Initiator Feeding Policies in Semi-Batch Free Radical Polymerization: A Monte Carlo Study
by
 , , and
, , and
Ali Seyedi
1, 
Mohammad Najafi
1,*,
Gregory T. Russell
2,* ,
,
Yousef Mohammadi
3,*,
Eduardo Vivaldo-Lima
4 and
and
Alexander Penlidis
5,* 1
Department of Polymer Engineering, School of Chemical Engineering, College of Engineering, University of Tehran, P.O. Box 11155-4563, Tehran 1417466191, Iran
2
School of Physical and Chemical Sciences, University of Canterbury, Private Bag 4800, Christchurch 8140, New Zealand
3
Petrochemical Research and Technology Company (NPC-rt), National Petrochemical Company (NPC), P.O. Box 14358-84711, Tehran 1993834557, Iran
4
Facultad de Química, Departamento de Ingeniería Química, Universidad Nacional Autónoma de México, CU, Mexico City 04510, Mexico
5
Department of Chemical Engineering, Institute for Polymer Research (IPR), University of Waterloo, Waterloo, ON N2L 3G1, Canada
*
Authors to whom correspondence should be addressed.
Processes 2020, 8(10), 1291; https://doi.org/10.3390/pr8101291
Submission received: 11 September 2020
/
Revised: 1 October 2020
/
Accepted: 3 October 2020
/
Published: 15 October 2020
(This article belongs to the Special Issue Modeling and Simulation of Polymerization Processes)
Abstract
:A Monte Carlo simulation algorithm is developed to visualize the impact of various initiator feeding policies on the kinetics of free radical polymerization. Three cases are studied: (1) general free radical polymerization using typical rate constants; (2) diffusion-controlled styrene free radical polymerization in a relatively small amount of solvent; and (3) methyl methacrylate free radical polymerization in solution. The number- and weight-average chain lengths, molecular weight distribution (MWD), and polymerization time were computed for each initiator feeding policy. The results show that a higher number of initiator shots throughout polymerization at a fixed amount of initiator significantly increases average molecular weight and broadens MWD. Similar results are also observed when most of the initiator is added at higher conversions. It is demonstrated that one can double the molecular weight of polystyrene and increase its dispersity by 50% through a four-shot instead of a single shot feeding policy. Similar behavior occurs in the case of methyl methacrylate, while the total time drops by about 5%. In addition, policies injecting initiator at high monomer conversions result in a higher unreacted initiator content in the final product. Lastly, simulation conversion-time profiles are in agreement with benchmark literature information for methyl methacrylate, which essentially validates the highly effective and flexible Monte Carlo algorithm developed in this work.
1. Introduction
Free radical polymerization is one of the most important polymerization techniques, contributing to the synthesis of about 50% of all the polymers produced worldwide. As a chain polymerization method, it is composed of initiation, propagation, chain transfer and termination steps [1]; this technique enjoys many advantages compared to other polymerization methods, such as ionic polymerization, making it very popular in industry. A number of advantages enhancing its robustness include lower sensitivity to impurities (although sensitive to oxygen, it is tolerant of water), broader range of monomers (almost all vinyl monomers), and milder reaction conditions [2]. Among the drawbacks of this method, one may mention failure to design well-defined macromolecules, which is usually embodied in ill-controlled molecular weight; broad molecular weight distribution; and weakly controlled monomer (sequence) distribution in the case of copolymerization. To overcome these disadvantages, new techniques based on specific feeding policies have been developed [3,4,5,6,7].
For polymerization in a batch reactor, all the monomers and initiators must be added to the reactor in the beginning of the polymerization. Therefore, the desired macromolecular microstructure—i.e., molecular weight average and distribution, branching index and distribution, copolymer composition (distribution), monomer sequence length distribution, etc.—cannot simply be controlled [8]. In particular, polymeric molecules that are formed earlier in a copolymerization may contain a high percentage of the more reactive monomer, while the chains produced later contain a high percentage of the remaining (less reactive) monomer. Since there is no steady state in a batch reactor, and also due to raw material variability, copolymer (and other) properties obtained may differ from batch to batch [9,10]. In a semi-batch reactor, at least one of the reactants is added over time; this mode helps design and produce macromolecules with a well-defined microstructure despite differences in the reactivity of the monomers. Another way of achieving a desired microstructure is to use predefined initiator feed policies [10,11,12].
In this context, Goto et al. studied the effect of tert-butyl hydroperoxide (BHP) initiator on the free radical polymerization of styrene initiated with styroxypiperidine and found that the addition of this initiator tripled the rate of the reaction without a significant influence on the molecular weight distribution and the number of polymer molecules [13]. A monomer feeding policy in a semi-batch atom transfer radical copolymerization (ATRP) showed that the copolymer composition distribution (CCD) could be precisely controlled by slowly feeding the monomers in a planned profile [9,14]. Wang et al. investigated four divinyl monomer feeding policies in the reversible addition-fragmentation (chain) transfer (RAFT) copolymerization of acrylamide and reported the effect of feeding policy on the microstructure and branching of star and hyperbranched chains produced [3]. Furthermore, Diaz-Camacho et al. [15] studied the semi-batch addition of initiator in the nitroxide-mediated polymerization of styrene, and modeled the system using the Predici software, but they slightly over-predicted the effect of semi-continuous addition on polymerization rate [16].
Simulation and modeling techniques have proven to be an extremely useful tool for predicting the microstructural characteristics of polymer chains during polymerization. Modeling of polymerization processes falls into the two categories of deterministic and stochastic modeling. The former involves writing mass balances for all the species present, leading to a set of differential (and algebraic) equations, the numerical solution of which leads to properties such as average molecular weights, average branching indicators, etc. either directly (with the method of moments) or indirectly (when chain lengths are treated individually). In the latter event, there is a very large number of equations that must be solved. This can be a major problem even with increasingly powerful computers used today [17]. A popular variant of the deterministic approach is the use of the method of moments. This method decreases the number of equations by rewriting the kinetically-derived population balances into moment balances [18].
Using mathematical modeling, Arzamendi et al. investigated three different monomer feeding policies in semi-batch emulsion copolymerization in order to optimize control over copolymer composition and minimize polymerization time [19]. In another study, the semi-batch polymerization of styrene, in a monomer and initiator starved-fed reactor, showed that the molecular weight and molecular weight distribution can be effectively controlled in this manner [20]. Pinto et al. optimized a cocktail of initiators in suspension polymerization of vinyl chloride using mathematical modeling and found that by using the optimized cocktail, process time could be significantly decreased compared with a typical single-initiator process [21]. In another work, a kinetic model was developed to consider the effect of a monomer feeding policy on the CCD in the mini-emulsion copolymerization of styrene and butyl acrylate; it was established that the proposed model was in good agreement with polymerization kinetics. Furthermore, several copolymers with a predesigned CCD were successfully synthesized using the presented model [22].
In contrast to deterministic approaches, stochastic approaches do not need to solve a system of differential equations. These approaches are usually based on the master chemical equation and require information about elementary reaction mechanisms and probabilities. This characteristic makes this technique invaluable for modeling complex polymer systems [18,23]. The kinetic Monte Carlo (KMC) algorithm is one of the stochastic approaches that provides an efficient method for investigating the microstructure and spatial topologies of polymer chains. It is also possible to simulate polymerization processes with complex molecular architectures and investigate macromolecular microstructures such as molecular weight distribution, copolymer composition (distribution), monomer sequence length (distribution), long-chain branching, short-chain branching, chain topology, etc. [23,24,25]
The effect of bi-functional initiators on free radical polymerization of styrene has been studied using Monte Carlo simulation, and it was found that using bi-functional initiators results in increased monomer conversion and molecular weight, and narrower molecular weight distribution compared with mono-functional initiators [26]. Tobita studied long-chain branching (LCB) and the distribution of branching density in poly(vinyl acetate) using this technique [27]. The Monte Carlo method was used in Prescott’s work studying the effect of chain-length-dependent termination on reversible deactivation radical polymerization (RDRP) using the reversible addition-fragmentation chain transfer (RAFT) process [28]. Using the Monte Carlo method with the Julia programming language, Pintos et al. simulated the kinetics of RAFT radical polymerization in batch mode, based on the theories of slow fragmentation, intermediate radical termination, and the termination of intermediate radicals with oligomers [29]. The effect of reactivity ratios and initial feed composition on copolymer microstructure was investigated for free radical copolymerization in a comprehensive study using a Monte Carlo simulation method. The model had the ability to illustrate changes in azeotropy and composition drift [30,31]. A high-performance Monte Carlo code was developed for simulating the kinetics of free radical and ATRP of styrene, considering chain-length dependent and diffusion-controlled termination. The obtained results were in close agreement with experimental data [32]. Saeb et al. used Monte-Carlo modeling to study the effect of co-monomer feeding policy on copolymer microstructure in metallocene catalyzed copolymerization of ethylene/1-hexene; this revealed the critical significance of the computerized feeding mode contrasted with the uncontrolled feeding mode [33]. A Monte Carlo method was also used to study the effect of hydrogen and co-catalyst concentration in ethylene polymerization using a Ziegler-Natta catalyst [34]. Finally, Najafi et al. studied four termination modes in styrene ATRP using a Monte Carlo approach [35].
