Comparative Physiological and Proteomic Analysis Reveals Different Involvement of Proteins during Artificial Aging of Siberian Wildrye Seeds

 , ,
, ,
Abstract
:1. Introduction
2. Results
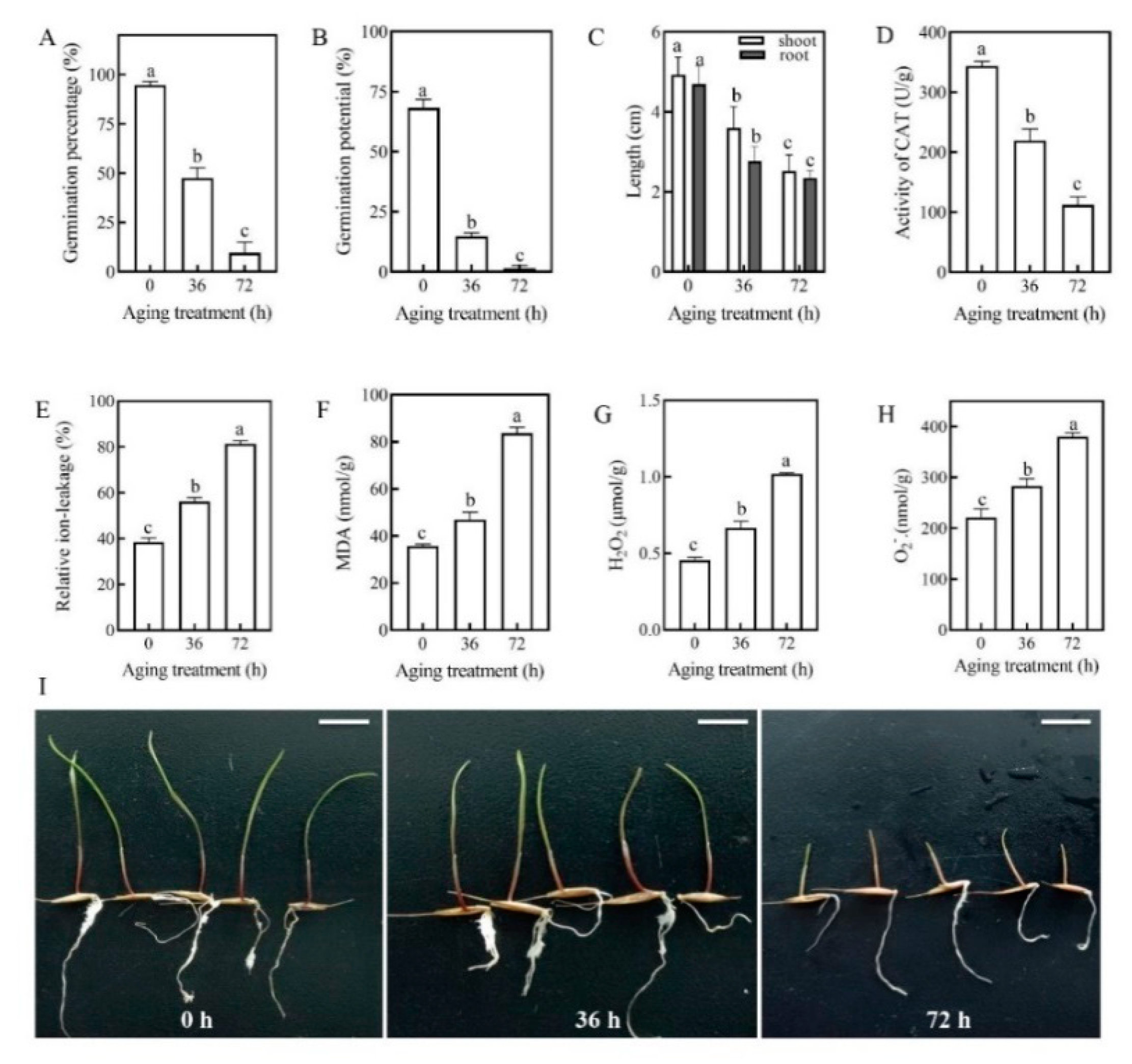
2.1. Seed Germination Characteristics and Physio-Biochemical Changes of Artificial Aging Treatments (AAT)
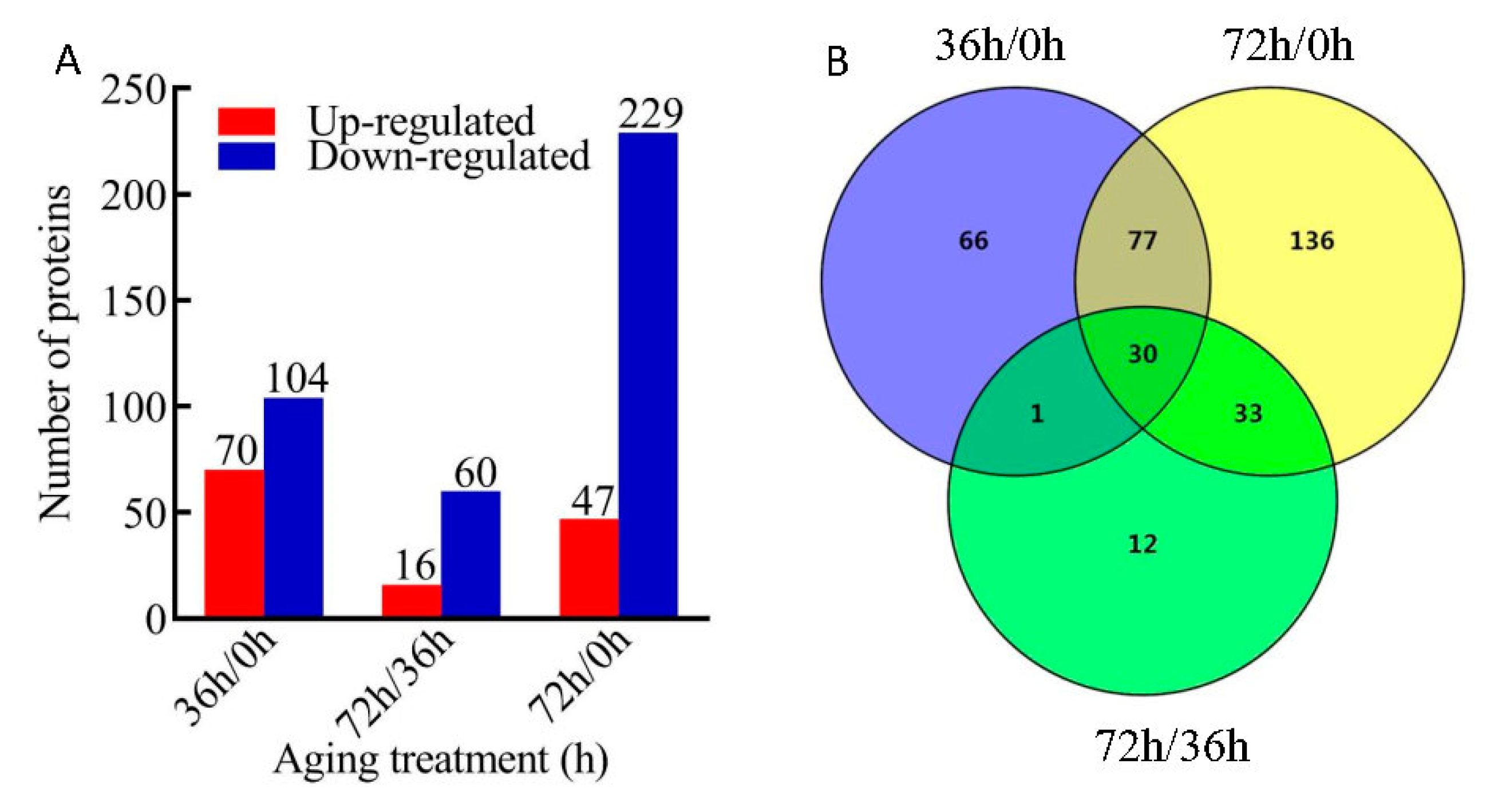
2.2. Changes in Protein Abundance after AAT
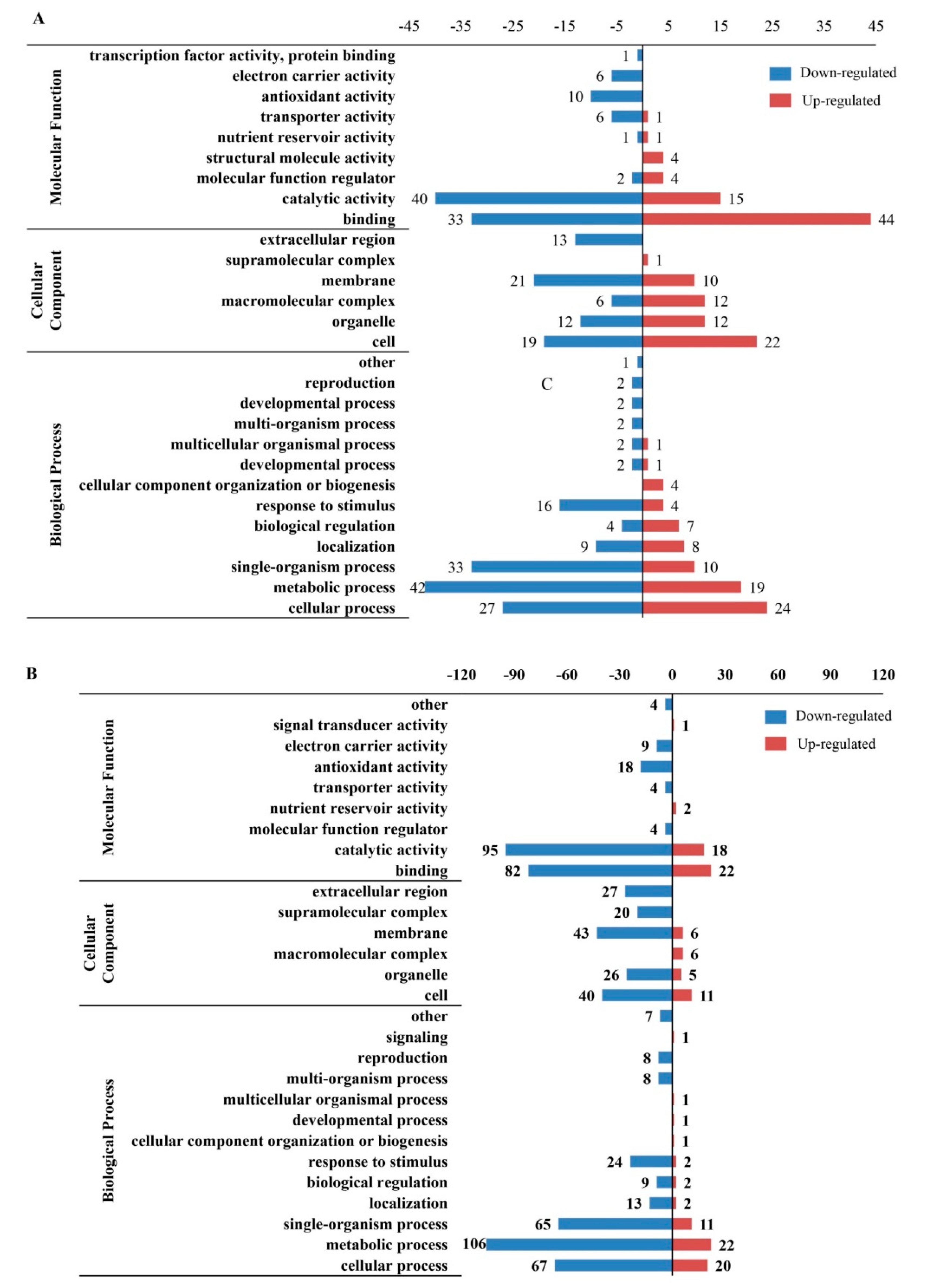
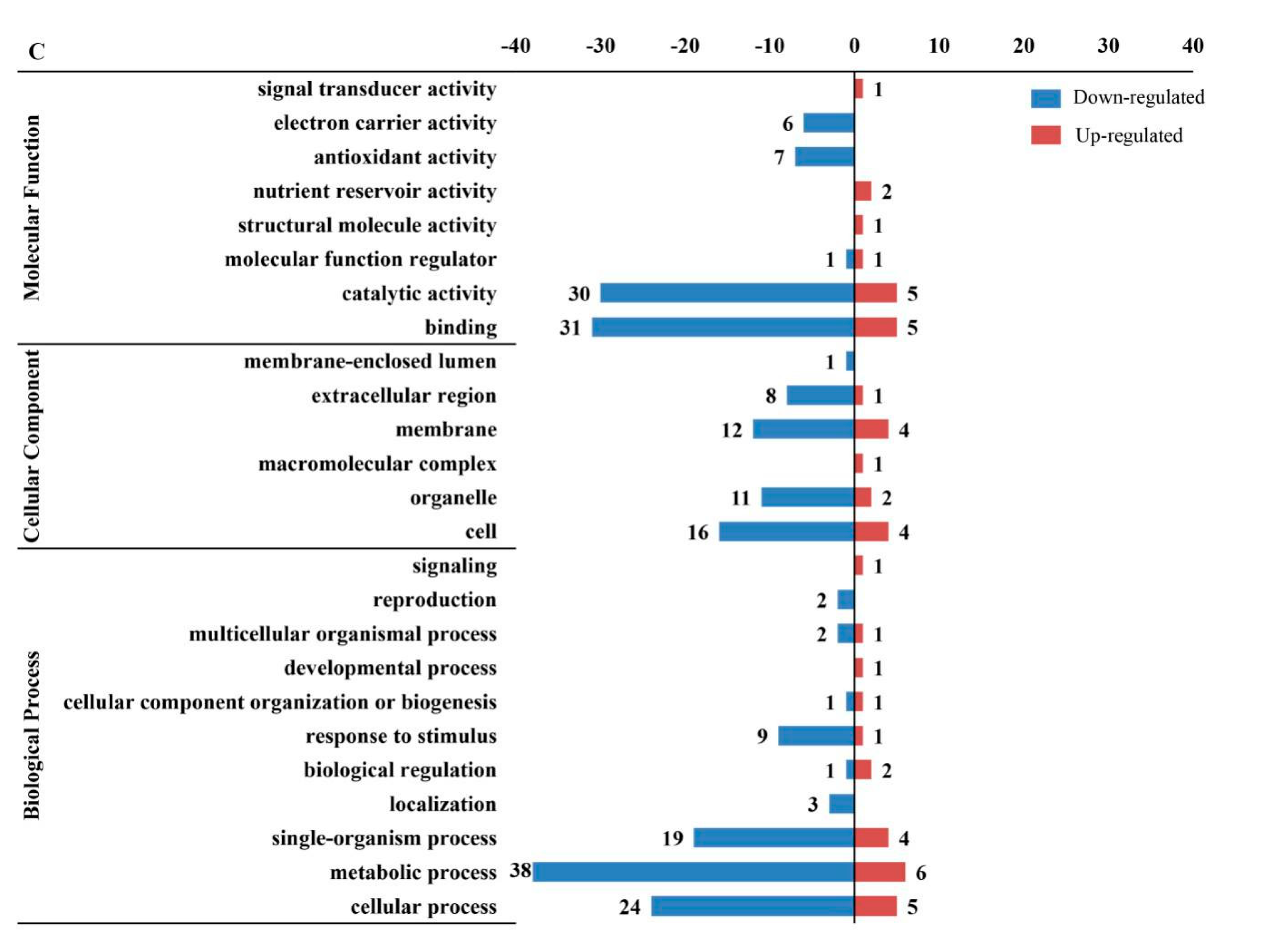
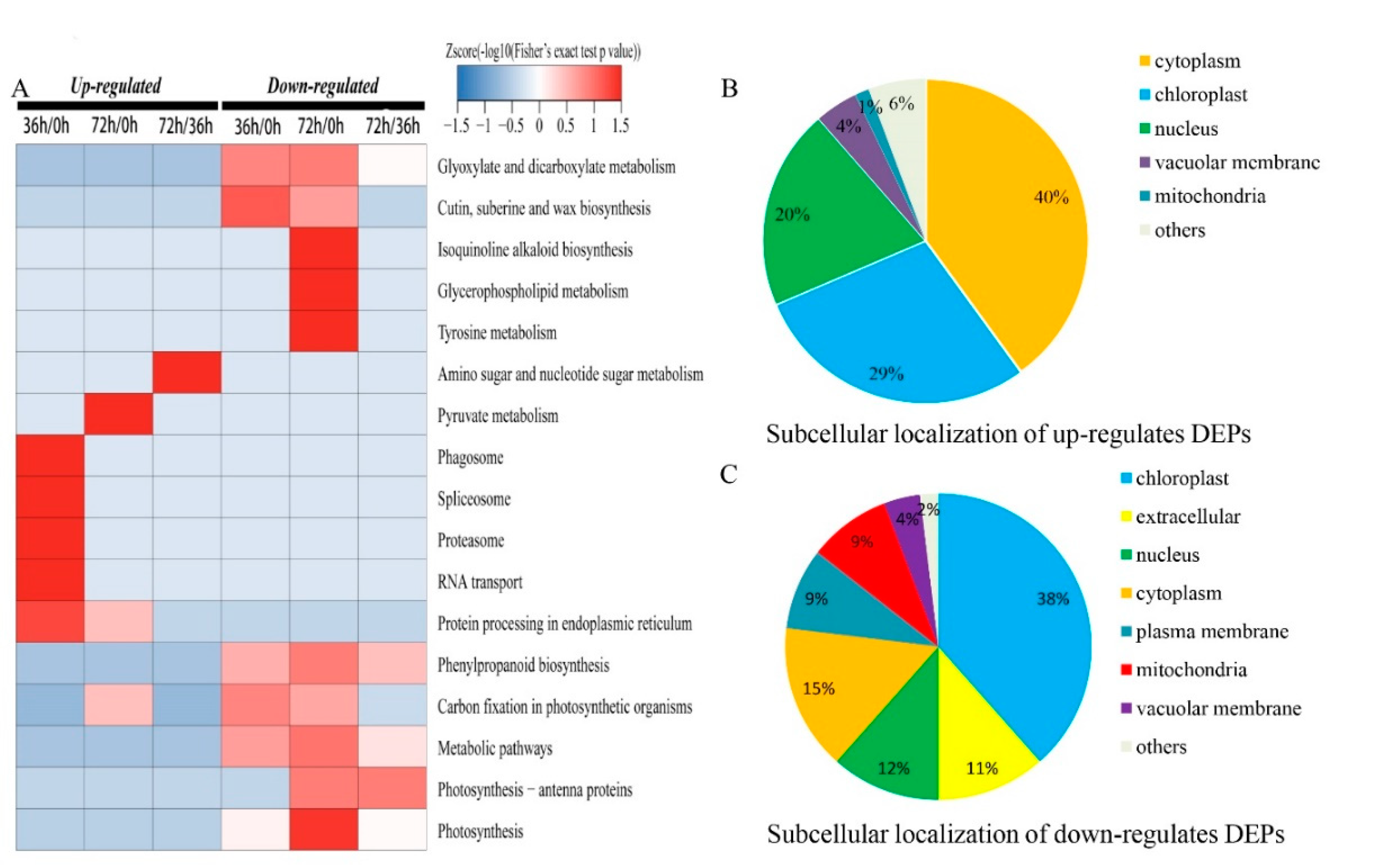
2.3. Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG) and Subcellular Location Analyses of Differentially Expressed Proteins (DEPs) in Response to Seed Aging
2.4. Key Proteins among DEPs in Response to AAT
2.5. Candidate Functional Protein Selections and Parallel Reaction Monitoring (PRM) Validation
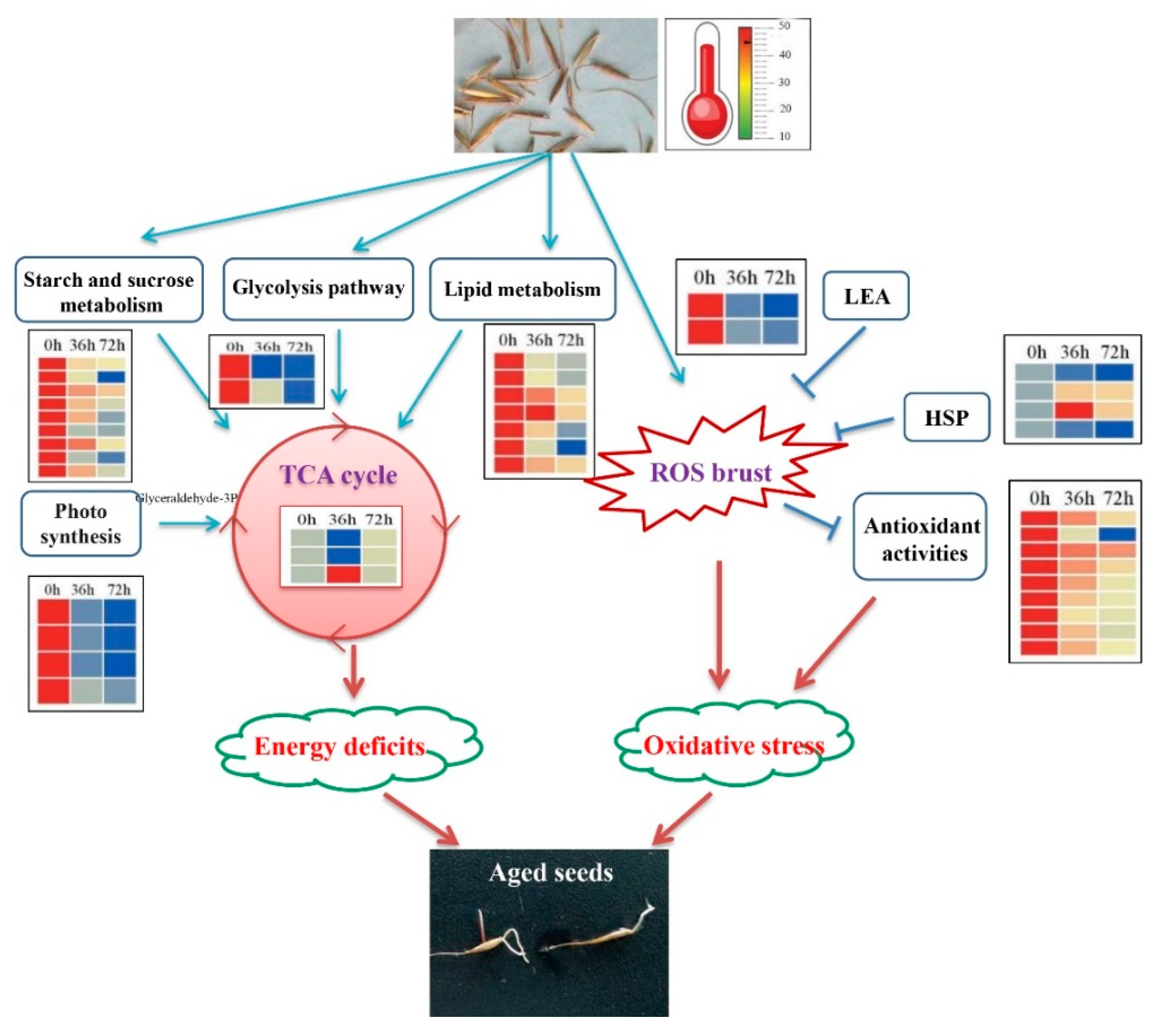
3. Discussion
3.1. Changes of Carbohydrate Metabolism-Related DEPs during AAT
3.2. Changes of Lipid Metabolism-Related Proteins during AAT
3.3. Antioxidant Activities Responded to AAT
3.4. The Role of Stress-Related Proteins during AAT
4. Materials and Methods
4.1. Plant Materials
4.2. Artificial Aging Treatment and Germination Test
4.3. Monitoring the Relative Ion-Leakage and Malondialdehyde Content
4.4. Measurement of Catalase (CAT) Activity and Reactive Oxygen Species (ROS) Content
4.5. Protein Extraction and Trypsin Digestion
4.6. Tandem Mass Tags (TMT) Labeling, High-Performance Mass Chromatography (HPLC) Fractionation and Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS) Analysis
4.7. MS/MS Database Search and Bioinformatics Analysis
4.8. Parallel Reaction Monitoring Validations
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ABA | abscisic acid |
| CAT | catalase |
| GA3 | gibberellin |
| GAPDH | glyceraldehyde triphosphate glyceraldehyde dehydrogenase |
| GO | Gene Ontology |
| GST | glutathione-S-transferase |
| H2O2 | hydrogen peroxide |
| Hsps | heat shock proteins |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| LC-MS/MS | liquid chromatography-tandem mass spectrometry |
| LEAs | late embryogenesis abundant proteins |
| LOX | lipoxygenase |
| MDA | malondialdehyde |
| superoxide anion | |
| -OH | hydroxyl radicals |
| PRM | parallel reaction monitoring |
| RIL | relative ion-leakage |
| ROS | reactive oxygen species |
| RuBisCO | Ribulose-1,5-bisphosphate carboxylase/oxygenase |
| SDH | succinate dehydrogenase |
| SOD | superoxide dismutase |
| TMT | tandem mass tags |
References
