Gut Microbiome in Children from Indigenous and Urban Communities in México: Different Subsistence Models, Different Microbiomes

 ,
,  , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Site
2.2. Sample Collection
2.3. Fecal DNA Extraction
2.4. 16S rRNA Gene Amplification and Sequencing
2.5. Analysis of the Sequence Data
2.6. Statistics Analyses
2.7. Ethics
3. Results
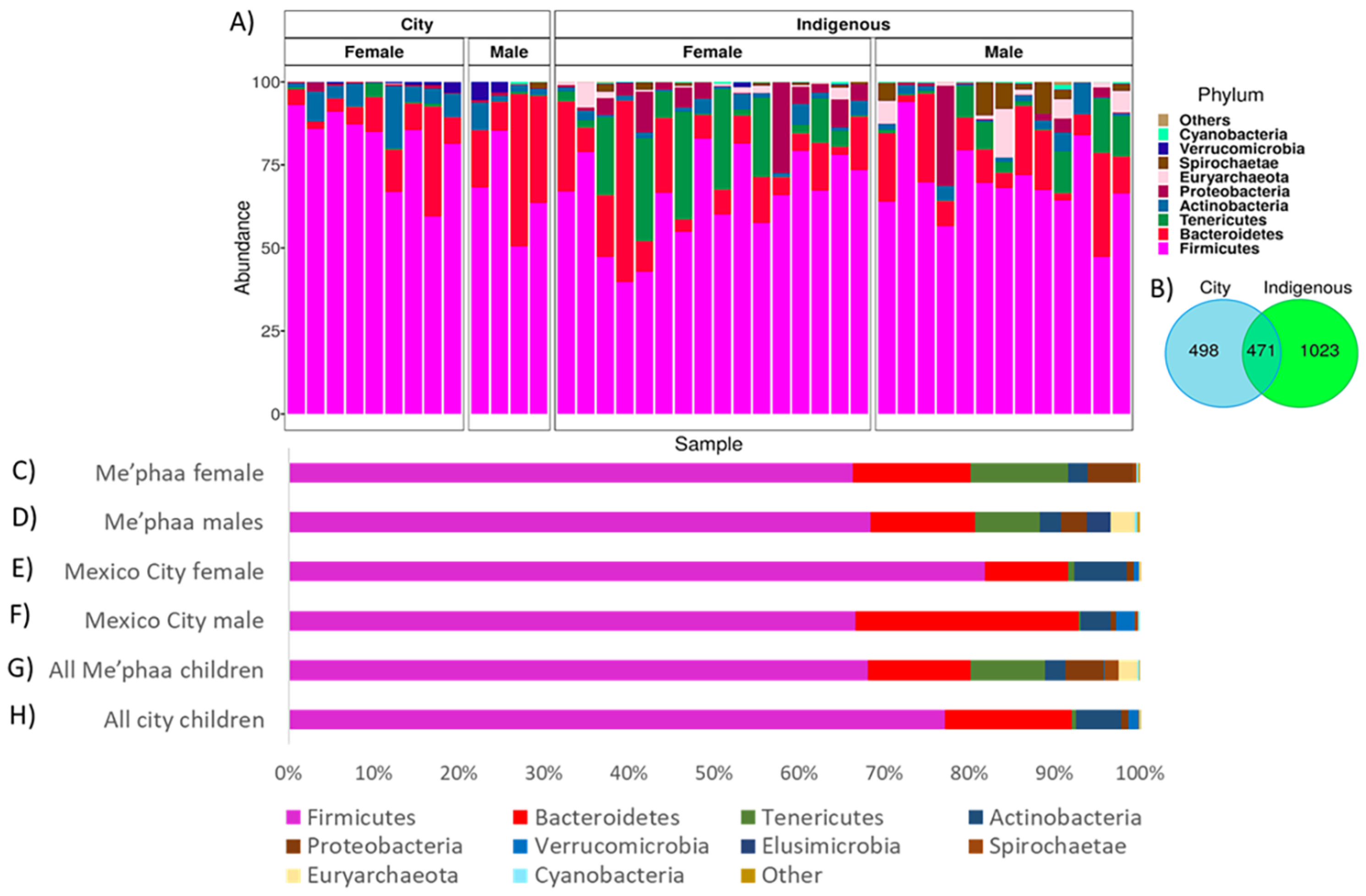
3.1. Microbial Taxonomic Comparison from México City and Me’phaa Communities
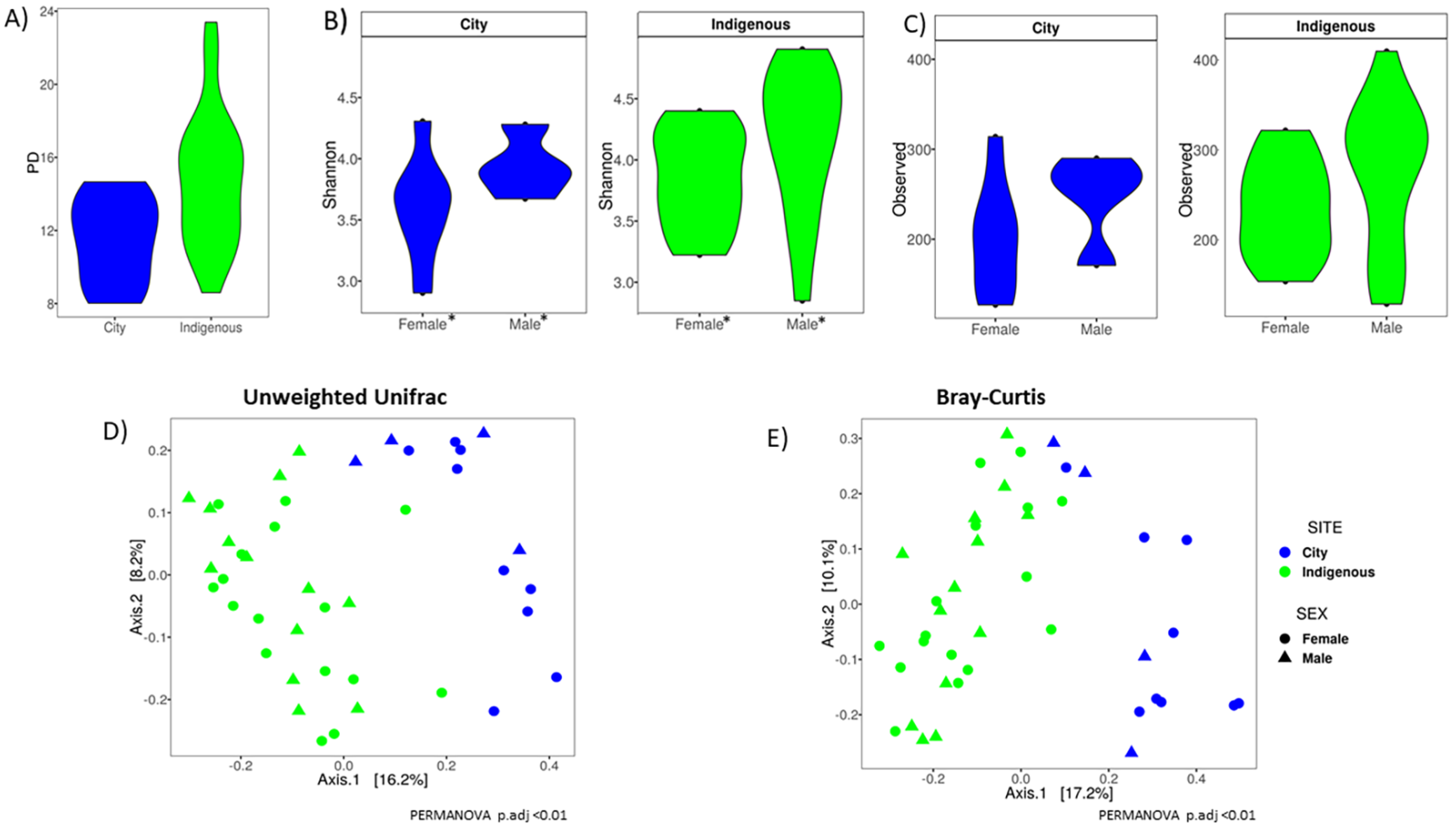
3.2. Alpha Diversity, Clustering Ordination, and Family/Genus Comparison of GM in Children from México City and Me’phaa Communities
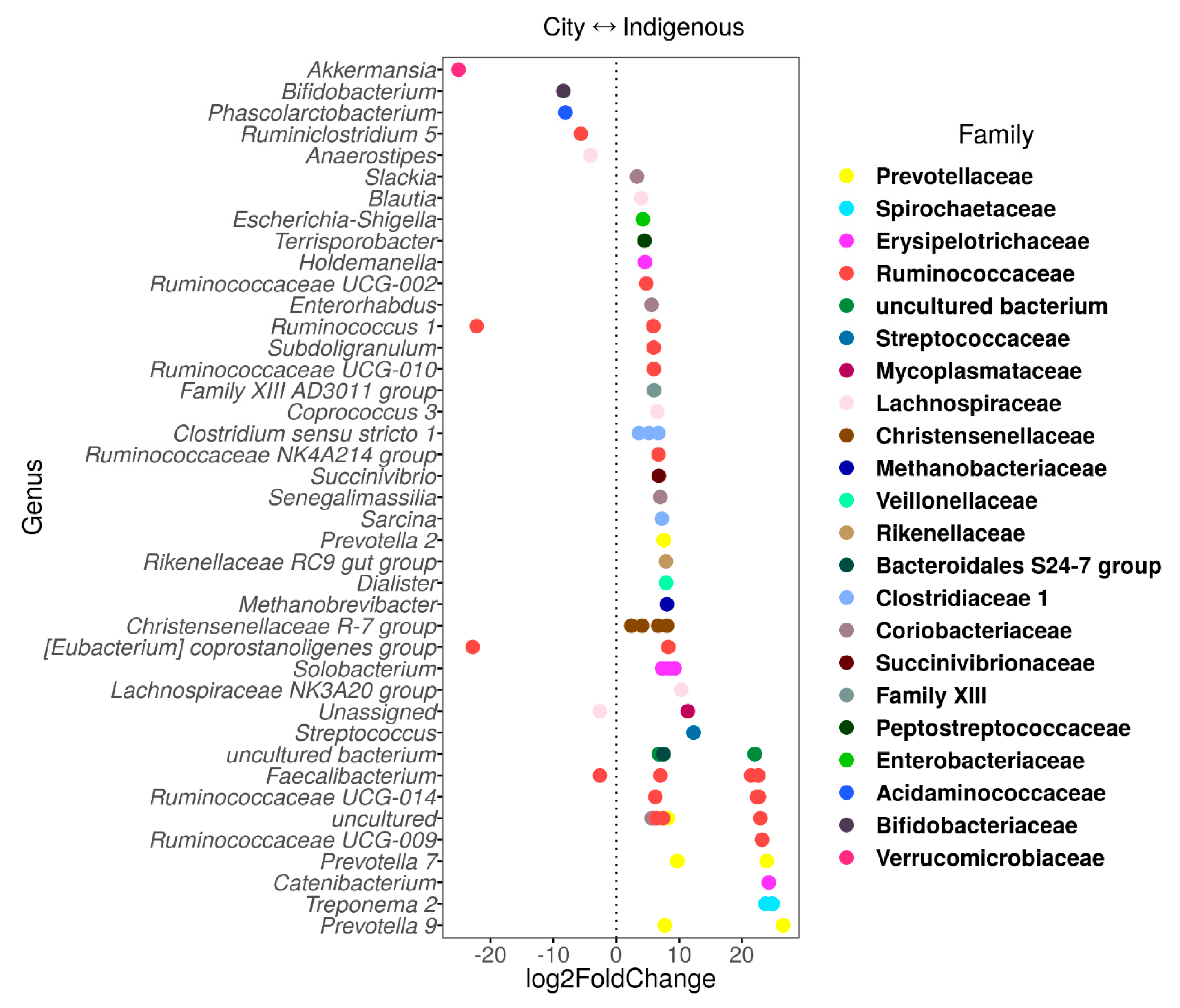
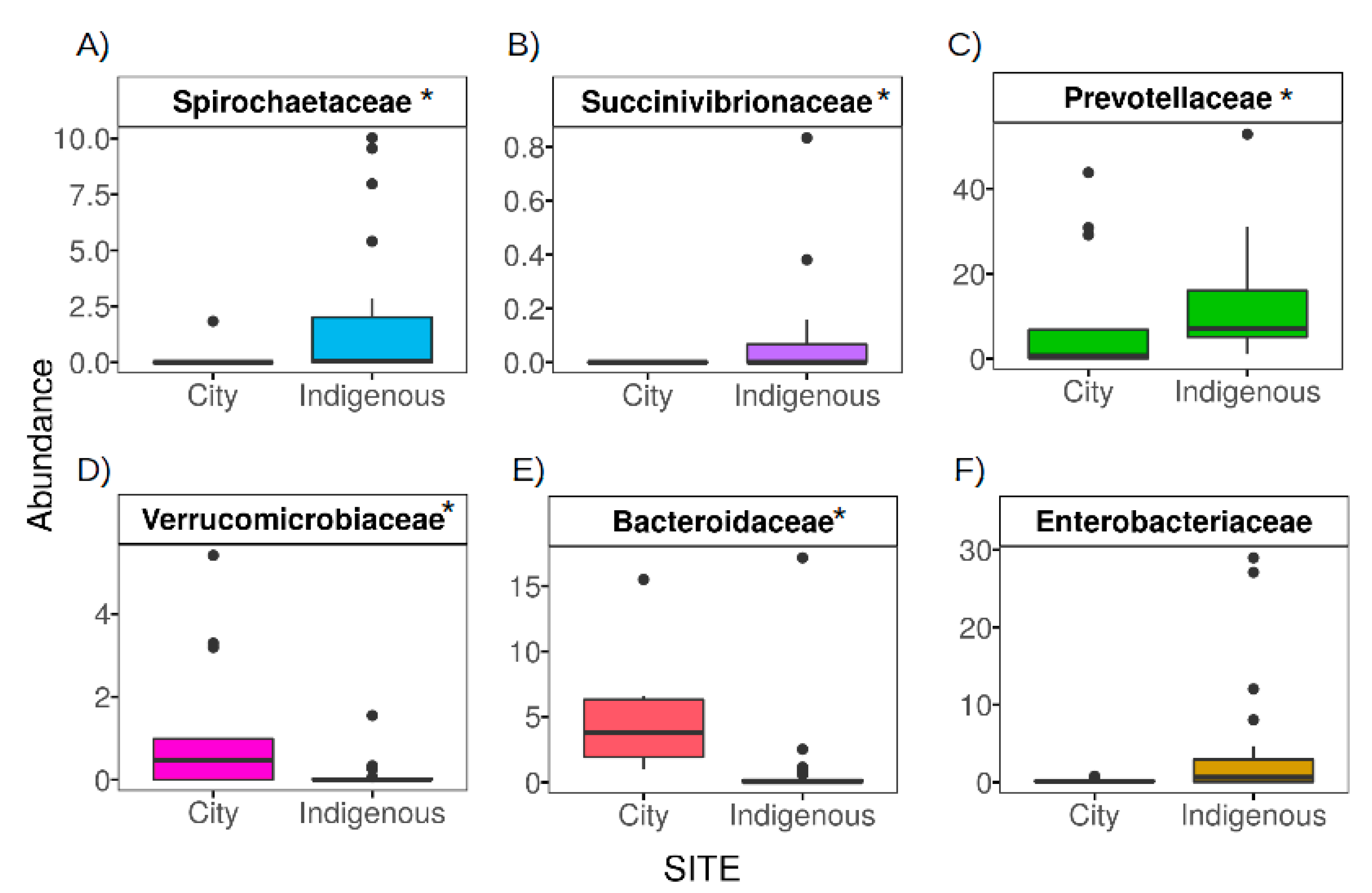
3.3. The Contrasting Relationship between VANISH and BloSSUM Groups between Indigenous and City Children
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sommer, F.; Bäckhed, F. The Gut Microbiota—Masters of Host Development and Physiology. Nat. Rev. Genet. 2013, 11, 227–238. [Google Scholar] [CrossRef]
- Rojo, D.; Méndez-García, C.; Raczkowska, B.A.; Bargiela, R.; Moya, A.; Ferrer, M.; Barbas, C. Exploring the Human Microbiome from Multiple Perspectives: Factors Altering Its Composition and Function. FEMS Microbiol. Rev. 2017, 41, 453–478. [Google Scholar] [CrossRef] [Green Version]
- Negi, S.; Das, D.K.; Pahari, S.; Nadeem, S.; Agrewala, J.N. Potential Role of Gut Microbiota in Induction and Regulation of Innate Immune Memory. Front. Immunol. 2019, 10, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guarner, F.; Malagelada, J.-R. Gut Flora in Health and Disease. Lancet 2003, 361, 512–519. [Google Scholar] [CrossRef]