The above is a critical selection of works on the simulation of the kinetics of free radical polymerization, in particular referring to feeding strategies and/or nature of initiation. To the very best of our knowledge, our work that follows is the first ever study using the Monte Carlo simulation method to investigate the effect of initiator feeding policies on a polymerization process and chain properties in semi-batch free radical polymerization. In this regard, our work is also complementary to the studies by Diaz-Camacho et al. [15] and Roa-Luna et al. [16] in that the approach in these two previous references was similar but derived using a commercial package (Predici) for a nitroxide-mediated polymerization system. In this context, we investigate three different cases: a general free radical polymerization with typical kinetic rate coefficient values (Case 1), a diffusion-controlled free radical polymerization of styrene (Case 2, low solvent fraction), and solution free radical polymerization of methyl methacrylate (Case 3, high solvent fraction). Thermal and chemical types of initiation, transfer reactions, and diffusion-control effects were all considered in the different case studies. In order to investigate the effect of initiator feeding policies during the course of the polymerization, three feeding strategies based on the number of shots (NOS), amount of shots (AOS), and time of shots (TOS) were considered in each case, and different indicators of polymerization performance, in terms of polymerization rate, polymer quality, molecular weight averages, and molecular weight distribution, were evaluated. We should state upfront that in these case studies, all being evaluated at the simulation/modeling level, mixing of the initiator shots in the polymerizing mixture is considered perfectly instantaneous and homogeneous.
2. Model Development
2.1. Kinetic Model
Monte Carlo algorithms were developed for three cases, as detailed in Table 1: general free radical polymerization; styrene free radical polymerization in solution (low fraction of solvent); and methyl methacrylate free radical polymerization in solution (high fraction of solvent). Table 1 lists the reactions for each case. In the three cases, the chemical initiation step is related to thermal decomposition of initiator, while thermal monomer self-initiation also contributes in styrene polymerization. Transfer to monomer, transfer to solvent, termination by combination, and termination by disproportionation may occur in each case. The kinetic parameters and constants required for the simulations are listed in Table 2.
2.2. Monte Carlo Simulation
The kinetic model based on the Monte Carlo algorithm is developed according to the probabilities of reactions. Due to the stochastic nature of Monte Carlo simulation, there may always be differences in the results obtained from each run. Hence, defining a proper simulation volume is vital not only because of its impact on the reliability and accuracy of the simulation results, but also due to its influence on the simulation performance and runtime. A larger simulation volume usually renders more accurate results, but it may lead to longer runtimes. The optimum sample size is specific to each polymerization system since it is greatly influenced by the reaction parameters and initial conditions [23,24]. The Monte Carlo computer code utilized in the current work employs Gillespie’s algorithm as its foundation [46]. The concentrations of the species can be related to the number of monomers in the simulation volume according to Equation (1):
where is the number of species and represents the concentration of species ; and stand for Avogadro’s number and reaction volume, respectively. In addition, the experimental rate constants () must be converted to Monte Carlo rate constants () according to Equations (2)–(5):
Unimolecular reaction:
Bimolecular reaction between different species:
Bimolecular reaction between identical species:
Termolecular reaction between identical species:
For example, the equation for rate of polymerization, , is:
where stands for the Monte Carlo propagation rate constant and and represent the number of (polymerizing) free radicals and monomer molecules, respectively. The probability of each reaction, , is defined by Equation (7):
where is the rate of reaction , is the total reaction rate of the system, and denotes the number of reaction channels. Equation 8 is used to determine the reaction channel which should happen at a given time:
where is the number of the selected reaction channel and denotes a random number in the (0,1] interval. An additional random number () should also be generated to determine the time interval () between two consecutive and sequential reactions (see Equation (9)):
The improved Mersenne Twister algorithm is embedded in the simulation code to generate random numbers [47]. The simulation code is written in Lazarus 2.0.10 and compiled into 64-bit executable code using FPC 3.2.0. The average runtime of the program is approximately 300 min at a simulation volume of 2.076 × 10–13 lit on a desktop computer equipped with Intel Core i7-3770K (3.50 GHz) and 32 GB of memory (2133 MHz) running Windows 7 Ultimate 64-bit operating system.
3. Results and Discussion
3.1. Initiator Feeding Policies
The simulation is performed in three scenarios according to three different feeding policies. In the first policy, the number of initiator shots injected into the system is varied. Four different feeding policies based on the number of shots (NOS) are simulated. In the second, the amount of initiators per shot is varied; four different feeding policies based on changing the amount of shots (AOS) are chosen. In the third scenario, the time at which the initiator is added to the reactor is varied, thus, four different feeding policies having various times of shots (TOS) are taken into account (Table 3). It is worth mentioning that the total amount of initiator used in each of the three scenarios is the same and equal to (of a regular initiator charge). In the NOS policy, the initiator is added to the reactor in one, two, four, and eight steps with equal fractions at distinct reaction conversions. In the AOS policy, the initiator is introduced to the reaction system at four different fractions at distinct conversions. In the feeding policy based on TOS, the initiator is injected at four equal fractions at various conversion levels.
3.2. General Free Radical Polymerization
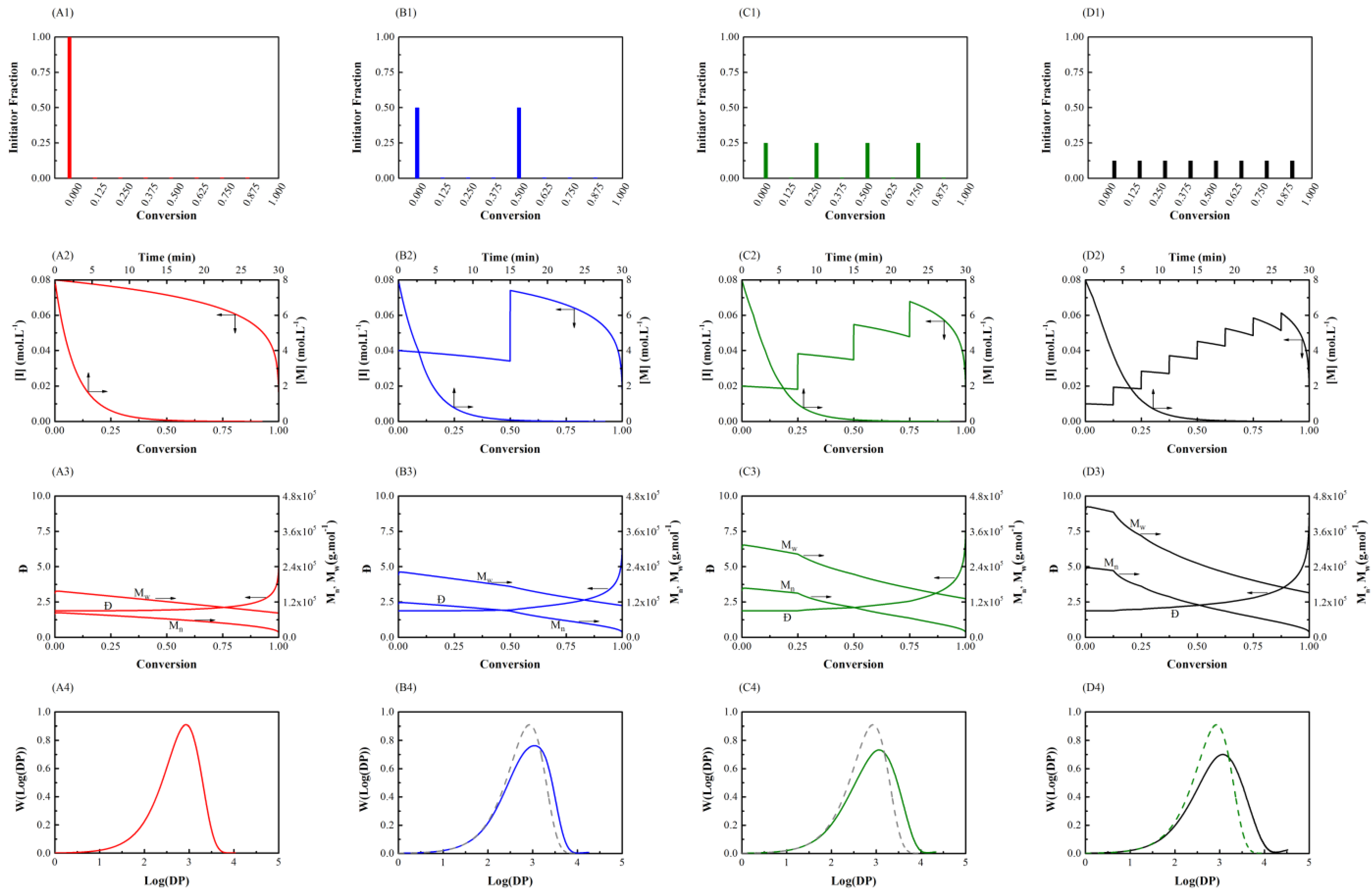
The results of varying the number of initiator shots are shown in Figure 1. In the initiator feeding policy for NOS-1 (A1), the entire amount of initiator is charged to the reactor at the beginning of the polymerization. Thus, the rate of monomer consumption in NOS-1 (A2) should be increased in comparison with NOS-2 (B2), NOS-3 (C2), and NOS-4 (D2). Increasing the number of shots, which approaches asymptotically the case of continuous feeding, results in a smaller slope in conversion-monomer curves (NOS-4 (D2)) in spite of decreasing the amount of initiator in each shot (NOS-1 (A2)). This indicates slower polymerization, and consequently, larger polymerization times. On the other hand, by distributing the concentration of the initiator at different conversions, because of a drop in the concentration of the initiator at the early stages of the polymerization, the molecular weight increases by changing feeding policy from NOS-1 (A3) to NOS-4 (D3). The same change causes a wider molecular weight distribution with a longer tail towards higher molecular weights. Therefore, the molecular weight distribution curves are shifted to the right by increasing the number of shots from NOS-1 (A4) to NOS-4 (D4). In fact, increasing the number of shots (NOS) leads to increasing number- and weight-average molecular weights (see third row) and a broader molecular weight distribution (MWD) (fourth row).
