
Abstract
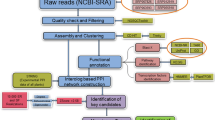
Modeling a protein functional network in concerned species is an efficient approach for identifying novel genes in certain biological pathways. Tea plant (Camellia sinensis) is an important commercial crop abundant in numerous characteristic secondary metabolites (e.g., polyphenols, alkaloids, alkaloids) that confer tea quality and health benefits. Decoding novel genes responsible for tea characteristic components is an important basis for applied genetic improvement and metabolic engineering. Herein, a high-quality protein functional network for tea plant (TeaPoN) was predicted using cross-species protein functional associations transferring and integration combined with a stringent biological network criterion control. TeaPoN contained 31,273 non-redundant functional interactions among 6,634 tea proteins (or genes), with general network topological properties such as scale-free and small-world. We revealed the modular organization of genes related to the major three tea characteristic components (theanine, caffeine, catechin) in TeaPoN, which served as strong evidence for the utility of TeaPoN in novel gene mining. Importantly, several case studies regarding gene identification for tea characteristic components were presented. To aid in the use of TeaPoN, a concise web interface for data deposit and novel gene screening was developed (http://teapon.wchoda.com). We believe that TeaPoN will serve as a useful platform for functional genomics studies associated with characteristic secondary metabolites in tea plant.





Similar content being viewed by others

References
Bader GD and Hogue CW 2003 An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinform. 4 2
Barab AL and Bonabeau E 2003 Scale-free networks. Sci. Am. 288 60–69
Barabasi AL and Oltvai ZN 2004 Network biology: Understanding the cell’s functional organization. Nat. Rev. Genet. 5 101–U115
Brbaklic L, Trkulja D, Kondicspika A and Kobiljski STB 2013 Detection of QTLs for important agronomical traitsin hexaploid wheat using association analysis. Czech J. Genet. Plant Breed. 49 1–8
Cantoro R, Crocco CD, Benecharnold RL and Rodríguez MVJJOEB 2013 In vitro binding of Sorghum bicolor transcription factors ABI4 and ABI5 to a conserved region of a GA 2-OXIDASE promoter: possible role of this interaction in the expression of seed dormancy. J. Exp. Bot. 64 5721–5735
Creighton TE 1993 Proteins: Structures and Molecular Properties (W. H. Freeman)
Ding YD, Chang JW, Guo J, Chen DJ, Li S et al. 2014 Prediction and functional analysis of the sweet orange protein-protein interaction network. BMC Plant Biol. 14 213
Gu H, Zhu P, Jiao Y, Meng Y and Chen M 2011 PRIN: a predicted rice interactome network. BMC Bioinform. 12 161
Higashi Y and Saito K 2013 Network analysis for gene discovery in plant-specialized metabolism. Plant Cell Environ. 36 1597–1606
Kim E, Hwang S and Lee I 2017 SoyNet: a database of co-functional networks for soybean Glycine max. Nucleic Acids Res. 45 D1082–D1089
Kim E, Kim H and Lee I 2013 JiffyNet: a web-based instant protein network modeler for newly sequenced species. Nucleic Acids Res. 41 W192–W197
Kim H, Kim BS, Shim JE, Hwang S, Yang S et al. 2017 TomatoNet: A genome-wide co-functional network for unveiling complex traits of tomato, a model crop for fleshy fruits. Mol. Plant 10 652–655
Kryukov GV and Gladyshev VN 2002 Mammalian selenoprotein gene signature: identification and functional analysis of selenoprotein genes using bioinformatics methods. Methods Enzymol. 347 84–100
Leonova IN and Budashkina EB 2017 The study of agronomical traits determining the productivity of the Triticum aestivum/Triticum timopheevii introgression lines with resistance to fungal diseases. Russ. J. Genet. Appl. Res. 7 299–307
