Two New Cases of Hypertrophic Cardiomyopathy and Skeletal Muscle Features Associated with ALPK3 Homozygous and Compound Heterozygous Variants

Abstract
:1. Introduction
2. Materials and Methods
2.1. Target and Whole Exome Sequencing
2.2. Cytogenetic Analysis
3. Results
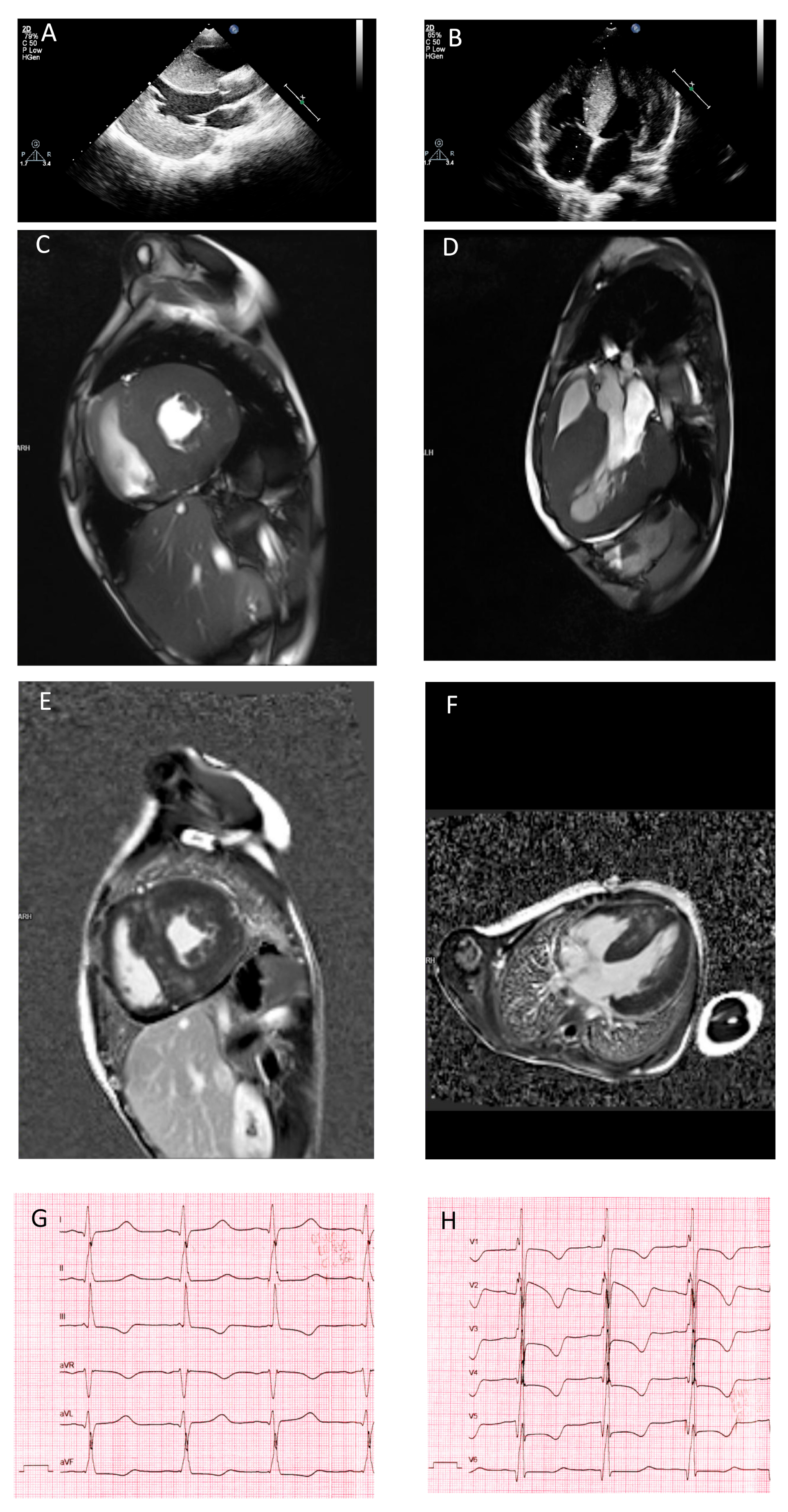
3.1. Clinical Presentation
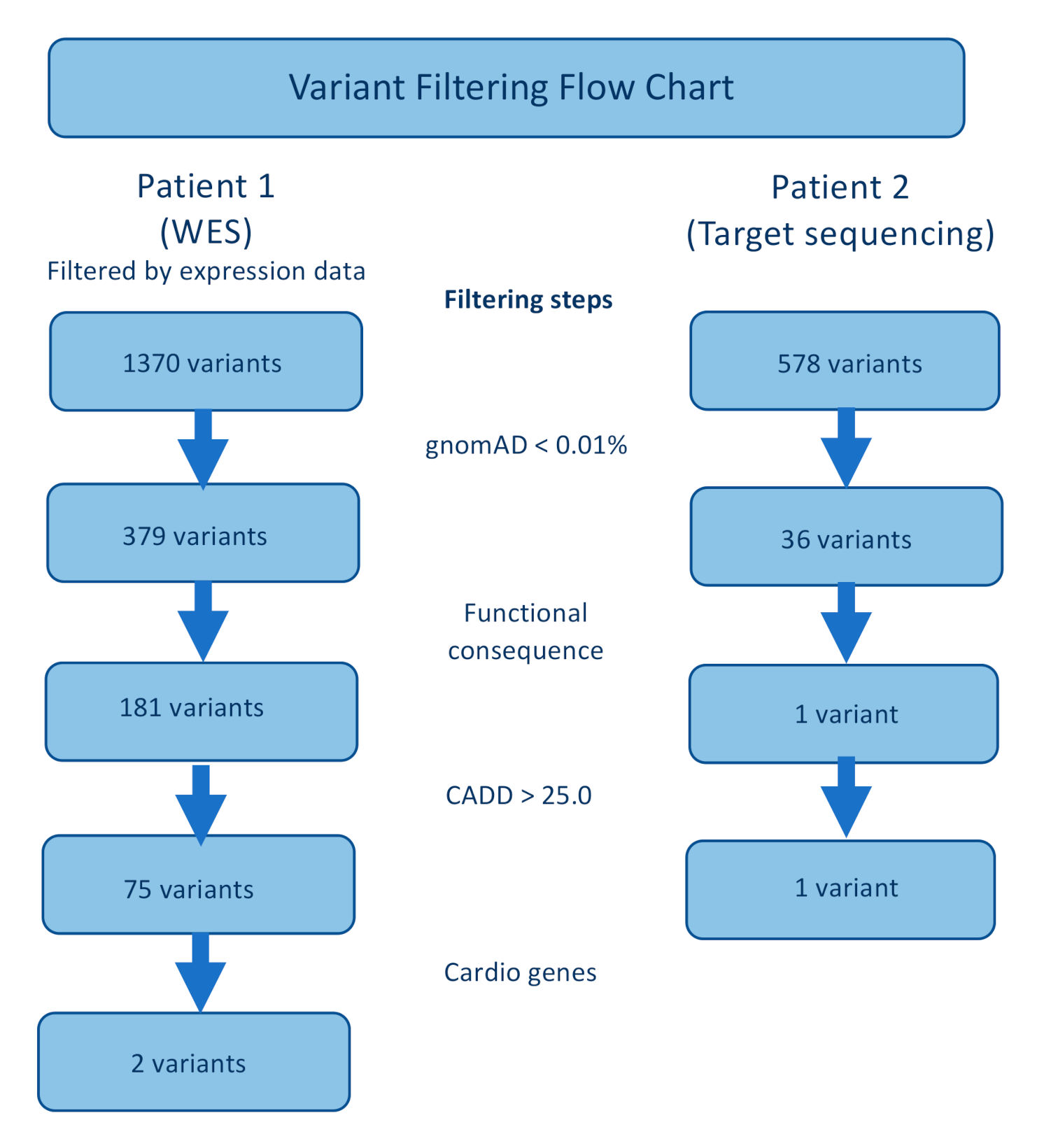
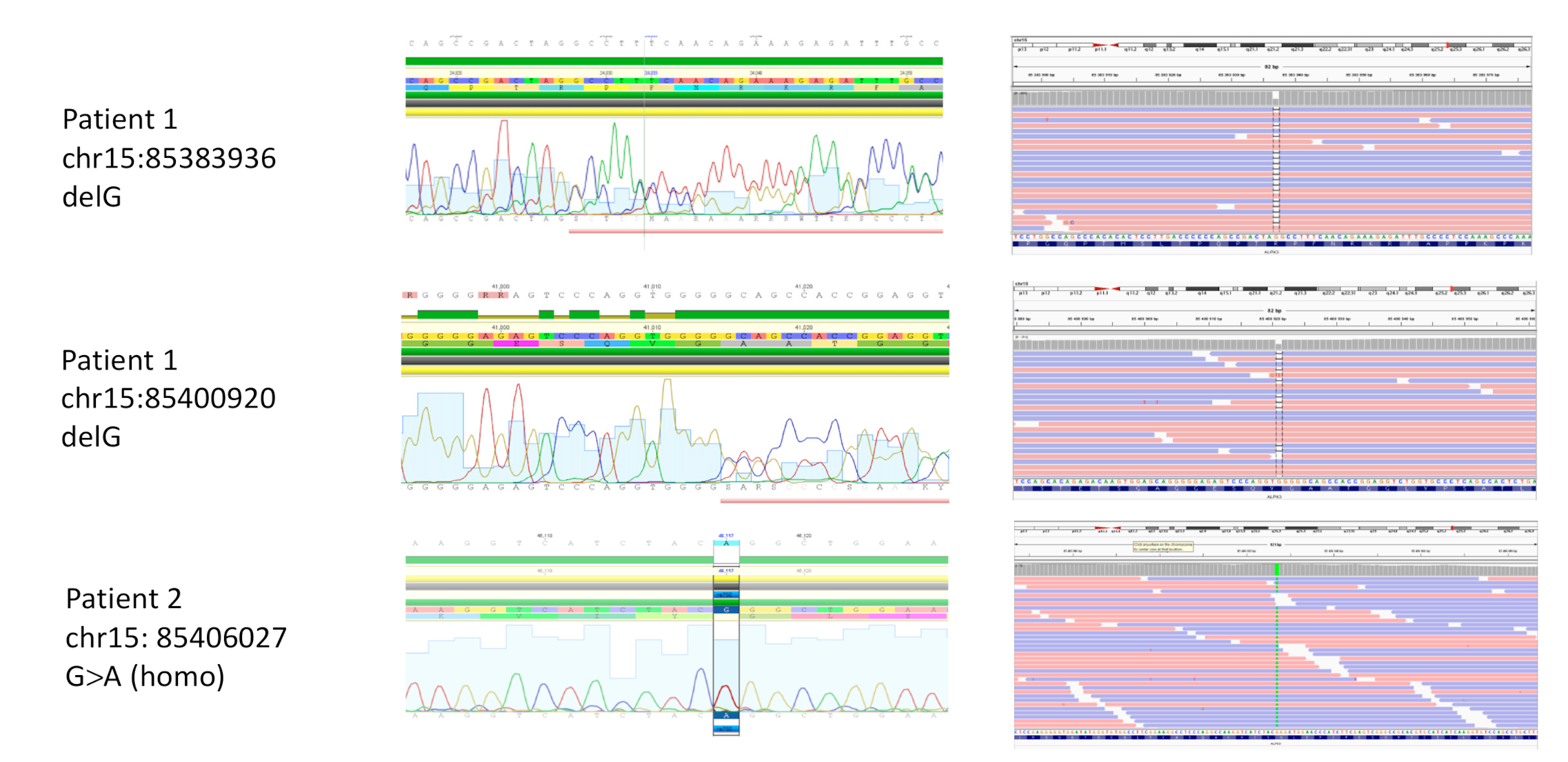
3.2. Sequencing and Genetic Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Kimura, A. Molecular genetics and pathogenesis of cardiomyopathy. J. Hum. Genet. 2016, 61, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.M.; Hsu, D.T.; Kantor, P.; Towbin, J.A.; Ware, S.M.; Colan, S.D.; Chung, W.K.; Jefferies, J.L.; Rossano, J.W.; Castleberry, C.D.; et al. Pediatric Cardiomyopathies. Circ. Res. 2017, 121, 855–873. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, J.E.; Hershberger, R.E. Genetic cardiomyopathies. Curr. Opin. Cardiol. 2018, 33, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Almomani, R.; Verhagen, J.M.A.; Herkert, J.C.; Brosens, E.; van Spaendonck-Zwarts, K.Y.; Asimaki, A.; van der Zwaag, P.A.; Frohn-Mulder, I.M.E.; Bertoli-Avella, A.M.; Boven, L.G.; et al. Biallelic Truncating Mutations in ALPK3 Cause Severe Pediatric Cardiomyopathy. J. Am. Coll. Cardiol. 2016, 67, 515–525. [Google Scholar] [CrossRef] [PubMed]