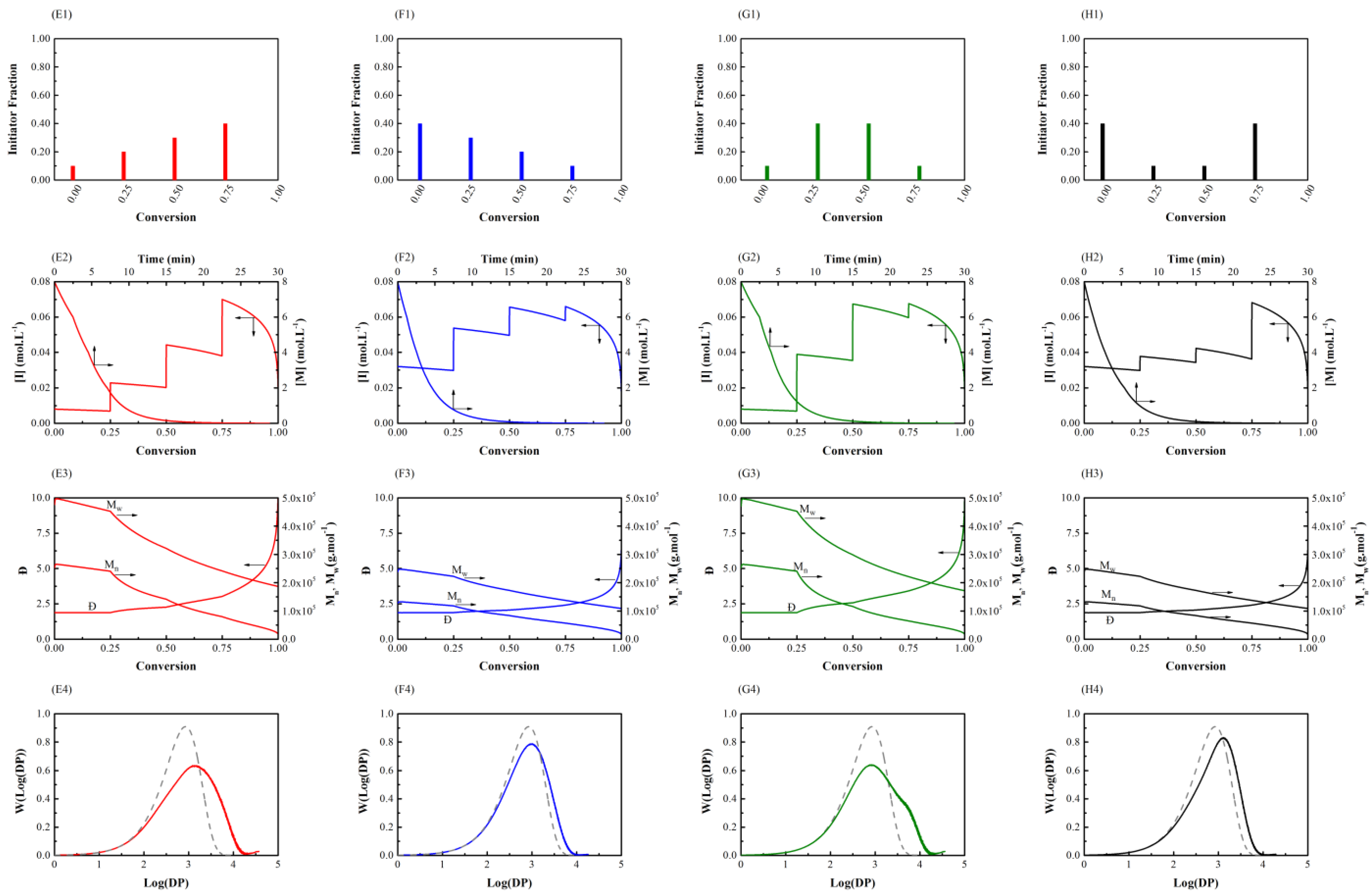
Figure 2 illustrates how varying the amount in initiator shots (AOS) affects chain microstructure. In the initiator feeding policy of AOS-1(E1), by feeding less initiator at the beginning of the polymerization, a higher molecular weight average is achieved (AOS-1 (E3)) compared with AOS-2 (F3), AOS-4 (H3), and the original reference simulation NOS-1 (A3) of Figure 1, where in all of which more initiator is added initially. Moreover, a much wider molecular weight distribution in comparison with NOS-1 (A4) is obtained. A similar result is obtained in AOS-3 (G1): a higher molecular weight and a much broader MWD (AOS-3 (G4)) result compared with the reference plot (NOS-1 (A4)). The rate of monomer consumption (graphs AOS-2 (F2) and AOS-4 (H2)) is increased (a steeper slope in conversion-time curves is seen) in comparison with AOS-1 (E2) and AOS-3 (G2) due to a higher fraction of initiator being injected at the beginning of the polymerization. By comparing AOS-3 (G1) with AOS-1 (E1), it is found that both policies have low initiator concentrations at the beginning, producing a large number of long chains, thereby creating a tail in high molecular weights (the right side of the distribution curve). The difference lies in the rate of initiator concentration increment, which is much more sudden in AOS-3; this leads to a larger number of short chains, thereby creating a low population of chains at log10DP ≈ 3.5 (see AOS-3 (G4)). By evaluating the graphs of AOS-2 (F4) and AOS-4 (H4), it is evident that initiator concentration is relatively high throughout the entire course of polymerization, which narrows the molecular weight distribution compared with AOS-1 (E4) and AOS-3 (G4). In contrast, AOS-2 (F2) and AOS-4 (H2) lead to higher conversions at shorter polymerization times; these policies act similarly to the reference case (graph (NOS-1 (A2)), while AOS-1 and AOS-3 raise molecular weight averages and broaden MWDs. In summary, increasing the fraction of initiator at later stages of the polymerization enhances number- and weight-average molecular weights and broadens molecular weight distribution by adding to the fractions of longer chains. In contrast, adding higher amounts of initiator at the beginning of the polymerization narrows MWD and reduces the average chain length.
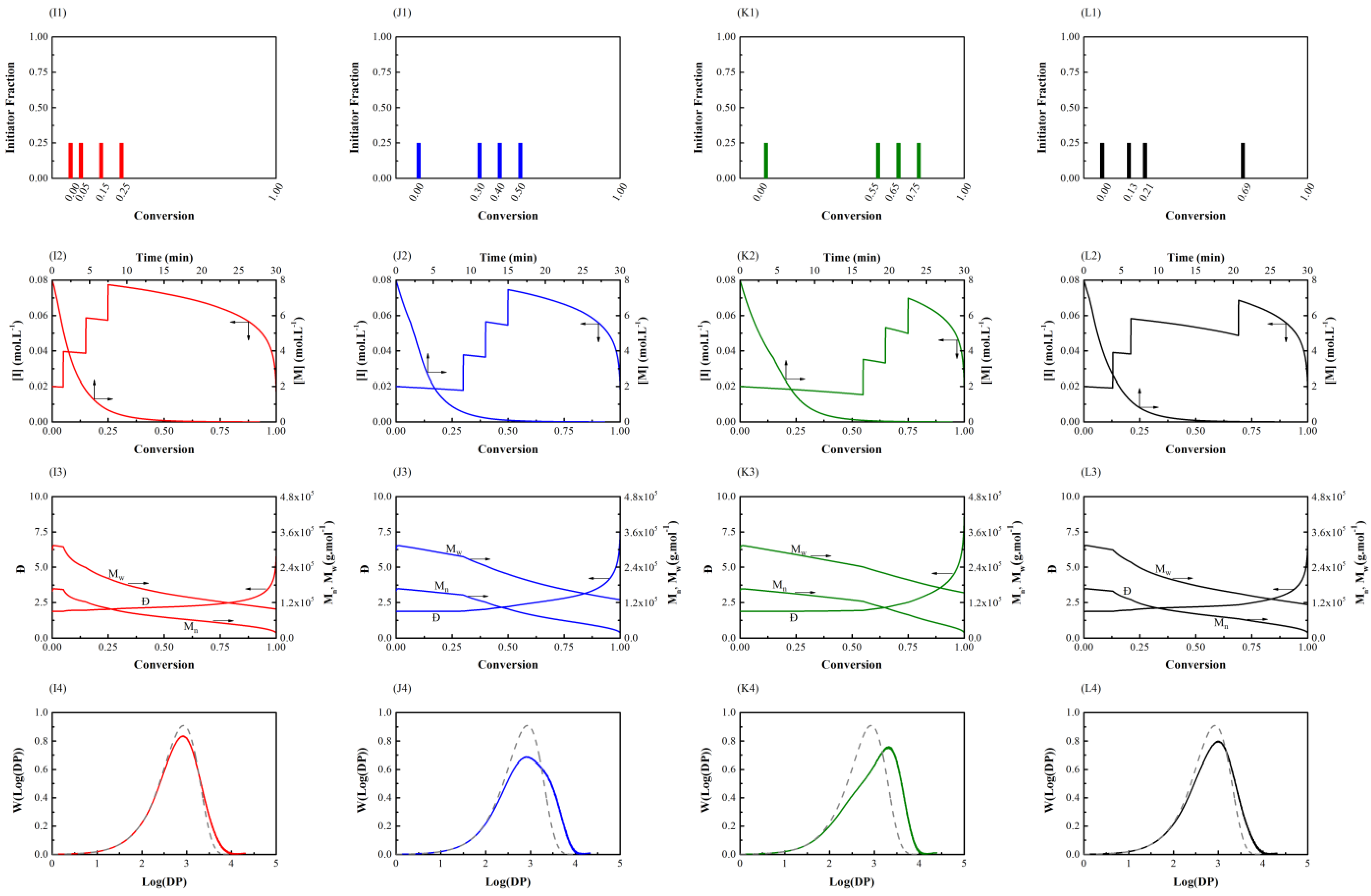
Figure 3 depicts the results of varying the reaction times at which initiator shots are added to the system. It is obvious that in the TOS-1 (I1) and TOS-4 (L1) cases, there is a high concentration of initiator throughout the polymerization. A similar result is realized when comparing with AOS-2 (F1) and AOS-4 (H1): there is a fairly narrow chain length distribution combined with higher polymerization rate. In TOS-2 (J1), initiator concentration is relatively low up to an extent of reaction of about 30%, creating a high population of long chains. Initiator added at later stages of the polymerization leads to shorter chains, which widens MWD compared with the NOS-1 (A4), TOS-1 (I4), and TOS-4 (L4) situations. In addition, the polymerization time needed to achieve a specific conversion is not much longer than that of the reference graph (NOS-1 (A2)). Therefore, a broader MWD without significantly sacrificing the polymerization rate is obtainable. Finally, in TOS-3 (K4), the peak in molecular weight distributions is shifted to the right compared with the reference graph (NOS-1 (A4)) and all other previously studied cases; this is attributed to relatively low initiator concentration up to 55% of conversion leading to a large number of long chains. A subsequent sudden rise in initiator content produces a large number of short chains, which generates a small shoulder in the distribution curve. Comparing with the reference graph (NOS-1 (A3)), weight-average molecular weight and dispersity are increased. Furthermore, the time for complete conversion to be reached—which one might regard as an indicator of productivity—drops. It is worth mentioning that number-average molecular weights are equal in all the policies because the total amount of initiator consumed is almost the same for all of the simulation cases.
