Redox Dependent Arsenic Occurrence and Partitioning in an Industrial Coastal Aquifer: Evidence from High Spatial Resolution Characterization of Groundwater and Sediments

 , , , and
, , , and 
Abstract
:1. Introduction
2. Materials and Methods
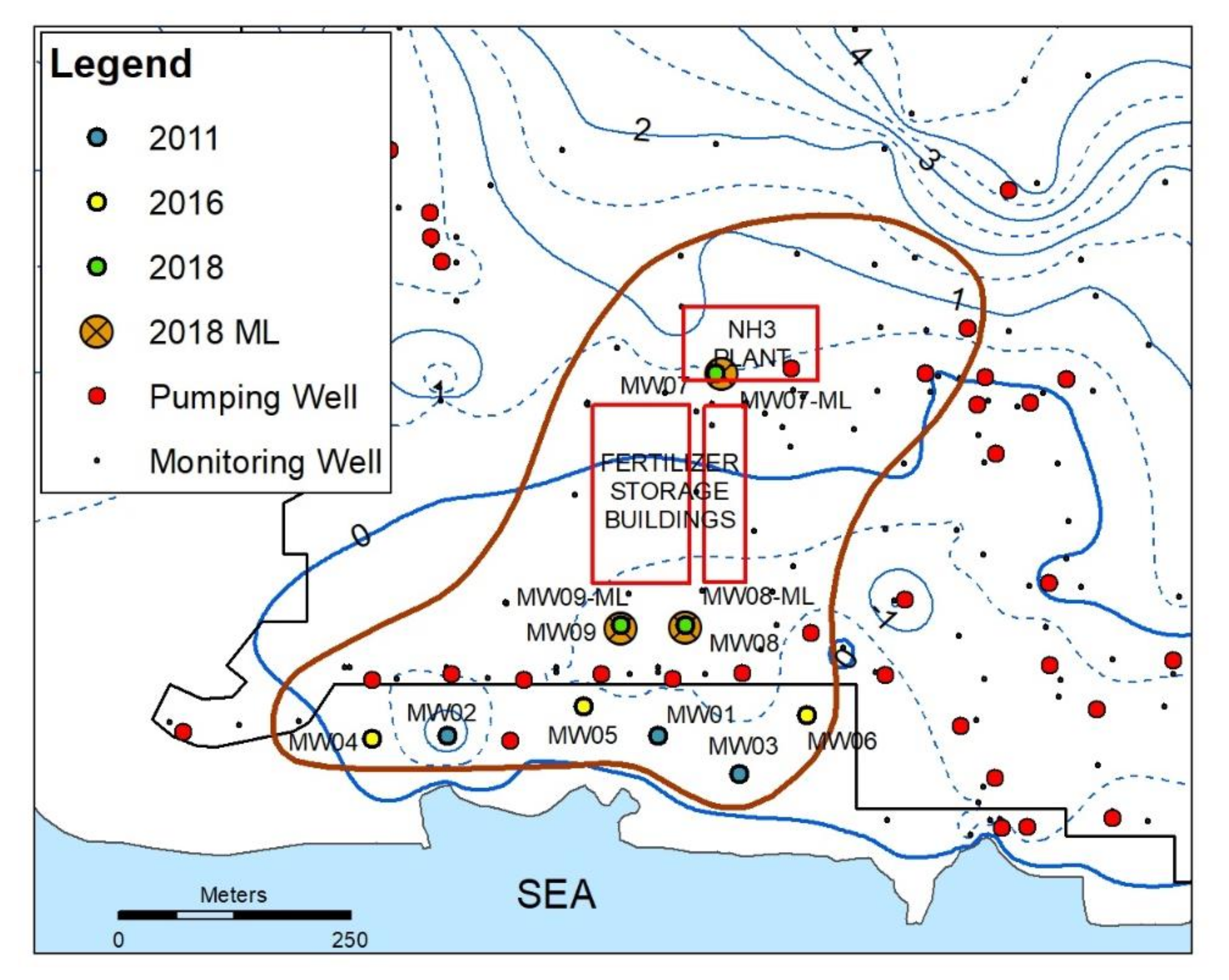
2.1. Site Description
2.2. Field Sampling
2.3. Laboratory Analyses
3. Results and Discussion
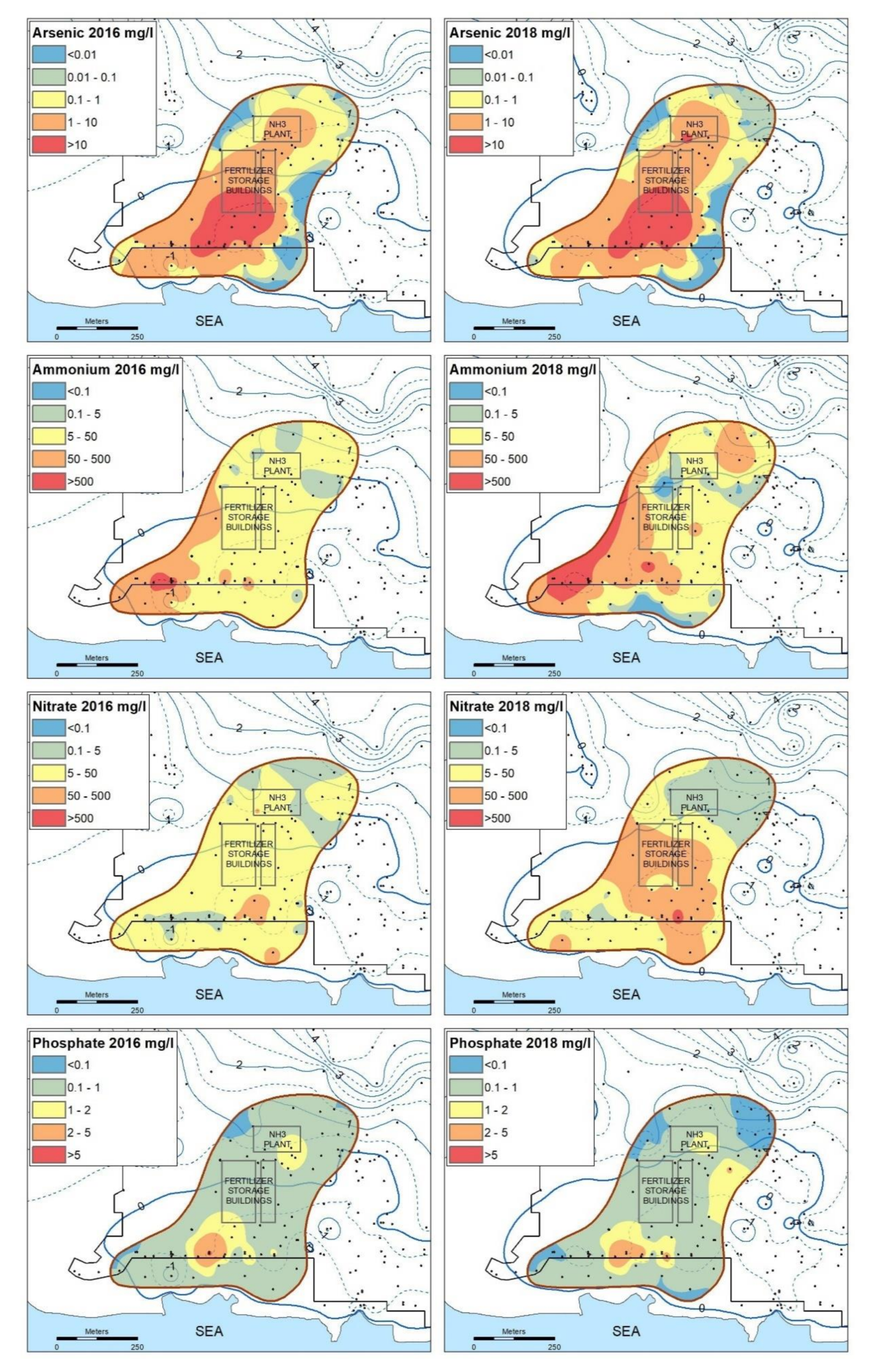
3.1. Groundwater Compositions
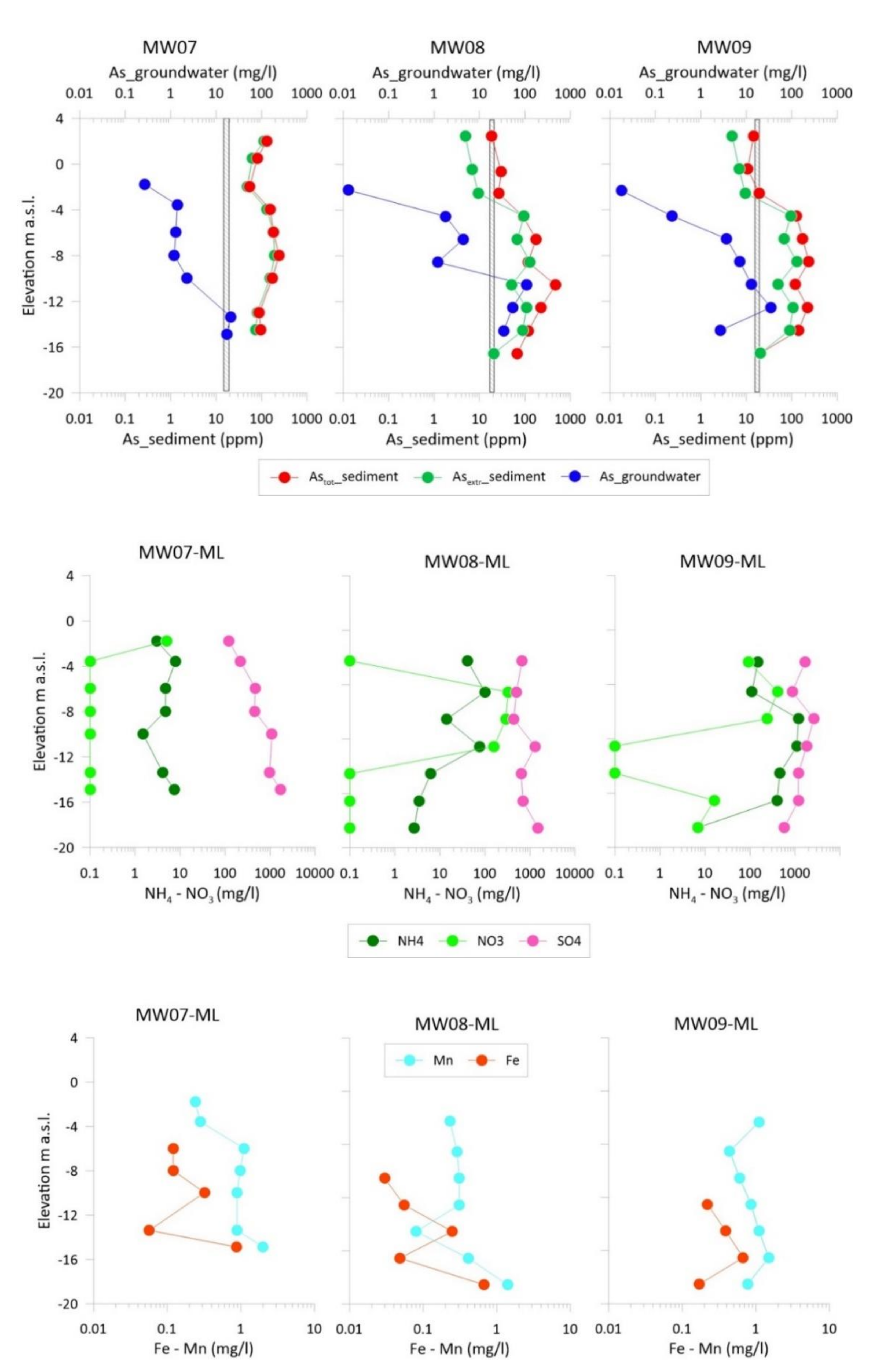
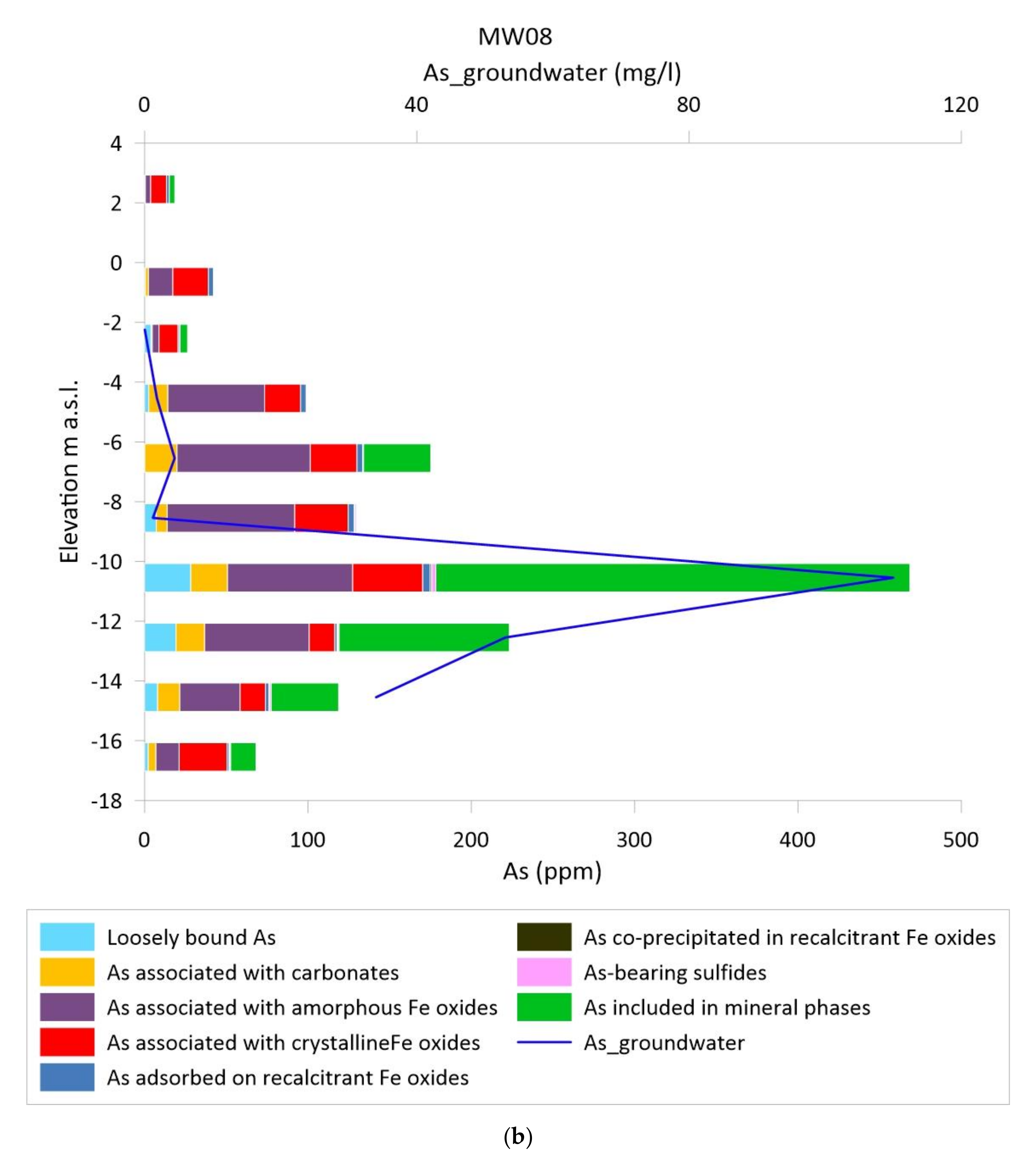
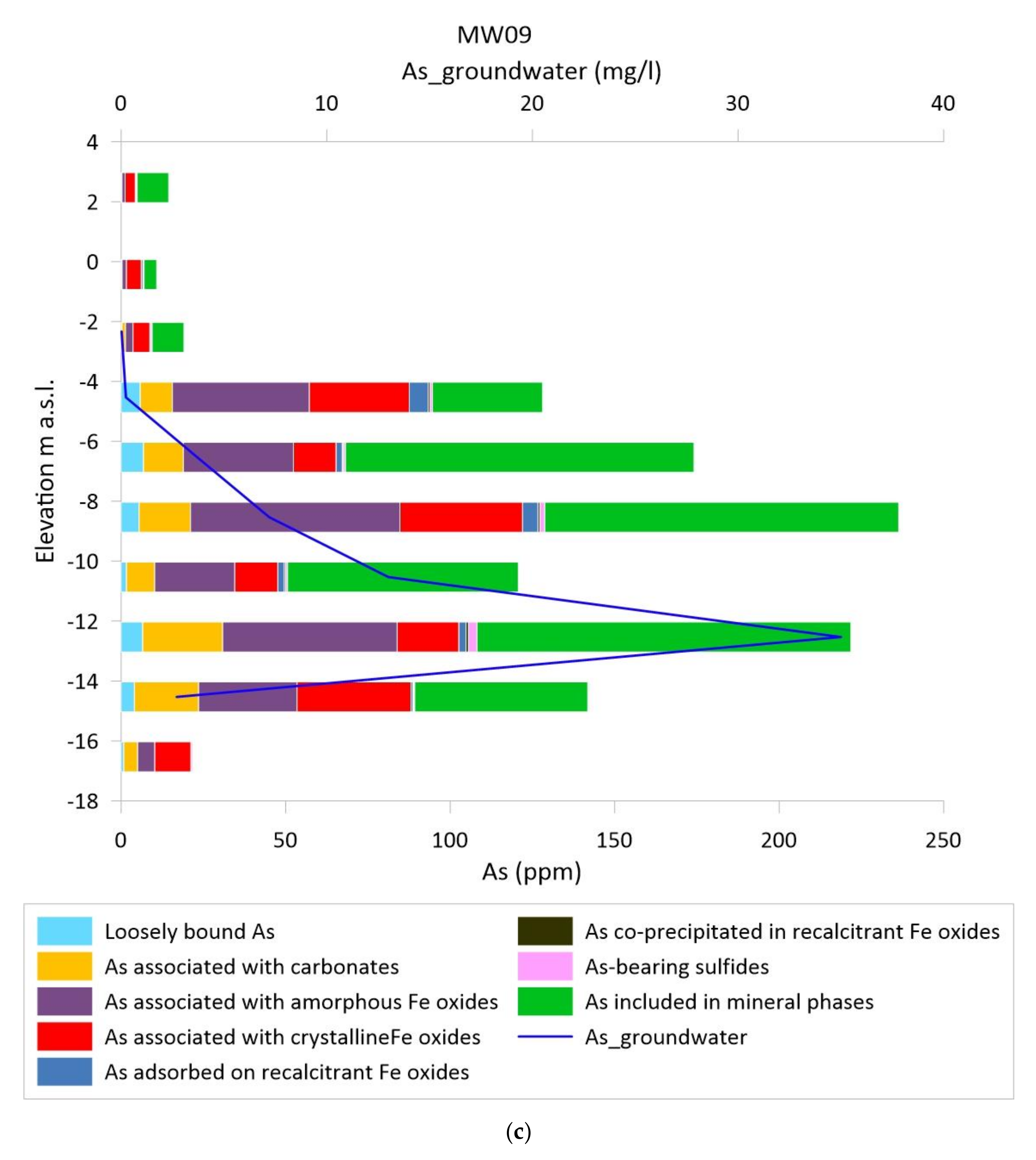
3.2. Vertical Distribution of As in Sediments and Groundwater
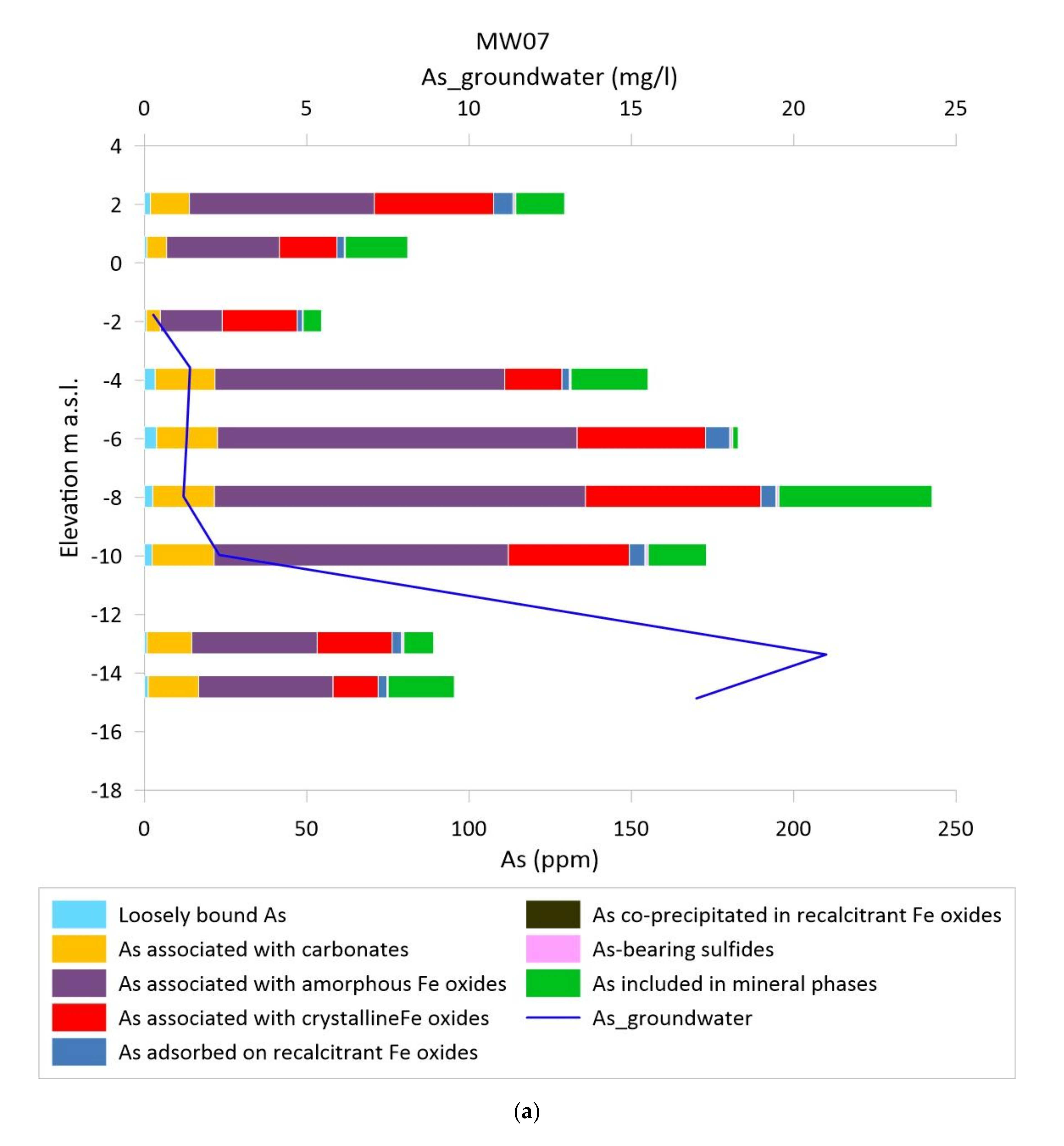
3.3. As Sequential Extraction
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Podgorski, J.; Berg, M. Global threat of arsenic in groundwater. Science 2020, 368, 845–850. [Google Scholar] [CrossRef] [PubMed]
