Single Cell ADNP Predictive of Human Muscle Disorders: Mouse Knockdown Results in Muscle Wasting



Abstract
:
1. Introduction
2. Materials and Methods
2.1. Human Muscle Single Cell Data Mining
2.2. Online Gene Expression Omnibus (GEO) Public Functional Genomics Data Repository, Human Muscle Diseases
2.3. Secondary Analysis of Tabula Muris Senis Single-Cell Data Set
2.4. Animals
2.4.1. Neuromuscular Junction (NMJ) Staining
2.4.2. Peptide Synthesis and Formulations
2.4.3. RNA Extraction and Quantitative Real-Time PCR
2.4.4. Virally-Delivered CRISPR-Mediated Adnp Knockdown in Muscle Tissue of Cas9 Mice: Cell Lines for Reagent Preparations
2.4.5. Single Guide RNA (sgRNA) Preparation and Plasmid Construction
2.4.6. Lentivirus Production and Injection
2.4.7. Behavioral Studies
2.4.8. Gait Analysis
2.4.9. Treadmill
2.4.10. Hot plate
2.5. Statistics
3. Results
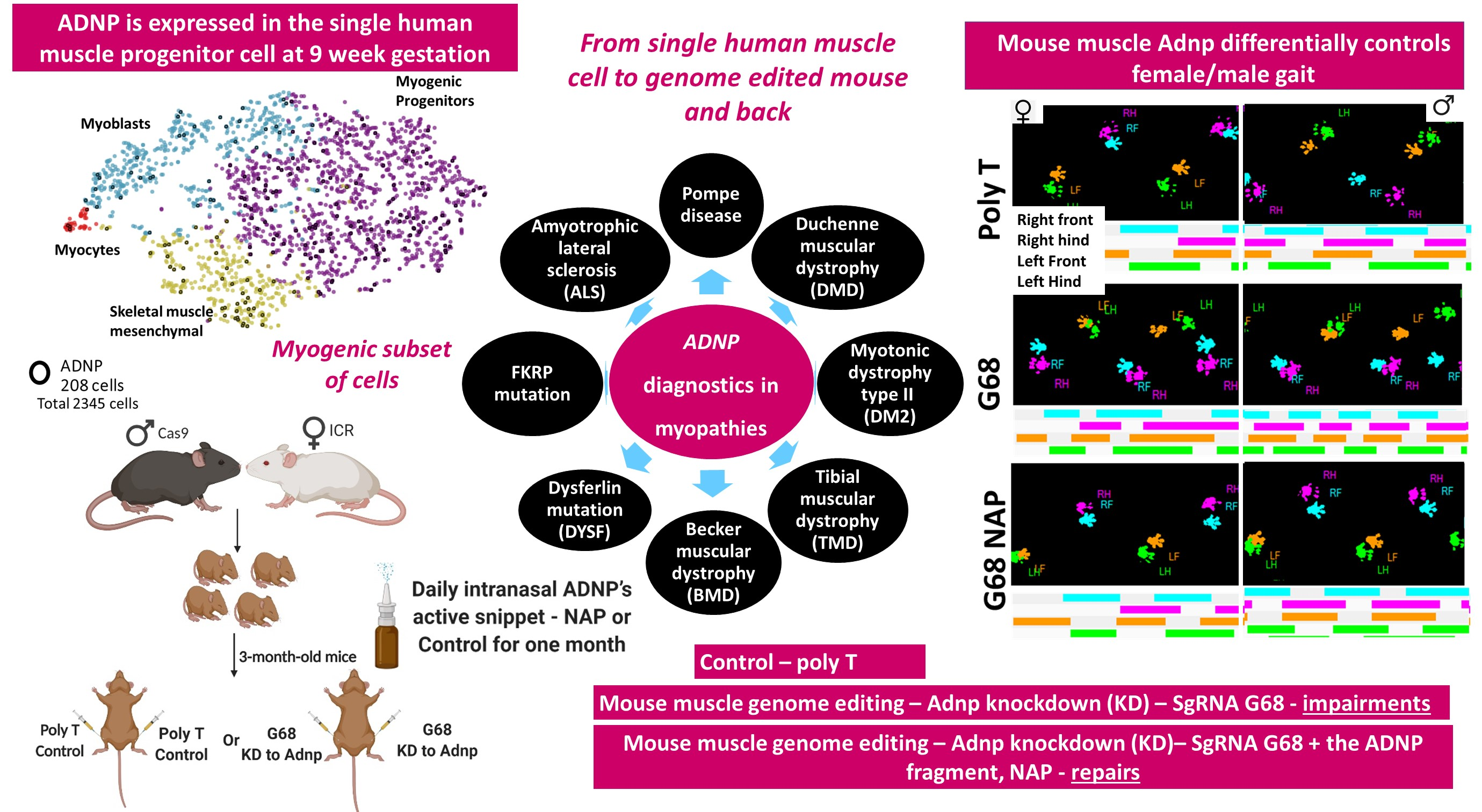
3.1. Human Single Cell ADNP Is Increased in Single Muscle Progenitor Cells
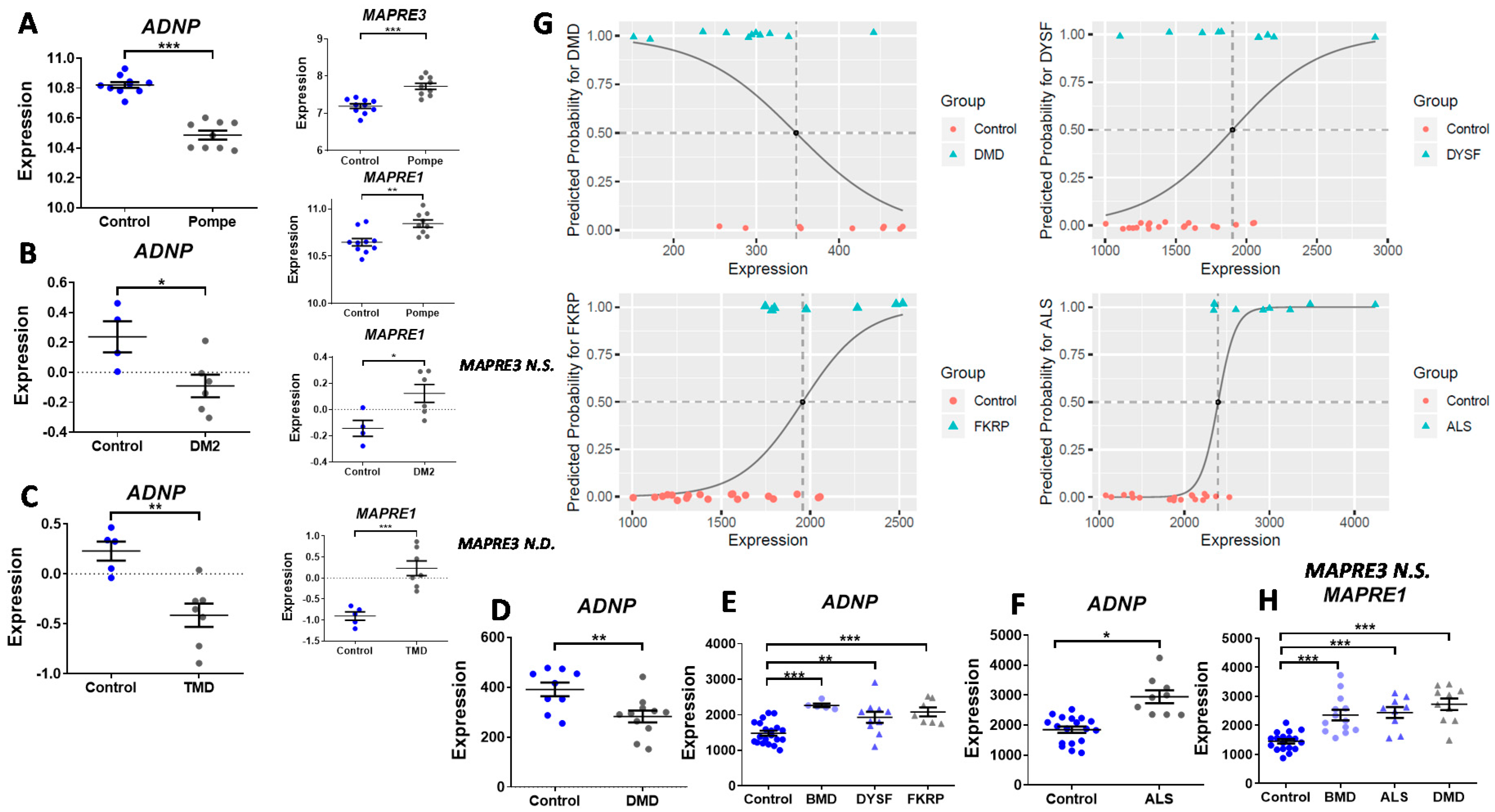
3.2. ADNP Expression Plays a Role in Childhood and Adult-Onset of Neuromuscular Disorders
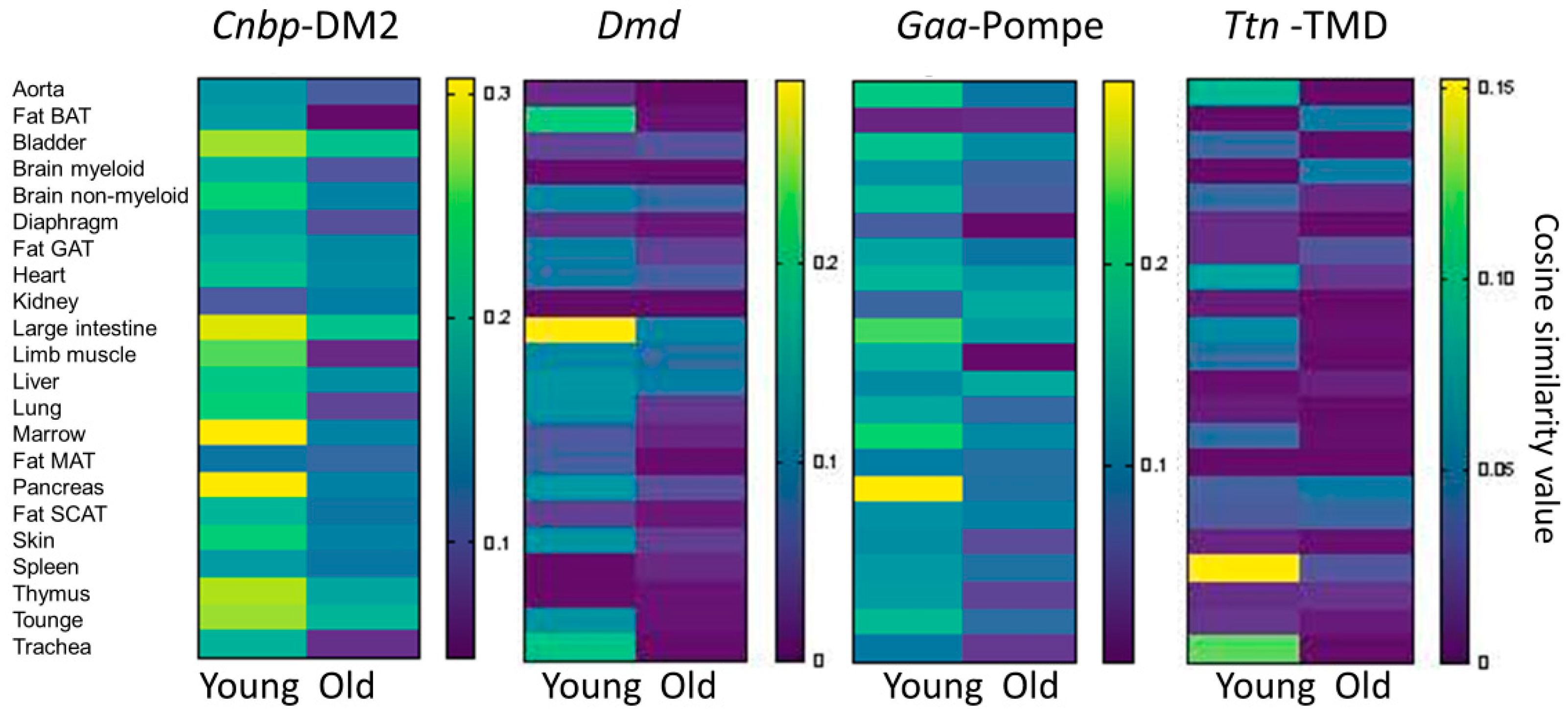
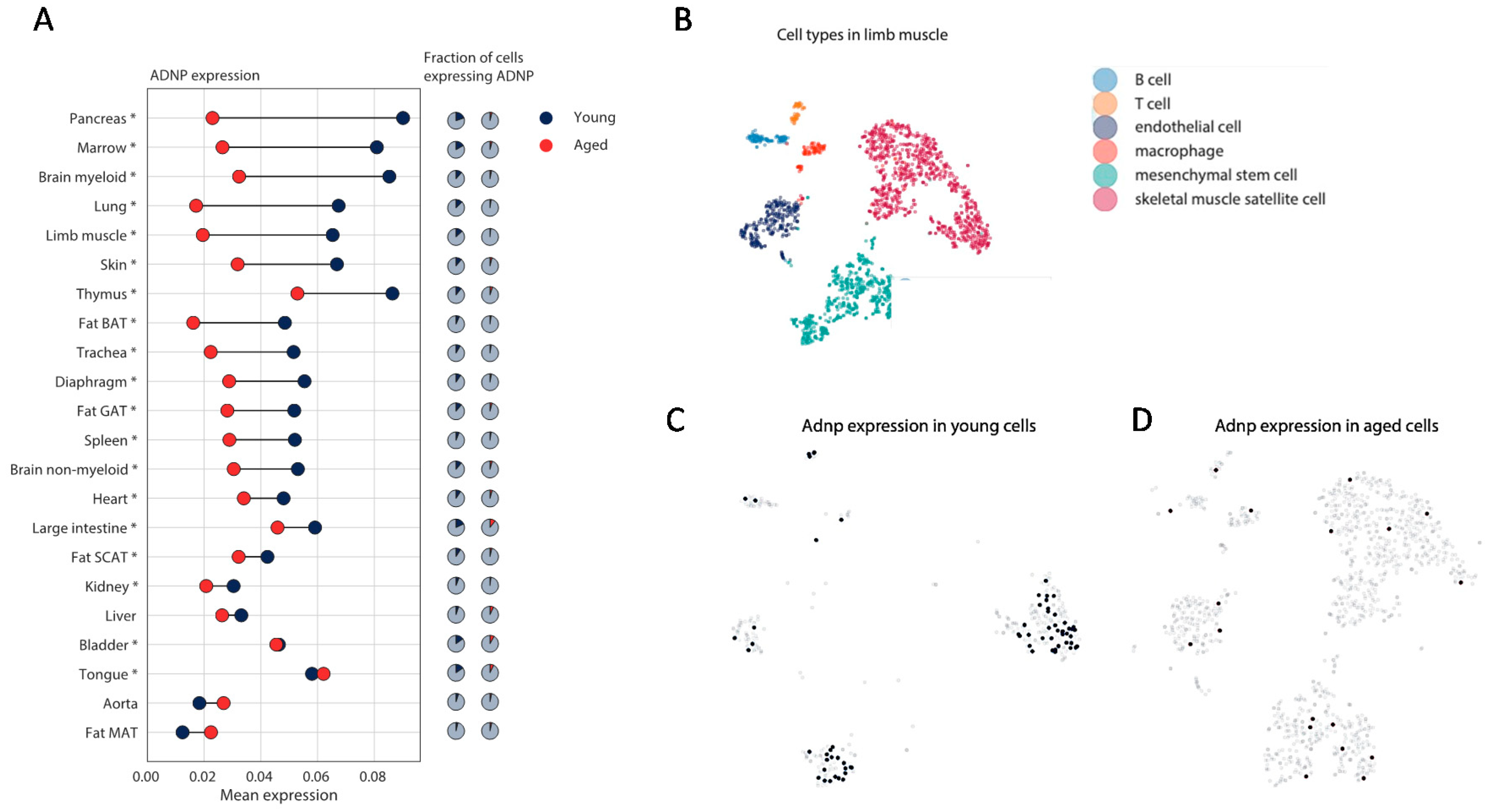
3.3. Mouse Single Cell Analysis Age-Dependently Correlates Adnp to Muscle Disease Genes
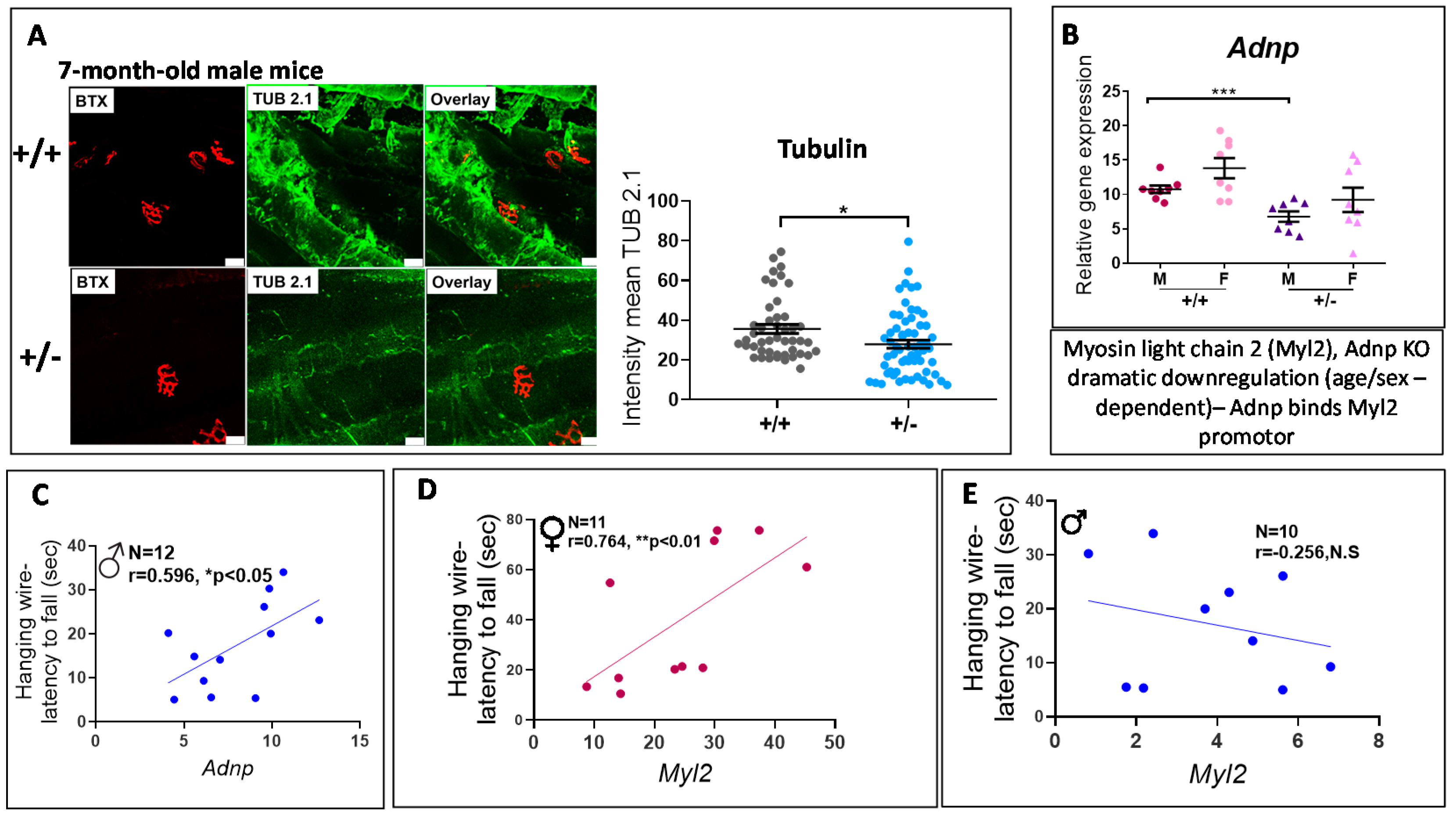
3.4. Adnp+/− Mice Display Neuromuscular Junction (NMJ) Disruption, Significantly Correlated with Behavioral Deficits
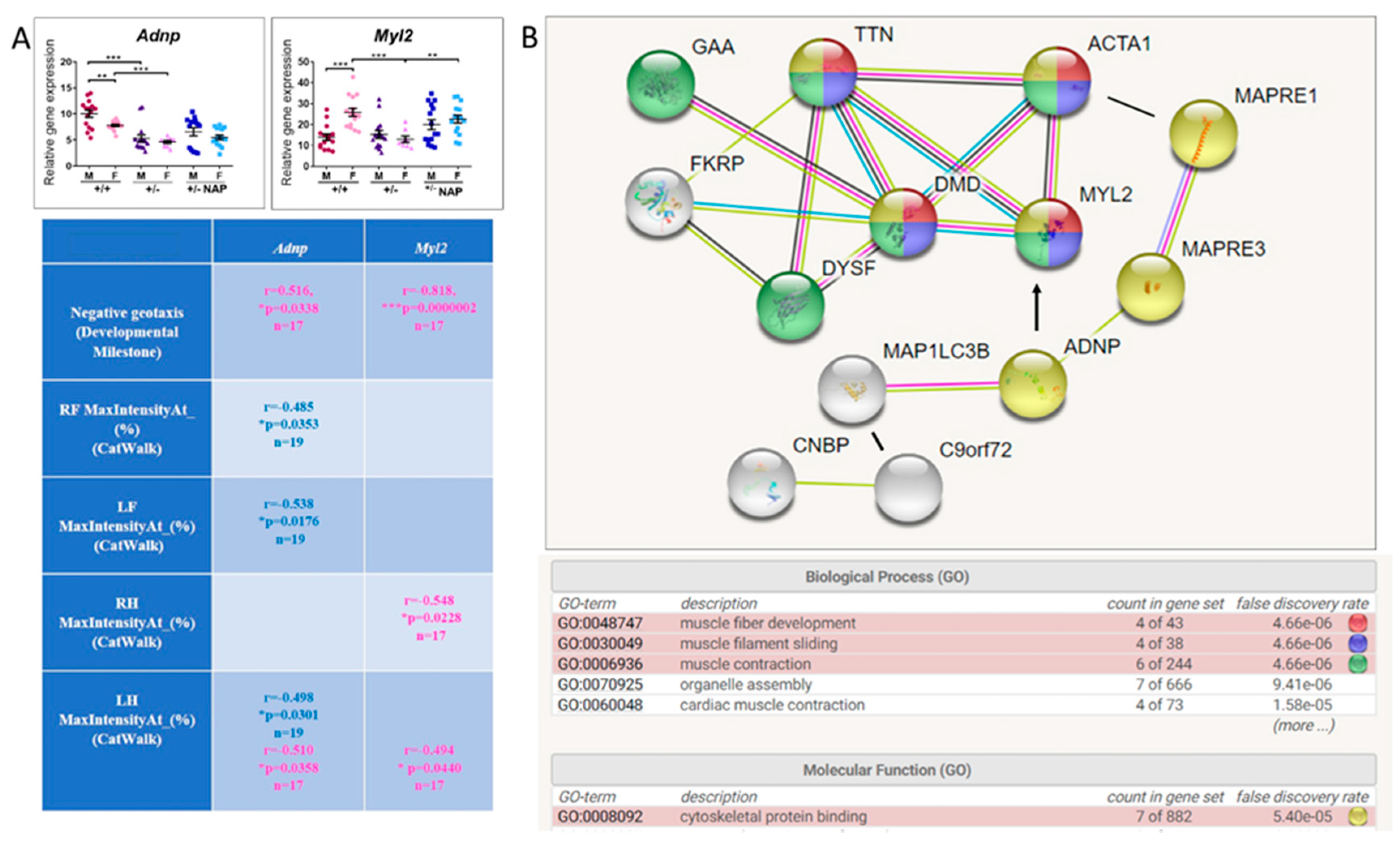
3.5. ADNP is Functionally Associated with Multiple Muscle Disease Proteins
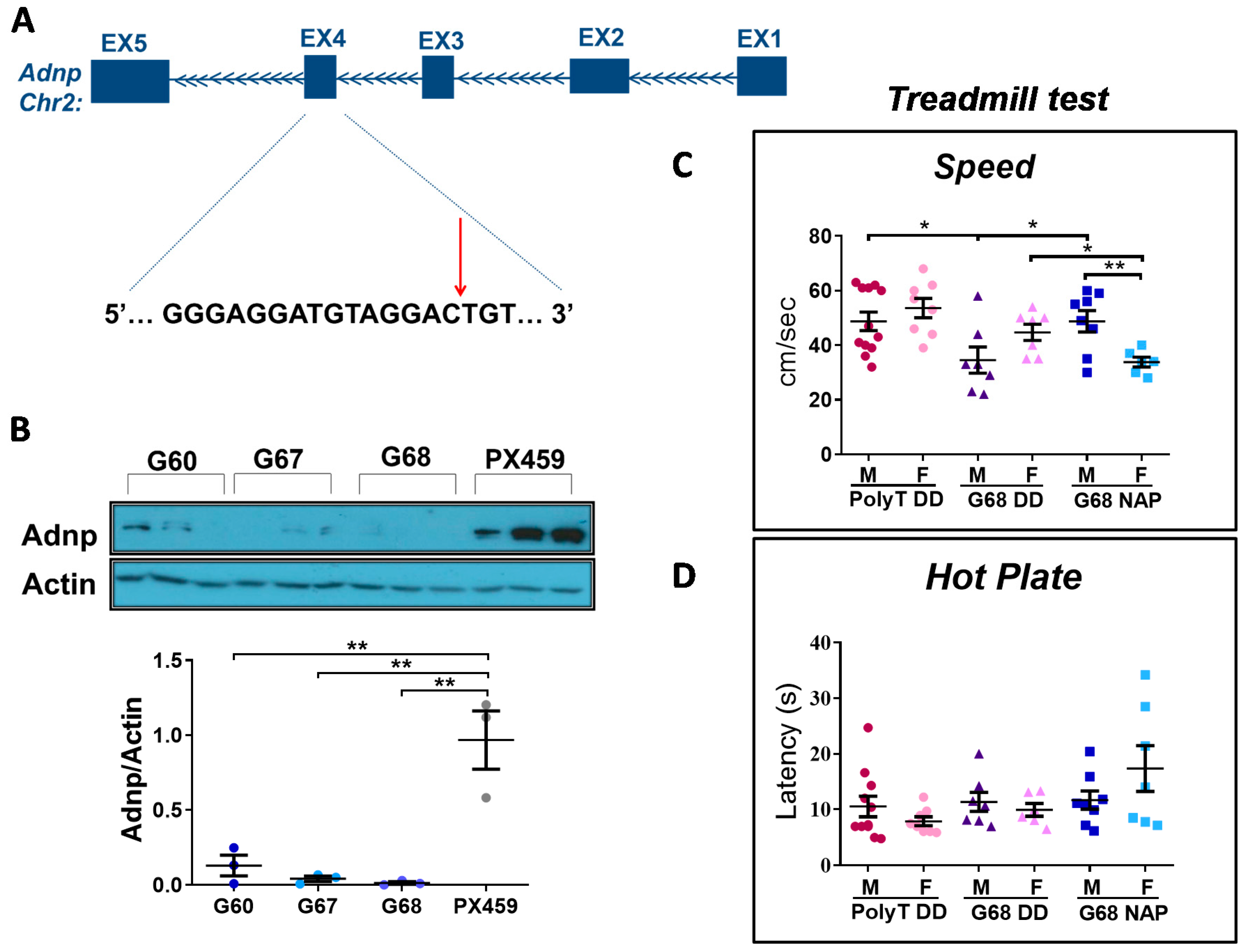
3.6. Choosing the Most Efficient Guide RNA for Muscle Adnp Knockdown
3.7. Adult Adnp Knockdown Male Mice Exhibit Aberrant Motor Performance in The Treadmill Test
3.8. Impaired Gait Parameters in Cas9 Female Mice Adnp Knockdown Are Ameliorated by NAP Treatment
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gozes, I.; Bassan, M.; Zamostiano, R.; Pinhasov, A.; Davidson, A.; Giladi, E.; Perl, O.; Glazner, G.W.; Brenneman, D.E. A novel signaling molecule for neuropeptide action: Activity-dependent neuroprotective protein. Ann. N. Y. Acad. Sci. 1999, 897, 125–135. [Google Scholar] [CrossRef]
- Zamostiano, R.; Pinhasov, A.; Gelber, E.; Steingart, R.A.; Seroussi, E.; Giladi, E.; Bassan, M.; Wollman, Y.; Eyre, H.J.; Mulley, J.C.; et al. Cloning and characterization of the human activity-dependent neuroprotective protein. J. Biol. Chem. 2001, 276, 708–714. [Google Scholar] [CrossRef] [Green Version]