Comparing all the policies, it is evident that the time required to complete the polymerization is different in various cases, in fact by up to 50%. The policies containing a higher initiator concentration at lower conversions show higher ‘performance,’ thus, there is a compromise between polymerization performance (polymerization time) and what is achieved in other polymer quality indicators, i.e., weight-average molecular weight and molecular weight distribution. As a result, the time interval between the shots plays a decisive role in the overall performance of the feeding policies based on the time of shots (TOS). If the initiator shots are injected close to each other, molecular weight average and dispersity become more uniform. Therefore, as the time intervals between the shots are reduced, the molecular weight distribution becomes narrower.
3.3. Styrene Free Radical Polymerization in Solution
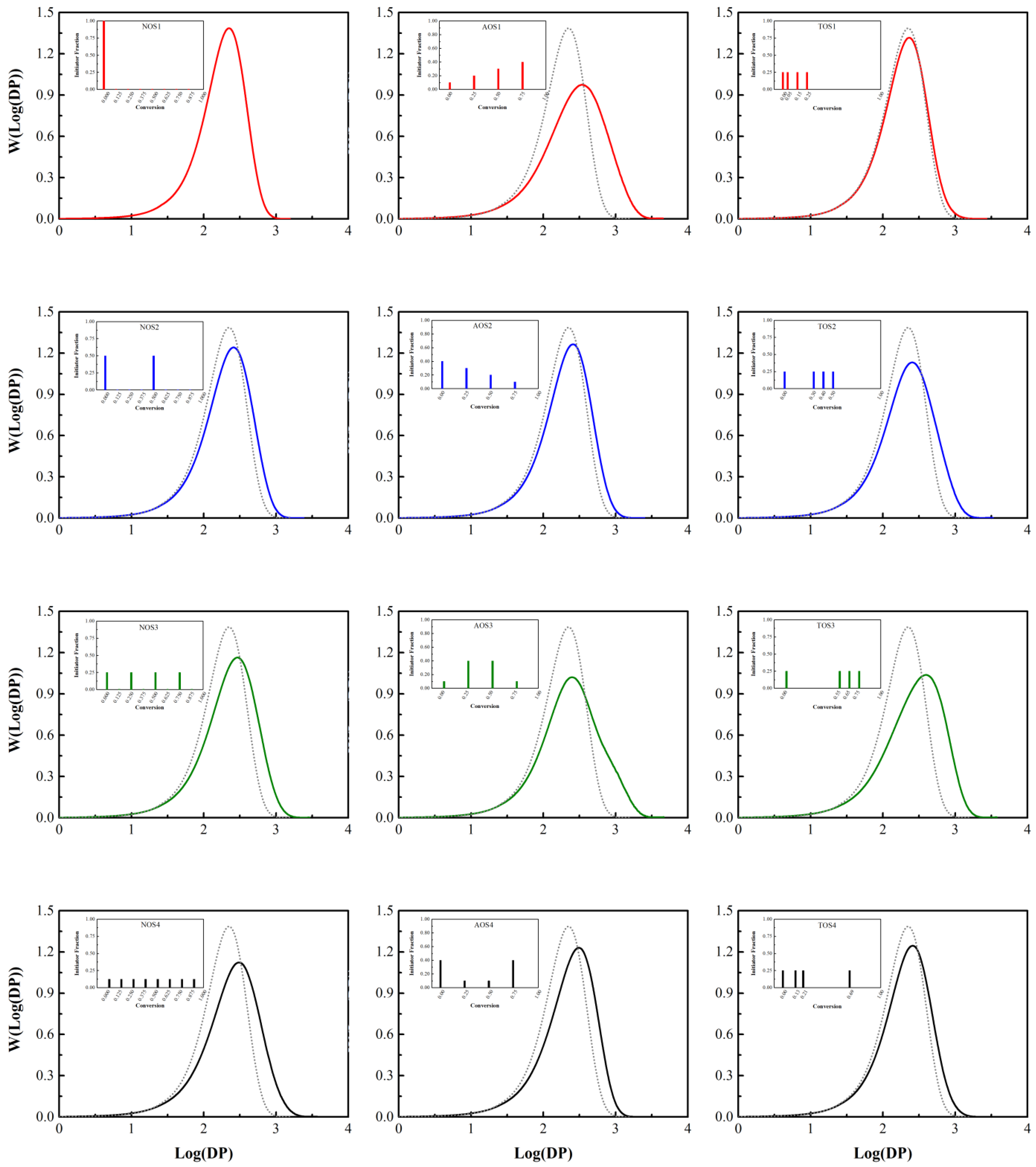
Table 4 presents results obtained from the Monte Carlo simulation of styrene free radical polymerization (Case 2 of Table 1 and Table 2; low fraction of solvent). By increasing the number of initiator shots (NOS) from one to eight, the weight-average molecular weight increases by 150%, while the polymerization time drops by 8%. In addition, by meticulously analyzing the results of the simulation, it is found that using the NOS policy not only increases the weight-average molecular weight, but also ‘tailors’ the molecular weight distribution to some extent, in that one can dial up the dispersity by altering the number of shots. Figure 4 probes this further by presenting the MWD in these scenarios of semi-batch free radical polymerization of styrene for the three different initiator feeding policies. Comparing NOS-1 with TOS-2 (first vs. third column of plots in Figure 4) reveals that changing the policy enables one to increase weight-average molecular weight and broaden the distribution to a moderate extent. Additionally, changing the policy to AOS-1 (middle column of plots in Figure 4) almost doubles the weight-average molecular weight and broadens the chain length distribution (a 150% increase in dispersity, Đ). The AOS-1 and AOS-3 policies provide a wider molecular weight distribution compared to the other policies, which is ascribed to the distribution of the initiator concentration over the course of the reaction. It may be stated that a policy leading to increased weight-average molecular weight usually results in a broadened MWD. In addition, there will be more unreacted initiator in the final polymerization mixture in policies in which an initiator shot is injected at the late stages of the reaction, namely NOS-4, AOS-1, AOS-4, and TOS-3; in all these cases, more than 10% of the initiator remains unreacted. This leads to a decrease in the number of monomer units allotted to each radical in the beginning of the polymerization and, consequently, the emergence of short chains, which is the cause of a widening molecular weight distribution. An increase in the concentration of the residual initiator at the end of the polymerization of styrene means less initiator decomposition over the course of the reaction, which leads to a higher fraction of long chains; as a result, molecular weight distribution widens, and average molecular weight and dispersity deviate from the reference graph (NOS-1 (A4)). However, unreacted (residual) initiator is undesirable for a number of reasons, both economical and operational (product quality).