- Satinder, A. Chapter 16—Lessons learned from water disasters. In Evaluating Water Quality to Prevent Future Disasters, 1st ed.; Satinder, A., Ed.; Academic Press: Cambridge, MA, USA, 2019; Volume 11, pp. 417–427. [Google Scholar]
- Mukherjee, A.; Sengupta, M.; Hossain, M.; Ahamed, S.; Das, B.; Nayak, B.; Lodh, D.; Rahman, M.M.; Chakraborti, D. Arsenic contamination in groundwater: A global perspective with emphasis on the Asian scenario. J. Health Popul. Nutr. 2006, 24, 142–163. [Google Scholar] [PubMed]
- Chakraborti, D.; Mukherjee, S.C.; Pati, S.; Sengupta, M.K.; Rahman, M.M.; Chowdhury, U.K.; Lodh, D.; Chanda, C.R.; Chakraborti, A.K.; Basu, G.K. Arsenic groundwater contamination in Middle Ganga Plain, Bihar, India: A future danger? Environ. Health Perspect. 2003, 111, 1194–1201. [Google Scholar] [CrossRef] [Green Version]
- Hu, Z.; Gao, S. Upper crustal abundances of trace elements: A revision and update. Chem. Geol. 2008, 253, 205–221. [Google Scholar] [CrossRef]
- Smith, D.B.; Solano, F.; Woodruff, L.G.; Cannon, W.F.; Ellefsen, K.J. Geochemical and Mineralogical Maps, with Interpretation, for Soils of the Conterminous United States; USGS: Denver, CO, USA, 2017.
- Van Geen, A.; Bostick, B.C.; Trang, P.T.K.; Lan, V.M.; Mai, N.-N.; Manh, P.D.; Viet, P.H.; Radloff, K.; Aziz, Z.; Mey, J.L.; et al. Retardation of arsenic transport through a Pleistocene aquifer. Nature 2013, 501, 204–207. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.; Culbertson, C.; Nielsen, M.; Schalk, C.; Johnson, C.; Marvinney, R.; Stute, M.; Zheng, Y. Flow and sorption controls of groundwater arsenic in individual boreholes from bedrock aquifers in central Maine, USA. Sci. Total Environ. 