Developing a Universal and Efficient Method for the Rapid Selection of Stable Fluorescent Protein-Tagged Pathogenic Vibrio Species


Abstract
:1. Introduction
2. Materials and Methods
2.1. Construction of RFP and eCFP Suicide Vectors
2.2. Plasmid Conjugation in Vibrio
2.3. PCR Confirmation of the Transconjugants
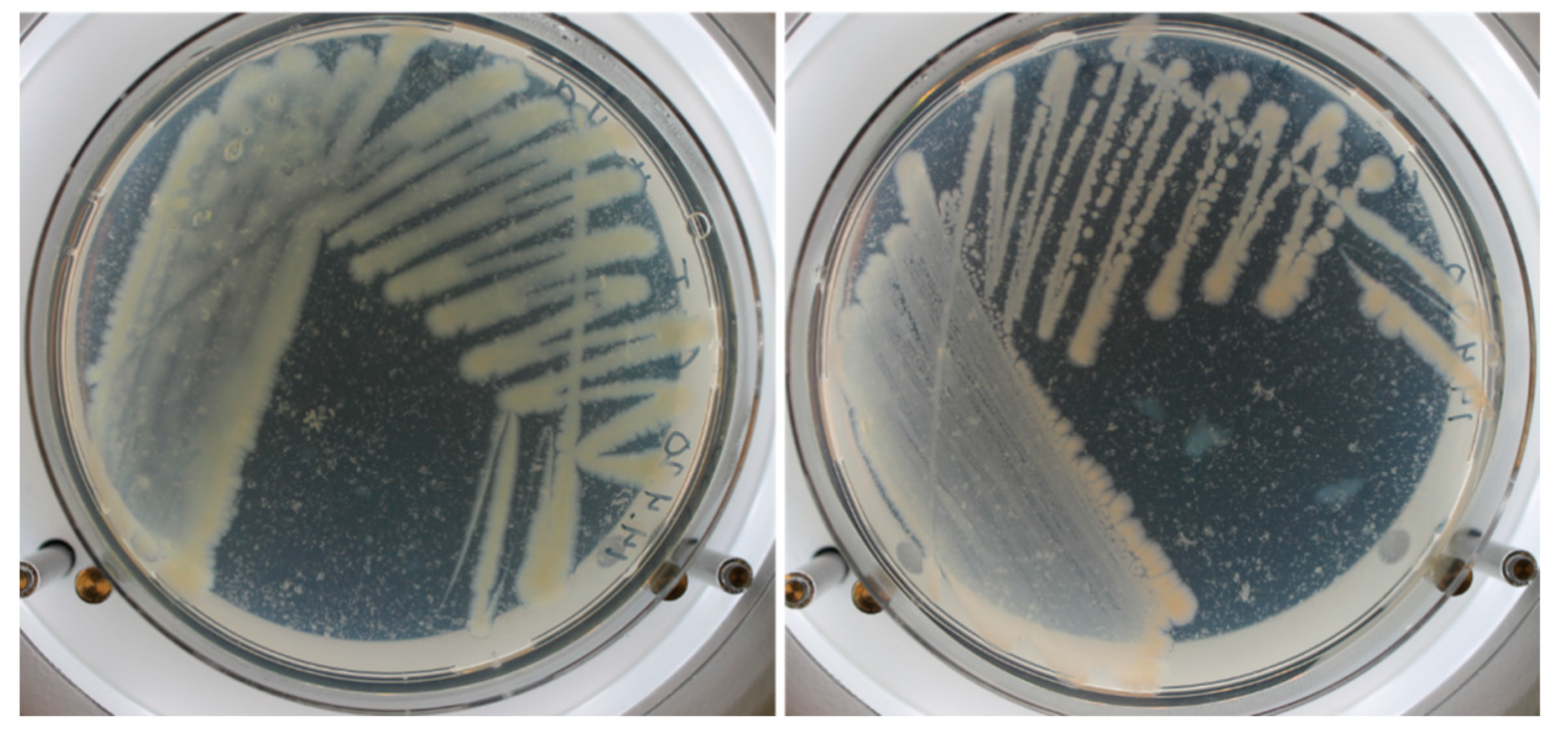
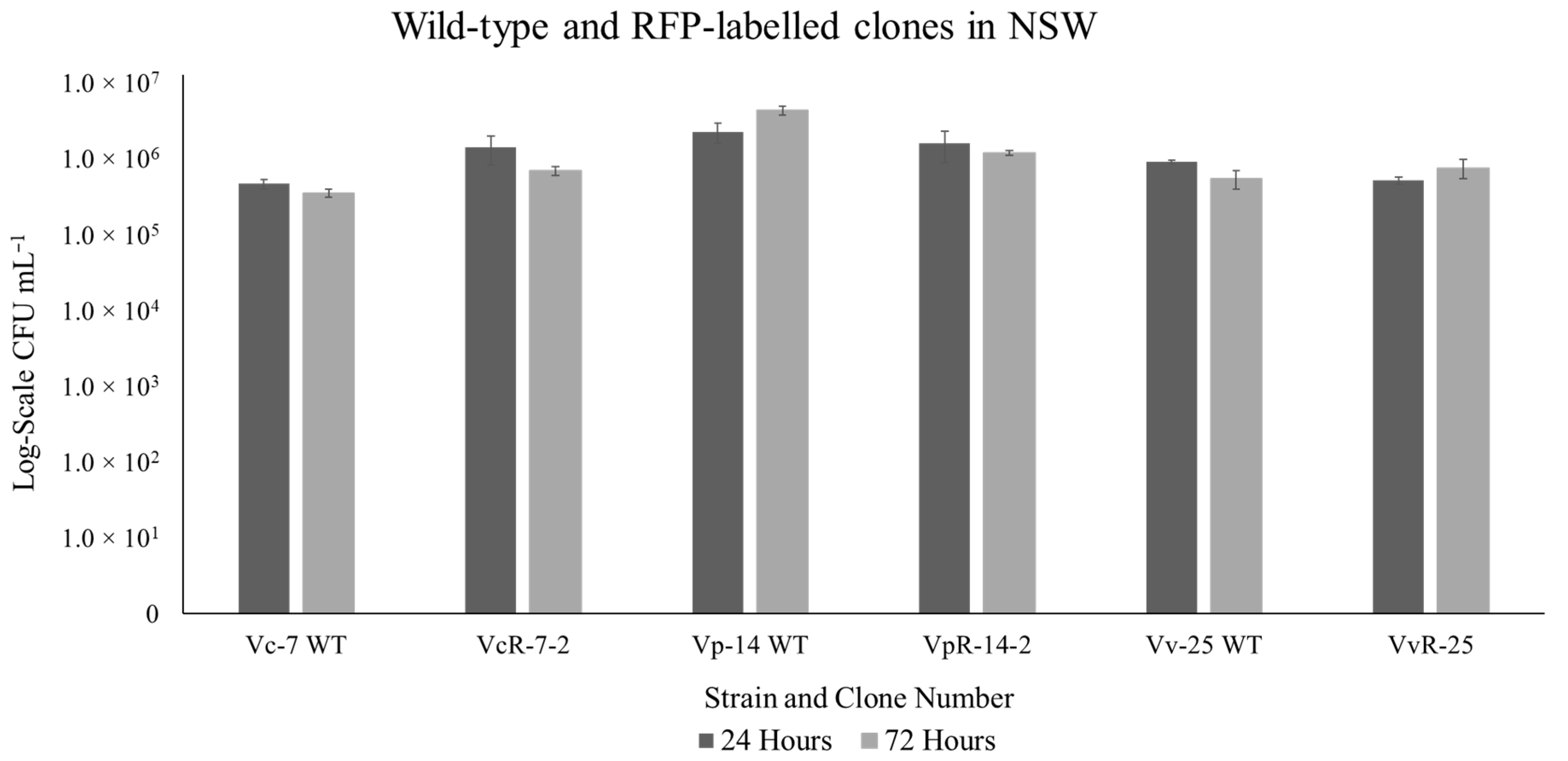
2.4. Comparative Analysis of RFP-Labelled Clones and the Wild-Type Strain
2.5. Determination of the Transposon Location in Selected Clones
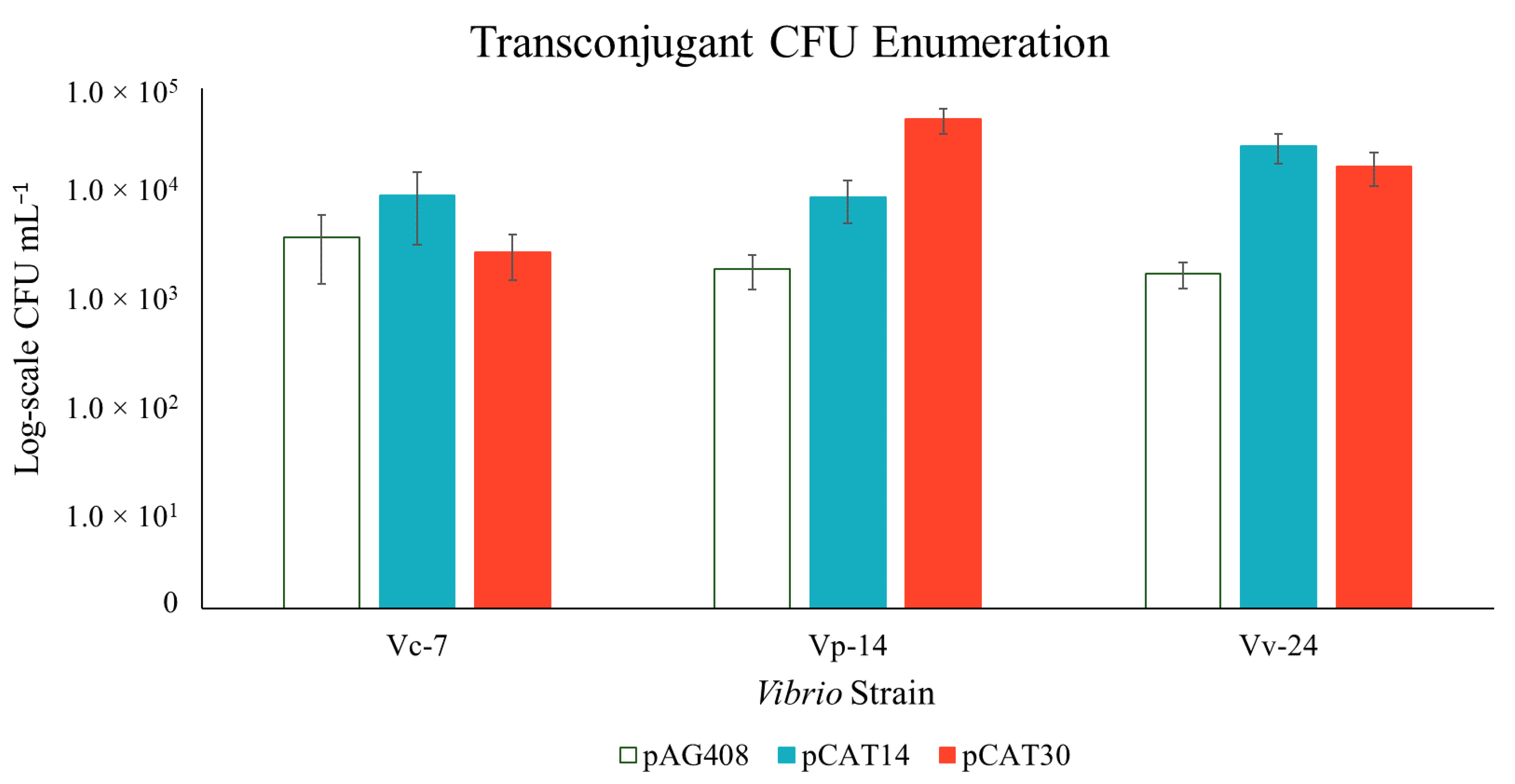
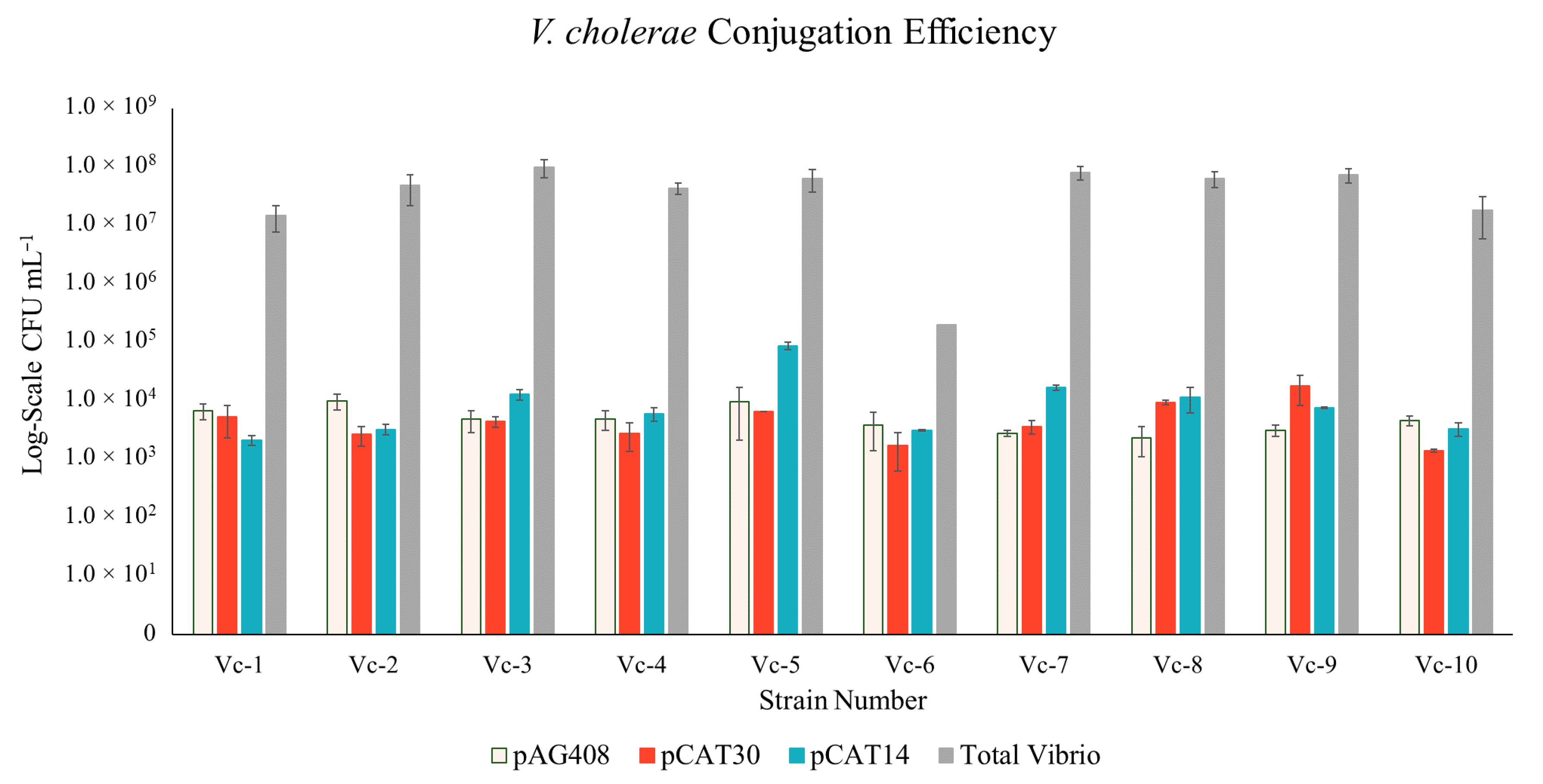
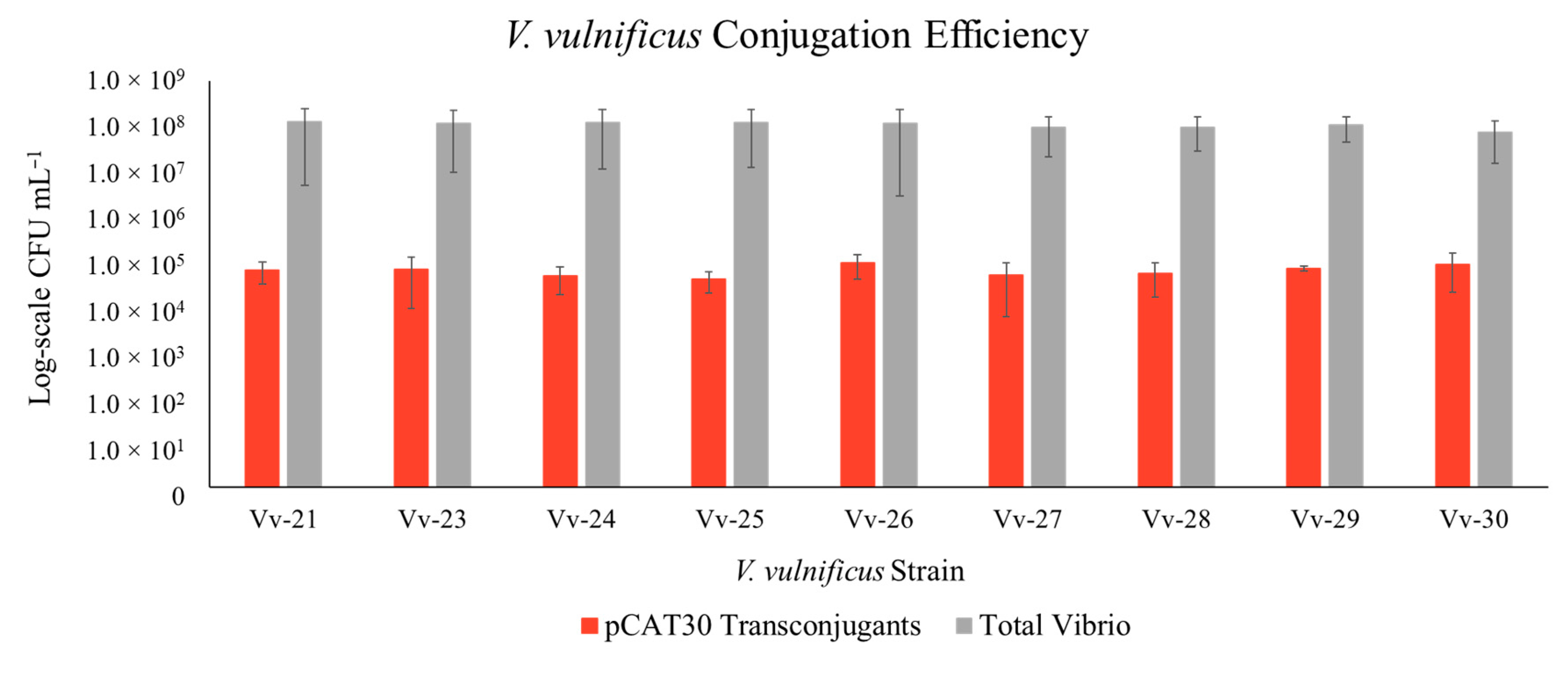
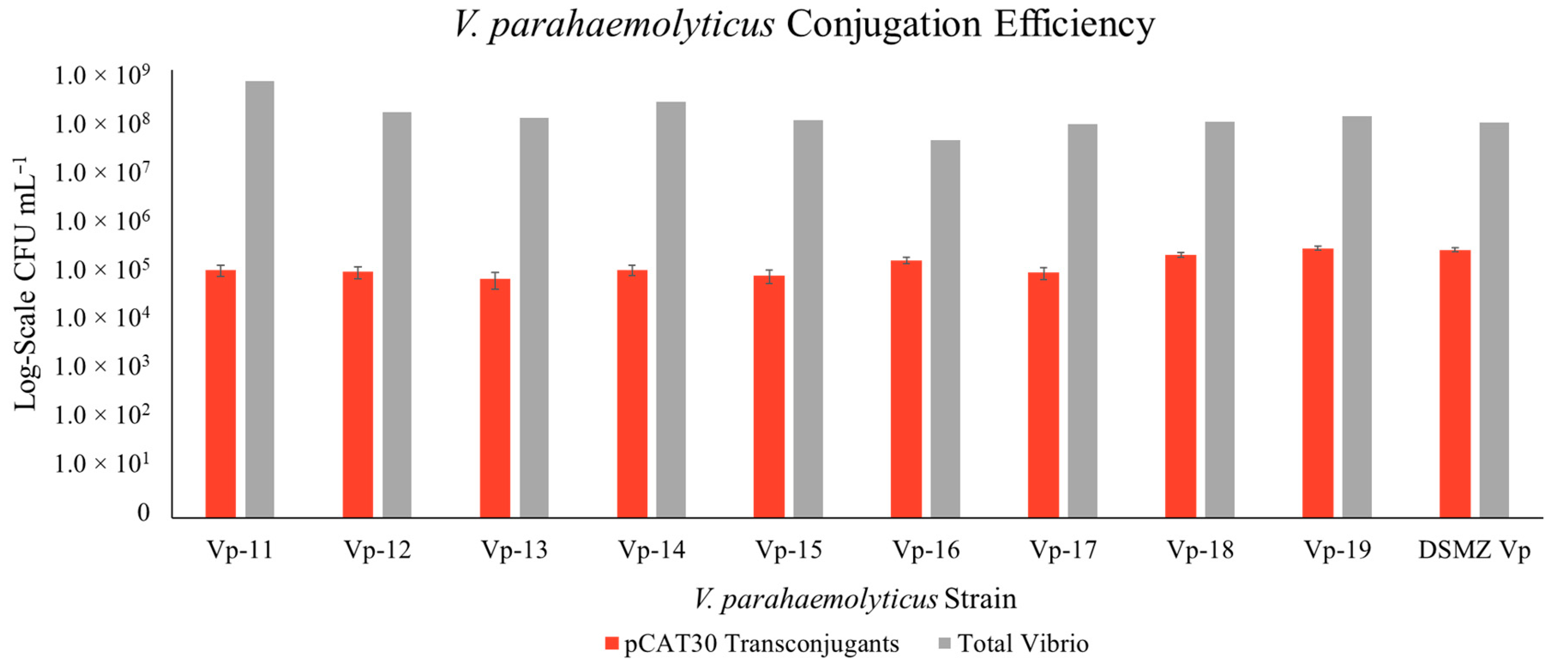
3. Results
3.1. Construction of New Suicide Vectors and Their Use in Vibrio Conjugation
3.2. Confirmation of the Transposon in the Vibrio Transconjugants
3.3. Transposon Insertion in Target Vibrio
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Norsworthy, A.N.; Visick, K.L. Gimme shelter: How Vibrio fischeri successfully navigates an animal’s multiple environments. Front. Microbiol. 2013, 4, 356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker-Austin, C.; Trinanes, J.A.; Salmenlinna, S.; Löfdahl, M.; Siitonen, A.; Taylor, N.G.H.; Martinez-Urtaza, J. Heat Wave—Associated Vibriosis, Sweden and Finland, 2014. Emerg. Infect. Dis. 2016, 22, 1216–1220. [Google Scholar] [CrossRef] [PubMed]
- Daniels, N.A.; MacKinnon, L.; Bishop, R.; Altekruse, S.; Ray, B.; Hammond, R.M.; Thompson, S.; Wilson, S.; Bean, N.H.; Griffin, P.M.; et al. Vibrio parahaemolyticus infections in the United States, 1973–1998. J. Infect. Dis. 2000, 181, 1661–1666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunn, A.K.; Millikan, D.S.; Adin, D.M.; Bose, J.L.; Stabb, E.V. New rfp- and pES213-Derived Tools for Analyzing Symbiotic Vibrio fischeri Reveal Patterns of Infection and lux Expression in Situ. Appl. Environ. Microbiol. 2006, 72, 802–810. [Google Scholar] [CrossRef] [Green Version]