- Zhou, W.; Chen, F.; Luo, X.; Dai, Y.; Yang, Y.; Zheng, C.; Yang, W.; Shu, K. A matter of life and death: Molecular, physiological, and environmental regulation of seed longevity. Plant Cell Environ. 2019, 43, 293–302. [Google Scholar] [CrossRef]
- Dantas, A.F.; Fascineli, M.L.; José, S.C.B.R.; Pádua, J.G.; Grisolia, C.K. Loss of genetic integrity in artificially aged seed lots of rice (Oryza sativa L.) and common bean (Phaseolus vulgaris L.). Mutat. Res. Genet. Toxicol. Environ. 2019, 846, 403080. [Google Scholar] [CrossRef]
- Liu, X.; Chen, Z.; Gao, Y.; Liu, Q.; Zhou, W.; Zhao, T.; Jiang, W.; Cui, X.; Cui, J.; Wang, Q. Combinative effects of Azospirillum brasilense inoculation and chemical priming on germination behavior and seedling growth in aged grass seeds. PLoS ONE 2019, 14, e0210453. [Google Scholar] [CrossRef] [Green Version]
- Yan, H.; Jia, S.; Mao, P. Melatonin priming alleviates aging-induced germination inhibition by regulating β-oxidation, protein translation, and antioxidant metabolism in oat (Avena sativa L.) seeds. Int. J. Mol. Sci. 2020, 21, 1898. [Google Scholar] [CrossRef] [Green Version]
- Parkhey, S.; Naithani, S.C.; Keshavkant, S. Protein metabolism during natural ageing in desiccating recalcitrant seeds of Shorea robusta. Acta Physiol. Plant. 2014, 36, 1649–1659. [Google Scholar] [CrossRef]
- Yin, X.; He, D.; Gupta, R.; Yang, P. Physiological and proteomic analyses on artificially aged Brassica napus seed. Front. Plant Sci. 2015, 6, 112. [Google Scholar] [CrossRef] [Green Version]
- Kurek, K.; Plitta-Michalak, B.; Ratajczak, E. Reactive oxygen species as potential drivers of the seed aging process. Plants 2019, 8, 174. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Wang, Q.; Karagić, D.; Liu, X.; Cui, J.; Gui, J.; Gu, M.; Gao, W. Effects of ultrasonication on increased germination and improved seedling growth of aged grass seeds of tall fescue and Russian wildrye. Sci. Rep. UK 2016, 6, 22403. [Google Scholar] [CrossRef] [Green Version]
- Khan, F.; Hussain, S.; Tanveer, M.; Khan, S.; Hussain, H.A.; Iqbal, B.; Geng, M. Coordinated effects of lead toxicity and nutrient deprivation on growth, oxidative status, and elemental composition of primed and non-primed rice seedlings. Environ. Sci. Pollut. Res. 2018, 25, 21185–21194. [Google Scholar] [CrossRef]
- Rajjou, L.; Lovigny, Y.; Groot, S.P.C.; Belghazi, M.; Job, C.; Job, D. Proteome-wide characterization of seed aging in Arabidopsis: A comparison between artificial and natural aging protocols. Plant Physiol. 2008, 148, 620–641. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Fu, H.; Zhou, X.; Chen, Z.; Luo, Y.; Cui, B.; Chen, G.; Liu, J. Comparative proteomic analysis of seed embryo proteins associated with seed storability in rice (Oryza sativa L) during natural aging. Plant Physiol. Bioch. 