- Van Sligtenhorst, I.; Ding, Z.-M.; Shi, Z.-Z.; Read, R.W.; Hansen, G.; Vogel, P. Cardiomyopathy in α-kinase 3 (ALPK3)-deficient mice. Vet. Pathol. 2012, 49, 131–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosoda, T.; Monzen, K.; Hiroi, Y.; Oka, T.; Takimoto, E.; Yazaki, Y.; Nagai, R.; Komuro, I. A novel myocyte-specific gene Midori promotes the differentiation of P19CL6 cells into cardiomyocytes. J. Biol. Chem. 2001, 276, 35978–35989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phelan, D.G.; Anderson, D.J.; Howden, S.E.; Wong, R.C.B.; Hickey, P.F.; Pope, K.; Wilson, G.R.; Pébay, A.; Davis, A.M.; Petrou, S.; et al. ALPK3-deficient cardiomyocytes generated from patient-derived induced pluripotent stem cells and mutant human embryonic stem cells display abnormal calcium handling and establish that ALPK3 deficiency underlies familial cardiomyopathy. Eur. Heart J. 2016, 37, 2586–2590. [Google Scholar] [CrossRef] [PubMed]
- Çağlayan, A.O.; Sezer, R.G.; Kaymakçalan, H.; Ulgen, E.; Yavuz, T.; Baranoski, J.F.; Bozaykut, A.; Harmanci, A.S.; Yalcin, Y.; Youngblood, M.W.; et al. ALPK3 gene mutation in a patient with congenital cardiomyopathy and dysmorphic features. Cold Spring Harb. Mol. Case Stud. 2017, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaouadi, H.; Kraoua, L.; Chaker, L.; Atkinson, A.; Delague, V.; Levy, N.; Benkhalifa, R.; Mrad, R.; Abdelhak, S.; Zaffran, S. Novel ALPK3 mutation in a Tunisian patient with pediatric cardiomyopathy and facio-thoraco-skeletal features. J. Hum. Genet. 2018, 63, 1077–1082. [Google Scholar] [CrossRef] [PubMed]