2015, 505, 1291–1307. [Google Scholar] [CrossRef] [Green Version]
- Bostick, B.; Harvey, C.; Stahl, M.; Oates, P.; Vi, L.; Nguyen, M.; Viet, P.; Pham, T.; Berg, M.; Stengel, C.; et al. Distribution of Arsenic Sulfides in Van Phuc, Vietnam, and Their Relationship to Aquifer Arsenic Concentrations. In Proceedings of the AGU Fall Meeting Abstracts, San Francisco, CA, USA, 13–17 December 2010. [Google Scholar]
- Fendorf, S.; Michael, H.A.; van Geen, A. Spatial and temporal variations of groundwater arsenic in South and Southeast Asia. Science 2010, 328, 1123–1127. [Google Scholar] [CrossRef] [Green Version]
- De Lemos, J.; Bostick, B.; Renshaw, C.; Stürup, S.; Feng, X. Landfill-stimulated iron reduction and arsenic release at the coakley superfund site (NH). Environ. Sci. Technol. 2006, 40, 67–73. [Google Scholar] [CrossRef]
- Alonso, D.L.; Pérez, R.; Okio, C.K.Y.A.; Castillo, E. Assessment of mining activity on arsenic contamination in surface water and sediments in southwestern area of Santurbán paramo, Colombia. J. Environ. Manag. 2020, 264, 110478. [Google Scholar] [CrossRef]
- Camacho, L.; Gutierrez, M.; Alarcón-Herrera, M.; Villalba, L.; Deng, S. Occurrence and treatment of arsenic in groundwater and soil in northern Mexico and southwestern USA. Chemosphere 2011, 83, 211–225. [Google Scholar] [CrossRef]
- U.S. Environmental Protection Agency. Arsenic Treatment Technologies for Soil, Waste, and Water; U.S. EPA/National Service Center for Environmental Publications: Cincinnati, OH, USA, 2002.