2016, 103, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Chen, Q.; Kong, L.; Xia, F.; Yan, H.; Zhu, Y.; Mao, P. Proteomic and physiological analysis of the response of oat (Avena sativa) seeds to heat stress under different moisture conditions. Front. Plant Sci. 2016, 7, 896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petla, B.P.; Kamble, N.U.; Kumar, M.; Verma, P.; Ghosh, S.; Singh, A.; Rao, V.; Salvi, P.; Kaur, H.; Saxena, S.C.; et al. Rice proteinl L-isoaspartyl methyltransferase isoforms differentially accumulate during seed maturation to restrict deleterious isoAsp and reactive oxygen species accumulation and are implicated in seed vigor and longevity. New Phytol. 2016, 211, 627–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawashima, Y.; Watanabe, E.; Umeyama, T.; Nakajima, D.; Hattori, M.; Honda, K.; Ohara, O. Optimization of data-independent acquisition mass spectrometry for deep and highly sensitive proteomic analysis. Int. J. Mol. Sci. 2019, 20, 5932. [Google Scholar] [CrossRef] [Green Version]
- Van Ulsen, P.; Kuhn, K.; Prinz, T.; Legner, H.; Schmid, P.; Baumann, C.; Tommassen, J. Identification of proteins of Neisseria meningitidis induced under iron-limiting conditions using the isobaric tandem mass tag (TMT) labeling approach. Proteomics (Weinheim) 2009, 9, 1771–1781. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, Y.; Xu, Y.; Zhang, X.; Peng, Y.; Ma, X.; Huang, L.; Yan, Y. Physiological and iTRAQ-based proteomic analyses reveal the function of spermidine on improving drought tolerance in white clover. J. Proteome Res. 2016, 15, 1563–1579. [Google Scholar] [CrossRef]
- Lv, Y.; Zhang, S.; Wang, J.; Hu, Y. Quantitative proteomic analysis of wheat seeds during artificial ageing and priming using the isobaric tandem mass tag labeling. PLoS ONE 2016, 11, e0162851. [Google Scholar] [CrossRef]
- Zhao, Z.; Liu, J.; Jia, R.; Bao, S.; Chen, X. Physiological and TMT-based proteomic analysis of oat early seedlings in response to alkali stress. J. Proteom. 2019, 193, 10–26. [Google Scholar] [CrossRef]
- Ma, X.; Zhang, X.; Zhou, Y.; Bai, S.; Liu, W. Assessing genetic diversity of Elymus sibiricus (Poaceae: Triticeae) populations from Qinghai-Tibet Plateau by ISSR markers. Biochem. Syst. Ecol. 2008, 36, 514–522. [Google Scholar] [CrossRef]
- Li, P.; Bai, S.; You, M.; Shen, Y. Effects of maturity stage and lactic acid bacteria on the fermentation quality and aerobic stability of Siberian wildrye silage. Food Sci. Nutr. 2016, 4, 664–670. [Google Scholar] [CrossRef]