- Al Senaidi, K.; Joshi, N.; Al-Nabhani, M.; Al-Kasbi, G.; Al Farqani, A.; Al-Thihli, K.; Al-Maawali, A. Phenotypic spectrum of ALPK3-related cardiomyopathy. Am. J. Med. Genet. A. 2019, 179, 1235–1240. [Google Scholar] [CrossRef] [PubMed]
- Herkert, J.C.; Verhagen, J.M.A.; Yotti, R.; Haghighi, A.; Phelan, D.G.; James, P.A.; Brown, N.J.; Stutterd, C.; Macciocca, I.; Leong, K.; et al. Expanding the clinical and genetic spectrum of ALPK3 variants: Phenotypes identified in pediatric cardiomyopathy patients and adults with heterozygous variants. Am. Heart J. 2020, 225, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Kostareva, A.; Kiselev, A.; Gudkova, A.; Frishman, G.; Ruepp, A.; Frishman, D.; Smolina, N.; Tarnovskaya, S.; Nilsson, D.; Zlotina, A.; et al. Genetic Spectrum of Idiopathic Restrictive Cardiomyopathy Uncovered by Next-Generation Sequencing. PLoS ONE 2016, 11, e0163362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiselev, A.; Vaz, R.; Knyazeva, A.; Khudiakov, A.; Tarnovskaya, S.; Liu, J.; Sergushichev, A.; Kazakov, S.; Frishman, D.; Smolina, N.; et al. De novo mutations in FLNC leading to early-onset restrictive cardiomyopathy and congenital myopathy. Hum. Mutat. 2018, 39, 1161–1172. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. BioRxiv 2020, 531210. [Google Scholar] [CrossRef] [PubMed]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sim, N.-L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Ref | Alt | Genotype | Variant Type | Amino Acid Change | Reference |
