Robust Dynamics of Synthetic Molecular Systems as a Consequence of Broken Symmetry


Faculty of Science, Hokkaido University, Hokkaido 060-0810, Japan
Symmetry 2020, 12(10), 1688; https://doi.org/10.3390/sym12101688
Submission received: 8 September 2020
/
Revised: 5 October 2020
/
Accepted: 10 October 2020
/
Published: 14 October 2020
(This article belongs to the Special Issue Chemical Symmetry Breaking)
Abstract
:The construction of molecular robot-like objects that imitate living things is an important challenge for current chemists. Such molecular devices are expected to perform their duties robustly to carry out mechanical motion, process information, and make independent decisions. Dissipative self-organization plays an essential role in meeting these purposes. To produce a micro-robot that can perform the above tasks autonomously as a single entity, a function generator is required. Although many elegant review articles featuring chemical devices that mimic biological mechanical functions have been published recently, the dissipative structure, which is the minimum requirement for mimicking these functions, has not been sufficiently discussed. This article aims to show clearly that dissipative self-organization is a phenomenon involving autonomy, robustness, mechanical functions, and energy transformation. Moreover, it reports the results of recent experiments with an autonomous light-driven molecular device that achieves all of these features. In addition, a chemical model of cell-amplification is also discussed to focus on the generation of hierarchical movement by dissipative self-organization. By reviewing this research, it may be perceived that mainstream approaches to synthetic chemistry have not always been appropriate. In summary, the author proposes that the integration of catalytic functions is a key issue for the creation of autonomous microarchitecture.

1. Introduction
It is one of the dreams of chemists to create life or its imitative system using a synthetic chemical method [1,2]. Life is thought to be a collection of nanomachines, and their cooperative behavior is capable of performing work continually in a stationary environment. The process termed ‘dissipative self-organization’ is one of the key processes involved in this framework [3]. On the other hand, typical inanimate objects in which modules are not self-organized can only perform work passively under transient environmental conditions. Here, work is broadly defined to denote the transfer of energy to the surroundings in a form other than thermal motion; the mechanical work this involves is defined in Section 2. The function of continuously developing in a stationary environment is defined as an autonomous function; autonomy is the realization of continuity by the internal factors of the system.
For example, fluorescent molecules emit light continuously in a photostationary state; catalyst molecules continuously convert reactive substrates into reaction products. The continuous function of these molecules (broadly defined as ‘work’) is derived from the fact that the molecules consist of self-organized nuclei and electrons. Focusing on such electronic characteristics at the molecular scale, a substantial number of chemical studies have been conducted. Because molecular structures effectively represent the self-organized forms of nuclei and electrons, chemists can predict and consider various functions by examining molecular structural formulas and calculating the density of their electron distributions, while studying phenomena from the perspective of these electronic properties.
This paper describes the autonomous function of molecular assembly generated by molecular self-organization. This concept realizes continuous work at a larger level than the molecular level. It is a concept of nanotechnology that is expected to be applied to material transfer devices and molecular robots and computers and to drive innovation in the field of energy research. Dissipative self-organization remains an attractive keyword for current chemistry. On the other hand, the present author is uncertain whether or not our community genuinely values the concept of dissipative self-organization. One reason for this doubt lies in the fact that the conceptual vocabulary and approaches of nonequilibrium thermodynamics are challenging for chemists who perform their experiments in a flask. Another reason is that clearly realized studies of dissipative self-organization in synthetic chemistry are limited in number. Besides, capturing a temporally developing phenomenon in a printed research paper is not easy. In addition, the terms of self-organization and self-assembly are frequently synonymous; in Japanese, for instance, they are the same word. In this paper, the basic concept of dissipative self-organization is defined in order to underline its importance to current chemical research, and recent developments in synthetic studies are reviewed. It should be noted that the origin of symmetry-breaking is not discussed in this paper.
2. Dissipative Self-Organization is a Concept for Autonomous Mechanical Work
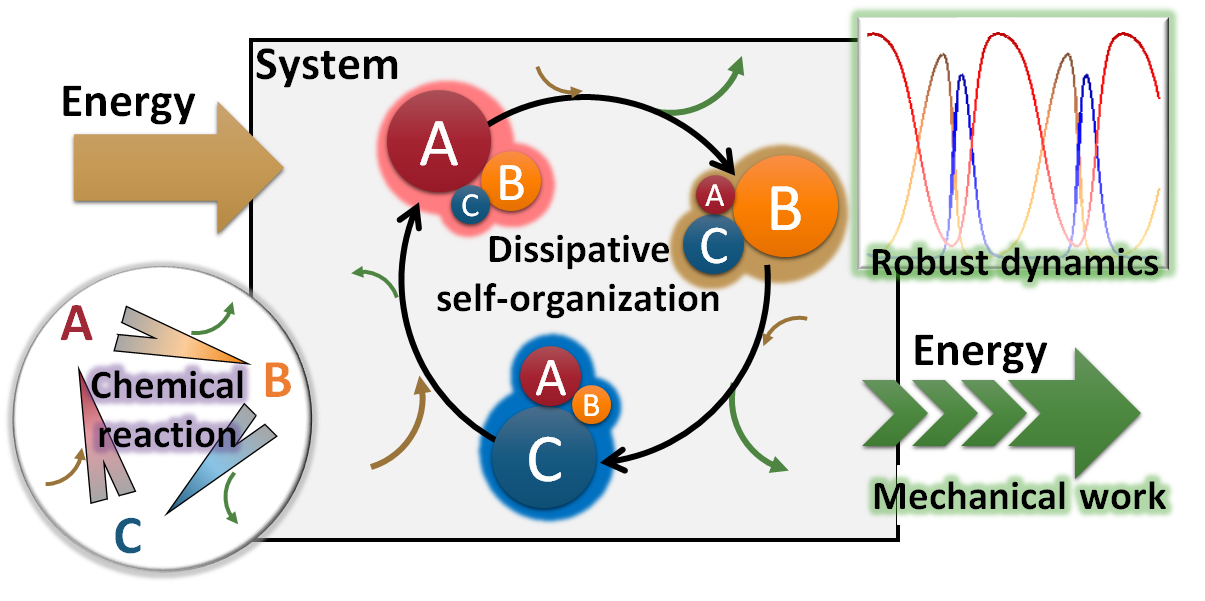
Atkins’ Physical Chemistry describes mechanical work as ‘the transfer of energy that makes use of organized motion in the surroundings’ [4] and distinguishes it from the transfer of energy to disordered thermal motion. In other words, to create molecular devices capable of performing useful work, it is necessary to provide the devices with proportions that will create organized movements in the surroundings (Figure 1a). Hence, a single synthetic molecular motor is too small to achieve beneficial mechanical work independently and, therefore, the motor molecule must either be positioned in a heterogeneous field or the molecules must be assembled [5]. This text considers the latter approach in detail. If a large number of molecules are merely assembled to increase the device’s size, the movement of molecules is time-averaged and the molecules show no macroscopic changes under a steady condition (Figure 1b). This state is referred to as a chemical or near-chemical equilibrium, and under it, the molecules cannot create organized movements in their surroundings. In other words, to create molecular devices capable of performing beneficial work, it is necessary to introduce ‘a mechanism in which the devices continually demonstrate macroscopic changes even if the environment is in a steady state’ (Figure 1c). This mechanism is referred to as dissipative self-organization. In short, it is a mechanism for generating macroscopic motions via movement at the molecular level. The generated motion also has the potential to create motion in a larger hierarchy. Furthermore, due to the ability of the self-continuous action to an external object, devices that exhibit dissipative self-organization can act as an oscillation generator to actuate the repetitive motion of equilibrial materials. The creation of such molecular technology will lead to the invention of autonomous molecular robots, autonomous molecular pumps, and autonomous molecular information processors.