- Root, A.R.; Vlassopoulos, D.; Rafferty, M.T.; Andrews, C.; O’Day, P.A. Speciation and natural attenuation of arsenic and iron in a tidally influenced shallow aquifer. Geochim. Cosmochim. Acta 2009, 73, 5528–5553. [Google Scholar] [CrossRef]
- Morin, G.; Calas, G. Arsenic in soils, mine tailings, and former industrial sites. Elements 2006, 2, 97–101. [Google Scholar] [CrossRef]
- Cancés, B.; Juillot, F.; Morin, G.; Laperche, V.; Alvarez, L.; Proux, O.; Hazemann, J.L.; Brown, G.E., Jr.; Calas, G. XAS Evidence of As(V) association with iron oxyhydroxides in a contaminated soil at a former arsenical pesticide processing plant. Environ. Sci. Technol. 2005, 39, 9398–9405. [Google Scholar] [CrossRef] [PubMed]
- Campos, V. Arsenic in groundwater affected by phosphate fertilizers at Sao Paulo, Brazil. Environ. Geol. 2002, 42, 83–87. [Google Scholar] [CrossRef]
- Giglioli, S.; Colombo, L.; Contestabile, P.; Musco, L.; Armiento, G.; Somma, R.; Vicinanza, D.; Azzellino, A. Source apportionment assessment of marine sediment contamination in a post-industrial area (Bagnoli, Naples). Water 2020, 12, 2181. [Google Scholar] [CrossRef]
- Dixit, S.; Hering, J.G. Comparison of arsenic (V) and arsenic (III) sorption onto iron oxide minerals: Implications for arsenic mobility. Environ. Sci. Technol. 2003, 37, 4182–4189. [Google Scholar] [CrossRef]
- Smedley, P.L.; Nicolli, H.B.; Macdonald, D.M.J.; Barros, A.J.; Tullio, J.O. Hydrogeochemistry of arsenic and other inorganic constituents in groundwaters from La Pampa, Argentina. Appl. Geochem. 2002, 17, 259–284. [Google Scholar] [CrossRef]
- Smedley, P.L.; Kinniburgh, D. Chapter 1. Source and Behaviour of Arsenic in Natural Waters. In United Nations Synthesis Report on Arsenic in Drinking Water; World Health Organization (WHO): Geneva, Switzerland, 2001. [Google Scholar]
- Prommer, H.; Sun, J.; Helm, L.; Rathi, B.; Siade, A.J.; Morris, R. Deoxygenation prevents arsenic mobilization during deepwell injection into sulfide-bearing aquifers. Environ. Sci. Technol. 2018, 52, 13801–13810. [Google Scholar] [CrossRef]
- Wallis, I.; Prommer, H.; Pichler, T.; Post, V.; Norton, S.B.; Annable, M.D.; Simmons, C.T. Process-based reactive transport model to quantify arsenic mobility during aquifer storage and recovery of potable water. Environmental Environ. Sci. Technol. 2011, 45, 6924–6931. [Google Scholar] [CrossRef]
- Wen, X.; Lu, J.; Wu, J.; Lin, Y.; Luo, Y. Influence of coastal groundwater salinization on the distribution and risks of heavy metals. Sci. Total Environ. 2019, 652, 267–277. [Google Scholar] [CrossRef]
- Jung, H.B.; Charette, M.A.; Zheng, Y. Field, laboratory, and modeling study of reactive transport of groundwater arsenic in a coastal aquifer. Environ. Sci. Technol. 2009, 43, 5333–5338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amarathunga, U.; Diyabalanage, S.; Bandara, U.G.C.; Chandrajith, R. Environmental factors controlling arsenic mobilization from sandy shallow coastal aquifer sediments in the Mannar Island, Sri Lanka. Appl. Geochem. 2019, 100, 152–159. [Google Scholar] [CrossRef]
- Morelli, G.; Rimondi, V.; Benvenuti, M.; Medas, D.; Costagliola, P.; Gasparon, M. Experimental simulation of arsenic desorption from Quaternary aquifer sediments following sea water intrusion. Appl. Geochem. 2017, 87, 176–187. [Google Scholar] [CrossRef]
- Liu, C.W.; Lu, K.L.; Kao, Y.H.; Wang, C.J.; Maji, S.K.; Lee, J.F. Identifying sources and controlling factors of arsenic release in saline groundwater aquifers. Hydrol. Earth Syst. Sci. 2014, 18, 1089–1103. [Google Scholar] [CrossRef] [Green Version]
- Le Monte, J.J.; Stuckey, J.W.; Sanchez, J.Z.; Tappero, R.; Rinklebe, J.; Sparks, D.L. Sea level rise induced arsenic release from historically contaminated coastal soils. Environ. Sci. Technol. 2017, 51, 5913–5922. [Google Scholar] [CrossRef]
- Sun, H. Mobilization of arsenic, lead, and mercury under conditions of sea water intrusion and road deicing salt application. J. Contam. Hydrol. 2015, 180, 12–24. [Google Scholar] [CrossRef]
- Misra, A.; Singh, A.; Suresh Babu, D.S.; Jain, V.; Verma, M.; Bansal, B.K.; Manish, K. Sediment and submarine groundwater discharge mediated arsenic flux into the Bay of Bengal, India: An Appraisal. Curr. Poll. Rep. 2020, 6, 206–216. [Google Scholar] [CrossRef]
- Qu, W.; Wang, C.; Luo, M.; Zheng, C.; Li, H. Distributions, quality assessments and fluxes of heavy metals carried by submarine groundwater discharge in different types of wetlands in Jiaozhou Bay, China. Mar. Pollut. Bull. 2020, 157, 111310. [Google Scholar] [CrossRef]