3.4. Free Radical Polymerization of Methyl Methacrylate in Solution
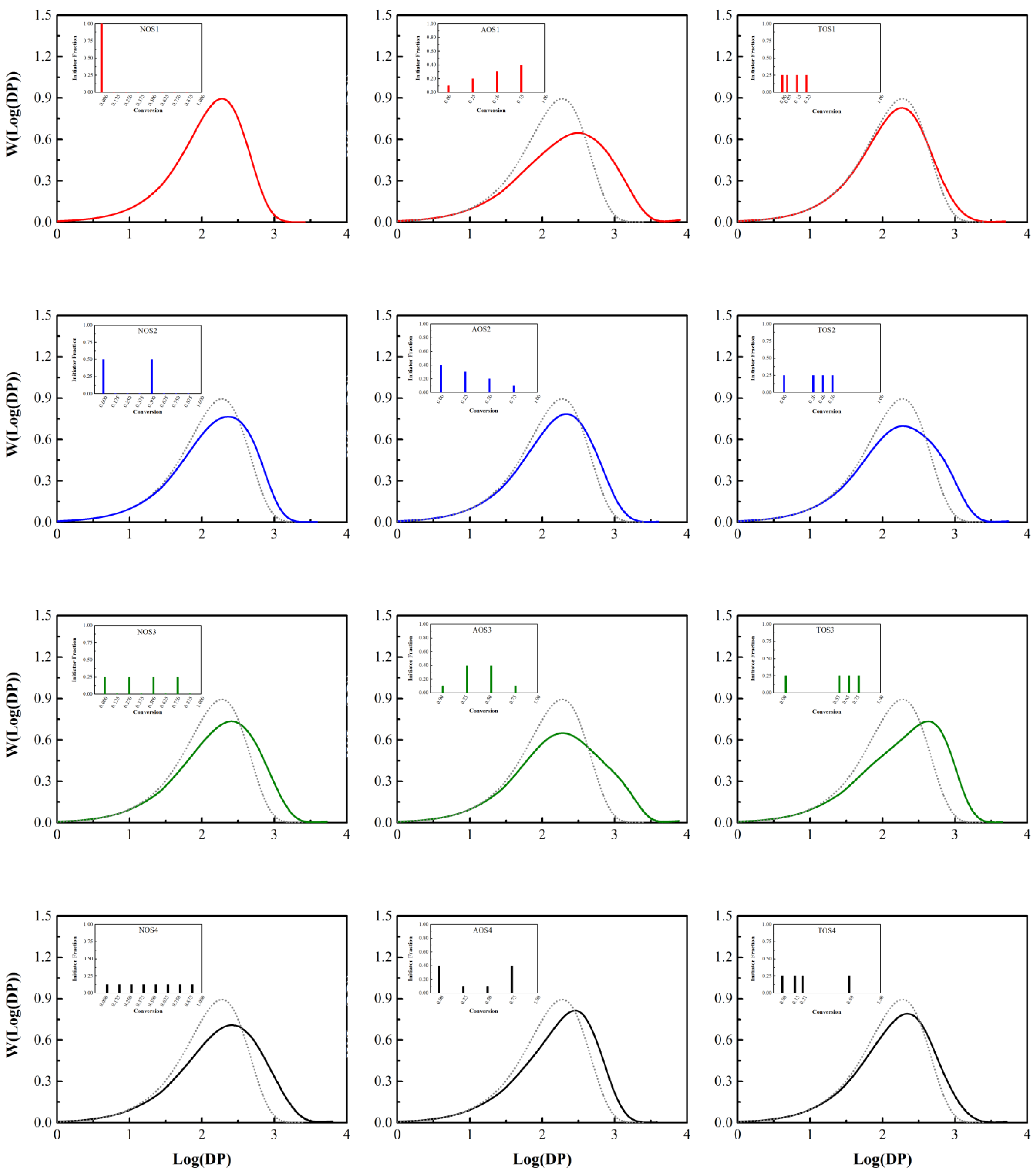
Results of the simulation of methyl methacrylate (MMA) free radical polymerization in solution (Case 3 of Table 1 and Table 2) are presented in Table 5 and Figure 5. The simulation captures the fact that the MWDs for MMA polymerization are broader compared with styrene (contrast the values of dispersity in Table 4 and Table 5, respectively).
According to the simulation results, increasing the number of shots (NOS) from 1 to 8 (first column of plots in Figure 5, from top to bottom) causes a 75% increase in weight-average molecular weight and a 65% increase in dispersity. The wider distributions (exhibiting a shoulder at high molecular weights) previously observed with the AOS-3 and TOS-3 policies (in Figure 4) are also present in MMA polymerization. Here, by changing the initiator feeding policy from NOS-1 to AOS-1, both weight-average molecular weight and dispersity are increased by a factor of 2. Additionally, the AOS-2 and AOS-4 policies provide a narrower MWD due to the higher fraction of initiator at lower conversions and the lower fraction of initiator at higher conversions. Within these policies, the molecular weight distribution is closer to the reference graph (NOS-1), and the time to achieve complete conversion is reduced. In NOS-4, AOS-1, and TOS-3 feeding policies, due to the higher fraction of initiator concentration at high conversions, a larger amount of initiator remains unreacted in comparison with the other policies. Therefore, the number of decomposed initiator molecules (produced chains) falls, thereby raising molecular weight and widening molecular weight distribution compared with the other policies. Overall, the later the initiator is introduced to the reaction system, the more heterogeneous the microstructure of the produced chains becomes. In fact, a lower fraction of initiator at the beginning of the polymerization generates fewer, but longer chains, thereby broadening chain length distribution, as one can observe from Table 5 and Figure 5.
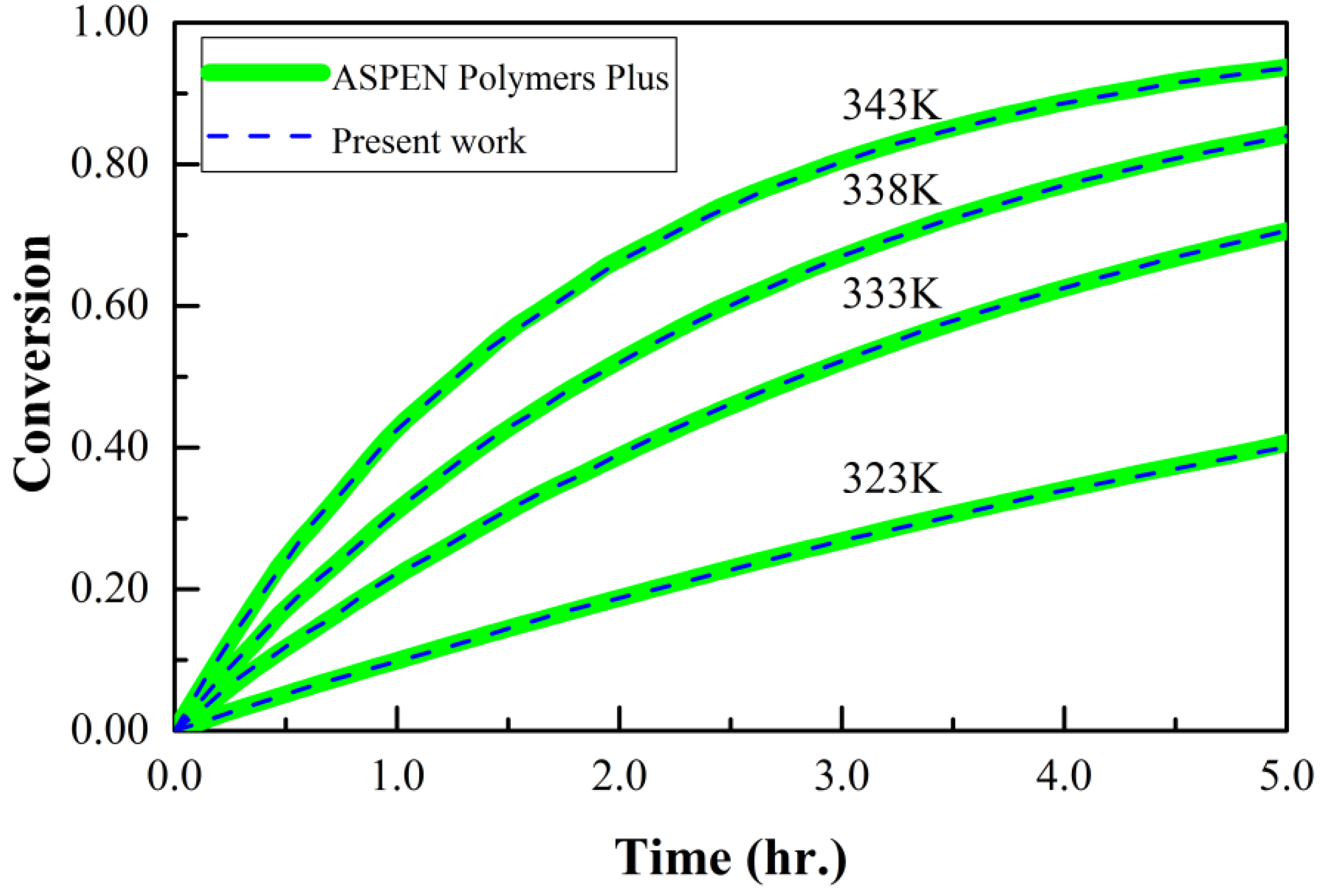
Finally, in order to ‘benchmark’ the simulation algorithm and coding developed herein, simulation data for MMA polymerization at four different temperatures were compared with ASPEN modeling data reported elsewhere [48], noting that the ASPEN modeling was already confirmed using experimental data [37]. As can be seen in Figure 6, the conversion-time results for NOS-1 are in complete agreement with the ASPEN data at all temperatures. Furthermore, as expected, raising the temperature increases the rate of monomer consumption.
4. Conclusions
This study introduces a new and remarkably simple way of tailoring the rate of polymer synthesis and the obtained MWD. Various initiator feeding policies are defined and evaluated to tune the chain microstructure in a free radical polymerization system. A general free radical polymerization with typical rate constant values (Case 1), free radical polymerization of styrene in solution (Case 2) with a lower solvent fraction, and free radical polymerization of methyl methacrylate in solution (Case 3, at a much higher solvent fraction) are simulated using a Monte Carlo method. By distributing the concentration of initiator at different conversion levels, because of the decrease in the concentration of the initiator at the beginning of the reaction, the molecular weight changes dramatically. Changing initiator feeding policy usually generates a wider molecular weight distribution with a longer tail at higher molecular weights. In addition, the policies in which lower amounts of initiator are injected at the beginning of the reaction produce a large number of long chains and create a tail at the high molecular weight portion of the MWDs. Similar results are achieved by the policies in which most of the initiator is added at high monomer conversions. The policies injecting higher initiator concentrations at lower conversions reach completion much faster. Therefore, there is a compromise between polymerization performance (rate of monomer consumption) and polymer quality, i.e., average molecular weight and molecular weight distribution. As a result, we can exploit an initiator feeding policy tuned for a specific polymerization system to strike a balance between productivity and quality/properties/polymer microstructure.