3. Dissipative Self-Organization for Robustness and Energy Conversion
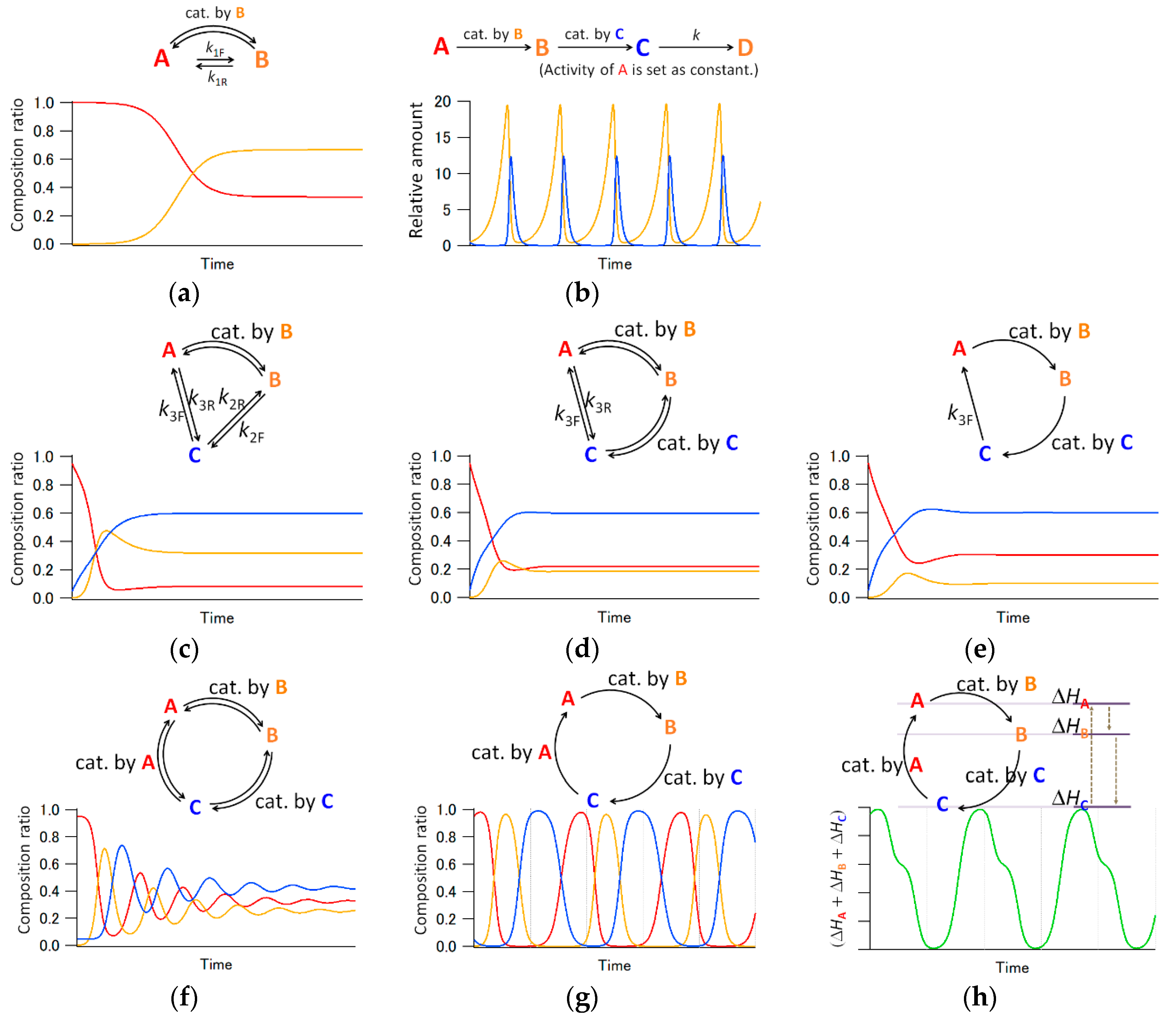
In chemistry, transient aspects of the system are generally described by reaction rate equations. Here, we hypothetically consider chemical reactions in a homogeneous solution, where (a) the symmetry of reactions is broken, and (b) the kinetic constants (ks) are consistent. The flaws of the hypothesis will be discussed below and in [6]. An autocatalytic reaction, in which the A → B reaction is accelerated by B, is introduced as a symmetry-broken reaction [7]. The reaction with the co-existence of a non-autocatalytic process, the kinetics equation of which is described in Appendix A, is shown in Figure 2a [6]. After introducing the logistic curve, general textbooks of nonlinear chemistry describe Lotka–Volterra-type dynamics: the open reaction of A → B → C → D with two autocatalytic steps demonstrates oscillatory behavior in composition (Figure 2b) [3,8]. On the other hand, we consider a triangular type reaction of A → B → C → A, aiming to distinguish the system’s boundary.
To simplify the discussion, we present here the simulation results while ignoring co-existing non-autocatalytic sub-routes within the autocatalytic steps [6]. In triangular-type reactions with one or two autocatalytic steps, it is easy to achieve equilibrium, as shown in Figure 2c,d. Even if we ignore the reverse processes, the reaction reaches equilibrium, as shown in Figure 2e. On the other hand, in reactions with three autocatalytic processes, periodic oscillations in the composition ratio are autonomously demonstrated, as shown in Figure 2f (taking into account reverse processes of autocatalytic paths) and Figure 2g (ignoring them). As described here, the kinetic equations indicate that multi-molecular oscillations may be generated by the contribution of plural nonlinear processes, and that the feature is emasculated by the coexistence of reverse processes. Note that autocatalysis is employed only because it is a well-known and easy-to-calculate nonlinear chemical process—other symmetry-broken phenomena, especially those with large nonlinearity and irreversibility, may contribute to realizing the oscillatory system.
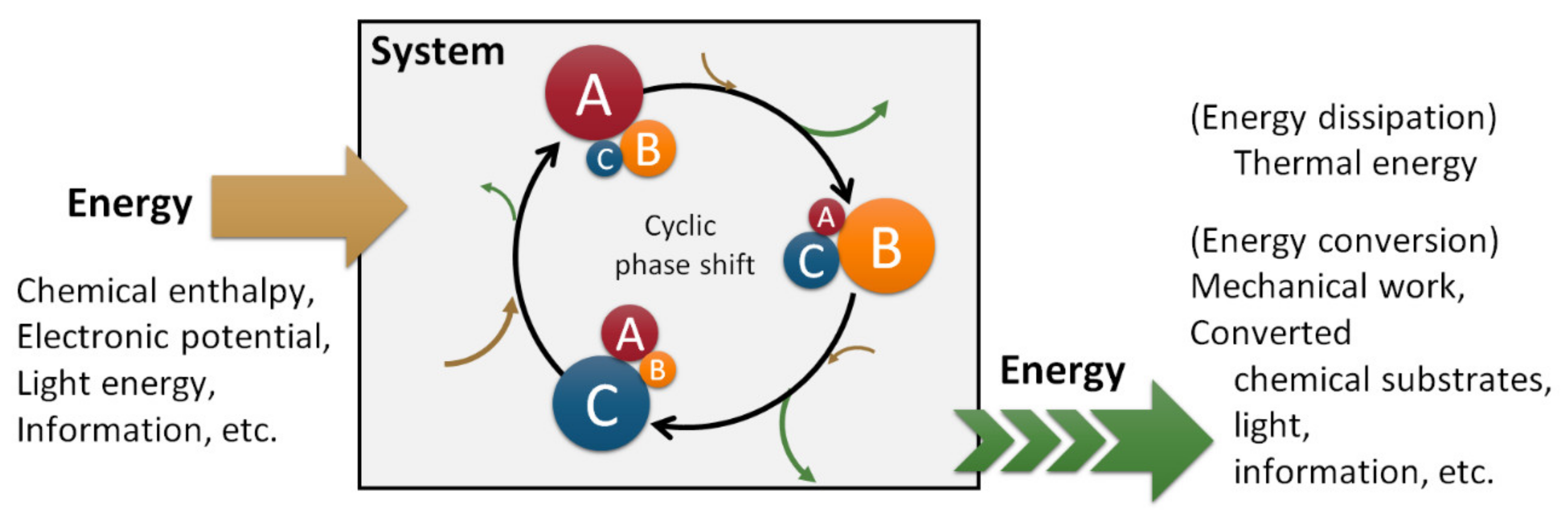
However, the enthalpy of the system varies with oscillation (Figure 2h). The actual system dissipates thermal energy outside the system when the enthalpy decreases, and the periodic behavior is relaxed and terminated. This fact suggests that a supply of convertible energy from outside the system is required to develop the oscillation. In summary, a system with autonomous multi-molecular oscillation can be realized as shown in Scheme 1. The reaction scheme is somewhat similar to a catalytic reaction and a fluorescence scheme; the difference is that the change is in the distribution of components or in the molecular-level structure, and that the nonlinear phenomena are involved in the scheme and act inter- or intra-molecularly.
