewett MC, Mrksich M (2020) Sequential glycosylation of proteins with substrate-specific N-Glycosyltransferases. ACS Cent Sci 6:144–154. https://doi.org/10.1021/acscentsci.9b00021
Liu F, Vijayakrishnan B, Faridmoayer A, Taylor TA, Parsons TB, Bernardes GJL, Kowarik M, Davis BG (2014) Rationally designed short polyisoprenol-linked PglB substrates for engineered polypeptide and protein N-glycosylation. J Am Chem Soc 136:566–569. https://doi.org/10.1021/ja409409h
MacDonald MR, McCourt DW, Krause JE (1988) Posttranslational processing of α-, β-, and γ-preprotachykinins. Cell-free translation and early posttranslational processing events. J Biol Chem 263:15176–15183
Madin K, Sawasaki T, Ogasawara T, Endo Y (2000) A highly efficient and robust cell-free protein synthesis system prepared from wheat embryos: Plants apparently contain a suicide system directed at ribosomes. Proc Natl Acad Sci USA 97:559–564. https://doi.org/10.1073/pnas.97.2.559
Maita N, Nyirenda J, Igura M, Kamishikiryo J, Kohda D (2010) Comparative structural biology of eubacterial and archaeal oligosaccharyltransferases. J Biol Chem 285:4941–4950. https://doi.org/10.1074/jbc.M109.081752
Martin RW, Des Soye BJ, Kwon YC, Kay J, Davis RG, Thomas PM, Majewska NI, Chen CX, Marcum RD, Weiss MG, Stoddart AE, Amiram M, Ranji Charna AK, Patel JR, Isaacs FJ, Kelleher NL, Hong SH, Jewett MC (2018) Cell-free protein synthesis from genomically recoded bacteria enables multisite incorporation of noncanonical amino acids. Nat Commun 9:1–9. https://doi.org/10.1038/s41467-018-03469-5
Matthews G, Colman A (1991) A highly efficient, cell-free translation/translocation system prepared from Xenopus eggs. Nucleic Acids Res 19:6405–6412
Merk H, Gless C, Maertens B, Gerrits M, Stiege W (2012) Cell-free synthesis of functional and endotoxin-free antibody Fab fragments by translocation into microsomes. Biotechniques 53:153–160. https://doi.org/10.2144/0000113904
Mikami S, Kobayashi T, Yokoyama S, Imataka H (2006) A hybridoma-based in vitro translation system that efficiently synthesizes glycoproteins. J Biotechnol 127:65–78. https://doi.org/10.1016/j.jbiotec.2006.06.018
Moremen KW, Tiemeyer M, Nairn AV (2014) Vertebrate protein glycosylation: diversity, synthesis andfunction. Nat Rev Mol Cell Biol 13:448–462. https://doi.org/10.1038/nrm3383.Vertebrate
Mrksich M (2008) Mass spectrometry of self-assembled monolayers: a new tool for molecular surface science. ACS Nano 2:7–18. https://doi.org/10.1021/nn7004156
Murakami M, Kiuchi T, Nishihara M, Tezuka K, Okamoto R, Izumi M, Kajihara Y (2016) Chemical synthesis of erythropoietin glycoforms for insights into the relationship between glycosylation pattern and bioactivity. Sci Adv 2:e1500678. https://doi.org/10.1126/sciadv.1500678
Murray CJ, Baliga R (2013) Cell-free translation of peptides and proteins: from high throughput screening to clinical production. Curr Opin Chem Biol 17:420–426. https://doi.org/10.1016/j.cbpa.2013.02.014
Naegeli A, Aebi M (2015) Current approaches to engineering N-linked protein glycosylation in bacteria. Glyco-Engineering. https://doi.org/10.1007/978-1-4939-2760-9_1
Naegeli A, Michaud G, Schubert M, Lin CW, Lizak C, Darbre T, Reymond JL, Aebi M (2014) Substrate specificity of cytoplasmic N-glycosyltransferase. J Biol Chem 289:24521–24532. https://doi.org/10.1074/jbc.M114.579326
Naegeli A, Neupert C, Fan YY, Lin CW, Poljak K, Papini AM, Schwarz F, Aebi M (2014) Molecular analysis of an alternative N-glycosylation machinery by functional transfer from Actinobacillus pleuropneumoniae to Escherichia coli. J Biol Chem 289:2170–2179. https://doi.org/10.1074/jbc.M113.524462