- Bassan, M.; Zamostiano, R.; Davidson, A.; Pinhasov, A.; Giladi, E.; Perl, O.; Bassan, H.; Blat, C.; Gibney, G.; Glazner, G.; et al. Complete sequence of a novel protein containing a femtomolar-activity-dependent neuroprotective peptide. J. Neurochem. 1999, 72, 1283–1293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinhasov, A.; Mandel, S.; Torchinsky, A.; Giladi, E.; Pittel, Z.; Goldsweig, A.M.; Servoss, S.J.; Brenneman, D.E.; Gozes, I. Activity-dependent neuroprotective protein: A novel gene essential for brain formation. Brain Res. Dev. Brain Res. 2003, 144, 83–90. [Google Scholar] [CrossRef]
- Van Dijck, A.; Vulto-van Silfhout, A.T.; Cappuyns, E.; van der Werf, I.M.; Mancini, G.M.; Tzschach, A.; Bernier, R.; Gozes, I.; Eichler, E.E.; Romano, C.; et al. Clinical Presentation of a Complex Neurodevelopmental Disorder Caused by Mutations in ADNP. Biol. Psychiatry 2019, 85, 287–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, J.; Cohen, D.; Herman, C.; Verloes, A.; Guinchat, V.; Diaz, L.; Cravero, C.; Mandel, A.; Gozes, I. Developmental Phenotype of the Rare Case of DJ Caused by a Unique ADNP Gene De Novo Mutation. J. Mol. Neurosci. MN 2019, 68, 321–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnett, A.B.; Rhoads, C.L.; Hoekzema, K.; Turner, T.N.; Gerdts, J.; Wallace, A.S.; Bedrosian-Sermone, S.; Eichler, E.E.; Bernier, R.A. The autism spectrum phenotype in ADNP syndrome. Autism Res. Off. J. Int. Soc. Autism Res. 2018, 11, 1300–1310. [Google Scholar] [CrossRef] [PubMed]
- Gozes, I.; Van Dijck, A.; Hacohen-Kleiman, G.; Grigg, I.; Karmon, G.; Giladi, E.; Eger, M.; Gabet, Y.; Pasmanik-Chor, M.; Cappuyns, E.; et al. Premature primary tooth eruption in cognitive/motor-delayed ADNP-mutated children. Transl. Psychiatry 2017, 7, e1043. [Google Scholar] [CrossRef] [PubMed]
- Mandel, S.; Rechavi, G.; Gozes, I. Activity-dependent neuroprotective protein (ADNP) differentially interacts with chromatin to regulate genes essential for embryogenesis. Dev. Biol. 2007, 303, 814–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapitansky, O.; Sragovich, S.; Jaljuli, I.; Hadar, A.; Giladi, E.; Gozes, I. Age and Sex-Dependent ADNP Regulation of Muscle Gene Expression Is Correlated with Motor Behavior: Possible Feedback Mechanism with PACAP. Int. J. Mol. Sci. 2020, 21, 6715. [Google Scholar] [CrossRef] [PubMed]
- Girard, B.M.; Campbell, S.E.; Beca, K.I.; Perkins, M.; Hsiang, H.; May, V.; Vizzard, M.A. Intrabladder PAC1 Receptor Antagonist, PACAP(6-38), Reduces Urinary Bladder Frequency and Pelvic Sensitivity in Mice Exposed to Repeated Variate Stress (RVS). J. Mol. Neurosci. MN 2020. [Google Scholar] [CrossRef]