- Yan, H.; Mao, P.; Sun, Y.; Li, M. Impacts of ascorbic acid on germination, antioxidant enzymes and ultrastructure of embryo cells of aged Elymus sibiricus seeds with different moisture contents. Int. J. Agric. Biol. 2015, 18, 176–183. [Google Scholar] [CrossRef]
- Yan, H.; Mao, P. Optimizing the accelerated ageing condition of Siberian wildrye seeds. Seed 2013, 32, 1–6. [Google Scholar]
- Xia, F.; Cheng, H.; Chen, L.; Zhu, H.; Mao, P.; Wang, M. Influence of exogenous ascorbic acid and glutathione priming on mitochondrial structural and functional systems to alleviate aging damage in oat seeds. BMC Plant Biol. 2020, 20, 104. [Google Scholar] [CrossRef] [Green Version]
- Huang, F.; Li, J.; Li, H.; Liu, L.; Shi, W.; Li, Z. Assessment of genetic integrity of Siberian wild rye (Elymus sibiricus L.) germplasm conserved ex situ and under accelerated aging using microsatellite markers. Genet. Resour. Crop. Evol. 2020, 67, 367–379. [Google Scholar] [CrossRef]
- Lehner, A.; Mamadou, N.; Poels, P.; Côme, D.; Bailly, C.; Corbineau, F. Changes in soluble carbohydrates, lipid peroxidation and antioxidant enzyme activities in the embryo during ageing in wheat grains. J. Cereal Sci. 2008, 47, 555–565. [Google Scholar] [CrossRef]
- Plaxton, W.C. The organization and regulation of plant glycolysis. Annu. Rev. Plant Phys. 1996, 47, 185–214. [Google Scholar] [CrossRef] [Green Version]
- He, D.; Yang, P. Proteomics of rice seed germination. Front. Plant Sci. 2013, 4, 246. [Google Scholar] [CrossRef] [Green Version]
- Kannababu, N.; Karivaratharaju, T.V. Effect of seed ageing on malate dehydrogenase and succinate dehydrogenase activity in seedling of sunflower. Indian J. Plant Physiol. 2000, 5, 397–399. [Google Scholar]
- Jardim, M.D.; Caverzan, A.; Rauber, R.; Ferreira, E.D.S.; Margis, P.M.; Galina, A. Succinate dehydrogenase (mitochondrial complex II) is a source of reactive oxygen species in plants and regulates development and stress responses. New Phytol. 2015, 208, 776–789. [Google Scholar] [CrossRef] [PubMed]
- Feussner, I.; Wasternack, C. The lipoxygenase pathway. Annu. Rev. Plant Biol. 2002, 53, 275–297. [Google Scholar] [CrossRef] [PubMed]
- Oenel, A.; Fekete, A.; Krischke, M.; Faul, S.C.; Gresser, G.; Havaux, M.; Mueller, M.J.; Berger, S. Enzymatic and non-enzymatic mechanisms contribute to lipid oxidation during seed aging. Plant Cell Physiol. 2017, 58, 925–933. [Google Scholar] [CrossRef] [PubMed]
- Mansouri-Far, C.; Goodarzian-Ghahfarokhi, M.; Saeidi, M.; Abdoli, M. Antioxidant enzyme activity and germination characteristics of different maize hybrid seeds during ageing. Environ. Exp. Biol. 2015, 13, 177–182. [Google Scholar]