Author Contributions
A.S., M.N. and Y.M. conceived the original idea (with discussions with G.T.R. and A.P.), designed and executed the computer programming, performed the simulations, analyzed the data, and wrote the original draft of the paper; G.R.T., A.P. and E.V.-L. read and corrected different versions of the manuscript, and provided extra references and discussion points.All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Chiefari, J.; Chong, Y.K.B.K.B.; Ercole, F.; Krstina, J.; Jeffery, J.; Le, T.P.T.P.T.; Mayadunne, R.T.A.T.A.; Meijs, G.F.F.; Moad, C.L.L.; Moad, G.; et al. Living Free-Radical Polymerization by Reversible Addition—Fragmentation Chain Transfer: The RAFT Process. Macromolecules 1998, 31, 5559–5562. [Google Scholar] [CrossRef]
- Noble, B.B.; Coote, M.L. First principles modelling of free-radical polymerisation kinetics. Int. Rev. Phys. Chem. 2013, 32, 467–513. [Google Scholar] [CrossRef]
- Wang, D.; Wang, W.-J.; Li, B.-G.; Zhu, S. Semibatch RAFT polymerization for branched polyacrylamide production: Effect of divinyl monomer feeding policies. AIChE J. 2013, 59, 1322–1333. [Google Scholar] [CrossRef]
- Lowe, A.B.; McCormick, C.L. Homogeneous controlled free radical polymerization in aqueous media. Aust. J. Chem. 2002, 55, 367–379. [Google Scholar] [CrossRef]
- Aydin, M.; Arsu, N.; Yagci, Y.; Jockusch, S.; Turro, N.J. Mechanistic study of photoinitiated free radical polymerization using thioxanthone thioacetic acid as one-component type II photoinitiator. Macromolecules 2005, 38, 4133–4138. [Google Scholar] [CrossRef]
- Crowley, T.J.; Choi, K.Y. Experimental studies on optimal molecular weight distribution control in a batch-free radical polymerization process. Chem. Eng. Sci. 1998, 53, 2769–2790. [Google Scholar] [CrossRef]
- Crowley, T.J.; Choi, K.Y. Discrete optimal control of molecular weight distribution in a batch free radical polymerization process. Ind. Eng. Chem. Res. 1997, 36, 3676–3684. [Google Scholar] [CrossRef]
- Kiparissides, C.; Krallis, A.; Meimaroglou, D.; Pladis, P.; Baltsas, A. From Molecular to Plant-Scale Modeling of Polymerization Processes: A Digital High-Pressure Low-Density Polyethylene Production Paradigm. Chem. Eng. Technol. 2010, 33, 1754–1766. [Google Scholar] [CrossRef]
- Wang, R.; Luo, Y.; Li, B.; Sun, X.; Zhu, S. Design and Control of Copolymer Composition Distribution in Living Radical Polymerization Using Semi-Batch Feeding Policies: A Model Simulation. Macromol. Theory Simul. 2006, 15, 356–368. [Google Scholar] [CrossRef]
- Zhang, M.; Ray, W.H. Modeling of “living” free-radical polymerization processes.I. Batch, semibatch, and continuous tank reactors. J. Appl. Polym. Sci. 2002, 86, 1630–1662. [Google Scholar] [CrossRef]
- Colombani, D. Chain-growth control in free radical polymerization. Prog. Polym. Sci. 1997, 22, 1649–1720. [Google Scholar] [CrossRef]
- Hamielec, A.E.; MacGregor, J.F.; Penlidis, A. Multicomponent free-radical polymerization in batch, semi-batch and continuous reactors. In Makromolekulare Chemie. Macromolecular Symposia; Hüthig & Wepf Verlag: Basel, Switzerland, 1987; Volume 10, pp. 521–570. [Google Scholar]
- Goto, A.; Fukuda, T. Effects of radical initiator on polymerization rate and polydispersity in nitroxide-controlled free radical polymerization. Macromolecules 1997, 30, 4272–4277. [Google Scholar] [CrossRef]
- Wang, R.; Luo, Y.; Li, B.-G.; Zhu, S. Control of gradient copolymer composition in ATRP using semibatch feeding policy. AIChE J. 2007, 53, 174–186. [Google Scholar] [CrossRef]
- Diaz-Camacho, F.; Lopez-Morales, S.; Vivaldo-Lima, E.; Saldivar-Guerra, E.; Vera-Graziano, R.; Alexandrova, L. Effect of Regime of Addition of Initiator on TEMPO-Mediated Polymerization of Styrene. Polym. Bull. 2004, 52, 339. [Google Scholar] [CrossRef]
- Roa-Luna, M.; Diaz-Barber, M.P.; Vivaldo-Lima, E.; Lona, L.M.F.; McManus, N.T.; Penlidis, A.; Macromol, J. Assessing the Importance of Diffusion-Controlled Effects on Polymerization Rate and Molecular Weight Development in Nitroxide-Mediated Radical Polymerization of Styrene. Sci. Pure Appl. Chem. 2007, 44, 192. [Google Scholar] [CrossRef]
- Mastan, E.; Zhu, S. Method of moments: A versatile tool for deterministic modeling of polymerization kinetics. Eur. Polym. J. 2015, 68, 139–160. [Google Scholar] [CrossRef]
- Mastan, E.; Li, X.; Zhu, S. Modeling and theoretical development in controlled radical polymerization. Prog. Polym. Sci. 2015, 45, 71–101. [Google Scholar] [CrossRef]
- Arzamendi, G.; Asua, J.M. Monomer addition policies for copolymer composition control in semicontinuous emulsion copolymerization. J. Appl. Polym. Sci. 1989, 38, 2019–2036. [Google Scholar] [CrossRef]
- Guiping, C.; Zhongnan, Z.; Huihui, L.; Minghua, Z. Molecular weight distribution of polystyrene produced in a starved feed reactor. Chinese J. Chem. Eng. 1999, 7, 205–213. [Google Scholar]
- Pinto, J.M.; Giudici, R. Optimization of a cocktail of initiators for suspension polymerization of vinyl chloride in batch reactors. Chem. Eng. Sci. 2001, 56, 1021–1028. [Google Scholar] [CrossRef]
- Li, X.; Wang, W.-J.; Weng, F.; Li, B.-G.; Zhu, S. Targeting copolymer composition distribution via model-based monomer feeding policy in semibatch RAFT mini-emulsion copolymerization of styrene and butyl acrylate. Ind. Eng. Chem. Res. 2014, 53, 7321–7332. [Google Scholar] [CrossRef]
- Meimaroglou, D.; Kiparissides, C. Review of Monte Carlo methods for the prediction of distributed molecular and morphological polymer properties. Ind. Eng. Chem. Res. 2014, 53, 8963–8979. [Google Scholar] [CrossRef]
- Brandão, A.L.T.; Soares, J.B.P.; Pinto, J.C.; Alberton, A.L. When polymer reaction engineers play dice: Applications of Monte Carlo models in PRE. Macromol. React. Eng. 2015, 9, 141–185. [Google Scholar] [CrossRef]
- Bruns, W.; Motoc, I.; O’Driscoll, K.F. Monte Carlo Applications in Polymer Science; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2012; Volume 27. [Google Scholar]
- Khubi-Arani, Z.; Salami-Kalajahi, M.; Najafi, M.; Roghani-Mamaqani, H.; Haddadi-Asl, V.; Ghafelebashi-Zarand, S.M. Simulation of styrene free radical polymerization over bi-functional initiators using Monte Carlo simulation method and comparison with mono-functional initiators. Polym. Sci. Ser. B 2010, 52, 184–192. [Google Scholar] [CrossRef]
- Tobita, H.; Hatanaka, K. Branched structure formation in free radical polymerization of vinyl acetate. J. Polym. Sci. Part B Polym. Phys. 1996, 34, 671–681. [Google Scholar] [CrossRef]
- Prescott, S.W. Chain-length dependence in living/controlled free-radical polymerizations: Physical manifestation and Monte Carlo simulation of reversible transfer agents. Macromolecules 2003, 36, 9608–9621. [Google Scholar] [CrossRef]
- Pintos, E.; Sarmoria, C.; Brandolin, A.; Asteasuain, M. Modeling of RAFT Polymerization Processes Using an Efficient Monte Carlo Algorithm in Julia. Ind. Eng. Chem. Res. 2016, 55, 8534–8547. [Google Scholar] [CrossRef]
- Mohammadi, Y.; Najafi, M.; Haddadi-Asl, V. Comprehensive study of free radical copolymerization using a Monte Carlo simulation method, 1. Macromol. Theory Simul. 2005, 14, 325–336. [Google Scholar] [CrossRef]
- Najafi, M.; Haddadi-Asl, V.; Mohammadi, Y. Application of the Monte Carlo simulation method to the investigation of peculiar free-radical copolymerization reactions: Systems with both reactivity ratios greater than unity (rA > 1 and rB > 1). J. Appl. Polym. Sci. 2007, 106, 4138. [Google Scholar] [CrossRef]
- Najafi, M.; Roghani-Mamaqani, H.; Haddadi-Asl, V.; Salami-Kalajahi, M. A simulation of kinetics and chain length distribution of styrene FRP and ATRP: Chain-length-dependent termination. Adv. Polym. Technol. 2011, 30, 257–268. [Google Scholar] [CrossRef]
- Saeb, M.R.; Mohammadi, Y.; Ahmadi, M.; Khorasani, M.M.; Stadler, F.J. A Monte Carlo-based feeding policy for tailoring microstructure of copolymer chains: Reconsidering the conventional metallocene catalyzed polymerization of $α$-olefins. Chem. Eng. J. 2015, 274, 169–180. [Google Scholar] [CrossRef]
- Salami-Kalajahi, M.; Najafi, M.; Haddadi-Asl, V. Application of Monte Carlo simulation method to polymerization kinetics over Ziegler--Natta catalysts. Int. J. Chem. Kinet. 2009, 41, 45–56. [Google Scholar] [CrossRef]