- Ghosh, T.S.; Gupta, S.S.; Bhattacharya, T.; Yadav, D.; Barik, A.; Chowdhury, A.; Das, B.; Mande, S.S.; Nair, G.B. Gut Microbiomes of Indian Children of Varying Nutritional Status. PLoS ONE 2014, 9, e95547. [Google Scholar] [CrossRef] [PubMed]
- Moens, E.; Veldhoen, M. Epithelial Barrier Biology: Good Fences Make Good Neighbours. Immunology 2011, 135, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Moya, A.; Ferrer, M. Functional Redundancy-Induced Stability of Gut Microbiota Subjected to Disturbance. Trends Microbiol. 2016, 24, 402–413. [Google Scholar] [CrossRef] [PubMed]
- Ramírez-Carrillo, E.; Gaona, O.; Nieto, J.; Sánchez-Quinto, A.; Cerqueda-García, D.; Falcon, L.I.; Rojas-Ramos, O.A.; González-Santoyo, I. Disturbance in Human Gut Microbiota Networks by Parasites and Its Implications in the Incidence of Depression. Sci. Rep. 2020, 10, 3680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falony, G.; Joossens, M.; Vieira-Silva, S.; Wang, J.; Darzi, Y.; Faust, K.; Kurilshikov, A.; Bonder, M.J.; Valles-Colomer, M.; Vandeputte, D.; et al. Population-Level Analysis of Gut Microbiome Variation. Science 2016, 352, 560–564. [Google Scholar] [CrossRef]
- Ursell, L.K.; Metcalf, J.L.; Parfrey, L.W.; Knight, R. Defining the Human Microbiome. Nutr. Rev. 2012, 70, S38–S44. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, E.; Zilber-Rosenberg, I. The Hologenome Concept of Evolution: Medical Implications. Rambam Maimonides Med. J. 2019, 10, e0005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonnenburg, E.D.; Sonnenburg, J.L. The Ancestral and Industrialized Gut Microbiota and Implications for Human Health. Nat. Rev. Genet. 