- Sragovich, S.; Ziv, Y.; Vaisvaser, S.; Shomron, N.; Hendler, T.; Gozes, I. The autism-mutated ADNP plays a key role in stress response. Transl. Psychiatry 2019, 9, 235. [Google Scholar] [CrossRef] [PubMed]
- Ivashko-Pachima, Y.; Hadar, A.; Grigg, I.; Korenkova, V.; Kapitansky, O.; Karmon, G.; Gershovits, M.; Sayas, C.L.; Kooy, R.F.; Attems, J.; et al. Discovery of autism/intellectual disability somatic mutations in Alzheimer’s brains: Mutated ADNP cytoskeletal impairments and repair as a case study. Mol. Psychiatry 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Guia, R.M.; Agerholm, M.; Nielsen, T.S.; Consitt, L.A.; Sogaard, D.; Helge, J.W.; Larsen, S.; Brandauer, J.; Houmard, J.A.; Treebak, J.T. Aerobic and resistance exercise training reverses age-dependent decline in NAD(+) salvage capacity in human skeletal muscle. Physiol. Rep. 2019, 7, e14139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivashko-Pachima, Y.; Sayas, C.L.; Malishkevich, A.; Gozes, I. ADNP/NAP dramatically increase microtubule end-binding protein-Tau interaction: A novel avenue for protection against tauopathy. Mol. Psychiatry 2017, 22, 1335–1344. [Google Scholar] [CrossRef] [PubMed]
- Mandel, S.; Spivak-Pohis, I.; Gozes, I. ADNP differential nucleus/cytoplasm localization in neurons suggests multiple roles in neuronal differentiation and maintenance. J. Mol. Neurosci. MN 2008, 35, 127–141. [Google Scholar] [CrossRef] [PubMed]
- Mandel, S.; Gozes, I. Activity-dependent neuroprotective protein constitutes a novel element in the SWI/SNF chromatin remodeling complex. J. Biol. Chem. 2007, 282, 34448–34456. [Google Scholar] [CrossRef] [Green Version]
- Ferrari, R.; de Llobet Cucalon, L.I.; Di Vona, C.; Le Dilly, F.; Vidal, E.; Lioutas, A.; Oliete, J.Q.; Jochem, L.; Cutts, E.; Dieci, G.; et al. TFIIIC Binding to Alu Elements Controls Gene Expression via Chromatin Looping and Histone Acetylation. Mol. Cell 2020, 77, 475–487.e11. [Google Scholar] [CrossRef] [Green Version]
- Kaaij, L.J.T.; Mohn, F.; van der Weide, R.H.; de Wit, E.; Buhler, M. The ChAHP Complex Counteracts Chromatin Looping at CTCF Sites that Emerged from SINE Expansions in Mouse. Cell 2019, 178, 1437–1451.e14. [Google Scholar] [CrossRef]
- Schirer, Y.; Malishkevich, A.; Ophir, Y.; Lewis, J.; Giladi, E.; Gozes, I. Novel marker for the onset of frontotemporal dementia: Early increase in activity-dependent neuroprotective protein (ADNP) in the face of Tau mutation. PLoS ONE 2014, 9, e87383. [Google Scholar] [CrossRef] [Green Version]