Napiórkowska M, Boilevin J, Sovdat T, Darbre T, Reymond J-L, Aebi M, Locher KP (2017) Molecular basis of lipid-linked oligosaccharide recognition and processing by bacterial oligosaccharyltransferase. Nat Struct Mol Biol 24:1100
Natarajan A, Jaroentomeechai T, Cabrera M, Mohammed J, Cox E, Young O, Shajahan A, Vilkhovoy M, Vadhin S, Varner J, Azadi P, DeLisa M (2020) Engineering orthogonal human O-linked glycoprotein biosynthesis in bacteria. Nat Chem Biol. https://doi.org/10.1038/s41589-020-0595-9
Nilsson KGI, Mandenlus C (1994) A carbohydrate biosensor surface for the detection of uropathogenic bacteria. Biotechnology 12:13–15
Nothaft H, Szymanski CM (2010) Protein glycosylation in bacteria: sweeter than ever. Nat Rev Microbiol 8:765–778. https://doi.org/10.1038/nrmicro2383
Nothaft H, Szymanski CM (2013) N -glycosylation: new perspectives and applications. J Biol Chem 288:6912–6920. https://doi.org/10.1074/jbc.R112.417857
Ollis AA, Chai Y, DeLisa MP (2015) Glyco-engineering: Methods and protocols. In: Castilho A (ed) Glyco-Engineering: Methods and Protocols. pp 1–439
Ollis AA, Chai Y, Natarajan A, Perregaux E, Jaroentomeechai T, Guarino C, Smith J, Zhang S, DeLisa MP (2015) Substitute sweeteners: diverse bacterial oligosaccharyltransferases with unique N-glycosylation site preferences. Sci Rep 5:15237. https://doi.org/10.1038/srep15237
Ollis AA, Zhang S, Fisher AC, Delisa MP (2014) Engineered oligosaccharyltransferases with greatly relaxed acceptor-site specificity. Nat Chem Biol 10:816–822. https://doi.org/10.1038/nchembio.1609
Oza JP, Aerni HR, Pirman NL, Barber KW, Ter Haar CM, Rogulina S, Amrofell MB, Isaacs FJ, Rinehart J, Jewett MC (2015) Robust production of recombinant phosphoproteins using cell-free protein synthesis. Nat Commun 6:8168. https://doi.org/10.1038/ncomms9168
Pan C, Sun P, Liu B, Liang H, Peng Z, Dong Y, Wang D, Liu X, Wang B, Zeng M, Wu J, Zhu L, Wang H (2016) Biosynthesis of conjugate vaccines using an O-linked glycosylation system. MBio. https://doi.org/10.1128/mBio.00443-16
Pardee K, Slomovic S, Nguyen PQ, Lee JW, Donghia N, Burrill D, Ferrante T, McSorley FR, Furuta Y, Vernet A, Lewandowski M, Boddy CN, Joshi NS, Collins JJ (2016) Portable, on-demand biomolecular manufacturing. Cell 167:248–259.e12. https://doi.org/10.1016/j.cell.2016.09.013
Parodi AJ (2000) Role of N-oligosaccharide endoplasmic reticulum processing reactions in glycoprotein folding and degradation. Biochem J 13:1–13
Passmore IJ, Andrejeva A, Wren BW, Cuccui J (2019) Cytoplasmic glycoengineering of Apx toxin fragments in the development of Actinobacillus pleuropneumoniae glycoconjugate vaccines. BMC Vet Res 15:1–13. https://doi.org/10.1186/s12917-018-1751-2
Perez JG, Stark JC, Jewett MC (2016) Cell-free synthetic biology: engineering beyond the cell. Cold Spring Harb Perspect Biol 8:a023853
Phanse Y, Carrillo-Conde BR, Ramer-Tait AE, Broderick S, Kong CS, Rajan K, Flick R, Mandell RB, Narasimhan B, Wannemuehler MJ (2014) A systems approach to designing next generation vaccines: Combining α-galactose modified antigens with nanoparticle platforms. Sci Rep 4:1–9. https://doi.org/10.1038/srep03775
Quast RB, Sonnabend A, Stech M, Wustenhagen DA, Kubick S (2016) High-yield cell-free synthesis of human EGFR by IRES-mediated protein translation in a continuous exchange cell-free reaction format. Sci Rep 6:1–10. https://doi.org/10.1038/srep30399
Ramezani-Rad M, Käufer NF, Hasilik A, Lochmann E-R (1985) In vitro studies with rough microsomes from Saccharomyces cerevisiae. Endocyt C Res 2:249–260
Rothblatt JA, Meyer DI (1986) Secretion in yeast: Reconstitution of the translocation and glycosylation of α-factor and invertase in a homologous cell-free system. Cell 44:619–628. https://doi.org/10.1016/0092-8674(86)90271-0