2019, 17, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Hollister, E.B.; Riehle, K.; Luna, R.A.; Weidler, E.M.; Rubio-Gonzales, M.; Mistretta, T.-A.; Raza, S.; Doddapaneni, H.V.; Metcalf, G.A.; Muzny, D.M.; et al. Structure and Function of the Healthy Pre-Adolescent Pediatric Gut Microbiome. Microbiome 2015, 3, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmer, C.; Bik, E.M.; DiGiulio, D.B.; Relman, D.A.; Brown, P.O. Development of the Human Infant Intestinal Microbiota. PLoS Biol. 2007, 5, e177. [Google Scholar] [CrossRef] [Green Version]
- Koenig, J.E.; Spor, A.; Scalfone, N.; Fricker, A.D.; Stombaugh, J.; Knight, R.; Angenent, L.T.; Ley, R. Succession of Microbial Consortia in the Developing Infant Gut Microbiome. Proc. Natl. Acad. Sci. USA 2010, 108, 4578–4585. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez, J.M.; Murphy, K.; Stanton, C.; Ross, R.P.; Kober, O.I.; Juge, N.; Avershina, E.; Rudi, K.; Narbad, A.; Jenmalm, M.C.; et al. The Composition of the Gut Microbiota Throughout Life, with an Emphasis on Early Life. Microb. Ecol. Health Dis. 2015, 26, 75. [Google Scholar] [CrossRef]
- Borre, Y.E.; O’Keeffe, G.W.; Clarke, G.; Stanton, C.; Dinan, T.G.; Cryan, J.F. Microbiota and Neurodevelopmental Windows: Implications for Brain Disorders. Trends Mol. Med. 2014, 20, 509–518. [Google Scholar] [CrossRef]
- De Filippo, C.; Cavalieri, D.; Di Paola, M.; Ramazzotti, M.; Poullet, J.B.; Massart, S.; Collini, S.; Pieraccini, G.; Lionetti, P. Impact of Diet in Shaping Gut Microbiota Revealed by a Comparative Study in Children from Europe and Rural Africa. Proc. Natl. Acad. Sci. USA 2010, 107, 14691–14696. [Google Scholar] [CrossRef] [Green Version]