- Malishkevich, A.; Amram, N.; Hacohen-Kleiman, G.; Magen, I.; Giladi, E.; Gozes, I. Activity-dependent neuroprotective protein (ADNP) exhibits striking sexual dichotomy impacting on autistic and Alzheimer’s pathologies. Transl. Psychiatry 2015, 5, e501. [Google Scholar] [CrossRef] [PubMed]
- Gkogkas, C.G.; Khoutorsky, A.; Ran, I.; Rampakakis, E.; Nevarko, T.; Weatherill, D.B.; Vasuta, C.; Yee, S.; Truitt, M.; Dallaire, P.; et al. Autism-related deficits via dysregulated eIF4E-dependent translational control. Nature 2013, 493, 371–377. [Google Scholar] [CrossRef] [Green Version]
- Oz, S.; Kapitansky, O.; Ivashco-Pachima, Y.; Malishkevich, A.; Giladi, E.; Skalka, N.; Rosin-Arbesfeld, R.; Mittelman, L.; Segev, O.; Hirsch, J.A.; et al. The NAP motif of activity-dependent neuroprotective protein (ADNP) regulates dendritic spines through microtubule end binding proteins. Mol. Psychiatry 2014, 19, 1115–1124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivashko-Pachima, Y.; Maor-Nof, M.; Gozes, I. NAP (davunetide) preferential interaction with dynamic 3-repeat Tau explains differential protection in selected tauopathies. PLoS ONE 2019, 14, e0213666. [Google Scholar] [CrossRef] [Green Version]
- Amram, N.; Hacohen-Kleiman, G.; Sragovich, S.; Malishkevich, A.; Katz, J.; Touloumi, O.; Lagoudaki, R.; Grigoriadis, N.C.; Giladi, E.; Yeheskel, A.; et al. Sexual divergence in microtubule function: The novel intranasal microtubule targeting SKIP normalizes axonal transport and enhances memory. Mol. Psychiatry 2016, 21, 1467–1476. [Google Scholar] [CrossRef] [PubMed]
- Esteves, A.R.; Gozes, I.; Cardoso, S.M. The rescue of microtubule-dependent traffic recovers mitochondrial function in Parkinson’s disease. Biochim. Biophys. Acta 2014, 1842, 7–21. [Google Scholar] [CrossRef] [Green Version]
- Merenlender-Wagner, A.; Malishkevich, A.; Shemer, Z.; Udawela, M.; Gibbons, A.; Scarr, E.; Dean, B.; Levine, J.; Agam, G.; Gozes, I. Autophagy has a key role in the pathophysiology of schizophrenia. Mol. Psychiatry 2015, 20, 126–132. [Google Scholar] [CrossRef] [Green Version]
- Hacohen-Kleiman, G.; Sragovich, S.; Karmon, G.; Gao, A.Y.L.; Grigg, I.; Pasmanik-Chor, M.; Le, A.; Korenkova, V.; McKinney, R.A.; Gozes, I. Activity-dependent neuroprotective protein deficiency models synaptic and developmental phenotypes of autism-like syndrome. J. Clin. Investig. 2018, 128, 4956–4969. [Google Scholar] [CrossRef] [Green Version]
- Meola, G.; Cardani, R. Myotonic dystrophy type 2 and modifier genes: An update on clinical and pathomolecular aspects. Neurol. Sci. Off. J. Ital. Neurol. Soc. Ital. Soc. Clin. Neurophysiol. 2017, 38, 535–546. [Google Scholar] [CrossRef]