Sachse R, Dondapati SK, Fenz SF, Schmidt T, Kubick S (2014) Membrane protein synthesis in cell-free systems: From bio-mimetic systems to bio-membranes. FEBS Lett. https://doi.org/10.1016/j.febslet.2014.06.007
Sachse R, Wüstenhagen D, Šamalíková M, Gerrits M, Bier F, Kubick S (2013) Synthesis of membrane proteins in eukaryotic cell-free systems. Eng Life Sci 13:39–48. https://doi.org/10.1002/elsc.201100235
Sauerzapfe B, Křenek K, Schmiedel J, Wakarchuk WW, Pelantová H, Křen V, Elling L (2009) Chemo-enzymatic synthesis of poly-N-acetyllactosamine (poly-LacNAc) structures and their characterization for CGL2-galectin-mediated binding of ECM glycoproteins to biomaterial surfaces. Glycoconj J 26:141–159. https://doi.org/10.1007/s10719-008-9172-2
Schäffer C, Messner P (2017) Emerging facets of prokaryotic glycosylation. FEMS Microbiol Rev 41:49–91
Schinn S, Broadbent A, Bradley WT, Bundy BC (2016) Protein synthesis directly from PCR: progress and applications of cell-free protein synthesis with linear DNA. N Biotechnol 33:480–487. https://doi.org/10.1016/j.nbt.2016.04.002
Schoborg JA, Hershewe J, Stark JC, Kightlinger W, Kath JE, Jaroentomeechai T, Natarajan A, DeLisa MP, Jewett MC (2017) A cell-free platform for rapid synthesis and testing of active oligosaccharyltransferases. Biotechnol Bioeng 115:739–750. https://doi.org/10.1002/bit.26502
Schwarz F, Aebi M (2011) Mechanisms and principles of N-linked protein glycosylation. Curr Opin Struct Biol 21:576–582. https://doi.org/10.1016/j.sbi.2011.08.005
Sethuraman N, Stadheim TA (2006) Challenges in therapeutic glycoprotein production. Curr Opin Biotechnol 17:341–346. https://doi.org/10.1016/j.copbio.2006.06.010
Shi X, Jarvis D (2007) Protein N-Glycosylation in the baculovirus-insect cell system. Curr Drug Targets 8:1116–1125. https://doi.org/10.2174/138945007782151360
Shimizu Y, Inoue A, Tomari Y, Suzuki T, Yokogawa T, Nishikawa K, Ueda T (2001) Cell-free translation reconstituted with purified components. Nat Biotechnol 19:751–755
Silverman AD, Karim AS, Jewett MC (2019) Cell-free gene expression: an expanded repertoire of applications. Nat Rev Genet. https://doi.org/10.1038/s41576-019-0186-3
Silverman AD, Kelley-Loughnane N, Lucks JB, Jewett MC (2019) Deconstructing cell-free extract preparation for in vitro activation of transcriptional genetic circuitry. ACS Synth Biol 8:403–414. https://doi.org/10.1021/acssynbio.8b00430
Smith MT, Berkheimer SD (2014) Lyophilized Escherichia coli -based cell-free systems for robust, high-density, long-term storage. Biotechniques 56:186–193. https://doi.org/10.2144/000114158
Solá RJ, Griebenow K (2010) Glycosylation of therapeutic proteins: An effective strategy to optimize efficacy. BioDrugs 24:9–21. https://doi.org/10.2165/11530550-000000000-00000
Des Soye BJ, Gerbasi VR, Thomas PM, Kelleher NL, Jewett MC (2019) A highly productive, one-pot cell-free protein synthesis platform based on genomically recoded Escherichia coli. Cell Chem Biol 26:1743–1754. https://doi.org/10.1016/j.chembiol.2019.10.008
Des Soye BJ, Patel JR, Isaacs FJ, Jewett MC (2015) Repurposing the translation apparatus for synthetic biology. Curr Opin Chem Biol 28:83–90
Stark JC, Huang A, Hsu KJ, Dubner RS, Forbrook J, Marshalla S, Rodriguez F, Washington M, Rybnicky GA, Nguyen PQ, Hasselbacher B, Jabri R, Kamran R, Koralewski V, Wightkin W, Martinez T, Jewett MC (2019) BioBits health: classroom activities exploring engineering, biology, and human health with fluorescent readouts. ACS Synth Biol 8:1001–1009. https://doi.org/10.1021/acssynbio.8b00381