- Agans, R.; Rigsbee, L.; Kenche, H.; Michail, S.; Khamis, H.J.; Paliy, O. Distal Gut Microbiota of Adolescent Children Is Different from That of Adults. FEMS Microbiol. Ecol. 2011, 77, 404–412. [Google Scholar] [CrossRef]
- Smits, S.A.; Leach, J.; Sonnenburg, E.D.; Gonzalez, C.G.; Lichtman, J.S.; Reid, G.; Knight, R.; Manjurano, A.; Changalucha, J.; Elias, J.E.; et al. Seasonal Cycling in the Gut Microbiome of the Hadza Hunter-Gatherers of Tanzania. Science 2017, 357, 802–806. [Google Scholar] [CrossRef] [Green Version]
- Ayeni, F.A.; Biagi, E.; Rampelli, S.; Fiori, J.; Soverini, M.; Audu, H.J.; Cristino, S.; Caporali, L.; Schnorr, S.L.; Carelli, V.; et al. Infant and Adult Gut Microbiome and Metabolome in Rural Bassa and Urban Settlers from Nigeria. Cell Rep. 2018, 23, 3056–3067. [Google Scholar] [CrossRef] [PubMed]
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P.; et al. Human Gut Microbiome Viewed across Age and Geography. Nature 2012, 486, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Jha, A.R.; Davenport, E.R.; Gautam, Y.; Bhandari, D.; Tandukar, S.; Ng, K.M.; Fragiadakis, G.K.; Holmes, S.; Gautam, G.P.; Leach, J.; et al. Gut Microbiome Transition across a Lifestyle Gradient in Himalaya. PLoS Biol. 2018, 16, e2005396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonnenburg, J.L.; Sonnenburg, E.D. Vulnerability of the Industrialized Microbiota. Science 2019, 366, eaaw9255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Instituto Nacional de los Pueblos Indígenas. 2019. Available online: https://www.gob.mx/inpi (accessed on 17 February 2020).