|---|---|---|---|---|---|
| G | A | Hom | Transition (acceptor site) | c.4736-1G > A | Almomani et al. [4] |
| C | T | Hom | Stopgain | c.3781C > T: p.R1261X | Almomani et al. [4] |
| G | A | Hom | Stopgain | c.5294G > A: p.W1765X | Almomani et al. [4] |
| G | A | Hom | Stopgain | c.3792G > A: p.W1264X | Phelan et al. [7] |
| C | - | Hom | Frameshift deletion | c.2018delC: p.Q675SfsX30 | Çağlayan et al. [8] |
| AA | - | Hom | Frameshift deletion | c.1531_1532delAA: p.K511RfsX12 | Jaouadi et al. [9] |
| G | A | Hom | Stopgain | c.639G > A: p.W213X | Al Senaidi et al. [10] |
| C | T | Comp het | Stopgian | c.1018C > T: p.Q340X | Herkert et al. [11] |
| G | A | Comp het | Nonsynonymous SNV | c.2434G > A: p.V812M | Herkert et al. [11] |
| C | - | Comp het | Frameshift deletion | c.4332delC: p.K1445RfsX29 | Herkert et al. [11] |
| G | - | Comp het | Framehsift deletion | c.541delG: p.A181PfsX130 | Herkert et al. [11] |
| C | T | Comp het | Nonsynonymous SNV | c.3439C > T: p.R1147W | Herkert et al. [11] |
| A | - | Comp het | Frameshift deletion | c.4997delA: p.N1666TfsX14 | Herkert et al. [11] |
| G | C | Comp het | Nonsynonymous SNV | c.4091G > C: p.G1364A | Herkert et al. [11] |
| G | C | Comp het | Transition (donor site) | c.5105 + 5G>C | Herkert et al. [11] |
| G | T | Comp het | Nonsynonymous SNV | c.597G > T: p.E199D | Herkert et al. [11] |
| G | T | Comp het | Nonsynonymous SNV | c.4888G > T: p.V1630F | Herkert et al. [11] |
| C | T | Hom | Transition (donor site) | c.3418C > T: p.Q1140X | Herkert et al. [11] |