- Yiu, E.M.; Kornberg, A.J. Duchenne muscular dystrophy. J. Paediatr. Child Health 2015, 51, 759–764. [Google Scholar] [CrossRef]
- Belanto, J.J.; Mader, T.L.; Eckhoff, M.D.; Strandjord, D.M.; Banks, G.B.; Gardner, M.K.; Lowe, D.A.; Ervasti, J.M. Microtubule binding distinguishes dystrophin from utrophin. Proc. Natl. Acad. Sci. USA 2014, 111, 5723–5728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capitanio, D.; Moriggi, M.; Torretta, E.; Barbacini, P.; De Palma, S.; Vigano, A.; Lochmuller, H.; Muntoni, F.; Ferlini, A.; Mora, M.; et al. Comparative proteomic analyses of Duchenne muscular dystrophy and Becker muscular dystrophy muscles: Changes contributing to preserve muscle function in Becker muscular dystrophy patients. J. Cachexiasarcopenia Muscle 2020, 11, 547–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrews, J.G.; Wahl, R.A. Duchenne and Becker muscular dystrophy in adolescents: Current perspectives. Adolesc. Healthmed. Ther. 2018, 9, 53–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohler, L.; Puertollano, R.; Raben, N. Pompe Disease: From Basic Science to Therapy. Neurother. J. Am. Soc. Exp. Neurother. 2018, 15, 928–942. [Google Scholar] [CrossRef] [Green Version]
- Savarese, M.; Sarparanta, J.; Vihola, A.; Udd, B.; Hackman, P. Increasing Role of Titin Mutations in Neuromuscular Disorders. J. Neuromuscul. Dis. 2016, 3, 293–308. [Google Scholar] [CrossRef] [Green Version]
- Bansal, D.; Miyake, K.; Vogel, S.S.; Groh, S.; Chen, C.C.; Williamson, R.; McNeil, P.L.; Campbell, K.P. Defective membrane repair in dysferlin-deficient muscular dystrophy. Nature 2003, 423, 168–172. [Google Scholar] [CrossRef] [PubMed]
- Gerin, I.; Ury, B.; Breloy, I.; Bouchet-Seraphin, C.; Bolsee, J.; Halbout, M.; Graff, J.; Vertommen, D.; Muccioli, G.G.; Seta, N.; et al. ISPD produces CDP-ribitol used by FKTN and FKRP to transfer ribitol phosphate onto alpha-dystroglycan. Nat. Commun. 2016, 7, 11534. [Google Scholar] [CrossRef]
- Masrori, P.; Van Damme, P. Amyotrophic lateral sclerosis: A clinical review. Eur. J. Neurol. 2020. [Google Scholar] [CrossRef]
- Xi, H.; Langerman, J.; Sabri, S.; Chien, P.; Young, C.S.; Younesi, S.; Hicks, M.; Gonzalez, K.; Fujiwara, W.; Marzi, J.; et al. A Human Skeletal Muscle Atlas Identifies the Trajectories of Stem and Progenitor Cells across Development and from Human Pluripotent Stem Cells. Cell Stem Cell 2020, 27, 181–185. [Google Scholar] [CrossRef]
- USCS Cell Browser. Available online: https://skeletal-muscle.cells.ucsc.edu (accessed on 15 August 2020).