Stark JC, Huang A, Nguyen PQ, Dubner RS, Hsu KJ, Ferrante TC, Anderson M, Kanapskyte A, Mucha Q, Packett JS, Patel P, Patel R, Qaq D, Zondor T, Burke J, Martinez T, Miller-berry A, Puppala A, Reichert K, Schmid M, Brand L, Hill LR, Chellaswamy JF, Faheem N, Fetherling S, Gong E, Gonzalzles EM, Granito T, Koritsaris J, Nguyen B, Ottman S (2019) BioBits TM Bright: A fluorescent synthetic biology education kit. Sci Adv 4:eaat5107
Stark JC, Jaroentomeechai T, Moeller TD, Dubner RS, Hsu KJ, Stevenson TC, DeLisa MP, Jewett MC (2019) On-demand, cell-free biomanufacturing of conjugate vaccines at the point-of-care. bioRxiv preprint. https://doi.org/10.1101/681841
Stech M, Nikolaeva O, Thoring L, Stöcklein WFM, Wüstenhagen DA, Hust M, Dübel S, Kubick S (2017) Cell-free synthesis of functional antibodies using a coupled in vitro transcription-translation system based on CHO cell lysates. Sci Rep 7:1–15. https://doi.org/10.1038/s41598-017-12364-w
Szymanski CM, Yao R, Ewing CP, Trust TJ, Guerry P (1999) Evidence for a system of general protein glycosylation in Campylobacter jejuni. Mol Microbiol 32:1022–1030
Tarui H, Imanishi S, Hara T (2000) A novel cell-free translation/glycosylation system prepared from insect cells. J Biosci Bioeng 90:508–514. https://doi.org/10.1016/S1389-1723(01)80031-1
Tarui H, Murata M, Tani I, Imanishi S, Nishikawa S, Hara T (2001) Establishment and characterization of cell-free translation/glycosylation in insect cell (Spodoptera frugiperda 21) extract prepared with high pressure treatment. Appl Microbiol Biotechnol 55:446–453. https://doi.org/10.1007/s002530000534
Techner JM, Kightlinger W, Lin L, Hershewe J, Ramesh A, Delisa MP, Jewett MC, Mrksich M (2020) High-throughput synthesis and analysis of intact glycoproteins using SAMDI-MS. Anal Chem 92:1963–1971. https://doi.org/10.1021/acs.analchem.9b04334
Tejwani V, Andersen MR, Nam JH, Sharfstein ST (2018) Glycoengineering in CHO cells: advances in systems biology. Biotechnol J 13:1–16. https://doi.org/10.1002/biot.201700234
Thoring L, Dondapati SK, Stech M, Wüstenhagen DA, Kubick S (2017) High-yield production of “difficult-to-express” proteins in a continuous exchange cell-free system based on CHO cell lysates. Sci Rep 7:1–15. https://doi.org/10.1038/s41598-017-12188-8
Thoring L, Wustenhagen DA, Borowiak M, Stech M, Sonnabend A, Kubick S (2016) Cell-free systems based on CHO cell lysates: Optimization strategies, synthesis of “difficult-to-express” proteins and future perspectives. PLoS ONE 11:1–21. https://doi.org/10.1371/journal.pone.0163670
Valderrama-Rincon JD, Fisher AC, Merritt JH, Fan Y, Reading CA, Chhiba K, Heiss C, Azadi P, Aebi M, DeLisa MP (2012) An engineered eukaryotic protein glycosylation pathyway in Escherichia coli. Nat Chem Biol 8:434. https://doi.org/10.1038/nchembio.92110.1038/NCHEMBIO.921
Vilkhovoy M, Adhikari A, Vadhin S (2020) The Evolution of cell free biomanufacturing. Processes https://doi.org/10.20944/preprints202003.0461.v1
Wacker M, Linton D, Hitchen PG, Nita-Lazar M, Haslam SM, North SJ, Panico M, Morris HR, Dell A, Wren BW (2002) N-linked glycosylation in Campylobacter jejuni and its functional transfer into E. coli. Science 298:1790–1793
Walter P, Blobel G (1981) Translocation of proteins across the endoplasmic reticulum. III. Signal recognition protein (SRP) causes signal-sequence dependent and site-specific arrest of chain elongation that is released by microsomal membranes. J Cell Biol 91:557–561
Wang L, Amin MN (2014) Review chemical and chemoenzymatic synthesis of glycoproteins for deciphering functions. Chem Biol 21:51–66. https://doi.org/10.1016/j.chembiol.2014.01.001
Wiedmann M, Wiedmann B, Voigt S, Wachter E, Müller HG, Rapoport TA (1988) Post-translational transport of proteins into microsomal membranes of Candida maltosa. EMBO J 7:1763–1768