| G | C | Hom | Nonsynonymous SNV | c.5155G > C: p.A1719P | Herkert et al. [11] |
| G | - | Comp het | Frameshift deletion | c.2033delG: p.R687fs | This study |
| G | - | Comp het | Frameshift deletion | c.3558delG: p.V1186fs | This study |
| G | A | Hom | Nonsynonymous SNV | c.G4897A: p.G1633R | This study |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jorholt, J.; Formicheva, Y.; Vershinina, T.; Kiselev, A.; Muravyev, A.; Demchenko, E.; Fedotov, P.; Zlotina, A.; Rygkov, A.; Vasichkina, E.; et al. Two New Cases of Hypertrophic Cardiomyopathy and Skeletal Muscle Features Associated with ALPK3 Homozygous and Compound Heterozygous Variants. Genes 2020, 11, 1201. https://doi.org/10.3390/genes11101201
Jorholt J, Formicheva Y, Vershinina T, Kiselev A, Muravyev A, Demchenko E, Fedotov P, Zlotina A, Rygkov A, Vasichkina E, et al. Two New Cases of Hypertrophic Cardiomyopathy and Skeletal Muscle Features Associated with ALPK3 Homozygous and Compound Heterozygous Variants. Genes. 2020; 11(10):1201. https://doi.org/10.3390/genes11101201
Chicago/Turabian StyleJorholt, John, Yulia Formicheva, Tatyana Vershinina, Artem Kiselev, Alexey Muravyev, Elena Demchenko, Petr Fedotov, Anna Zlotina, Anton Rygkov, Elena Vasichkina, and et al. 2020. "Two New Cases of Hypertrophic Cardiomyopathy and Skeletal Muscle Features Associated with ALPK3 Homozygous and Compound Heterozygous Variants" Genes 11, no. 10: 1201. https://doi.org/10.3390/genes11101201