- Palermo, A.T.; Palmer, R.E.; So, K.S.; Oba-Shinjo, S.M.; Zhang, M.; Richards, B.; Madhiwalla, S.T.; Finn, P.F.; Hasegawa, A.; Ciociola, K.M.; et al. Transcriptional response to GAA deficiency (Pompe disease) in infantile-onset patients. Mol. Genet. Metab. 2012, 106, 287–300. [Google Scholar] [CrossRef]
- Haslett, J.N.; Sanoudou, D.; Kho, A.T.; Han, M.; Bennett, R.R.; Kohane, I.S.; Beggs, A.H.; Kunkel, L.M. Gene expression profiling of Duchenne muscular dystrophy skeletal muscle. Neurogenetics 2003, 4, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Screen, M.; Jonson, P.H.; Raheem, O.; Palmio, J.; Laaksonen, R.; Lehtimaki, T.; Sirito, M.; Krahe, R.; Hackman, P.; Udd, B. Abnormal splicing of NEDD4 in myotonic dystrophy type 2: Possible link to statin adverse reactions. Am. J. Pathol. 2014, 184, 2322–2332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dadgar, S.; Wang, Z.; Johnston, H.; Kesari, A.; Nagaraju, K.; Chen, Y.W.; Hill, D.A.; Partridge, T.A.; Giri, M.; Freishtat, R.J.; et al. Asynchronous remodeling is a driver of failed regeneration in Duchenne muscular dystrophy. J. Cell Biol. 2014, 207, 139–158. [Google Scholar] [CrossRef]
- Bakay, M.; Wang, Z.; Melcon, G.; Schiltz, L.; Xuan, J.; Zhao, P.; Sartorelli, V.; Seo, J.; Pegoraro, E.; Angelini, C.; et al. Nuclear envelope dystrophies show a transcriptional fingerprint suggesting disruption of Rb-MyoD pathways in muscle regeneration. Brain J. Neurol. 2006, 129, 996–1013. [Google Scholar] [CrossRef]
- Vulih-Shultzman, I.; Pinhasov, A.; Mandel, S.; Grigoriadis, N.; Touloumi, O.; Pittel, Z.; Gozes, I. Activity-dependent neuroprotective protein snippet NAP reduces tau hyperphosphorylation and enhances learning in a novel transgenic mouse model. J. Pharmacol. Exp. Ther. 2007, 323, 438–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mollinedo, P.; Kapitansky, O.; Gonzalez-Lamuno, D.; Zaslavsky, A.; Real, P.; Gozes, I.; Gandarillas, A.; Fernandez-Luna, J.L. Cellular and animal models of skin alterations in the autism-related ADNP syndrome. Sci. Rep. 2019, 9, 736. [Google Scholar] [CrossRef]
- Maimon, R.; Ionescu, A.; Bonnie, A.; Sweetat, S.; Wald-Altman, S.; Inbar, S.; Gradus, T.; Trotti, D.; Weil, M.; Behar, O.; et al. miR126-5p Downregulation Facilitates Axon Degeneration and NMJ Disruption via a Non-Cell-Autonomous Mechanism in ALS. J. Neurosci. Off. J. Soc. Neurosci. 2018, 38, 5478–5494. [Google Scholar] [CrossRef] [Green Version]
- Gozes, I.; Barnstable, C.J. Monoclonal antibodies that recognize discrete forms of tubulin. Proc. Natl. Acad. Sci. USA 1982, 79, 2579–2583. [Google Scholar] [CrossRef] [Green Version]
- Alcalay, R.N.; Giladi, E.; Pick, C.G.; Gozes, I. Intranasal administration of NAP, a neuroprotective peptide, decreases anxiety-like behavior in aging mice in the elevated plus maze. Neurosci. Lett. 2004, 361, 128–131. [Google Scholar] [CrossRef]
- Sragovich, S.; Malishkevich, A.; Piontkewitz, Y.; Giladi, E.; Touloumi, O.; Lagoudaki, R.; Grigoriadis, N.; Gozes, I. The autism/neuroprotection-linked ADNP/NAP regulate the excitatory glutamatergic synapse. Transl. Psychiatry 2019, 9, 2. [Google Scholar] [CrossRef] [Green Version]
- Dangoor, D.; Biondi, B.; Gobbo, M.; Vachutinski, Y.; Fridkin, M.; Gozes, I.; Rocchi, R. Novel glycosylated VIP analogs: Synthesis, biological activity, and metabolic stability. J. Pept. Sci. Off. Publ. Eur. Pept. Soc. 2008, 14, 321–328. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- CRISPOR. Available online: http://crispor.tefor.net/ (accessed on 15 August 2018).
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [Green Version]
- Weintraub, A.S.; Li, C.H.; Zamudio, A.V.; Sigova, A.A.; Hannett, N.M.; Day, D.S.; Abraham, B.J.; Cohen, M.A.; Nabet, B.; Buckley, D.L.; et al. YY1 Is a Structural Regulator of Enhancer-Promoter Loops. Cell 2017, 171, 1573–1588.e28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kutner, R.H.; Zhang, X.Y.; Reiser, J. Production, concentration and titration of pseudotyped HIV-1-based lentiviral vectors. Nat. Protoc. 2009, 4, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Kutner, R.H.; Puthli, S.; Marino, M.P.; Reiser, J. Simplified production and concentration of HIV-1-based lentiviral vectors using HYPERFlask vessels and anion exchange membrane chromatography. BMC Biotechnol. 2009, 9, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kyriakou, E.I.; van der Kieft, J.G.; de Heer, R.C.; Spink, A.; Nguyen, H.P.; Homberg, J.R.; van der Harst, J.E. Automated quantitative analysis to assess motor function in different rat models of impaired coordination and ataxia. J. Neurosci. Methods 2016, 268, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Zhang, J.; Walczak, P.; Bulte, J.W.M. Quantification of motor neuron loss and muscular atrophy in ricin-induced focal nerve injury. J. Neurosci. Methods 2018, 308, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Marques-Aleixo, I.; Santos-Alves, E.; Mariani, D.; Rizo-Roca, D.; Padrao, A.I.; Rocha-Rodrigues, S.; Viscor, G.; Torrella, J.R.; Ferreira, R.; Oliveira, P.J.; et al. Physical exercise prior and during treatment reduces sub-chronic doxorubicin-induced mitochondrial toxicity and oxidative stress. Mitochondrion 2015, 20, 22–33. [Google Scholar] [CrossRef]
- al-Hachim, G.M.; al-Khatim, A.S. Prenatal effects of aqueous plastic extract on offspring. Fetal Diagn. 1997, 12, 28–31. [Google Scholar] [CrossRef]
- Vincent, A.E.; Rosa, H.S.; Alston, C.L.; Grady, J.P.; Rygiel, K.A.; Rocha, M.C.; Barresi, R.; Taylor, R.W.; Turnbull, D.M. Dysferlin mutations and mitochondrial dysfunction. Neuromuscul. Disord. NMD 2016, 26, 782–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, E.C.; Alrowaished, S.S.; Kilroy, E.A.; Crooks, E.S.; Drinkert, D.M.; Karunasiri, C.M.; Belanger, J.J.; Khalil, A.; Kelley, J.B.; Henry, C.A. NAD+ improves neuromuscular development in a zebrafish model of FKRP-associated dystroglycanopathy. Skelet. Muscle 2019, 9, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaum, N.; Lehallier, B.; Hahn, O.; Palovics, R.; Hosseinzadeh, S.; Lee, S.E.; Sit, R.; Lee, D.P.; Losada, P.M.; Zardeneta, M.E.; et al. Ageing hallmarks exhibit organ-specific temporal signatures. Nature 2020, 583, 596–602. [Google Scholar] [CrossRef] [PubMed]
- Tabula Muris, C. A single-cell transcriptomic atlas characterizes ageing tissues in the mouse. Nature 2020, 583, 590–595. [Google Scholar] [CrossRef]
- String. Available online: https://string-db.org/ (accessed on 15 July 2020).