This temporal pattern in a multi-molecular system under continuous energy supply and periodic energy dissipation is referred to as a dissipative structure; while making cyclic transitions (A → B → C → A) at the molecular level, periodic behaviors are generated as a molecular ensemble, the time scale of which is larger than molecular-level conversion. Although we cannot predict when a chemical reaction happens at the molecular level, we can predict when the macroscopic change occurs by observing the robust temporal pattern. Even if the reaction solution is reduced by half or is frozen temporarily in the middle of the reaction, the autonomous oscillation is robustly maintained. These features are characteristic of dissipative structures.
In a homogeneous system, the variation of the system’s enthalpy is dissipated as thermal energy because the system cannot form organized motion. Regarding devices with shapes (such as molecular assemblies), variation in enthalpy can be used to apply mechanical work to the surroundings. This energy conversion function at the multi-molecular scale is the essential attraction of dissipative self-organization with molecular self-assembly.
4. Research Aiming Mechanical Work with Energy Conversion
Molecular machines and motors, which are defined as molecules that are expected to convert supplied energy into mechanical forces and motion, have attracted considerable attention among chemists [9,10,11,12,13,14]. Molecules that reversibly alter their equilibrium structures, triggered by shifts in external conditions, are known as molecular machines, distinguishing them from molecular motors, a term denoting molecules that maintain their symmetry-broken motion under the continuous supply of energy [12,13]. In contrast to the molecular machines, the symmetry-broken motion of molecular motors may perform autonomous work in molecular-level space if in contact with another object. Feringa’s research into nano-cars may provide one example of this, although his team’s experiment was conducted using an intermittent electric current rather than continuous light waves [15]. Photo-induced ion or electron transportation across membranes [16,17] provides further instances of this phenomenon [5]. However, to attain more significant autonomous work at the multi-molecular scale, the concept of dissipative self-organization is required. Even if we gather molecular motors and supply energy, the motions are time-averaged and the energy is dissipated as thermal energy. In fact, according to Feringa’s report, the motion of a glass-rod on a molecular motor-containing liquid crystal was terminated despite the continuous energy supply [18,19], similar to the system employing molecular machines [20,21]. Therefore, his collaborators are currently directing their efforts to the design of multi-molecular systems where the motors are synchronized to realize mechanical work at the macroscopic level [22].
As mentioned here and elsewhere recently [14,23], the chemical challenge to active work lies in the creation of macroscopic and self-continuous motion. However, one successful example has already been published [24]. This approach, which did not use a chiral molecule, differed from the strategies of other chemists. Molecular-level chirality (e.g., chirality due to an asymmetric form of carbon) is not required for macroscopic objects to move, whereas symmetry-breaking is a critical factor in their motion. Herein, the autonomous flipping and swimming of a submillimeter-size molecular-assembly is introduced [24,25,26].
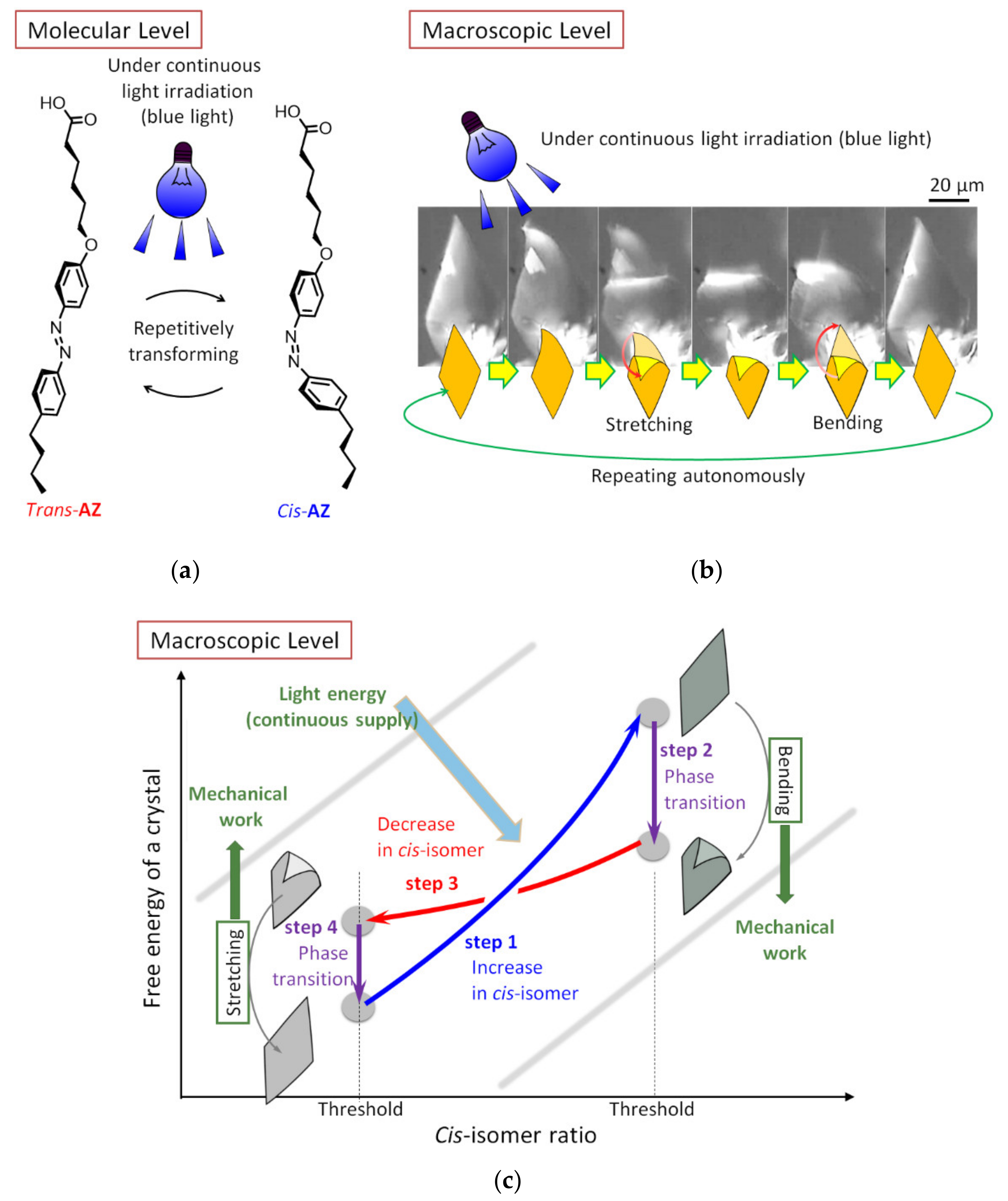
The submillimeter-size assembly of an achiral azobenzene derivative (AZ) showed an autonomous bending-unbending motion under continuous light irradiation (Figure 3). The photochromic compound repetitively changes its molecular structure between trans- and cis-isomers under light irradiation. Ordinarily, repetitive photoisomerization yields a mixture of isomers, the composition ratio of which is constant and depends on the photoreaction rates. However, in the study, a phase transition occurred before the ratio achieved the equilibrium value, switching the reaction rates. Coupling the sets of photoisomerization and its induced phase transition led to a periodic transition consisting of an increase in the cis-isomer, a morphological change, a decrease in the cis-isomer, and then to a morphological recovery [24,27]. The enthalpy also changed periodically with the cyclic process [25]; the cycle enabled autonomous energy conversion from light to mechanical work. At the bending motion, another asymmetry defines the direction of the transformation. According to the single-crystal analysis [25], the space group of the assembly was P1, and it was assumed that the asymmetry concerns the direction of bending. In addition, some break in symmetry is required to produce an autonomous swimming motion: if the bending-and-unbending motion is spatially reciprocal, it is impossible for the assembly to swim in a Newtonian fluid, as the Scallop theorem indicates [28]. The directional swimming of the assembly indicated that the morphological changes occurred in a non-reciprocal manner [26]. Furthermore, the authors also demonstrated signal-dependent pattern formation, a concept that can be applied to molecular-based information processors [25].