- Black, C.A. An Autosegmental Analysis of Me’phaa (Tlapanec) Noun Inflection. SIL México. 2004. Available online: https://mexico.sil.org/es/resources/archives/2860 (accessed on 17 February 2020).
- Camacho, Z. Montaña de Guerrero Pobreza y Militarización. Revista Contralínea. 2007. Available online: https://www.contralinea.com.mx/archivo/2007/enero/htm/montana_guerrero_militares.htm (accessed on 20 February 2020).
- Miramontes, O.; DeSouza, O.; Hernández, D.; Ceccon, E. Non-Lévy mobility patterns of Mexican Me’phaa peasants searching for fuel wood. Hum. Ecol. 2012, 40, 167–174. [Google Scholar] [CrossRef] [Green Version]
- Borda-Niño, M.; Santiago, M.C.; Hernández-Muciño, D.; Muciño-Muciño, M. Restauración Productiva en la Práctica: El caso de las Comunidades Indígenas Me’phaa de la Montaña de Guerrero. Available online: https://www.researchgate.net/profile/Diego_Hernandez-Mucino/publication/309681903_Restauracion_productiva_en_la_practica_el_caso_de_las_comunidades_indigenas_Me_Phaa_de_La_Montana_de_Guerrero_Mexico/links/581d04b608ae40da2cab4244/Restauracion-productiva-en-la-practica-el-caso-de-las-comunidades-indigenas-Me-Phaa-de-La-Montana-de-Guerrero-Mexico.pdf (accessed on 17 February 2020).
- United Nations Development Programme (PNUD). Índice de Desarrollo Humano Para las Entidades Federativas, México 2015. El PNUD en México. 2012. Available online: https://www.mx.undp.org/content/mexico/es/home/library/poverty/indice-de-desarrollo-humano-para-las-entidades-federativas--mexi.html (accessed on 17 February 2020).