- Stabb, E.V.; Ruby, E.G. RP4-based plasmids for conjugation between Escherichia coli and members of the Vibrionaceae. Methods Enzymol. 2002, 358, 413–426. [Google Scholar]
- Zeaiter, Z.; Mapelli, F.; Crotti, E.; Borin, S. Methods for the genetic manipulation of marine bacteria. Electron. J. Biotechnol. 2018, 33, 17–28. [Google Scholar] [CrossRef]
- Hamashima, H.; Nakano, T.; Tamura, S.; Arai, T. Genetic Transformation of Vibrio parahaemolyticus, Vibrio alginolyticus, and Vibrio cholerae Non O-1 with Plasmid DNA by Electroporation. Microbiol. Immunol. 1990, 34, 703–708. [Google Scholar] [CrossRef]
- Klevanskaa, K.; Bier, N.; Stingl, K.; Strauch, E.; Hertwig, S. PVv3, a New Shuttle Vector for Gene Expression in Vibrio vulnificus. Appl. Environ. Microbiol. 2014, 80, 1477–1481. [Google Scholar] [CrossRef]
- Marcus, H.; Ketley, J.M.; Kaper, J.B.; Holmes, R.K. Effects of DNase production, plasmid size, and restriction barriers on transformation of Vibrio cholerae by electroporation and osmotic shock. FEMS Microbiol. Lett. 1990, 68, 149–154. [Google Scholar] [CrossRef]
- Weinstock, M.; Hesek, E.; Wilson, C.; Gibson, D. Vibrio natriegens as a fast-growing host for molecular biology. Nat. Methods 2016, 13. [Google Scholar] [CrossRef]
- Chen, Y.; Dai, J.; Morris, J.G.; Johnson, J.A. Genetic analysis of the capsule polysaccharide (K antigen) and exopolysaccharide genes in pandemic Vibrio parahaemolyticus O3:K6. BMC Microbiol. 2010, 10, 274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gulig, P.A.; Tucker, M.S.; Thiaville, P.C.; Joseph, J.L.; Brown, R.N. USER Friendly Cloning Coupled with Chitin-Based Natural Transformation Enables Rapid Mutagenesis of Vibrio vulnificus. Appl. Environ. Microbiol. 2009, 75, 4936–4949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marvig, R.L.; Blokesch, M. Natural transformation of Vibrio cholerae as a tool-optimizing the procedure. BMC Microbiol. 2010, 10, 155. [Google Scholar] [CrossRef] [Green Version]