Applying stimuli-responsive materials, oscillatory motions, and directional motions has also been reported [29,30,31,32,33,34,35,36,37,38,39,40,41]. Stimuli-responsive materials generally terminate their motion when the systems reach equilibrium. The oscillations in those studies were motivated by the coupling of the directionality of external energy-supply and the time-delayed transition of materials [27,39]. Therefore, we can anticipate that the motions are characterized and controllable by the external conditions, but they lack robustness—for example, adjustment of the energy source position seriously affects the mechanical behavior [27]. Some researchers wrongly regard this feature as an instance of taxis, but it actually arises from a lack of self-excitation. On the other hand, dissipative self-organized materials possess the feature of autonomy. The sustainability and directionality of the movements are guaranteed by the internal factors of the materials. The series of studies of BZ-hydrogels, which are hydrogels periodically repeating swelling and unswelling through the Belousov–Zhabotinsky (BZ) reaction, clearly indicated the superiorities of dissipative self-organization for mechanical device applications [42,43].
5. Research into Self-Developing Molecular Systems: The Chemical Model of Cell Amplification
Cell amplification is also intrinsic to living things [1,2,44,45,46]. The pioneering study of the chemical model of cell amplification was reported by Luisi [47,48]. The team monitored the increase in oleate assembly in water following the addition of oleic anhydride and concluded that the autocatalytic hydrolysis of the anhydride occurred in the oleate assembly to form oleic acid [48]. Inspired by the study, Sugawara’s group designed a system where a membrane molecule formed from a bipolar amphiphile via hydrolysis, catalyzed by a molecule embedded in the vesicular membrane, and, thereby, captured the growth-and-division dynamics of vesicular assemblies [49,50]. In addition, they succeeded in demonstrating the amplification of DNA via a polymerase chain reaction within the vesicles [51,52]. Furthermore, the group firstly realized autocatalytic vesicular amplification systems, via organic synthesis [53,54]. Following these studies, Devaraj’s group [55] also reported the production of an autocatalytic membrane amplification system, in which a molecular catalyst formed spontaneously.
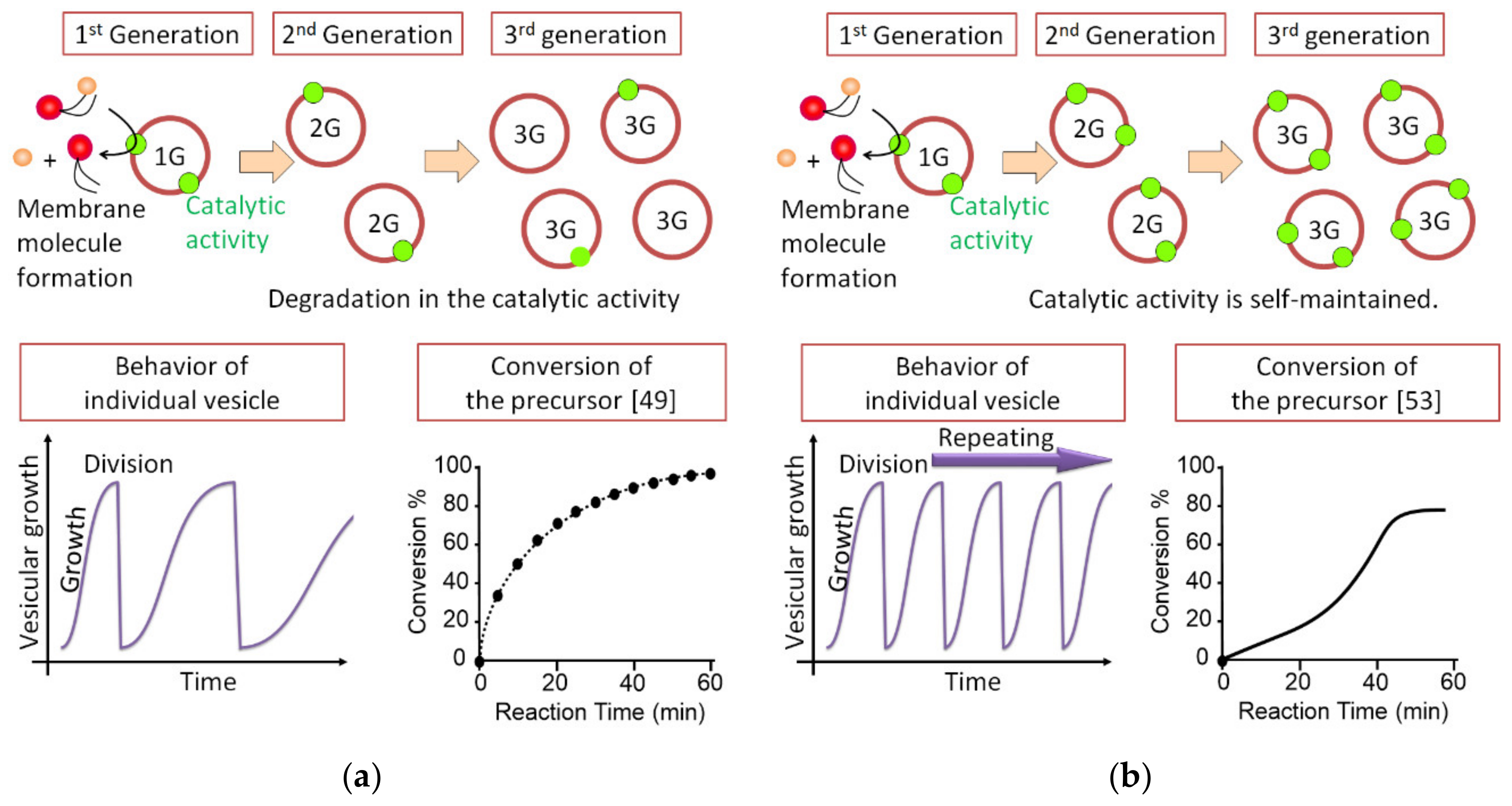
Considering the intrinsic feature of cell amplification, the concept of autocatalysis is fundamental [56,57], and all self-replications occur in an autocatalytic manner [7,58,59,60,61,62,63,64,65,66,67,68,69,70,71]. In the vesicular self-reproduction system without autocatalysis, the generated vesicles were different from the original ones—the catalytic ratio decreased proportionally in each generation, and the system tended to reach equilibrium, even if the precursor for the membrane molecule was continuously supplied (Figure 4a) [49,50,52]. This behavior is the same as inanimate systems. On the other hand, in the autocatalytic system, the catalytic ability is maintained across generations. When the precursor is supplied continuously or an excess amount of precursor is presented, or under both conditions, the individual vesicle repeats its growth and division periodically (Figure 4b) [53,54,55].