- Najafi, M.; Roghani-Mamaqani, H.; Salami-Kalajahi, M.; Haddadi-Asl, V. A comprehensive Monte Carlo simulation of styrene atom transfer radical polymerization. Chinese J. Polym. Sci. 2010, 28, 483–497. [Google Scholar] [CrossRef]
- Fu, Y.; Cunningham, M.F.; Hutchinson, R.A. Modeling of Nitroxide-Mediated Semibatch Radical Polymerization. Macromol. React. Eng. 2007, 1, 243–252. [Google Scholar] [CrossRef]
- Ellis, M.F.; Taylor, T.W.; Gonzalez, V.; Jensen, K.F. Estimation of the molecular weight distribution in batch polymerization. AIChE J. 1988, 34, 1341–1353. [Google Scholar] [CrossRef]
- Buback, M.; Sandmann, J. Pressure and temperature dependence of the decomposition rate of aliphatic tert-butyl peroxyesters. Zeitschrift für Phys. Chemie 2000, 214, 583. [Google Scholar] [CrossRef]
- Buback, M.; Wittkowski, L. Acid-Induced Decomposition of Di-tert-butyl Peroxide in n-Heptane Solution up to High Temperatures and Pressures. Zeitschrift für Phys. Chemie 1999, 210, 61–81. [Google Scholar] [CrossRef]
- Hui, A.W.; Hamielec, A.E. Thermal polymerization of styrene at high conversions and temperatures. An experimental study. J. Appl. Polym. Sci. 1972, 16, 749–769. [Google Scholar] [CrossRef]
- Buback, M.; Gilbert, R.G.; Hutchinson, R.A.; Klumperman, B.; Kuchta, F.-D.; Manders, B.G.; O’Driscoll, K.F.; Russell, G.T.; Schweer, J. Critically evaluated rate coefficients for free-radical polymerization, 1. Propagation rate coefficient for styrene. Macromol. Chem. Phys. 1995, 196, 3267–3280. [Google Scholar] [CrossRef] [Green Version]
- Buback, M.; Kuchta, F.-D. Variation of the propagation rate coefficient with pressure and temperature in the free-radical bulk polymerization of styrene. Macromol. Chem. Phys. 1995, 196, 1887–1898. [Google Scholar] [CrossRef]
- Brandrup, J.; Immergut, E.H.; Grulke, E.A. Polymer Handbook, 4th ed.; Wiley: New York, NY, USA, 1999; ISBN 0471166286. [Google Scholar]
- Chen, C.-Y.; Wu, Z.-Z.; Kuo, J.-F. Determination of the mode of termination in free-radical copolymerization. Polym. Eng. Sci. 1987, 27, 553–557. [Google Scholar] [CrossRef]
- Schmidt, A.D.; Ray, W.H. The dynamic behavior of continuous polymerization reactors—I: Isothermal solution polymerization in a CSTR. Chem. Eng. Sci. 1981, 36, 1401–1410. [Google Scholar] [CrossRef]
- Gillespie, D.T. Exact stochastic simulation of coupled chemical reactions. J. Phys. Chem. 1977, 81, 2340–2361. [Google Scholar] [CrossRef]
- Matsumoto, M.; Nishimura, T. Mersenne twister: A 623-dimensionally equidistributed uniform pseudo-random number generator. ACM Trans. Model. Comput. Simul. 1998, 8, 3–30. [Google Scholar] [CrossRef] [Green Version]
- AspenTech Methyl Methacrylate Polymerization in Ethyl Acetate. Aspen Polymer Plus User Guide. Examples Application Case B (3rd ed.). 1981. Available online: https://sites.chemengr.ucsb.edu/~ceweb/courses/che184b/aspenplus/PolymersPlusUserGuideVolume2.pdf (accessed on 10 September 2020).
Figure 1.
Semi-batch general free radical polymerization (Case 1) using different number of shots (NOS) feeding policies with initial values = 8.00 mol L–1 and = 0.08 mol L–1. First row: initiator feeding policy for NOS-1 (A1), NOS-2 (B1), NOS-3 (C1), and NOS-4 (D1). Second row: total monomer and initiator conversion for NOS-1 (A2), NOS-2 (B2), NOS-3 (C2), and NOS-4 (D2). Third row: number- and weight-average molecular weight and dispersity versus conversion for NOS-1 (A3), NOS-2 (B3), NOS-3 (C3), and NOS-4 (D3). Fourth row: final molecular weight distribution for NOS-1 (A4), NOS-2 (B4), NOS-3 (C4), and NOS-4 (D4) (solid line), the latter three also presenting the A4 result (dashed line) for comparison.
Figure 1.
Semi-batch general free radical polymerization (Case 1) using different number of shots (NOS) feeding policies with initial values = 8.00 mol L–1 and = 0.08 mol L–1. First row: initiator feeding policy for NOS-1 (A1), NOS-2 (B1), NOS-3 (C1), and NOS-4 (D1). Second row: total monomer and initiator conversion for NOS-1 (A2), NOS-2 (B2), NOS-3 (C2), and NOS-4 (D2). Third row: number- and weight-average molecular weight and dispersity versus conversion for NOS-1 (A3), NOS-2 (B3), NOS-3 (C3), and NOS-4 (D3). Fourth row: final molecular weight distribution for NOS-1 (A4), NOS-2 (B4), NOS-3 (C4), and NOS-4 (D4) (solid line), the latter three also presenting the A4 result (dashed line) for comparison.

Figure 2.
Semi-batch general free radical polymerization (Case 1) using different amount of shots (AOS) feeding policies with initial values = 8.00 mol L–1 and = 0.08 mol L–1. First row: initiator feeding policy for AOS-1 (E1), AOS-2 (F1), AOS-3 (G1),and AOS-4 (H1). Second row: total monomer and initiator consumption versus conversion for AOS-1 (E2), AOS-2 (F2), AOS-3 (G2), and AOS-4 (H2). Third row: number- and weight-average molecular weight and dispersity versus conversion for AOS-1 (E3), AOS-2 (F3), AOS-3 (G3), and AOS-4 (H3). Fourth row: final molecular weight distribution for AOS-1 (E4), AOS-2 (F4), AOS-3 (G4), and AOS-4 (H4) (solid line), where the NOS-1 (A4) result (dashed line from Figure 1) is also presented for comparison.
Figure 2.
Semi-batch general free radical polymerization (Case 1) using different amount of shots (AOS) feeding policies with initial values = 8.00 mol L–1 and = 0.08 mol L–1. First row: initiator feeding policy for AOS-1 (E1), AOS-2 (F1), AOS-3 (G1),and AOS-4 (H1). Second row: total monomer and initiator consumption versus conversion for AOS-1 (E2), AOS-2 (F2), AOS-3 (G2), and AOS-4 (H2). Third row: number- and weight-average molecular weight and dispersity versus conversion for AOS-1 (E3), AOS-2 (F3), AOS-3 (G3), and AOS-4 (H3). Fourth row: final molecular weight distribution for AOS-1 (E4), AOS-2 (F4), AOS-3 (G4), and AOS-4 (H4) (solid line), where the NOS-1 (A4) result (dashed line from Figure 1) is also presented for comparison.

Figure 3.
Semi-batch general free radical polymerization (Case 1) using different (TOS) feeding policies with initial values = 8.00 mol L–1 and = 0.08 mol L–1. First row: initiator feeding policy for TOS-1 (I1), TOS-2 (J1), TOS-3 (K1), and TOS-4 (L1). Second row: total monomer and initiator consumption versus conversion for TOS-1 (I2), TOS-2 (J2), TOS-3 (K2), and TOS-4 (L2). Third row: number-average and weight-average molecular weight and dispersity versus conversion for TOS-1 (I3), TOS-2 (J3), TOS-3 (K3), and TOS-4 (L3). Fourth row: molecular weight distribution for TOS-1 (I4), TOS-2 (J4), TOS-3 (K4), and TOS-4 (L4) (solid line), where the NOS-1 (A4) result (dashed line from Figure 1) is also presented for comparison.
Figure 3.
Semi-batch general free radical polymerization (Case 1) using different (TOS) feeding policies with initial values = 8.00 mol L–1 and = 0.08 mol L–1. First row: initiator feeding policy for TOS-1 (I1), TOS-2 (J1), TOS-3 (K1), and TOS-4 (L1). Second row: total monomer and initiator consumption versus conversion for TOS-1 (I2), TOS-2 (J2), TOS-3 (K2), and TOS-4 (L2). Third row: number-average and weight-average molecular weight and dispersity versus conversion for TOS-1 (I3), TOS-2 (J3), TOS-3 (K3), and TOS-4 (L3). Fourth row: molecular weight distribution for TOS-1 (I4), TOS-2 (J4), TOS-3 (K4), and TOS-4 (L4) (solid line), where the NOS-1 (A4) result (dashed line from Figure 1) is also presented for comparison.

Figure 4.
Final molecular weight distributions in semi-batch free radical polymerization of styrene (Case 2) with = 6.06 mol L–1, = 2.54 mol L–1, = 0.075 mol L–1, and a temperature of 138 °C for the three different initiator feeding policies (insets). Solvent mole fraction about 30%; S refers to solvent. Left column: NOS; middle column: AOS; right column: TOS. For comparison, the top left (NOS-1) result is presented in every other plot as a dotted line.
Figure 4.
Final molecular weight distributions in semi-batch free radical polymerization of styrene (Case 2) with = 6.06 mol L–1, = 2.54 mol L–1, = 0.075 mol L–1, and a temperature of 138 °C for the three different initiator feeding policies (insets). Solvent mole fraction about 30%; S refers to solvent. Left column: NOS; middle column: AOS; right column: TOS. For comparison, the top left (NOS-1) result is presented in every other plot as a dotted line.