- Yin, G.; Xin, X.; Song, C.; Chen, X.; Zhang, J.; Wu, S.; Li, R.; Liu, X.; Lu, X. Activity levels and expression of antioxidant enzymes in the ascorbate-glutathione cycle in artificially aged rice seed. Plant Physiol. Biochem. 2014, 80, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Susumu, H.; Katsutomo, S.; Hiroyuki, I.; Yuko, O.; Hirokazu, M. A large family of class III plant peroxidases. Plant Cell Physiol. 2001, 42, 462–468. [Google Scholar]
- Gill, R.S.; Gupta, A.K.; Taggar, G.K.; Taggar, M.S. Review article: Role of oxidative enzymes in plant defenses against insect herbivory. Acta Phytopathol. Acad. Sci. Hung. 2010, 45, 277–290. [Google Scholar] [CrossRef]
- Xia, F.; Wang, X.; Li, M.; Mao, P. Mitochondrial structural and antioxidant system responses to aging in oat (Avena sativa L.) seeds with different moisture contents. Plant Physiol. Biochem. 2015, 94, 122–129. [Google Scholar] [CrossRef]
- Jiang, M.; Zhang, J. Effect of abscisic acid on active oxygen species, antioxidative defence system and oxidative damage in leaves of maize seedlings. Plant Cell Physiol. 2001, 42, 1265–1273. [Google Scholar] [CrossRef]
- Parkhey, S.; Naithani, S.C.; Keshavkant, S. ROS production and lipid catabolism in desiccating Shorea robusta seeds during aging. Plant Physiol. Biochem. 2012, 57, 261–267. [Google Scholar] [CrossRef]
- Hernández Estévez, I.; Rodríguez Hernández, M. Plant Glutathione S-transferases: An overview. Plant Genet. Resour. C 2020, 23, 100233. [Google Scholar]
- Liu, D.; Liu, Y.; Rao, J.; Wang, G.; Li, H.; Ge, F.; Chen, C. Overexpression of the glutathione S-transferase gene from Pyrus pyrifolia fruit improves tolerance to abiotic stress in transgenic tobacco plants. Mol. Biol. 2013, 47, 515–523. [Google Scholar] [CrossRef]
- Kissoudis, C.; Kalloniati, C.; Flemetakis, E.; Madesis, P.; Labrou, N.E.; Tsaftaris, A.; Nianiou-Obeidat, I. Stress-inducible GmGSTU4 shapes transgenic tobacco plants metabolome towards increased salinity tolerance. Acta Physiol. Plant. 2015, 37, 128. [Google Scholar] [CrossRef]
- Cheng, H.; Ma, X.; Jia, S.; Li, M.; Mao, P. Transcriptomic analysis reveals the changes of energy production and AsA-GSH cycle in oat embryos during seed ageing. Plant Physiol. Biochem. 2020, 153, 40–52. [Google Scholar] [CrossRef] [PubMed]
- Rajjou, L.C.; Debeaujon, I. Seed longevity: Survival and maintenance of high germination ability of dry seeds. Comptes Rendus Biol. 2008, 331, 796–805. [Google Scholar] [CrossRef]
- Wang, X.; Zhu, H.; Jin, G.; Liu, H.; Wu, W.; Zhu, J. Genome-scale identification and analysis of LEA genes in rice (Oryza sativa L.). Plant Sci. 2007, 172, 414–420. [Google Scholar] [CrossRef]