- Meibom, K.L.; Blokesch, M.; Dolganov, N.A.; Wu, C.-Y.; Schoolnik, G.K. Chitin induces natural competence in Vibrio cholerae. Science 2005, 310, 1824–1827. [Google Scholar] [CrossRef]
- Aune, T.E.V.; Aachmann, F.L. Methodologies to increase the transformation efficiencies and the range of bacteria that can be transformed. Appl. Microbiol. Biotechnol. 2010, 85, 1301–1313. [Google Scholar] [CrossRef]
- Le Roux, F.; Binesse, J.; Saulnier, D.; Mazel, D. Construction of a Vibrio splendidus Mutant Lacking the Metalloprotease Gene vsm by Use of a Novel Counterselectable Suicide Vector. Appl. Environ. Microbiol. 2007, 73, 777–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawabe, T.; Fukui, Y.; Stabb, E.V. Simple conjugation and outgrowth procedures for tagging vibrios with GFP, and factors affecting the stable expression of the gfp tag. Lett. Appl. Microbiol. 2006, 43, 514–522. [Google Scholar] [CrossRef]
- Travers, M.-A.; Barbou, A.; Le Goïc, N.; Huchette, S.; Paillard, C.; Koken, M. Construction of a stable GFP-tagged Vibrio harveyi strain for bacterial dynamics analysis of abalone infection. FEMS Microbiol. Lett. 2008, 289, 34–40. [Google Scholar] [CrossRef] [Green Version]
- Gobi, N.; Malaikozhundan, B.; Sekar, V.; Shanthi, S.; Vaseeharan, B.; Jayakumar, R.; Nazar, A.K. GFP tagged Vibrio parahaemolyticus Dahv2 infection and the protective effects of the probiotic Bacillus licheniformis Dahb1 on the growth, immune and antioxidant responses in Pangasius hypophthalmus. Fish Shellfish Immunol. 2016, 52, 230–238. [Google Scholar] [CrossRef]
- Nyholm, S.V.; Stabb, E.V.; Ruby, E.G.; McFall-Ngai, M.J. Establishment of an animal–bacterial association: Recruiting symbiotic vibrios from the environment. Proc. Natl. Acad. Sci. USA 2000, 97, 10231–10235. [Google Scholar] [CrossRef] [Green Version]
- Cabello, F.C.; Espejo, R.; Hernandez, M.C.; Rioseco, M.L.; Ulloa, J.; Vergara, J.A. Vibrio parahaemolyticus O3:K6 Epidemic Diarrhea, Chile, 2005. Emerg. Infect. Dis. 2007, 13, 655–656. [Google Scholar] [CrossRef] [PubMed]