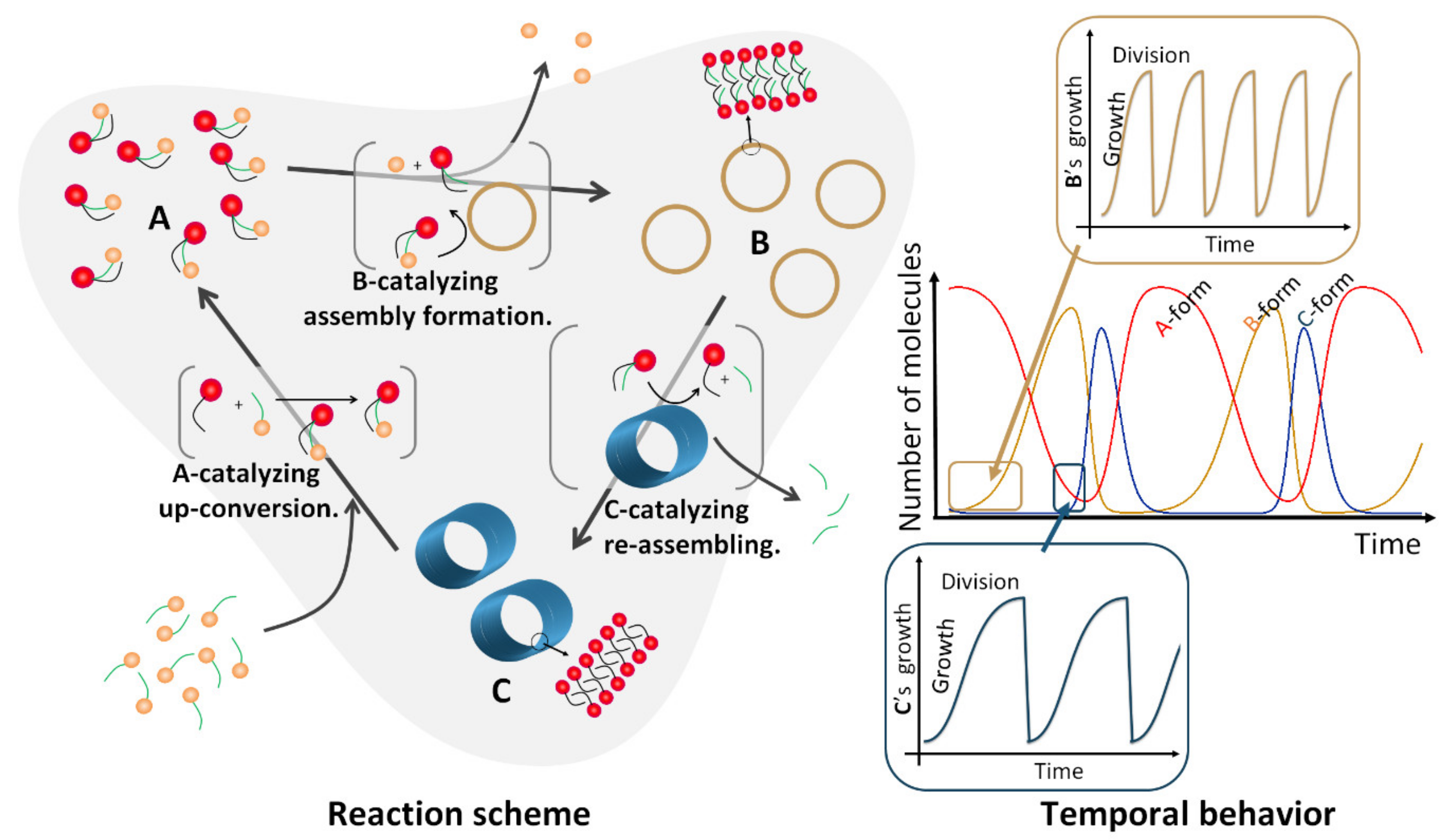
Besides, as Fletcher comments on his autocatalytic micellar system [72,73], a cyclic reaction with an autocatalytic vesicular formation maintains vesicle numbers in an open system; this scheme is represented in Figure 2a,c. Developing this concept, we would like to consider the existence of one additional autocatalytic reaction: the system where the vesicle becomes prey to a larger self-assembly (the consumption ratio is proportional to the number of the larger assembly) and the larger self-assembly decomposes. The reaction equation of the system is complicated because of the diversity in molecular numbers in each vesicle. Nevertheless, it can be expected that the behavior can be represented by the Lotka–Volterra model (if the decomposition is exhausted to the exterior of the dispersion; Figure 2b) or the similar model shown in Section 3 (if the component is recycled autocatalytically by accepting energy from outside the system; Figure 2f,g). Under such conditions, a spatially and temporally larger oscillation than the vesicular growth-division periodicity will be generated (Figure 5). This predator–prey concept is linked to the robustness of nature in an overall sense.
6. Conclusions and Perspectives
Dissipative self-organization is the concept of the cooperation of dynamics to form larger dynamic systems [3]. At the nano level, the combination of thermal fluctuation, a symmetry-breaking phenomenon, and a supply of energy results in the formation of a tiny but organized dynamic system. Once organized dynamics are generated, more robustness and more significant organized motion are formed if symmetry-breaking processes are involved. Through iterative self-organization, a highly hierarchical molecular world is constructed. The symmetry-breaking phenomena contribute to making the system differ from the equilibrium structure. Here, a supply of convertible energy is required to realize self-organization, and both the reception and the dissipation of the energy occur autonomously. The concept of dissipative self-organization is directly linked to the chemical energy conversion to achieve autonomous mechanical functions.
We chemists ordinarily encounter self-organized phenomena. If we consider a molecule to be a thermodynamic system composed of electrons and nuclei, the system demonstrates self-organized behavior whether the molecule is termed a machine or not. However, such systems are too small to perform mechanical work, and the assembly of molecules is, thus, proposed to meet this function. However, controlling symmetry-breaking at the multi-molecular level is a complex task; the artificial system of a molecular assembly tends to form its equilibrial structure. The BZ reaction that was applied to mechanical gel [42,43] and the recent azobenzene-assembly system [24,25,26] provide successful examples. The creation of chemically fueled mechanical work is particularly challenging; if we outdistance the system from the thermal fluctuation level by consuming the dilute energy of chemical compounds, we have to recruit effective ratcheting phenomena.
A recently proposed concept, ‘dissipative self-assembly’, seems to share the same issues [74,75,76,77,78,79]. However, from the author’s standpoint, the claims of dissipative self-assembly are still unclear and weak in the aspect of the distancing from equilibrium; most of the papers reported consecutive reactions where the intermedium products were assemblies and claimed that the assembled state can be maintained through continuous substance supply [72,73,80,81,82,83,84,85,86,87]. The structure of such nonequilibrium systems is not a dissipative structure, according to Prigogine [3]. In this context, the supplied energy is consumed to sustain the assembly, but not for autonomous tasks [88,89]. To imitate living dynamics clearly, we have to employ one or more symmetry-breaking effects to replace the system from the equilibrium structure at the tentative state to a nonequilibrium resume [90]; according to Chapter 7 in reference 3 and the references therein, at least a cubic nonlinearity in the rate equations is required to destabilize the thermodynamic branch.
However, a negative attitude to the current difficulties in imitating the intrinsic features of living dynamics is misplaced. One of the reasons for the struggle to reproduce autonomous dynamics is that chemists attempt to create mechanical materials using switchable molecules, so-called molecular machines. Such molecules are convenient for chemists because their status is easily controlled. Yet in living things, it is not switching motions but continuous motions that are able to do power- autonomous work. In other words, enzymes work with ratcheting mechanisms to prevent the system from attaining equilibrium. By imitating the function of enzymes [91,92,93]—molecular-recognition with nonlinear adaptation and repetitive transformation with energy conversion—and by assembling the synthesized products [23], the author believes that we chemists will attain breakthroughs in nanotechnology; indeed, research based on this perspective has recently begun.
Funding
This research was funded by Japan society for the promotion of science (JSPS) KAKENHI (Scientific Research on Innovative Areas), Grant Numbers JP18H05423 “Molecular Engine”, JP17H05346 “Coordination Asymmetry”, and JP20H04622 “Discrete Geometric Analysis for Materials Design”.
Acknowledgments
The author thanks Professor Sadamu Takeda and Dr. Goro Maruta for their fruitful discussions.
Conflicts of Interest
The author declares no conflict of interest.
Appendix A
Each graph in Figure 2 was the result of numerical analyses of Equations (A1)–(A7). The symbols of χs, ks, and t, denote the molar fractions, kinetic constants, and time, respectively. The term kXY indicates the kinetic constant of the autocatalytic step of X→Y. The calculated timespan was from 0 to 1000 (arbitrary unit). The ‘N’s denote the relative number of portions. To solve the ordinary differential equation, the ODE45 method was used and operated by Matlab 2020a software provided by The Mathworks Inc., Natick, MA, USA. For Figure 2a the following equations were used:
For the demonstration, 0.02 and 0.01 were assigned for kAB and kBA, respectively, 1 × 10−5 was assigned for k1F and k1R, and 1 was assigned for the initial molar fraction of A. For Figure 2b the following equations were used:
For the visualization, 0.03, 0.02, and 0.1 were assigned for kAB, kBC, and kCD, respectively, and 0.5 was assigned for the initial numbers of B and C. For Figure 2c–g the following equations were used:
Each value for the calculations is indicated in Table A1.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table A1.