- Hundertmark, M.; Buitink, J.; Leprince, O.; Hincha, D.K. The reduction of seed-specific dehydrins reduces seed longevity in Arabidopsis thaliana. Seed Sci. Res. 2011, 21, 165–173. [Google Scholar] [CrossRef]
- Kalemba, E.A.M.; Pukacka, S.A. Possible roles of LEA proteins and HSPs in seed protection:a short review. Biol. Lett. 2007, 44, 3–16. [Google Scholar]
- Jungkunz, I.; Link, K.; Vogel, F.; Voll, L.M.; Sonnewald, S.; Sonnewald, U. AtHsp70-15-deficient Arabidopsis plants are characterized by reduced growth, a constitutive cytosolic protein response and enhanced resistance to TuMV. Plant J. 2011, 66, 983–995. [Google Scholar] [CrossRef]
- Wang, R.; Zhang, Y.; Kieffer, M.; Yu, H.; Kepinski, S.; Estelle, M. HSP90 regulates temperature-dependent seedling growth in Arabidopsis by stabilizing the auxin co-receptor F-box protein TIR1. Nat. Commun. 2016, 7, 10269. [Google Scholar] [CrossRef]
- Queitsch, C.; Hong, S.W.; Vierling, E.; Lindquist, S. Heat shock protein 101 plays a crucial role in thermotolerance in Arabidopsis. Plant Cell 2000, 12, 479–492. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.; He, D.; Teng, H.; Chena, L.; Song, H.; Huang, Q. Physiological and proteomic analyses of coix seed aging during storage. Food Chem. 2018, 260, 82–89. [Google Scholar] [CrossRef]
- Tabita, F.R.; Hanson, T.E.; Li, H.; Satagopan, S.; Chan, S. Function, structure, and evolution of the RubisCO-like proteins and their RubisCO homologs. Microbiol. Mol. Biol. Rev. 2008, 71, 576–599. [Google Scholar] [CrossRef] [Green Version]
- Wei, L.; Wang, Q.; Xin, Y.; Lu, Y.; Xu, J. Enhancing photosynthetic biomass productivity of industrial oleaginous microalgae by overexpression of RuBisCO activase. Algal Res. 2017, 27, 366–375. [Google Scholar] [CrossRef]
- Leitao, L.; Bethenod, O.; Biolley, J.P. The impact of ozone on juvenile maize (Zea mays L.) plant photosynthesis: Effects on vegetative biomass, pigmentation, and carboxylases (PEPc and Rubisco). Plant Biol. 2007, 9, 478–488. [Google Scholar] [CrossRef]
- Gong, B.; Wen, D.; Vandenlangenberg, K.; Wei, M.; Yang, F.; Shi, Q.; Wang, X. Comparative effects of NaCl and NaHCO3 stress on photosynthetic parameters, nutrient metabolism, and the antioxidant system in tomato leaves. Sci. Hortic. Amst. 2013, 157, 1–12. [Google Scholar] [CrossRef]
- Xu, X.; Liu, T.; Yang, J.; Chen, L.; Liu, B.; Wang, L.; Jin, Q. The first whole-cell proteome and lysine-acetylome-based comparison between trichophyton rubrum conidial and mycelial stages. J. Proteome Res. 2018, 17, 1436–1451. [Google Scholar] [CrossRef]