Li CF, Zhu Y, Yu Y, Zhao QY, Wang SJ et al. 2015 Global transcriptome and gene regulation network for secondary metabolite biosynthesis of tea plant (Camellia sinensis). BMC Genomics 16 560
Mao C, Lu S, Lv B, Zhang B, Shen J et al. 2017 A rice NAC transcription factor promotes leaf senescence via ABA biosynthesis. Plant Physiol. 174 1747–1763
Milenkovic T, Memisevic V and Ganesan AK 2010 Systems-level cancer gene identification from protein interaction network topology applied to melanogenesis-related functional genomics data. J. R. Soc. Interface 7 423–437
Newman ME 2006 Modularity and community structure in networks. Proc. Natl. Acad. Sci. 103 8577–8582
Newman MEJ and Watts DJ 1999 Renormalization group analysis of the small-world network model. Phys. Lett. A 263 341–346
Park J and Barabási AL 2007 Distribution of node characteristics in complex networks. Proc. Natl. Acad. Sci. USA 104 17916–17920
Romanov N, Kuhn M, Aebersold R, Ori A, Beck M et al. 2019 Disentangling genetic and environmental effects on the proteotypes of individuals. Cell 177 1308–1318
Romero-Campero FJ, Ignacio PH, Eva LR, Romero JM and Federico V 2016 ChlamyNET: aChlamydomonasgene co-expression network reveals global properties of the transcriptome and the early setup of key co-expression patterns in the green lineage. BMC Genomics 17 227
Shi CYJBG 2011 Deep sequencing of the Camellia sinensis transcriptome revealed candidate genes for major metabolic pathways of tea-specific compounds. BMC Genomics 12 131
Shim JE, Lee T and Lee I 2017 From sequencing data to gene functions: co-functional network approaches. Anim. Cells Syst. 21 77–83
Tai Y, Liu C, Yu S, Hua Y, Sun J et al. 2018 Gene co-expression network analysis reveals coordinated regulation of three characteristic secondary biosynthetic pathways in tea plant (Camellia sinensis). BMC Genomics 19 616
Thirumalaikumar VP, Devkar V, Mehterov N, Ali S, Ozgur R et al. 2017 NAC transcription factor JUNGBRUNNEN1 enhances drought tolerance in tomato. Plant. Biotechnol. J. 16 354–366
Visscher PM, Wray NR, Zhang Q, Sklar P, Mccarthy MI et al. 2017 10 Years of GWAS discovery: biology, function, and translation. Am. J. Hum. Genet. 101 5–22
Wei C, Yang H, Wang S, Zhao J, Liu C et al. 2018 Draft genome sequence of Camellia sinensis var. sinensis provides insights into the evolution of the tea genome and tea quality. Proc. Natl. Acad. Sci. USA 115 201719622
Wolfe CJ, Kohane IS and Butte AJ 2005 Systematic survey reveals general applicability of “guilt-by-association” within gene coexpression networks. BMC Bioinform. 6 227
Xin L, Lihua Y, Xianyao Z, Miaoping Z, Yan L et al. 2013 Transgenic wheat expressing Thinopyrum intermedium MYB transcription factor TiMYB2R-1 shows enhanced resistance to the take-all disease. J. Exp. Bot. 64 2243–2253
Zhang S, Xuan H, Zhang L, Fu S, Wang Y et al. 2016 TBC2health: a database of experimentally validated health-beneficial effects of tea bioactive compounds. Brief. Bioinform. 18 830–836
Zhang S, Zhang L, Tai Y, Wang X, Ho C-T et al. 2018 Gene discovery of characteristic metabolic pathways in the tea plant (Camellia sinensis) using ‘Omics’-based network approaches: a future perspective. Front. Plant Sci. 9 https://doi.org/10.3389/fpls.2018.00480
Zhang Z, Lin Y, Gao S, Zhou S, Guan J et al. 2009 Trapping in scale-free networks with hierarchical organization of modularity. Phys. Rev. E 80 051120
Acknowledgments
Funding was provided by National Natural Science Foundation of China (Grant Nos. 31270714 and 31301248).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Corresponding editor: Sreenivas Chavali
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Zhang, S., Ma, Y., Zhang, R. et al. A predicted protein functional network aids in novel gene mining for characteristic secondary metabolites in tea plant (Camellia sinensis). J Biosci 45, 129 (2020). https://doi.org/10.1007/s12038-020-00101-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12038-020-00101-x