Values assigned to each parameter for the numerical analysis.
| Figure 2c | Figure 2d | Figure 2e | Figure 2f | Figure 2g | |
|---|---|---|---|---|---|
| kAB | 0.1 | 0.1 | 0.1 | 0.1 | 0.1 |
| kBA | 0.001 | 0.01 | 0 | 0.005 | 0 |
| kBC | 0 | 0.05 | 0.05 | 0.08 | 0.08 |
| kCB | 0 | 0.005 | 0 | 0.004 | 0 |
| kCA | 0 | 0 | 0 | 0.06 | 0.06 |
| kAC | 0 | 0 | 0 | 0.003 | 0 |
| k2F | 0.01 | 0 | 0 | 0 | 0 |
| k2R | 0.001 | 0 | 0 | 0 | 0 |
| k3F | 0.005 | 0.01 | 0.01 | 0 | 0 |
| k3R | 0.005 | 0.01 | 0 | 0 | 0 |
| χB (initial) | 0.001 | 0.001 | 0.001 | 0.001 | 0.001 |
| χC (initial) | 0.049 | 0.049 | 0.049 | 0.049 | 0.049 |
References
- Yewdall, N.A.; Mason, A.F.; van Hest, J.C.M. The hallmarks of living systems: Towards creating artificial cells. Interface Focus 2018, 8, 20180023. [Google Scholar] [CrossRef] [Green Version]
- Ashkenasy, G.; Hermans, T.M.; Otto, S.; Taylor, A.F. Systems chemistry. Chem. Soc. Rev. 2017, 46, 2543–2554. [Google Scholar] [CrossRef] [PubMed]
- Nicolis, G.; Prigogine, I. Self-Organization in Nonequilibrium Systems; John Wiley & Sons, Inc.: Toronto, ON, Canada, 1977. [Google Scholar]
- Atkins, P.; de Paula, J. Internal Energy. In Atkins’ Physcial Chemistry, 10th ed.; Chapter 2; Oxford University Press: Oxford, UK, 2014; pp. 64–74. [Google Scholar]
- Kageyama, Y. Interplay of Photoisomerization and Phase Transition Events Provide a Working Supramolecular Motor. In Photosynergetic Responses in Molecules and Molecular Aggregates; Miyasaka, H., Matsuda, K., Abe, J., Kawai, T., Eds.; Chapter 26; Springer: Singapore, 2020. [Google Scholar] [CrossRef]
- The severity of the impact of inverse reactions or coexisting subreactions on molecular collective behavior depends on the spatio-temporal size of the system under consideration. While microscopic reversibility is important for consideration of thermal/mechanical chemical dynamics at the molecular level, there is less necessity to consider reverse processes if the system already has large spatio-temporal structures, such as those which occur in cell amplification. The purpose of the first half of this section is to show that oscillatory phenomena may be generated in an open system due to the involvement of three nonlinear processes. The assumption introduced in the main text is permissible within the context of the objective.
- Hanopolskyi, A.I.; Smaliak, V.A.; Novichkov, A.I.; Semenov, S.N. Autocatalysis: Kinetics, Mechanisms and Design. ChemSystemsChem 2020. [Google Scholar] [CrossRef]
- Haken, H. Synergetics—An Introduction; Springer: Berlin/Heidelberg, Germany, 1978. (In Japanese) [Google Scholar]
- Kay, E.R.; Leigh, D.A.; Zerbetto, F. Synthetic molecular motors and mechanical machines. Angew. Chem. Int. Ed. 2007, 46, 72–191. [Google Scholar] [CrossRef] [PubMed]
- Silvi, S.; Venturi, M.; Credi, A. Light operated molecular machines. Chem. Commun. 2011, 47, 2483–2489. [Google Scholar] [CrossRef]
- Coskun, A.; Banaszak, M.; Astumian, R.D.; Stoddart, J.F.; Grzybowski, B.A. Great expectations: Can artificial molecular machines deliver on their promise? Chem. Soc. Rev. 2012, 41, 19–30. [Google Scholar] [CrossRef]
- Kassem, S.; van Leeuwen, T.; Lubbe, A.S.; Wilson, M.R.; Feringa, B.L.; Leigh, D.A. Artificial molecular motors. Chem. Soc. Rev. 2017, 46, 2592–2621. [Google Scholar] [CrossRef] [Green Version]
- Dattler, D.; Fuks, G.; Heiser, J.; Moulin, E.; Perrot, A.; Yao, X.; Giuseppone, N. Design of Collective Motions from Synthetic Molecular Switches, Rotors, and Motors. Chem. Rev. 2020, 120, 310–433. [Google Scholar] [CrossRef] [Green Version]
- Aprahamian, I. The Future of Molecular Machines. ACS Cent. Sci. 2020, 6, 347–358. [Google Scholar] [CrossRef] [Green Version]
- Kudernac, T.; Ruangsupapichat, N.; Parschau, M.; Macia, B.; Katsonis, N.; Harutyunyan, S.R.; Ernst, K.H.; Feringa, B.L. Electrically driven directional motion of a four-wheeled molecule on a metal surface. Nature 2011, 479, 208–211. [Google Scholar] [CrossRef]
- Steinberg-Yfrach, G.; Liddell, P.A.; Hung, S.-C.; Moore, A.L.; Gust, D.; Moore, T.A. Conversion of light energy to proton potential in liposomes by artificial photosynthetic reaction centres. Nature 1997, 385, 239–241. [Google Scholar] [CrossRef]
- Mizushima, T.; Yoshida, A.; Harada, A.; Yoneda, Y.; Minatani, T.; Murata, S. Pyrene-sensitized electron transport across vesicle bilayers: Dependence of transport efficiency on pyrene substituents. Org. Biomol. Chem. 2006, 4, 4336–4344. [Google Scholar] [CrossRef] [PubMed]
- Eelkema, R.; Pollard, M.M.; Vicario, J.; Katsonis, N.; Ramon, B.S.; Bastiaansen, C.W.M.; Broer, D.J.; Feringa, B.L. Nanomotor rotates microscale objects. Nature 2006, 440, 163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eelkema, R.; Pollard, M.M.; Katsonis, N.; Vicario, J.; Broer, D.J.; Feringa, B.L. Rotational Reorganization of Doped Cholesteric Liquid Crystalline Films. J. Am. Chem. Soc. 2006, 128, 14397–14407. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Tamaoki, N. Photoresponsive Chiral Dopants: Light-Driven Helicity Manipulation in Cholesteric Liquid Crystals for Optical and Mechanical Functions. ChemPhotoChem 2019, 3, 284–303. [Google Scholar] [CrossRef]
- Thomas, R.; Yoshida, Y.; Akasaka, T.; Tamaoki, N. Influence of a Change in Helical Twisting Power of Photoresponsive Chiral Dopants on Rotational Manipulation of Micro-Objects on the Surface of Chiral Nematic Liquid Crystalline Films. Chem. Eur. J. 2012, 18, 12337–12348. [Google Scholar] [CrossRef]
- Katsonis, N. Cooperative action of molecular motors in mechanically active soft materials. In Proceedings of the 1st International Symposium on Molecular Engine, Chiba University, Chiba, Japan, 8 January 2019. [Google Scholar]
- Astumian, R.D. How molecular motors work—Insights from the molecular machinist’s toolbox: The Nobel prize in Chemistry 2016. Chem. Sci. 2017, 8, 840–845. [Google Scholar] [CrossRef] [Green Version]
- Ikegami, T.; Kageyama, Y.; Obara, K.; Takeda, S. Dissipative and Autonomous Square-Wave Self-Oscillation of a Macroscopic Hybrid Self-Assembly under Continuous Light Irradiation. Angew. Chem. Int. Ed. 2016, 55, 8239–8243. [Google Scholar] [CrossRef]
- Kageyama, Y.; Ikegami, T.; Satonaga, S.; Obara, K.; Sato, H.; Takeda, S. Light-Driven Flipping of Azobenzene Assemblies-Sparse Crystal Structures and Responsive Behaviour to Polarised Light. Chem. Eur. J. 2020, 26, 10759–10768. [Google Scholar] [CrossRef]