- Goodier, J.L.; Soares, A.O.; Pereira, G.C.; DeVine, L.R.; Sanchez, L.; Cole, R.N.; Garcia-Perez, J.L. C9orf72-associated SMCR8 protein binds in the ubiquitin pathway and with proteins linked with neurological disease. Acta Neuropathol. Commun. 2020, 8, 110. [Google Scholar] [CrossRef]
- Wei, C.; Stock, L.; Schneider-Gold, C.; Sommer, C.; Timchenko, N.A.; Timchenko, L. Reduction of Cellular Nucleic Acid Binding Protein Encoded by a Myotonic Dystrophy Type 2 Gene Causes Muscle Atrophy. Mol. Cell. Biol. 2018, 38. [Google Scholar] [CrossRef] [Green Version]
- Gao, Z.; Herrera-Carrillo, E.; Berkhout, B. Delineation of the Exact Transcription Termination Signal for Type 3 Polymerase III. Mol. Therapy. Nucleic Acids 2018, 10, 36–44. [Google Scholar] [CrossRef] [Green Version]
- Sragovich, S.; Merenlender-Wagner, A.; Gozes, I. ADNP Plays a Key Role in Autophagy: From Autism to Schizophrenia and Alzheimer’s Disease. Bioessays News Rev. Mol. Cell. Dev. Biol. 2017, 39. [Google Scholar] [CrossRef]
- Lukas, Z.; Falk, M.; Feit, J.; Soucek, O.; Falkova, I.; Stefancikova, L.; Janousova, E.; Fajkusova, L.; Zaoralkova, J.; Hrabalkova, R. Sequestration of MBNL1 in tissues of patients with myotonic dystrophy type 2. Neuromuscul. Disord. NMD 2012, 22, 604–616. [Google Scholar] [CrossRef]
- Byers, T.J.; Beggs, A.H.; McNally, E.M.; Kunkel, L.M. Novel actin crosslinker superfamily member identified by a two step degenerate PCR procedure. FEBS Lett. 1995, 368, 500–504. [Google Scholar] [CrossRef] [Green Version]
- Miao, Z.; Ali, A.; Hu, L.; Zhao, F.; Yin, C.; Chen, C.; Yang, T.; Qian, A. Microtubule actin cross-linking factor 1, a novel potential target in cancer. Cancer Sci. 2017, 108, 1953–1958. [Google Scholar] [CrossRef] [Green Version]
- Lane, T.R.; Fuchs, E.; Slep, K.C. Structure of the ACF7 EF-Hand-GAR Module and Delineation of Microtubule Binding Determinants. Structure 2017, 25, 1130–1138.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aboonq, M.S.; Vasiliou, S.A.; Haddley, K.; Quinn, J.P.; Bubb, V.J. Activity-dependent neuroprotective protein modulates its own gene expression. J. Mol. Neurosci. MN 2012, 46, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Jouroukhin, Y.; Ostritsky, R.; Assaf, Y.; Pelled, G.; Giladi, E.; Gozes, I. NAP (davunetide) modifies disease progression in a mouse model of severe neurodegeneration: Protection against impairments in axonal transport. Neurobiol. Dis. 2013, 56, 79–94. [Google Scholar] [CrossRef] [PubMed]
- Kodama, A.; Karakesisoglou, I.; Wong, E.; Vaezi, A.; Fuchs, E. ACF7: An essential integrator of microtubule dynamics. Cell 2003, 115, 343–354. [Google Scholar] [CrossRef] [Green Version]
- Konno, T.; Ross, O.A.; Teive, H.A.G.; Slawek, J.; Dickson, D.W.; Wszolek, Z.K. DCTN1-related neurodegeneration: Perry syndrome and beyond. Parkinsonism Relat. Disord. 2017, 41, 14–24. [Google Scholar] [CrossRef] [Green Version]
- Grigg, I.; Ivashko-Pachima, Y.; Hait, T.A.; Korenkova, V.; Touloumi, O.; Lagoudaki, R.; Van Dijck, A.; Marusic, Z.; Anicic, M.; Vukovic, J.; et al. Tauopathy in the young autistic brain: Novel biomarker and therapeutic target. Transl. Psychiatry 2020, 10, 228. [Google Scholar] [CrossRef]
- CTCFBSDB 2.0: A database for CTCF binding sites and genome organization. Available online: http://insulatordb.uthsc.edu/ (accessed on 15 July 2020).