- Suarez, A.; Güttler, A.; Strätz, M.; Staendner, L.H.; Timmis, K.N.; Guzmán, C.A. Green fluorescent protein-based reporter systems for genetic analysis of bacteria including monocopy applications. Gene 1997, 196, 69–74. [Google Scholar] [CrossRef]
- Sonnenschein, E.C.; Gärdes, A.; Seebah, S.; Torres-Monroy, I.; Grossart, H.-P.; Ullrich, M.S. Development of a genetic system for Marinobacter adhaerens HP15 involved in marine aggregate formation by interacting with diatom cells. J. Microbiol. Methods 2011, 87, 176–183. [Google Scholar] [CrossRef]
- Herrero, M.; de Lorenzo, V.; Timmis, K.N. Transposon vectors containing non-antibiotic resistance selection markers for cloning and stable chromosomal insertion of foreign genes in gram-negative bacteria. J. Bacteriol. 1990, 172, 6557–6567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujino, T.; Okuno, Y.; Nakada, D.; Aoyama, A.; Fukai, K.; Mukai, T.; Ueho, T. On the bacteriological examination of shirasu-food poisoning. Med. J. Osaka Univ. 1953, 4, 299–304. [Google Scholar]
- Stahl, A. Interaction of the Marine Bacterium Marinobacter Adhaerens HP15 with the Diatom Thalassiosira Weissflogii Analyzed by Proteomics Approaches. Ph.D. Thesis, Jacobs University Bremen, Bremen, Germany, 2016. [Google Scholar]
- Lambertsen, L.; Sternberg, C.; Molin, S. Mini-Tn7 transposons for site-specific tagging of bacteria with fluorescent proteins. Environ. Microbiol. 2004, 6, 726–732. [Google Scholar] [CrossRef] [Green Version]
- Gonzales, M.F.; Brooks, T.; Pukatzki, S.U.; Provenzano, D. Rapid protocol for preparation of electrocompetent Escherichia coli and Vibrio cholerae. JoVE J. Vis. Exp. 2013, e50684. [Google Scholar]
- Neogi, S.B.; Chowdhury, N.; Asakura, M.; Hinenoya, A.; Haldar, S.; Saidi, S.M.; Kogure, K.; Lara, R.J.; Yamasaki, S. A highly sensitive and specific multiplex PCR assay for simultaneous detection of Vibrio cholerae, Vibrio parahaemolyticus and Vibrio vulnificus. Lett. Appl. Microbiol. 2010, 51, 293–300. [Google Scholar] [CrossRef]