- Kim, H.J.; Lin, D.; Lee, H.J.; Li, M.; Liebler, D.C. Quantitative profiling of protein tyrosine kinases in human cancer cell lines by multiplexed parallel reaction monitoring assays. Mol. Cell. Proteom. 2016, 15, 682–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathway | Accession No. | Protein Name | Log2 (Fold Change) | ||
|---|---|---|---|---|---|
| 36 h/0 h | 72 h/0 h | 72 h/36 h | |||
| glycolysis pathway | A0A1D5TTT1 | Glyceraldehyde-3-phosphate dehydrogenase | −1.154c | −1.109 | 0.045 |
| A0A1D5YL69 | Glyceraldehyde-3-phosphate dehydrogenase | −0.545 | −1.092 | −0.546 | |
| tricarboxylic acid cycle (TCA) | A0A1D6CTY7 | Succinate dehydrogenase subunit 6 | −0.369 | −0.444 | −0.075 |
| A0A1D6D5I2 | Acyl-CoA-binding protein-like | −0.379 | −0.307 | 0.072 | |
| W5FCI5 | Phosphoenolpyruvate carboxylase 2 | 0.516 | 0.561 | 0.045 | |
| starch and sucrose metabolism | W5B7S6 | Probable UDP-arabinose 4-epimerase 2 | −0.431 | −0.484 | −0.052 |
| Q1ERG1 | Endo-beta-1,3-glucanase | −0.528 | −1.234 | −0.706 | |
| A0A1D5UYF9 | Glucan endo-1,3-beta-glucosidase 3-like isoform X1 | −0.25 | −0.387 | −0.136 | |
| W5EDL3 | Fructose-bisphosphate aldolase | −0.345 | −0.576 | −0.23 | |
| A0A1D5Z7K1 | Predicted: beta-glucosidase BoGH3B-like | −0.318 | −0.802 | −0.484 | |
| A0A1D6S663 | Beta-glucosidase 31-like | −0.667 | −0.76 | −0.093 | |
| A0A1D5ZSY5 | Glucan endo-1,3-beta-glucosidase 4-like isoform X2 | −0.165 | −0.465 | −0.299 | |
| A0A1D5TQZ5 | Serine/threonine-protein kinase | −0.666 | −1.005 | −0.339 | |
| A0A1D6DGU9 | Glucan endo-1,3-beta-glucosidase GII-like | −0.3 | −0.589 | −0.289 | |
| lipid metabolism | A0A1D6CGK6 | Lipoxygenase | −0.536 | −0.664 | −0.128 |
| A0A1D6CX16 | Non-specific lipid transfer protein GPI-anchored 1 | −0.49 | −0.697 | −0.207 | |
| A0A077RZ37 | Glycerophosphodiester phosphodiesterase GDPD6-like | −0.175 | −0.424 | −0.248 | |
| A0A0A0S1W1 | 12-oxo-phytodienoic acid reductase 2 | 0.01 | −0.396 | −0.405 | |
| A0A1B5GFV1 | Caleosin | −0.378 | −0.828 | −0.45 | |
| A0A1B5GFV6 | Caleosin | −0.548 | −1.207 | −0.659 | |
| A0A1D6B2R1 | Diacylglycerol kinase | −0.294 | −0.406 | −0.113 | |
| antioxidant activities | A0A1D5V3N8 | Peroxidase | −0.353 | −0.743 | −0.391 |
| A0A1D5WDA8 | Peroxidase | −0.946 | −3.095 | −2.148 | |
| A0A1D6CQN7 | Peroxidase | −0.269 | −0.387 | −0.118 | |
| A0A1D6CV94 | Peroxidase | −0.396 | −0.717 | −0.321 | |
| W4ZYX8 | Peroxidase | −0.448 | −0.901 | −0.453 | |
| A0A1D5WT84 | Peroxidase | −0.623 | −0.896 | −0.273 | |
| W5FEC7 | Peroxidase | −0.783 | −0.923 | −0.14 | |
| W5G6B5 | Peroxidase | −0.575 | −0.997 | −0.422 | |
| A0A1D6CAM6 | Probable glutathione S-transferase BZ2 | −0.507 | −0.855 | −0.348 | |
| anti-stress protein | A0A024CKY0 | LEA protein | −0.555 | −0.683 | −0.128 |