- UNFPA. SWOP Report 2019. UNFPA—United Nations Population Fund. 2019. Available online: https://www.unfpa.org/es/swop-2019 (accessed on 18 February 2020).
- Allaband, C.; McDonald, D.; Vázquez-Baeza, Y.; Minich, J.J.; Tripathi, A.; Brenner, D.A.; Loomba, R.; Smarr, L.; Sandborn, W.J.; Schnabl, B.; et al. Microbiome 101: Studying, Analyzing, and Interpreting Gut Microbiome Data for Clinicians. Clin. Gastroenterol. Hepatol. 2018, 17, 218–230. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Lauber, C.L.; A Walters, W.; Berg-Lyons, D.; Huntley, J.; Fierer, N.; Owens, S.M.; Betley, J.; Fraser, L.; Bauer, M.; et al. Ultra-High-Throughput Microbial Community Analysis on the Illumina HiSeq and MiSeq Platforms. ISME J. 2012, 6, 1621–1624. [Google Scholar] [CrossRef] [Green Version]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; A Johnson, A.J.; Holmes, S. DADA2: High-Resolution Sample Inference from Illumina Amplicon Data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahe, F. VSEARCH: A Versatile Open Source Tool for Metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA Ribosomal RNA Gene Database Project: Improved Data Processing and Web-Based Tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef]
- Katoh, K.; Asimenos, G.; Toh, H. Multiple Alignment of DNA Sequences with MAFFT. Adv. Struct. Saf. Stud. 2009, 537, 39–64. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma Powers Differential Expression Analyses for RNA-Sequencing and Microarray Studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 31. [Google Scholar] [CrossRef] [Green Version]
- Holm, S. A simple sequentially rejective multiple test procedure. Scand. J. Stat. 1979, 6, 65–70. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing. 2018. Available online: https://www.gbif.org/es/tool/81287/r-a-language-and-environment-for-statistical-computing (accessed on 19 February 2020).
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Package ‘Vegan’ Title Community Ecology Package Version 2.5-6. 2019. Available online: https://cran.r-project.org/web/packages/vegan/vegan.pdf (accessed on 10 February 2020).
- Venables, W.N.; Ripley, B.D. Modern Applied Statistics with S; Springer: Cham, Switzerland, 2002. [Google Scholar] [CrossRef]
- Rigby, R.; Stasinopoulos, D.M. Generalized Additive Models for Location, Scale and Shape (with Discussion). J. R. Stat. Soc. Ser. C 2005, 54, 507–554. [Google Scholar] [CrossRef] [Green Version]
- Segata, N. Gut Microbiome: Westernization and the Disappearance of Intestinal Diversity. Curr. Biol. 2015, 25, R611–R613. [Google Scholar] [CrossRef] [Green Version]
- Clemente, J.C.; Pehrsson, E.C.; Blaser, M.J.; Sandhu, K.; Gao, Z.; Wang, B.; Magris, M.; Hidalgo, G.; Contreras, M.; Noya-Alarcón, Ó.; et al. The Microbiome of Uncontacted Amerindians. Sci. Adv. 2015, 1, e1500183. [Google Scholar] [CrossRef] [Green Version]
- Pontzer, H.; Raichlen, D.A.; Wood, B.M.; Mabulla, A.Z.P.; Racette, S.B.; Marlowe, F.W. Hunter-Gatherer Energetics and Human Obesity. PLoS ONE 2012, 7, e40503. [Google Scholar] [CrossRef]
- Anwesh, M.; Kumar, K.V.; Nagarajan, M.; Chander, M.P.; Kartick, C.; Paluru, V. Elucidating the Richness of Bacterial Groups in the Gut of Nicobarese Tribal Community—Perspective on Their Lifestyle Transition. Anaerobe 2016, 39, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Martínez, I.; Stegen, J.C.; Maldonado-Gomez, M.X.; Eren, A.M.; Siba, P.M.; Greenhill, A.R.; Walter, J. The Gut Microbiota of Rural Papua New Guineans: Composition, Diversity Patterns, and Ecological Processes. Cell Rep. 2015, 11, 527–538. [Google Scholar] [CrossRef] [Green Version]
- Martínez, I.; Lattimer, J.M.; Hubach, K.L.; Case, J.C.; Yang, J.; Weber, C.G.; Louk, J.A.; Rose, D.J.; Kyureghian, G.; Peterson, D.A.; et al. Gut Microbiome Composition Is Linked to Whole Grain-Induced Immunological Improvements. ISME J. 2012, 7, 269–280. [Google Scholar] [CrossRef] [Green Version]
- Sarmiento-Silva, S. Tlapanecos de Guerrero. Proyecto Perfiles Indígenas de México. Documento de Trabajo. 2001. Available online: https://www.aacademica.org/salomon.nahmad.sitton/58.pdf (accessed on 12 February 2020).