- Blanco, L.P.; DiRita, V.J. Bacterial-associated cholera toxin and GM1 binding are required for transcytosis of classical biotype Vibrio cholerae through an in vitro M cell model system. Cell. Microbiol. 2006, 8, 982–998. [Google Scholar] [CrossRef] [Green Version]
- Kovach, M.; Phillips, R.; Elzer, P.; Roop, R.; Perterson, K. pBBR1MCS: A broad-host-range cloning vector. BioTechniques 1994, 16, 800–802. [Google Scholar]
- Berg, D.E. Transposon Tn5. In Mobile DNA; Howe, M.M., Ed.; American Society for Microbiology: Washington, DC, USA, 1989; pp. 185–210. [Google Scholar]
- Stretton, S.; Techkarnjanaruk, S.; McLennan, A.M.; Goodman, A.E. Use of green fluorescent protein to tag and investigate gene expression in marine bacteria. Appl. Environ. Microbiol. 1998, 64, 2554–2559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newland, J.W.; Green, B.A.; Holmes, R.K. Transposon-mediated mutagenesis and recombination in Vibrio cholerae. Infect. Immun. 1984, 45, 428–432. [Google Scholar] [CrossRef] [Green Version]
- Leveau, J.H.J.; Lindow, S.E. Predictive and Interpretive Simulation of Green Fluorescent Protein Expression in Reporter Bacteria. J. Bacteriol. 2001, 183, 6752–6762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rang, C.; Galen, J.E.; Kaper, J.B.; Chao, L. Fitness cost of the green fluorescent protein in gastrointestinal bacteria. Can. J. Microbiol. 2003, 49, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Koch, B.; Jensen, L.E.; Nybroe, O. A panel of Tn7-based vectors for insertion of the gfp marker gene or for delivery of cloned DNA into Gram-negative bacteria at a neutral chromosomal site. J. Microbiol. Methods 2001, 45, 187–195. [Google Scholar] [CrossRef]
- Drake, S.L.; Elhanafi, D.; Bang, W.; Drake, M.A.; Green, D.P.; Jaykus, L.A. Validation of a Green Fluorescent Protein-Labeled Strain of Vibrio vulnificus for Use in the Evaluation of Postharvest Strategies for Handling of Raw Oysters. Appl. Environ. Microbiol. 2006, 72, 7205–7211. [Google Scholar] [CrossRef] [Green Version]