| A0A1D5ZWT3 | 11 kDa LEA protein-like | −0.487 | −0.561 | −0.073 | |
| F4Y590 | Heat shock protein 90 | 0.51 | 0.288 | −0.222 | |
| Q9SPH4 | Heat shock protein 101 | 0.448 | 0.418 | −0.03 | |
| W5FD68 | Chaperone protein ClpB1-like isoform X1 | 0.89 | 0.436 | −0.454 | |
| A0A1D6DGU9 | Heat shock 70 kDa protein 14-like | 0.407 | 0.155 | −0.252 | |
| A0A1D5Z4N3 | Ribulose bisphosphate carboxylase small chain | −1.115 | −1.428 | −0.313 | |
| Protein Accession | Protein Description | 36 h/0 h Ratio (TMT) | 36 h/0 h Ratio (PRM) | 72 h/0 h Ratio (TMT) | 72 h/0 h Ratio (PRM) |
|---|---|---|---|---|---|
| A0A1D6B2D8 | Carboxypeptidase | 0.84 | 0.82 | 0.73 | 0.67 |
| W5AC28 | Glutathione-S-transferase DHAR2-like | 0.69 | 0.64 | 0.74 | 0.81 |
| Q8GVD3 | Thioredoxin | 0.74 | 0.67 | 0.79 | 0.72 |
| A0A1D6RL87 | 26S proteasome non-ATPase Regulatory subunit 1 homolog A-like isoform X4 | 1.37 | 1.50 | 1.24 | 1.46 |
| Q2PCD2 | Non-specific lipid-transfer protein | 0.81 | 0.87 | 0.75 | 0.72 |
| A0A1D5WTN1 | Predicted: transmembrane protein 214-B | 0.89 | 0.77 | 0.76 | 0.73 |
| A0A1D5Z9W3 | Elongation factor 2-like | 0.84 | 1.34 | 0.73 | 1.31 |
| P11383 | Ribulose bisphosphate carboxylase large chain | 0.34 | 0.16 | 0.21 | 0.09 |
| P12112 | Adenosine triphosphate (ATP) synthase subunit alpha, chloroplastic | 0.93 | 1.08 | 0.76 | 0.84 |
| F4Y590 | Heat shock protein 90 | 1.42 | 1.56 | 1.22 | 1.40 |
| A0A1D5ZWT3 | 11 kDa late embryogenesis abundant protein-like | 0.71 | 0.72 | 0.68 | 0.71 |
| A0A1D6C2V8 | Transmembrane protein 120 homolog | 0.86 | 0.77 | 0.75 | 0.63 |
| W5D591 | Small ubiquitin-related modifier | 0.79 | 0.79 | 0.67 | 0.65 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lei, X.; Liu, W.; Zhao, J.; You, M.; Xiong, C.; Xiong, Y.; Xiong, Y.; Yu, Q.; Bai, S.; Ma, X. Comparative Physiological and Proteomic Analysis Reveals Different Involvement of Proteins during Artificial Aging of Siberian Wildrye Seeds. Plants 2020, 9, 1370. https://doi.org/10.3390/plants9101370
Lei X, Liu W, Zhao J, You M, Xiong C, Xiong Y, Xiong Y, Yu Q, Bai S, Ma X. Comparative Physiological and Proteomic Analysis Reveals Different Involvement of Proteins during Artificial Aging of Siberian Wildrye Seeds. Plants. 2020; 9(10):1370. https://doi.org/10.3390/plants9101370
Chicago/Turabian StyleLei, Xiong, Wenhui Liu, Junming Zhao, Minghong You, Chaohui Xiong, Yi Xiong, Yanli Xiong, Qingqing Yu, Shiqie Bai, and Xiao Ma. 2020. "Comparative Physiological and Proteomic Analysis Reveals Different Involvement of Proteins during Artificial Aging of Siberian Wildrye Seeds" Plants 9, no. 10: 1370. https://doi.org/10.3390/plants9101370