- Schnorr, S.L.; Candela, M.; Rampelli, S.; Centanni, M.; Consolandi, C.; Basaglia, G.; Turroni, S.; Biagi, E.; Peano, C.; Severgnini, M.; et al. Gut Microbiome of the Hadza Hunter-Gatherers. Nat. Commun. 2014, 5, 3654. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Zhang, M.; Pang, X.; Zhao, Y.; Wang, L.; Zhao, L. Structural Resilience of the Gut Microbiota in Adult Mice under High-Fat Dietary Perturbations. ISME J. 2012, 6, 1848–1857. [Google Scholar] [CrossRef] [PubMed]
- Murphy, E.A.; Velázquez, K.T.; Herbert, K.M. Influence of High-Fat Diet on Gut Microbiota. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 515–520. [Google Scholar] [CrossRef]
- Alou, M.T.; Lagier, J.-C.; Raoult, D. Diet Influence on the Gut Microbiota and Dysbiosis Related to Nutritional Disorders. Hum. Microbiome J. 2016, 1, 3–11. [Google Scholar] [CrossRef] [Green Version]
- Arumugam, M.; Raes, J.; Pelletier, E.; Le Paslier, D.; Yamada, T.; Mende, D.R.; Fernandes, G.R.; Tap, J.; Bruls, T.; Batto, J.-M.; et al. Enterotypes of the Human Gut Microbiome. Nature 2011, 473, 174–180. [Google Scholar] [CrossRef]
- Méndez-Salazar, E.O.; Ortiz-López, M.G.; Granados-Silvestre, M.D.L.Á.; Palacios-González, B.; Menjivar, M. Altered Gut Microbiota and Compositional Changes in Firmicutes and Proteobacteria in Mexican Undernourished and Obese Children. Front. Microbiol. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Morán-Ramos, S.; Lopez-Contreras, B.E.; Vela-Amieva, M.; Gomez-Perez, F.J.; Velázquez-Cruz, R.; Salmerón, J.; Reyes-Castillo, Z.; Aguilar-Salinas, C.A.; Canizales-Quinteros, S.; Villarruel-Vazquez, R.; et al. Environmental and Intrinsic Factors Shaping Gut Microbiota Composition and Diversity and Its Relation to Metabolic Health in Children and Early Adolescents: A Population-Based Study. Gut Microbes 2020, 11, 900–917. [Google Scholar] [CrossRef]
- Cayrou, C.; Sambe, B.; Armougom, F.; Raoult, D.; Drancourt, M. Molecular Diversity of the Planctomycetesin the Human Gut Microbiota in France and Senegal. APMIS 2013, 121, 1082–1090. [Google Scholar] [CrossRef]
- Van Niftrik, L.; Devos, D.P. Editorial: Planctomycetes-Verrucomicrobia-Chlamydiae Bacterial Superphylum: New Model Organisms for Evolutionary Cell Biology. Front. Microbiol. 2017, 8. [Google Scholar] [CrossRef]
- Angelakis, E.; Bachar, D.; Yasir, M.; Musso, D.; Djossou, F.; Gaborit, B.; Brah, S.; Diallo, A.; Ndombe, G.M.; Mediannikov, O.; et al. Treponema Species Enrich the Gut Microbiota of Traditional Rural Populations but Are Absent from Urban Individuals. New Microbes New Infect. 2018, 27, 14–21. [Google Scholar] [CrossRef]
- Youssef, N.H.; Farag, I.F.; Rinke, C.; Hallam, S.J.; Woyke, T.; Elshahed, M.S. In Silico Analysis of the Metabolic Potential and Niche Specialization of Candidate Phylum Latescibacteria (WS3). PLoS ONE 2015, 10, e0127499. [Google Scholar] [CrossRef]
- Momper, L.; Aronson, H.S.; Amend, J.P. Genomic Description of Candidatus Abyssubacteria, a Novel Subsurface Lineage within the Candidate Phylum Hydrogenedentes. Front. Microbiol. 2018, 9. [Google Scholar] [CrossRef]
- Rahman, N.A.; Parks, D.H.; VanWonterghem, I.; Morrison, M.; Tyson, G.W.; Hugenholtz, P. A Phylogenomic Analysis of the Bacterial Phylum Fibrobacteres. Front. Microbiol. 2016, 6. [Google Scholar] [CrossRef] [Green Version]
- Kindaichi, T.; Yamaoka, S.; Uehara, R.; Ozaki, N.; Ohashi, A.; Albertsen, M.; Nielsen, J.L. Phylogenetic Diversity and Ecophysiology of Candidate Phylum Saccharibacteria in Activated Sludge. FEMS Microbiol. Ecol. 2016, 92, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Bervoets, L.; Van Hoorenbeeck, K.; Kortleven, I.; Van Noten, C.; Hens, N.; Vael, C.; Goossens, H.; Desager, K.N.; Vankerckhoven, V. Differences in Gut Microbiota Composition Between Obese and Lean Children: A Cross-Sectional Study. Gut Pathog. 2013, 5, 10. [Google Scholar] [CrossRef] [Green Version]
- Riva, A.; Borgo, F.C.; Lassandro, C.; Verduci, E.; Morace, G.; Borghi, E.; Berry, D. Pediatric Obesity Is Associated with an Altered Gut Microbiota and Discordant Shifts in F Irmicutes Populations. Environ. Microbiol. 2016, 19, 95–105. [Google Scholar] [CrossRef]