- Luo, P.; He, X.; Liu, Q.; Hu, C. Developing Universal Genetic Tools for Rapid and Efficient Deletion Mutation in Vibrio Species Based on Suicide T-Vectors Carrying a Novel Counterselectable Marker, vmi480. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [Green Version]
- Partridge, S.R.; Brown, H.J.; Stokes, H.W.; Hall, R.M. Transposons Tn1696 and Tn21 and their integrons In4 and In2 have independent origins. Antimicrob. Agents Chemother. 2001, 45, 1263–1270. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | ID Number | Strain Number | Isolation Source | Source |
|---|---|---|---|---|
| E. coli CC118 (λpir) | - | - | - | [24] |
| E. coli PIR-1 | - | - | - | Thermofischer Scientific, Cat. no. C101010 |
| E. coli ST18 | - | - | - | [23] |
| E. coli S17-1 | - | - | - | M.F. Alexeyev |
| V. cholerae | Vc-1 | VN-03213 | North Sea Water | this study |
| V. cholerae | Vc-2 | VN-04051 | Clinical isolate | this study |
| V. cholerae | Vc-3 | VN-04216 | North Sea Water | this study |
| V. cholerae | Vc-4 | VN-04219 | North Sea Water | this study |
| V. cholerae | Vc-5 | VN-04223 | North Sea Water | this study |
| V. cholerae | Vc-6 | VN-04226 | North Sea Water | this study |
| V. cholerae | Vc-7 | VN-04233 | North Sea Water | this study |
| V. cholerae | Vc-8 | VN-04261 | North Sea Water | this study |
| V. cholerae | Vc-9 | VN-10012 | unknown | this study |
| V. cholerae | Vc-10 | VN-10013 | unknown | this study |
| V. parahaemolyticus | Vp-11 | VN-02509 | North Sea Phytoplankton | this study |
| V. parahaemolyticus | Vp-12 | VN-02537 | North Sea Zooplankton | this study |
| V. parahaemolyticus | Vp-13 | VN-02538 | North Sea Mytilus edulis | this study |
| V. parahaemolyticus | Vp-14 | VN-02989 | North Sea Water | this study |
| V. parahaemolyticus | Vp-15 | VN-03220 | North Sea Water | this study |