Figure 5.
Final molecular weight distributions in semi-batch solution free radical polymerization of methyl methacrylate (Case 3) with = 1.0 mol L–1, = 3.40 mol L–1, = 0.02288 mol L–1, and a temperature of 70 °C for the three different initiator feeding policies (insets). Solvent mole fraction about 75%; S refers to solvent. Left column: NOS; middle column: AOS; right column: TOS. For comparison, the top left (NOS-1) result is presented in every other plot as a dotted line.
Figure 5.
Final molecular weight distributions in semi-batch solution free radical polymerization of methyl methacrylate (Case 3) with = 1.0 mol L–1, = 3.40 mol L–1, = 0.02288 mol L–1, and a temperature of 70 °C for the three different initiator feeding policies (insets). Solvent mole fraction about 75%; S refers to solvent. Left column: NOS; middle column: AOS; right column: TOS. For comparison, the top left (NOS-1) result is presented in every other plot as a dotted line.

Figure 6.
‘Benchmarking’ of simulation data by comparison with ASPEN modeling: conversion versus time data for MMA polymerization with NOS-1 initiation policy.
Figure 6.
‘Benchmarking’ of simulation data by comparison with ASPEN modeling: conversion versus time data for MMA polymerization with NOS-1 initiation policy.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Elementary reactions in the cases considered in this work: general free radical polymerization (Case 1), styrene free radical polymerization in solution (Case 2) [36], and methyl methacrylate free radical polymerization in solution (Case 3) [37].
| Case 1: General Free Radical Polymerization | |
| Initiator decomposition | |
| Chain initiation | |
| Propagation | |
| Termination by combination | |
| Termination by disproportionation | |
| Case 2: Free Radical Polymerization of Styrene | |
| Chemical Initiation | |
| Thermal Initiation | |
| Propagation | |
| Termination by combination | |
| Termination by disproportionation | |
| Chain transfer to monomer | |
| Chain transfer to solvent | |
| Case 3: Free Radical Polymerization of Methyl Methacrylate | |
| Chemical Initiation | |
| Propagation | |
| Termination by disproportionation | |
| Chain transfer to monomer | |
| Chain transfer to solvent | |
Table 2.
Rate parameters used in the simulations.
| Parameters | Value/Expression | References |
|---|---|---|
| Case 1: General Free Radical Polymerization | ||
| f | 0.50 | This work |
| kd (s−1) | 10−3 | This work |
| ki (L.mol−1.s−1) | 104 | This work |
| Kp (L.mol−1.s−1) | 104 | This work |
| ktc (L.mol−1.s−1) | 108 | This work |
| ktd (L.mol−1.s−1) | 108 | This work |
| Case 2: Free Radical Polymerization of Styrene | ||
| f | 0.50 | [38,39] |
| kd (s−1) | 6.78 × 1015 exp(−17,714.0 / T) | [38,39] |
| kthermal (L2.mol−2.s−1) | 2.19 × 105 exp(−13,800.0 / T) | [40] |
| Kp (L.mol−1.s−1) | 4.266 × 107 exp(−3910.0 / T) | [41] |
| kt,0 (L.mol−1.s−1) | 3.82 × 109 exp(−958.0 / T) | [42] |
| KtrM (L.mol−1.s−1) | 2.31 × 106 exp(−6377.0 / T) | [40] |
| KtrS (L.mol−1.s−1) | 1.8 | [43] |
| Kt (L.mol−1.s−1) | [10] | |
| ktc (L.mol−1.s−1) | 0.01kt | [44] |
| ktd (L.mol−1.s−1) | 0.99kt | [44] |
| Note: wp is the polymer weight fraction. | ||
| Case 3: Free Radical Polymerization of Methyl Methacrylate | ||
| f | 0.50 | [45] |
| kd (s−1) | 1.2525 × 1014 exp(−14,770.0 / T) | [43] |
| Kp (L.mol−1.s−1) | 4.92 × 105 exp(−2191.0 / T) | [45] |
| ktrM (L.mol−1.s−1) | 7.177 × 109 exp(−9036.0 / T) | [43] |
| ktrS (L.mol−1.s−1) | 4.673 × 108 exp(−7902.0 / T) | [43] |
| ktd (L.mol−1.s−1) | 9.8 × 107 exp(−353.0 / T) | [45] |
Table 3.
Initiator feeding policy configurations with the same total amount of initiator.
| Number of Shots | Initiator Fraction |
| NOS-1 | One step (1.000) |
| NOS-2 | Two steps (0.500, 0.500) |
| NOS-3 | Four steps (0.250, 0.250, 0.250, 0.250) |
| NOS-4 | Eight steps (0.125, 0.125, 0.125, 0.125, 0.125, 0.125, 0.125, 0.125) |
| Amount of Shots | Initiator Fraction |
| AOS-1 | (0.10, 0.20, 0.30, 0.40) |
| AOS-2 | (0.40, 0.30, 0.20, 0.10) |
| AOS-3 | (0.10, 0.40, 0.40, 0.10) |
| AOS-4 | (0.40, 0.10, 0.10, 0.40) |
| Time of Shots | Conversion of Each Shot (Initiator Fraction is Equal to 0.25 for Each Step) |
| TOS-1 | (0.00, 0.05, 0.15, 0.25) |
| TOS-2 | (0.00, 0.30, 0.40, 0.50) |
| TOS-3 | (0.00, 0.55, 0.65, 0.75) |
| TOS-4 | (0.00, random, random, random) |
Table 4.
Simulated data of free radical polymerization of styrene (Case 2) according to various initiator feeding policies (NOS, AOS, and TOS as per Table 3, with an equal total amount of initiator).
Table 4.
Simulated data of free radical polymerization of styrene (Case 2) according to various initiator feeding policies (NOS, AOS, and TOS as per Table 3, with an equal total amount of initiator).
| Policy | Reaction Time at Conversion 100% (min) | Đ | Unreacted Initiator (%) | |
|---|---|---|---|---|
| Number of Shots | ||||
| NOS-1 | 42 | 22,000 | 2.1 | 3.5 |
| NOS-2 | 39 | 27,000 | 2.4 | 6.7 |
| NOS-3 | 39 | 31,000 | 2.7 | 9.3 |
| NOS-4 | 39 | 34,000 | 2.9 | 11 |
| Amount of Shots | ||||
| AOS-1 | 42 | 41,000 | 3.4 | 12.3 |
| AOS-2 | 39 | 27,000 | 2.4 | 6.6 |
| AOS-3 | 43 | 37,000 | 3.2 | 8.3 |
| AOS-4 | 37 | 30,000 | 2.6 | 10.6 |
| Time of Shots | ||||
| TOS-1 | 44 | 24,000 | 2.3 | 3.7 |
| TOS-2 | 40 | 30,000 | 2.7 | 7.4 |
| TOS-3 | 41 | 38,000 | 3.2 | 12.8 |
| TOS-4 | 40 | 27,000 | 2.5 | 6.4 |
Table 5.
Simulated data of free radical polymerization of methyl methacrylate in solution (Case 3) according to the various initiator feeding policies (NOS, AOS, and TOS of Table 3, with an equal total amount of initiator).
Table 5.
Simulated data of free radical polymerization of methyl methacrylate in solution (Case 3) according to the various initiator feeding policies (NOS, AOS, and TOS of Table 3, with an equal total amount of initiator).
| Policy | Reaction Time at Conversion 100% (min) | Đ | Unreacted Initiator (%) | |
|---|---|---|---|---|
| Number of Shots | ||||
| NOS-1 | 1700 | 19,000 | 4.2 | 7.4 |
| NOS-2 | 1650 | 24,500 | 5.3 | 9.5 |
| NOS-3 | 1600 | 30,000 | 6.2 | 11.2 |
| NOS-4 | 1600 | 33,000 | 7.0 | 12.5 |
| Amount of Shots | ||||
| AOS-1 | 1600 | 40,000 | 8.4 | 13.3 |
| AOS-2 | 1600 | 25,000 | 5.4 | 9.5 |
| AOS-3 | 1650 | 37,000 | 7.9 | 11.0 |
| AOS-4 | 1600 | 27,000 | 5.7 | 11.6 |
| Time of Shots | ||||
| TOS-1 | 1700 | 22,000 | 4.9 | 8.1 |
| TOS-2 | 1650 | 29,000 | 6.3 | 9.9 |
| TOS-3 | 1600 | 34,000 | 7.2 | 13.1 |
| TOS-4 | 1630 | 25,500 | 5.5 | 9.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Seyedi, A.; Najafi, M.; Russell, G.T.; Mohammadi, Y.; Vivaldo-Lima, E.; Penlidis, A. Initiator Feeding Policies in Semi-Batch Free Radical Polymerization: A Monte Carlo Study. Processes 2020, 8, 1291. https://doi.org/10.3390/pr8101291
AMA Style
Seyedi A, Najafi M, Russell GT, Mohammadi Y, Vivaldo-Lima E, Penlidis A. Initiator Feeding Policies in Semi-Batch Free Radical Polymerization: A Monte Carlo Study. Processes. 2020; 8(10):1291. https://doi.org/10.3390/pr8101291
Chicago/Turabian StyleSeyedi, Ali, Mohammad Najafi, Gregory T. Russell, Yousef Mohammadi, Eduardo Vivaldo-Lima, and Alexander Penlidis. 2020. "Initiator Feeding Policies in Semi-Batch Free Radical Polymerization: A Monte Carlo Study" Processes 8, no. 10: 1291. https://doi.org/10.3390/pr8101291
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.