- Bai, J.; Hu, Y.; Bruner, D.W. Composition of Gut Microbiota and Its Association with Body Mass Index and Lifestyle Factors in a Cohort of 7-18 Years Old Children from the American Gut Project. Pediatr. Obes. 2018, 14, e12480. [Google Scholar] [CrossRef]
- Indiani, C.M.D.S.P.; Rizzardi, K.F.; Castelo, P.M.; Ferraz, L.F.C.; Darrieux, M.; Parisotto, T.M. Childhood Obesity and Firmicutes/Bacteroidetes Ratio in the Gut Microbiota: A Systematic Review. Child. Obes. 2018, 14, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Waters, J.L.; Ley, R. The Human Gut Bacteria Christensenellaceae Are Widespread, Heritable, and Associated with Health. BMC Biol. 2019, 17, 83. [Google Scholar] [CrossRef] [PubMed]
- Cirstea, M.; Radisavljevic, N.; Finlay, B.B. Good Bug, Bad Bug: Breaking through Microbial Stereotypes. Cell Host Microbe 2018, 23, 10–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gareau, M.; Silva, M.; Perdue, M. Pathophysiological Mechanisms of Stress-Induced Intestina Damage. Curr. Mol. Med. 2008, 8, 274–281. [Google Scholar] [CrossRef]
- Gomez, A.; Petrzelkova, K.J.; Burns, M.B.; Yeoman, C.J.; Amato, K.R.; Vlčková, K.; Modry, D.; Todd, A.; Robinson, C.A.J.; Remis, M.J.; et al. Gut Microbiome of Coexisting BaAka Pygmies and Bantu Reflects Gradients of Traditional Subsistence Patterns. Cell Rep. 2016, 14, 2142–2153. [Google Scholar] [CrossRef] [Green Version]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet Rapidly and Reproducibly Alters the Human Gut Microbiome. Nature 2013, 505, 559–563. [Google Scholar] [CrossRef] [Green Version]
- Turnbaugh, P.J.; Ridaura, V.K.; Faith, J.J.; Rey, F.E.; Knight, R.; Gordon, J.I. The Effect of Diet on the Human Gut Microbiome: A Metagenomic Analysis in Humanized Gnotobiotic Mice. Sci. Transl. Med. 2009, 1, 6ra14. [Google Scholar] [CrossRef] [Green Version]
- Cantarel, B.L.; Lombard, V.; Henrissat, B. Complex Carbohydrate Utilization by the Healthy Human Microbiome. PLoS ONE 2012, 7, e28742. [Google Scholar] [CrossRef] [Green Version]
- El Kaoutari, A.; Armougom, F.; Gordon, J.I.; Raoult, D.; Henrissat, B. The Abundance and Variety of Carbohydrate-Active Enzymes in the Human Gut Microbiota. Nat. Rev. Genet. 2013, 11, 497–504. [Google Scholar] [CrossRef]
- Horwood, P.F.; Tarantola, A.; Goarant, C.; Matsui, M.; Klement, E.; Umezaki, M.; Navarro, S.; Greenhill, A.R. Health Challenges of the Pacific Region: Insights from History, Geography, Social Determinants, Genetics, and the Microbiome. Front. Immunol. 2019, 10, 10. [Google Scholar] [CrossRef]
- Zmora, N.; Zeevi, D.; Korem, T.; Segal, E.; Elinav, E. Taking It Personally: Personalized Utilization of the Human Microbiome in Health and Disease. Cell Host Microbe 2016, 19, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Instituto Mexicano del Seguro Social. Guía Para el Cuidado de la Salud. 2018. Available online: http://www.imss.gob.mx/sites/all/statics/salud/guias_salud/2018/guia-salud-ninas-ninos-2018.pdf (accessed on 15 February 2020).





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sánchez-Quinto, A.; Cerqueda-García, D.; Falcón, L.I.; Gaona, O.; Martínez-Correa, S.; Nieto, J.; G-Santoyo, I. Gut Microbiome in Children from Indigenous and Urban Communities in México: Different Subsistence Models, Different Microbiomes. Microorganisms 2020, 8, 1592. https://doi.org/10.3390/microorganisms8101592
Sánchez-Quinto A, Cerqueda-García D, Falcón LI, Gaona O, Martínez-Correa S, Nieto J, G-Santoyo I. Gut Microbiome in Children from Indigenous and Urban Communities in México: Different Subsistence Models, Different Microbiomes. Microorganisms. 2020; 8(10):1592. https://doi.org/10.3390/microorganisms8101592
Chicago/Turabian StyleSánchez-Quinto, Andrés, Daniel Cerqueda-García, Luisa I. Falcón, Osiris Gaona, Santiago Martínez-Correa, Javier Nieto, and Isaac G-Santoyo. 2020. "Gut Microbiome in Children from Indigenous and Urban Communities in México: Different Subsistence Models, Different Microbiomes" Microorganisms 8, no. 10: 1592. https://doi.org/10.3390/microorganisms8101592