| V. parahaemolyticus | Vp-16 | VN-03257 | North Sea Water | this study |
| V. parahaemolyticus | Vp-17 | VN-03653 | North Sea Crassostrea gigas | this study |
| V. parahaemolyticus | Vp-18 | VN-03747 | North Sea Crassostrea gigas | this study |
| V. parahaemolyticus | Vp-19 | VN-03857 | English Channel | this study |
| V. parahaemolyticus | Vp-20 | VN-04006 | North Sea Water | this study |
| V. parahaemolyticus | Vp-DSMZ | DSM-11058 | United Kingdom | [25] |
| V. vulnificus | Vv-21 | VN-03373 | North Sea Water | this study |
| V. vulnificus | Vv-23 | VN-03451 | North Sea Water | this study |
| V. vulnificus | Vv-24 | VN-03467 | North Sea Water | this study |
| V. vulnificus | Vv-25 | VN-03949 | North Sea Water | this study |
| V. vulnificus | Vv-26 | VN-02813 | North Sea Water | this study |
| V. vulnificus | Vv-27 | VN-03110 | North Sea, Phytoplankton | this study |
| V. vulnificus | Vv-28 | VN-03369 | North Sea Water | this study |
| V. vulnificus | Vv-29 | VN-03478 | North Sea Water | this study |
| V. vulnificus | Vv-30 | VN-03478 | North Sea Water | this study |
| Vector Name | Description | Relevant Characteristics | Study |
|---|---|---|---|
| pBBR1MCS4-eCFP.cat | Source of the eCFP and CmR genes | eCFP, CmR, ApR | [26,27] |
| pBBR1MCS4-dsRedExpress.cat | Source of the DsRedExpress and CmR genes | DsRedExpress, CmR, ApR | [26,27] |
| pAG408 | Suicide vector, requires presence of λpir-1 gene in host to replicate | GFP, KmR, GmR, ApR | [22] |
| pAG-SC | Backbone of pAG408, with the atpE, GFP, and KmR regions excised | GmR, ApR | this study |
| pCAT14 | eCFP and CmR genes cloned into pAG-SC | eCFP, CmR, GmR, ApR | this study |
| pCAT30 | DsRedExpress and CmR genes cloned into pAG-SC | DsRedExpress, CmR, GmR, ApR | this study |
| pBluescriptII SK+ | Cloning vector for sequencing of the transposon | ApR | - |
| ML_04 | Transposon region from pCAT14 cloned into pBluescriptII SK- | eCFP, CmR, GmR, ApR | this study |