Wilding KM, Hunt JP, Wilkerson JW, Funk PJ, Swensen RL, Carver WC, Christian ML, Bundy BC (2019) Endotoxin-free E. coli-based cell-free protein synthesis: pre-expression endotoxin removal approaches for on-demand cancer therapeutic production. Biotechnol J 14:1–6. https://doi.org/10.1002/biot.201800271
Wright TH, Bower BJ, Chalker JM, Bernardes GJL, Wiewiora R, Ng W-L, Raj R, Faulkner S, Vallée MRJ, Phanumartwiwath A, Coleman OD, Thézénas M-L, Khan M, Galan SRG, Lercher L, Schombs MW, Gerstberger S, Palm-Espling ME, Baldwin AJ, Kessler BM, Claridge TDW, Mohammed S, Davis BG (2016) Posttranslational mutagenesis: A chemical strategy for exploring protein side-chain diversity. Science. https://doi.org/10.1126/science.aag1465
Zawada JF, Yin G, Steiner AR, Yang J, Naresh A, Roy SM, Gold DS, Heinsohn HG, Murray CJ (2011) Microscale to manufacturing scale-up of cell-free cytokine production-a new approach for shortening protein production development timelines. Biotechnol Bioeng 108:1570–1578. https://doi.org/10.1002/bit.23103
Zemella A, Thoring L, Hoffmeister C, Kubick S (2015) Cell-free protein synthesis: pros and cons of prokaryotic and eukaryotic systems. ChemBioChem 16:2420–2431. https://doi.org/10.1002/cbic.201500340
Zemella A, Thoring L, Hoffmeister C, Šamalíková M, Ehren P, Wüstenhagen DA, Kubick S (2018) Cell-free protein synthesis as a novel tool for directed glycoengineering of active erythropoietin. Sci Rep 8:1–12. https://doi.org/10.1038/s41598-018-26936-x
Zhang P, Woen S, Wang T, Liau B, Zhao S, Chen C, Yang Y, Song Z, Wormald MR, Yu C, Rudd PM (2016) Challenges of glycosylation analysis and control: an integrated approach to producing optimal and consistent therapeutic drugs. Drug Discov Today 21:740–765. https://doi.org/10.1016/j.drudis.2016.01.006
Zheng K, Bantog C, Bayer R (2011) The impact of glycosylation on monoclonal antibody conformation and stability. MAbs 3:568–576. https://doi.org/10.4161/mabs.3.6.17922
Acknowledgements
M.C.J. acknowledges support from the Defense Threat Reduction Agency Grant HDTRA1-15-10052/P00001, the DARPA 1000 Molecules Program HR0011-15-C-0084, the Air Force Research Laboratory Center of Excellence Grant FA8650-15-2-5518, the David and Lucile Packard Foundation, and the Camille Dreyfus Teacher-Scholar Program. This project was also supported in part by a fellowship award awarded to J.M.H. through the National Defense Science and Engineering (NDSEG) Fellowship Program, sponsored by the Air Force Research Laboratory, the Office of Naval Research, and the Army Research Office. J.M.H. also received support through the Ryan Fellowship awarded by Northwestern University. The U.S. Government is authorized to reproduce and distribute reprints for Governmental purposes notwithstanding any copyright notation thereon. The views and conclusions contained herein are those of the authors and should not be interpreted as necessarily representing the official policies or endorsements, either expressed or implied, of DARPA, Defense Threat Reduction Agency, or the U.S. Government.
Author information
Authors and Affiliations
Contributions
The authors contributed to all aspects of the article.
Corresponding author
Ethics declarations
Conflict of interest
M.C.J. has a financial interest in Design Pharmaceuticals Inc. and SwiftScale Biologics. M.C.J.’s interests are reviewed and managed by Northwestern University in accordance with their conflict of interest policies. All other authors declare no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Hershewe, J., Kightlinger, W. & Jewett, M.C. Cell-free systems for accelerating glycoprotein expression and biomanufacturing. J Ind Microbiol Biotechnol 47, 977–991 (2020). https://doi.org/10.1007/s10295-020-02321-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10295-020-02321-4