- Wang, Y.; Jiao, J.; Cherry, J. Occurrence and geochemical behavior of arsenic in a coastal aquifer-aquitard system of the Pearl River Delta, China. Environ. Sci. Technol. 2012, 427, 286–297. [Google Scholar] [CrossRef]
- Mastrocicco, M.; Colombani, N.; Sbarbati, C.; Petitta, M. Assessing the effect of saltwater intrusion on petroleum hydrocarbons plumes via numerical modelling. Water Air Soil Pollut. 2012, 223, 4417–4427. [Google Scholar] [CrossRef]
- Sun, J.; Bostick, B.C.; Mailloux, B.J.; Jamieson, J.; Yan, B.; Pitiranggon, M.; Chillrud, S.N. Arsenic mobilization from iron oxides in the presence of oxalic acid under hydrodynamic conditions. Chemosphere 2018, 212, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Keimowitz, A.R.; Zheng, Y.; Chillrud, S.N.; Mailloux, B.; Jung, H.B.; Stute, M.H.; Simpson, J. Arsenic redistribution between sediments and water near a highly contaminated source. Environ. Sci. Technol. 2005, 39, 8606–8613. [Google Scholar] [CrossRef] [PubMed]
- Srithongkul, C.; Wongsaipun, S.; Krongchai, C.; Santasup, C.; Kittiwachana, S. Investigation of mobility and bioavailability of arsenic in agricultural soil after treatment by various soil amendments using sequential extraction procedure and multivariate analysis. Catena 2019, 181, 104084. [Google Scholar] [CrossRef]
- Hudson-Edwards, K.; Houghton, S.; Osborn, A. Extraction and analysis of arsenic in soils and sediments. TrAC Trends Anal. Chem. 2004, 23, 745–752. [Google Scholar] [CrossRef]
- Wenzel, W.W.; Kirchbaumer, N.; Stingeder, G.; Prohaska, T.; Lombi, E.; Domy, C.A. Arsenic fractionation in soils using an improved sequential extraction procedure. Anal. Chim. Acta 2001, 436, 309–323. [Google Scholar] [CrossRef]
- Biswas, A.; Gustafsson, J.P.; Neidhardt, H.; Halder, D.; Kundu, A.K.; Chatterjee, D.; Berner, Z.; Bhattacharya, P. Role of competing ions in the mobilization of arsenic in groundwater of Bengal Basin: Insight from surface complexation modeling. Water Res. 2014, 55, 30–39. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Mulligan, C.N. Effect of natural organic matter on arsenic release from soils and sediments to groundwater. Environ. Geochem. Health 2006, 28, 197–214. [Google Scholar] [CrossRef]
- Colombani, N.; Mastrocicco, M.; Prommer, H.; Sbarbati, C.; Petitta, M. Fate of arsenic, phosphate and ammonium plumes in a coastal aquifer affected by saltwater intrusion. J. Contam. Hydrol. 2015, 179, 116–131. [Google Scholar] [CrossRef]
- Stopelli, E.; Duyen, V.T.; Mai, T.T.; Trang, P.T.; Viet, P.H.; Lightfoot, A.; Kipfer, R.; Schneider, M.; Eiche, E.; Kontny, A.; et al. Spatial and temporal evolution of groundwater arsenic contamination in the Red River delta, Vietnam: Interplay of mobilisation and retardation processes. Sci. Total Environ. 2020, 717, 137143. [Google Scholar] [CrossRef]
- Aullon Alcaine, A.; Schulz, C.; Bundschuh, J.; Jacks, G.; Thunvik, R.; Gustafsson, J.; Mörth, C.M.; Sracek, O.; Ahmad, A.; Bhattacharya, P. Hydrogeochemical controls on the mobility of arsenic, fluoride and other geogenic co-contaminants in the shallow aquifers of northeastern La Pampa Province in Argentina. Sci. Total Environ. 2020, 715, 136671. [Google Scholar] [CrossRef]
- Van Herreweghe, S.; Swennen, R.; Vandecasteele, C.; Cappuyns, V. Solid phase speciation of arsenic by sequential extraction in standard reference materials and industrially contaminated soil samples. Environ. Pollut. 2003, 122, 323–342. [Google Scholar] [CrossRef]
- Mastrocicco, M.; Sbarbati, C.; Colombani, N.; Petitta, M. Efficiency verification of a horizontal flow barrier via flowmeter tests and multilevel sampling. Hydrol. Process. 2013, 27, 2414–2421. [Google Scholar] [CrossRef]
- Sbarbati, C.; Colombani, N.; Mastrocicco, M.; Aravena, R.; Petitta, M. Performance of different assessment methods to evaluate contaminant sources and fate in a coastal aquifer. Environ. Sci. Pollut. Res. 2015, 22, 15536–15548. [Google Scholar] [CrossRef] [PubMed]
- Arlotti, D.; Bozzini, R.; Petitta, M.; Mancini, M.; Pianu, M.; Alberto Blanco, G.; Sbarbati, C.; Colombo, R. Control and dynamic management of a sixty-seven wells hydraulic barrier. Chem. Eng. Trans. 2012, 28, 247–252. [Google Scholar]
- Sbarbati, C.; Colombani, N.; Mastrocicco, M.; Petitta, M.; Aravena, R. Reactive and mixing processes governing ammonium and nitrate coexistence in a polluted coastal aquifer. Geosciences 2018, 8, 210. [Google Scholar] [CrossRef] [Green Version]
- USEPA. Test Methods for Evaluating Solid Waste, Physical/Chemical Methods; U.S. Environmental Protection Agency: Washington, DC, USA, 2006.