- Geiger, R.C.; Taylor, W.; Glucksberg, M.R.; Dean, D.A. Cyclic stretch-induced reorganization of the cytoskeleton and its role in enhanced gene transfer. Gene Ther. 2006, 13, 725–731. [Google Scholar] [CrossRef] [Green Version]
- Polanco, M.J.; Parodi, S.; Piol, D.; Stack, C.; Chivet, M.; Contestabile, A.; Miranda, H.C.; Lievens, P.M.; Espinoza, S.; Jochum, T.; et al. Adenylyl cyclase activating polypeptide reduces phosphorylation and toxicity of the polyglutamine-expanded androgen receptor in spinobulbar muscular atrophy. Sci. Transl. Med. 2016, 8, 370ra181. [Google Scholar] [CrossRef]
- Gozes, I.; Iram, T.; Maryanovsky, E.; Arviv, C.; Rozenberg, L.; Schirer, Y.; Giladi, E.; Furman-Assaf, S. Novel tubulin and tau neuroprotective fragments sharing structural similarities with the drug candidate NAP (Davuentide). J. Alzheimers Dis. 2014, 40 (Suppl. 1), S23–S36. [Google Scholar] [CrossRef]
- Malishkevich, A.; Marshall, G.A.; Schultz, A.P.; Sperling, R.A.; Aharon-Peretz, J.; Gozes, I. Blood-Borne Activity-Dependent Neuroprotective Protein (ADNP) is Correlated with Premorbid Intelligence, Clinical Stage, and Alzheimer’s Disease Biomarkers. J. Alzheimers Dis. 2016, 50, 249–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunkel, L.M. To dystrophin and beyond: An interview with Louis Kunkel. Dis. Models Mech. 2019, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnett, A.B.; Beighley, J.S.; Kurtz-Nelson, E.C.; Hoekzema, K.; Wang, T.; Bernier, R.A.; Eichler, E.E. Developmental Predictors of Cognitive and Adaptive Outcomes in Genetic Subtypes of Autism Spectrum Disorder. Autism Res. 2020. [Google Scholar] [CrossRef]
- Dresner, E.; Agam, G.; Gozes, I. Activity-dependent neuroprotective protein (ADNP) expression level is correlated with the expression of the sister protein ADNP2: Deregulation in schizophrenia. Eur. Neuropsychopharmacol. J. Eur. Coll. Neuropsychopharmacol. 2011, 21, 355–361. [Google Scholar] [CrossRef]
- Caballero-Garrido, E.; Pena-Philippides, J.C.; Galochkina, Z.; Erhardt, E.; Roitbak, T. Characterization of long-term gait deficits in mouse dMCAO, using the CatWalk system. Behav. Brain Res. 2017, 331, 282–296. [Google Scholar] [CrossRef] [PubMed]
- Simoes, G.F.; Benitez, S.U.; Oliveira, A.L. Granulocyte colony-stimulating factor (G-CSF) positive effects on muscle fiber degeneration and gait recovery after nerve lesion in MDX mice. Brain Behav. 2014, 4, 738–753. [Google Scholar] [CrossRef]
- Hamers, F.P.; Koopmans, G.C.; Joosten, E.A. CatWalk-assisted gait analysis in the assessment of spinal cord injury. J. Neurotrauma 2006, 23, 537–548. [Google Scholar] [CrossRef]
- Deumens, R.; Jaken, R.J.; Marcus, M.A.; Joosten, E.A. The CatWalk gait analysis in assessment of both dynamic and static gait changes after adult rat sciatic nerve resection. J. Neurosci. Methods 2007, 164, 120–130. [Google Scholar] [CrossRef]
- Wooley, C.M.; Sher, R.B.; Kale, A.; Frankel, W.N.; Cox, G.A.; Seburn, K.L. Gait analysis detects early changes in transgenic SOD1(G93A) mice. Muscle Nerve 2005, 32, 43–50. [Google Scholar] [CrossRef] [Green Version]
- Amende, I.; Kale, A.; McCue, S.; Glazier, S.; Morgan, J.P.; Hampton, T.G. Gait dynamics in mouse models of Parkinson’s disease and Huntington’s disease. J. Neuroeng. Rehabil. 2005, 2, 20. [Google Scholar] [CrossRef] [Green Version]
- Carter, R.J.; Lione, L.A.; Humby, T.; Mangiarini, L.; Mahal, A.; Bates, G.P.; Dunnett, S.B.; Morton, A.J. Characterization of progressive motor deficits in mice transgenic for the human Huntington’s disease mutation. J. Neurosci. Off. J. Soc. Neurosci. 1999, 19, 3248–3257. [Google Scholar] [CrossRef] [Green Version]
- Domanskyi, A.; Geissler, C.; Vinnikov, I.A.; Alter, H.; Schober, A.; Vogt, M.A.; Gass, P.; Parlato, R.; Schutz, G. Pten ablation in adult dopaminergic neurons is neuroprotective in Parkinson’s disease models. FASEB J. 2011, 25, 2898–2910. [Google Scholar] [CrossRef] [PubMed]
- Glajch, K.E.; Fleming, S.M.; Surmeier, D.J.; Osten, P. Sensorimotor assessment of the unilateral 6-hydroxydopamine mouse model of Parkinson’s disease. Behav. Brain Res. 2012, 230, 309–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benchaouir, R.; Meregalli, M.; Farini, A.; D’Antona, G.; Belicchi, M.; Goyenvalle, A.; Battistelli, M.; Bresolin, N.; Bottinelli, R.; Garcia, L.; et al. Restoration of human dystrophin following transplantation of exon-skipping-engineered DMD patient stem cells into dystrophic mice. Cell Stem Cell 2007, 1, 646–657. [Google Scholar] [CrossRef] [Green Version]
- Puzzo, D.; Raiteri, R.; Castaldo, C.; Capasso, R.; Pagano, E.; Tedesco, M.; Gulisano, W.; Drozd, L.; Lippiello, P.; Palmeri, A.; et al. CL316,243, a beta3-adrenergic receptor agonist, induces muscle hypertrophy and increased strength. Sci. Rep. 2016, 5, 37504. [Google Scholar] [CrossRef] [Green Version]
- Waning, D.L.; Mohammad, K.S.; Reiken, S.; Xie, W.; Andersson, D.C.; John, S.; Chiechi, A.; Wright, L.E.; Umanskaya, A.; Niewolna, M.; et al. Excess TGF-beta mediates muscle weakness associated with bone metastases in mice. Nat. Med. 2015, 21, 1262–1271. [Google Scholar] [CrossRef]
- Yue, F.; Bi, P.; Wang, C.; Li, J.; Liu, X.; Kuang, S. Conditional Loss of Pten in Myogenic Progenitors Leads to Postnatal Skeletal Muscle Hypertrophy but Age-Dependent Exhaustion of Satellite Cells. Cell Rep. 2016, 17, 2340–2353. [Google Scholar] [CrossRef] [Green Version]
- Gadalla, K.K.; Ross, P.D.; Riddell, J.S.; Bailey, M.E.; Cobb, S.R. Gait analysis in a Mecp2 knockout mouse model of Rett syndrome reveals early-onset and progressive motor deficits. PLoS ONE 2014, 9, e112889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furman, S.; Hill, J.M.; Vulih, I.; Zaltzman, R.; Hauser, J.M.; Brenneman, D.E.; Gozes, I. Sexual dimorphism of activity-dependent neuroprotective protein in the mouse arcuate nucleus. Neurosci. Lett. 2005, 373, 73–78. [Google Scholar] [CrossRef]
- Kapitansky, O.; Giladi, E.; Jaljuli, I.; Bereswill, S.; Heimesaat, M.M.; Gozes, I. Microbiota changes associated with ADNP deficiencies: Rapid indicators for NAP (CP201) treatment of the ADNP syndrome and beyond. J. Neural Transm. 2020, 127, 251–263. [Google Scholar] [CrossRef] [Green Version]
- Meyfour, A.; Ansari, H.; Pahlavan, S.; Mirshahvaladi, S.; Rezaei-Tavirani, M.; Gourabi, H.; Baharvand, H.; Salekdeh, G.H. Y Chromosome Missing Protein, TBL1Y, May Play an Important Role in Cardiac Differentiation. J. Proteome Res. 2017, 16, 4391–4402. [Google Scholar] [CrossRef] [PubMed]
- Boxer, A.L.; Lang, A.E.; Grossman, M.; Knopman, D.S.; Miller, B.L.; Schneider, L.S.; Doody, R.S.; Lees, A.; Golbe, L.I.; Williams, D.R.; et al. Davunetide in patients with progressive supranuclear palsy: A randomised, double-blind, placebo-controlled phase 2/3 trial. Lancet Neurol. 2014, 13, 676–685. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Brown Adipose | Pancreas | Large Intestine | Lung | |||||
|---|---|---|---|---|---|---|---|---|
| Cell type | Mesenchymal stem cell | Endothelial cell | Polypeptide cell | Goblet cell | Epithelial cell | bronchial smooth muscle cell | ||
| Gene | Dmd | C9orf72 | Cnbp | Fkrp | Ttn | Gaa | Fkrp | Dysf |
| Adnp correlation (young) | 0.539 | 0.643 | 0.547 | 0.402 | 0.392 | 0.545 | 0.577 | 0.383 |
| Adnp correlation (old) | 0 | 0 | 0 | 0 | 0 | 0 | 0.497 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kapitansky, O.; Karmon, G.; Sragovich, S.; Hadar, A.; Shahoha, M.; Jaljuli, I.; Bikovski, L.; Giladi, E.; Palovics, R.; Iram, T.; et al. Single Cell ADNP Predictive of Human Muscle Disorders: Mouse Knockdown Results in Muscle Wasting. Cells 2020, 9, 2320. https://doi.org/10.3390/cells9102320
Kapitansky O, Karmon G, Sragovich S, Hadar A, Shahoha M, Jaljuli I, Bikovski L, Giladi E, Palovics R, Iram T, et al. Single Cell ADNP Predictive of Human Muscle Disorders: Mouse Knockdown Results in Muscle Wasting. Cells. 2020; 9(10):2320. https://doi.org/10.3390/cells9102320
Chicago/Turabian StyleKapitansky, Oxana, Gidon Karmon, Shlomo Sragovich, Adva Hadar, Meishar Shahoha, Iman Jaljuli, Lior Bikovski, Eliezer Giladi, Robert Palovics, Tal Iram, and et al. 2020. "Single Cell ADNP Predictive of Human Muscle Disorders: Mouse Knockdown Results in Muscle Wasting" Cells 9, no. 10: 2320. https://doi.org/10.3390/cells9102320