- Obara, K.; Kageyama, Y.; Takeda, S. Swimming motion of micro-sized thin crystals with autonomous and rapid flipping under blue light irradiation. In Proceedings of the 99th Annual Meeting of Chemical Society of Japan, 2I7-50, Konan University, Kobe, Japan, 16–19 March 2019. [Google Scholar]
- Kageyama, Y. Light-Powered Self-Sustainable Macroscopic Motion for the Active Locomotion of Materials. ChemPhotoChem 2019, 3, 327–336. [Google Scholar] [CrossRef] [Green Version]
- Purcell, E.M. Life at low Reynolds number. Am. J. Phys. 1977, 45, 3–11. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Chang, L.; Hu, Y.; Huang, M.; Ji, Q.; Lu, P.; Liu, J.; Chen, W.; Wu, Y. An Autonomous Soft Actuator with Light-Driven Self-Sustained Wavelike Oscillation for Phototactic Self-Locomotion and Power Generation. Adv. Funct. Mater 2020, 30, 1908842. [Google Scholar] [CrossRef]
- Serak, S.; Tabiryan, N.; Vergara, R.; White, T.J.; Vaia, R.A.; Bunning, T.J. Liquid crystalline polymer cantilever oscillators fueled by light. Soft Matter 2010, 6, 779–783. [Google Scholar] [CrossRef]
- White, T.J.; Tabiryan, N.V.; Serak, S.V.; Hrozhyk, U.A.; Tondiglia, V.P.; Koerner, H.; Vaia, R.A.; Bunning, T.J. A high frequency photodriven polymer oscillator. Soft Matter 2008, 4, 1796–1798. [Google Scholar] [CrossRef]
- Lee, K.M.; Smith, M.L.; Koerner, H.; Tabiryan, N.; Vaia, R.A.; Bunning, T.J.; White, T.J. Photodriven, Flexural-Torsional Oscillation of Glassy Azobenzene Liquid Crystal Polymer Networks. Adv. Funct. Mater 2011, 21, 2913–2918. [Google Scholar] [CrossRef]
- Vantomme, G.; Gelebart, A.H.; Broer, D.J.; Meijer, E.W. A four-blade light-driven plastic mill based on hydrazone liquid-crystal networks. Tetrahedron 2017, 73, 4963–4967. [Google Scholar] [CrossRef]
- Gelebart, A.H.; Vantomme, G.; Meijer, E.W.; Broer, D.J. Mastering the Photothermal Effect in Liquid Crystal Networks: A General Approach for Self-Sustained Mechanical Oscillators. Adv. Mater. 2017, 29, 1606712. [Google Scholar] [CrossRef] [Green Version]
- Gelebart, A.H.; Jan Mulder, D.; Varga, M.; Konya, A.; Vantomme, G.; Meijer, E.W.; Selinger, R.L.B.; Broer, D.J. Making waves in a photoactive polymer film. Nature 2017, 546, 632–636. [Google Scholar] [CrossRef] [Green Version]
- Pilz da Cunha, M.; Peeketi, A.R.; Mehta, K.; Broer, D.J.; Annabattula, R.K.; Schenning, A.; Debije, M.G. A self-sustained soft actuator able to rock and roll. Chem. Commun. 2019, 55, 11029–11032. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.; Guo, S.; Tong, X.; Xia, H.; Zhao, Y. Tunable Photocontrolled Motions Using Stored Strain Energy in Malleable Azobenzene Liquid Crystalline Polymer Actuators. Adv. Mater. 2017, 29, 1606467. [Google Scholar] [CrossRef]
- Ge, F.; Yang, R.; Tong, X.; Camerel, F.; Zhao, Y. A Multifunctional Dye-doped Liquid Crystal Polymer Actuator: Light-Guided Transportation, Turning in Locomotion, and Autonomous Motion. Angew. Chem. Int. Ed. 2018, 57, 11758–11763. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Lahikainen, M.; Liu, L.; Ahmed, Z.; Wani, O.M.; Wang, M.; Yang, H.; Priimagi, A. Light-fuelled freestyle self-oscillators. Nat. Commun. 2019, 10, 5057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uchida, E.; Azumi, R.; Norikane, Y. Light-induced crawling of crystals on a glass surface. Nat. Commun. 2015, 6, 7310. [Google Scholar] [CrossRef] [PubMed]
- Yamada, M.; Kondo, M.; Mamiya, J.; Yu, Y.; Kinoshita, M.; Barrett, C.; Ikeda, T. Photomobile polymer materials: Towards light-driven plastic motors. Angew. Chem. Int. Ed. 2008, 47, 4986–4988. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, R. Development of self-oscillating polymers and gels with autonomous function. Polym. J. 2010, 42, 777–789. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, R.; Ueki, T. Evolution of self-oscillating polymer gels as autonomous polymer systems. NPG Asia Mater. 2014, 6, e107. [Google Scholar] [CrossRef] [Green Version]
- Szostak, J.W.; Bartel, D.P.; Luisi, P.L. Synthesizing life. Nature 2001, 409, 387–390. [Google Scholar] [CrossRef]
- Stano, P.; Luisi, P.L. Achievements and open questions in the self-reproduction of vesicles and synthetic minimal cells. Chem. Commun. 2010, 46, 3639–3653. [Google Scholar] [CrossRef]
- Lancet, D.; Segre, D.; Kahana, A. Twenty Years of “Lipid World”: A Fertile Partnership with David Deamer. Life 2019, 9, 77. [Google Scholar] [CrossRef] [Green Version]
- Bachmann, P.A.; Luisi, P.L.; Lang, J. Autocatalytic self-replicating micelles as models for prebiotic structures. Nature 1992, 357, 57–59. [Google Scholar] [CrossRef]
- Walde, P.; Wick, R.; Fresta, M.; Mangone, A.; Luisi, P.L. Autopoietic Self-Reproduction of Fatty Acid Vesicles. J. Am. Chem. Soc. 1994, 116, 11649–11654. [Google Scholar] [CrossRef]
- Takakura, K.; Sugawara, T. Membrane dynamics of a myelin-like giant multilamellar vesicle applicable to a self-reproducing system. Langmuir 2004, 20, 3832–3834. [Google Scholar] [CrossRef] [PubMed]
- Toyota, T.; Takakura, K.; Kageyama, Y.; Kurihara, K.; Maru, N.; Ohnuma, K.; Kaneko, K.; Sugawara, T. Population study of sizes and components of self-reproducing giant multilamellar vesicles. Langmuir 2008, 24, 3037–3044. [Google Scholar] [CrossRef] [PubMed]
- Shohda, K.-I.; Tamura, M.; Kageyama, Y.; Suzuki, K.; Suyama, A.; Sugawara, T. Compartment size dependence of performance of polymerase chain reaction inside giant vesicles. Soft Matter 2011, 7, 3750. [Google Scholar] [CrossRef]
- Kurihara, K.; Tamura, M.; Shohda, K.; Toyota, T.; Suzuki, K.; Sugawara, T. Self-reproduction of supramolecular giant vesicles combined with the amplification of encapsulated DNA. Nat. Chem. 2011, 3, 775–781. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Kageyama, Y.; Kurihara, K.; Takakura, K.; Murata, S.; Sugawara, T. Autocatalytic membrane-amplification on a pre-existing vesicular surface. Chem. Commun. 2010, 46, 8791–8793. [Google Scholar] [CrossRef]
- Matsuo, M.; Ohyama, S.; Sakurai, K.; Toyota, T.; Suzuki, K.; Sugawara, T. A sustainable self-reproducing liposome consisting of a synthetic phospholipid. Chem. Phys. Lipids 2019, 222, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Hardy, M.D.; Yang, J.; Selimkhanov, J.; Cole, C.M.; Tsimring, L.S.; Devaraj, N.K. Self-reproducing catalyst drives repeated phospholipid synthesis and membrane growth. Proc. Natl. Acad. Sci. USA 2015, 112, 8187–8192. [Google Scholar] [CrossRef] [Green Version]