- Keon, N.E.; Swartz, C.H.; Brabander, D.J.; Harvey, C.; Hemond, H.F. Validation of an arsenic sequential extraction method for evaluating mobility in sediments. Environ. Sci. Technol. 2001, 35, 2778–2784. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Chillrud, S.N.; Mailloux, B.J.; Bostick, B.C. In situ magnetite formation and longterm arsenic immobilization under advective flow conditions. Environ. Sci. Technol. 2016, 50, 10162–10171. [Google Scholar] [CrossRef] [Green Version]
- Barbieri, M.; Nigro, A.; Petitta, M. Groundwater mixing in the discharge area of San Vittorino Plain (Central Italy): Geochemical characterization and implication for drinking uses. Environ. Earth Sci. 2017, 76, 393. [Google Scholar] [CrossRef]
- Nordstrom, D.; Wilde, F. Reduction-6.5 Oxidation Potential (Electrode Method); U.S. Geological Survey TWRI Book 9; U.S. Geological Survey: Reston, VA, USA, 1998.
- Hutchinson, M.F.; Xu, T.; Stein, J.A. Recent progress in the ANUDEM elevation gridding procedure. Geomorphometry 2011, 2011, 19–22. [Google Scholar]
- Bostick, B.C.; Fendorf, S.; Brown, G.E. In situ analysis of thioarsenite complexes in neutral to alkaline arsenic sulphide solutions. Miner. Mag. 2005, 69, 781–795. [Google Scholar] [CrossRef]
- Manno, E.; Vassallo, M.; Varrica, D.; Dongarrà, G.; Hauser, S. Hydrogeochemistry and water balance in the coastal wetland area of “Biviere di Gela”, Sicily, Italy. Water Air Soil Pollut. 2007, 178, 179–193. [Google Scholar] [CrossRef]
- Guo, H.; Zhang, D.; Wen, D.; Wu, Y.; Ni, P.; Jiang, Y.; Guo, Q.; Li, F.; Zheng, H.; Zhou, Y. Arsenic mobilization in aquifers of the Southwest Songnen basin, P.R. China: Evidences from chemical and isotopic characteristics. Sci. Total. Environ. 2014, 490, 590–602. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Stute, M.; van Geen, A.; Gavrieli, I.; Dhar, R.; Simpson, H.J.; Schlosser, P.; Ahmed, K.M. Redox control of arsenic mobilization in Bangladesh groundwater. Appl. Geochem. 2004, 19, 201–214. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Step | Extractant and Time | Target Fe Phase | Target As Phase |
|---|---|---|---|
| 1 | 1 mol/L magnesium chloride, pH 7, 2 h, one repetition | Exchangeable Fe | Loosely bound As |
| 2 * | 1 mol/L sodium acetate adjusted to pH 4.5 with acetic acid, 24 h, one repetition | Fe carbonates (siderite) | As associated with carbonates, weakly bound As |
| 3 * | 1 mol/L hydroxylamine-hydrochloride in 25% v/v acetic acid, 48 h, one repetition | Amorphous Fe(III) oxides (ferrihydrite) | As associated with amorphous Fe oxides |
| 4 * | 50 g/L sodium dithionite, pH 4.8 with acetic acid/sodium citrate, 2 h, one repetition | Crystalline Fe(III) oxides (goethite and hematite) | As associated with crystalline Fe oxides |
| 5 | 1 mol/L sodium phosphate, pH 5, 16 h & 24 h, one repetition for each time period | -- | As adsorbed on recalcitrant Fe oxides |
| 6 * | 0.2 mol/L ammonium oxalate/0.17 mol/L oxalic acid, 6 h, one repetition | Recalcitrant Fe oxides (magnetite) | As co-precipitated in recalcitrant Fe oxides |
| 7 * | 16 mol/L nitric acid, 2 h, one repetition | Fe(II) sulfides (mackinawite and pyrite) | As-bearing sulfides |
| MW07 | Kd | MW08 | Kd | MW09 | Kd |
|---|---|---|---|---|---|
| Depth m a.s.l. | mL/g | Depth m a.s.l. | mL/g | Depth m a.s.l. | mL/g |
| −2.0 | 201.9 | −2.5 | 2019.2 | −2.5 | 1058.9 |
| −4.0 | 110.8 | −4.5 | 50.5 | −4.5 | 557.0 |
| −6.0 | 140.7 | −6.5 | 39.8 | −6.5 | 47.1 |
| −8.0 | 202.2 | −8.5 | 97.2 | −8.5 | 32.8 |
| −10.0 | 75.3 | −10.5 | 4.3 | −10.5 | 9.3 |
| −13.0 | 4.2 | −12.5 | 4.2 | −12.5 | 6.3 |
| −14.5 | 5.6 | −14.5 | 3.5 | −14.5 | 52.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sbarbati, C.; Barbieri, M.; Barron, A.; Bostick, B.; Colombani, N.; Mastrocicco, M.; Prommer, H.; Passaretti, S.; Zheng, Y.; Petitta, M. Redox Dependent Arsenic Occurrence and Partitioning in an Industrial Coastal Aquifer: Evidence from High Spatial Resolution Characterization of Groundwater and Sediments. Water 2020, 12, 2932. https://doi.org/10.3390/w12102932
Sbarbati C, Barbieri M, Barron A, Bostick B, Colombani N, Mastrocicco M, Prommer H, Passaretti S, Zheng Y, Petitta M. Redox Dependent Arsenic Occurrence and Partitioning in an Industrial Coastal Aquifer: Evidence from High Spatial Resolution Characterization of Groundwater and Sediments. Water. 2020; 12(10):2932. https://doi.org/10.3390/w12102932
Chicago/Turabian StyleSbarbati, Chiara, Maurizio Barbieri, Alyssa Barron, Benjamin Bostick, Nicolò Colombani, Micòl Mastrocicco, Henning Prommer, Stefania Passaretti, Yan Zheng, and Marco Petitta. 2020. "Redox Dependent Arsenic Occurrence and Partitioning in an Industrial Coastal Aquifer: Evidence from High Spatial Resolution Characterization of Groundwater and Sediments" Water 12, no. 10: 2932. https://doi.org/10.3390/w12102932