| ML_6.2 | Transposon region from pCAT30 cloned into pBluescriptII SK- | DsRedExpress, CmR, GmR, ApR | this study |
| ML_02 | Transposon region from VcR-7-2 (clone 120) inserted into pBluescriptII SK- | DsRedExpress, CmR, GmR, ApR | this study |
| ML_12 | Transposon region from VvR-21 (clone150) inserted into pBluescriptII SK- | DsRedExpress, CmR, GmR, ApR | this study |
| Primer Name | Sequence 5′→3′ | Target | Product Size | Source | Annealing Temperature |
|---|---|---|---|---|---|
| pAG Tr FW | GGG GTA CCC CGC TCG AGT AAT TTA CCA A | pAG408 transposon | 1500 bp | [22] | 57 °C |
| pAG Tr RV | GGG GTA CCC CGC TTT TTA GAC ATC TAA ATC | ||||
| Vc ToxR 403F | GAA GCT GCT CAT GAC ATC | V. cholerae toxR gene | 275 bp | [29] | 55 °C |
| Vc ToxR 678R | AAG ATC AAG GGT GGT TAT TC | ||||
| Vp ToxR 325F | TGT ACT GTT GAA CGC CTA A | V. parahaemolyticus toxR gene | 503 bp | ||
| Vp ToxR 828R | CAC GTT CTC ATA CGA GTG | ||||
| VvhA 870F | CAC TCA ACT ATC GTG CAC G | V. vulnificus hemolysinA gene | 366 bp | ||
| VvhA 1236R | ACA CTG TTC GAC TGT GAG | ||||
| T3 | AAT TAA CCC TCA CTA AAG GG | Cloning region in pBluescriptII | varies | - | 50 °C |
| T7 | TAA TAC GAC TCA CTA TAG GG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thorstenson, C.A.; Ullrich, M.S. Developing a Universal and Efficient Method for the Rapid Selection of Stable Fluorescent Protein-Tagged Pathogenic Vibrio Species. J. Mar. Sci. Eng. 2020, 8, 804. https://doi.org/10.3390/jmse8100804
Thorstenson CA, Ullrich MS. Developing a Universal and Efficient Method for the Rapid Selection of Stable Fluorescent Protein-Tagged Pathogenic Vibrio Species. Journal of Marine Science and Engineering. 2020; 8(10):804. https://doi.org/10.3390/jmse8100804
Chicago/Turabian StyleThorstenson, Candice A., and Matthias S. Ullrich. 2020. "Developing a Universal and Efficient Method for the Rapid Selection of Stable Fluorescent Protein-Tagged Pathogenic Vibrio Species" Journal of Marine Science and Engineering 8, no. 10: 804. https://doi.org/10.3390/jmse8100804