- Bissette, A.J.; Fletcher, S.P. Mechanisms of autocatalysis. Angew. Chem. Int. Ed. 2013, 52, 12800–12826. [Google Scholar] [CrossRef]
- Kosikova, T.; Philp, D. Exploring the emergence of complexity using synthetic replicators. Chem. Soc. Rev. 2017, 46, 7274–7305. [Google Scholar] [CrossRef] [Green Version]
- Soai, K.; Shibata, T.; Morioka, H.; Choji, K. Asymmetric autocatalysis and amplification of enantiomeric excess of a chiral molecule. Nature 1995, 378, 767–768. [Google Scholar] [CrossRef]
- Tjivikua, T.; Ballester, P.; Rebek, J. Self-replicating system. J. Am. Chem. Soc. 1990, 112, 1249–1250. [Google Scholar] [CrossRef]
- Rotello, V.; Hong, J.I.; Rebek, J. Sigmoidal growth in a self-replicating system. J. Am. Chem. Soc. 1991, 113, 9422–9423. [Google Scholar] [CrossRef]
- Wintner, E.A.; Conn, M.M.; Rebek, J. Self-Replicating Molecules: A Second Generation. J. Am. Chem. Soc. 1994, 116, 8877–8884. [Google Scholar] [CrossRef]
- Chen, J.; Korner, S.; Craig, S.L.; Lin, S.; Rudkevich, D.M.; Rebek, J., Jr. Chemical amplification with encapsulated reagents. Proc. Natl. Acad. Sci. USA 2002, 99, 2593–2596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamioka, S.; Ajami, D.; Rebek, J., Jr. Autocatalysis and organocatalysis with synthetic structures. Proc. Natl. Acad. Sci. USA 2010, 107, 541–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zielinski, W.S.; Orgel, L.E. Autocatalytic synthesis of a tetranucleotide analogue. Nature 1987, 327, 346–347. [Google Scholar] [CrossRef]
- Orgel, L.E. Molecular replication. Nature 1992, 358, 203–209. [Google Scholar] [CrossRef]
- Von Kiedrowski, G.; Wlotzka, B.; Helbing, J.; Matzen, M.; Jordan, S. Parabolic Growth of a Self-Replicating Hexadeoxynucleotide Bearing a 3′-5′-Phosphoamidate Linkage. Angew. Chem. Int. Ed. 1991, 30, 423–426. [Google Scholar] [CrossRef]
- Terfort, A.; von Kiedrowski, G. Self-Replication by Condensation of 3-Aminobenzamidines and 2-Formylphenoxyacetic Acids. Angew. Chem. Int. Ed. 1992, 31, 654–656. [Google Scholar] [CrossRef]
- Achilles, T.; von Kiedrowski, G. A Self-Replicating System from Three Starting Materials. Angew. Chem. Int. Ed. 1993, 32, 1198–1201. [Google Scholar] [CrossRef]
- Sadownik, J.W.; Mattia, E.; Nowak, P.; Otto, S. Diversification of self-replicating molecules. Nat. Chem. 2016, 8, 264–269. [Google Scholar] [CrossRef] [PubMed]
- Nowak, P.; Colomb-Delsuc, M.; Otto, S.; Li, J.W. Template-Triggered Emergence of a Self-Replicator from a Dynamic Combinatorial Library. J. Am. Chem. Soc. 2015, 137, 10965–10969. [Google Scholar] [CrossRef] [PubMed]
- Leonetti, G.; Otto, S. Solvent Composition Dictates Emergence in Dynamic Molecular Networks Containing Competing Replicators. J. Am. Chem. Soc. 2015, 137, 2067–2072. [Google Scholar] [CrossRef] [PubMed]
- Colomer, I.; Morrow, S.M.; Fletcher, S.P. A transient self-assembling self-replicator. Nat. Commun. 2018, 9, 2239. [Google Scholar] [CrossRef] [Green Version]
- Morrow, S.M.; Colomer, I.; Fletcher, S.P. A chemically fuelled self-replicator. Nat. Commun. 2019, 10, 1011. [Google Scholar] [CrossRef]
- Della Sala, F.; Neri, S.; Maiti, S.; Chen, J.L.Y.; Prins, L.J. Transient self-assembly of molecular nanostructures driven by chemical fuels. Curr. Opin. Biotechnol. 2017, 46, 27–33. [Google Scholar] [CrossRef] [Green Version]
- Van Rossum, S.A.P.; Tena-Solsona, M.; van Esch, J.H.; Eelkema, R.; Boekhoven, J. Dissipative out-of-equilibrium assembly of man-made supramolecular materials. Chem. Soc. Rev. 2017, 46, 5519–5535. [Google Scholar] [CrossRef]
- De, S.; Klajn, R. Dissipative Self-Assembly Driven by the Consumption of Chemical Fuels. Adv. Mater. 2018, 30, e1706750. [Google Scholar] [CrossRef]
- Ragazzon, G.; Prins, L.J. Energy consumption in chemical fuel-driven self-assembly. Nat. Nanotechnol. 2018, 13, 882–889. [Google Scholar] [CrossRef] [Green Version]
- Leng, Z.; Peng, F.; Hao, X. Chemical-Fuel-Driven Assembly in Macromolecular Science: Recent Advances and Challenges. ChemPlusChem 2020, 85, 1190–1199. [Google Scholar] [CrossRef] [PubMed]
- Fialkowski, M.; Bishop, K.J.; Klajn, R.; Smoukov, S.K.; Campbell, C.J.; Grzybowski, B.A. Principles and implementations of dissipative (dynamic) self-assembly. J. Phys. Chem. B 2006, 110, 2482–2496. [Google Scholar] [CrossRef]
- Boekhoven, J.; Brizard, A.M.; Kowlgi, K.N.; Koper, G.J.; Eelkema, R.; van Esch, J.H. Dissipative self-assembly of a molecular gelator by using a chemical fuel. Angew. Chem. Int. Ed. 2010, 49, 4825–4828. [Google Scholar] [CrossRef] [PubMed]
- Maiti, S.; Fortunati, I.; Ferrante, C.; Scrimin, P.; Prins, L.J. Dissipative self-assembly of vesicular nanoreactors. Nat. Chem. 2016, 8, 725–731. [Google Scholar] [CrossRef]
- Hao, X.; Sang, W.; Hu, J.; Yan, Q. Pulsating Polymer Micelles via ATP-Fueled Dissipative Self-Assembly. ACS Macro. Lett. 2017, 6, 1151–1155. [Google Scholar] [CrossRef]
- Riess, B.; Boekhoven, J. Applications of Dissipative Supramolecular Materials with a Tunable Lifetime. ChemNanoMat 2018, 4, 710–719. [Google Scholar] [CrossRef]
- Bal, S.; Das, K.; Ahmed, S.; Das, D. Chemically Fueled Dissipative Self-Assembly that Exploits Cooperative Catalysis. Angew. Chem. Int. Ed. 2019, 58, 244–247. [Google Scholar] [CrossRef]
- Wanzke, C.; Jussupow, A.; Kohler, F.; Dietz, H.; Kaila, V.R.I.; Boekhoven, J. Dynamic Vesicles Formed By Dissipative Self-Assembly. ChemSystemsChem 2019, 2, e1900044. [Google Scholar] [CrossRef]
- Post, E.A.J.; Fletcher, S.P. Controlling the Kinetics of Self-Reproducing Micelles by Catalyst Compartmentalization in a Biphasic System. J. Org. Chem. 2019, 84, 2741–2755. [Google Scholar] [CrossRef] [Green Version]
- Post, E.A.J.; Fletcher, S.P. Dissipative self-assembly, competition and inhibition in a self-reproducing protocell model. Chem. Sci. 2020, 11, 9434–9442. [Google Scholar] [CrossRef]
- Penocchio, E.; Rao, R.; Esposito, M. Thermodynamic efficiency in dissipative chemistry. Nat. Commun. 2019, 10, 3865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koper, G.J.M.; Boekhoven, J.; Hendriksen, W.E.; van Esch, J.H.; Eelkema, R.; Pagonabarraga, I.; Rubí, J.M.; Bedeaux, D. The Lost Work in Dissipative Self-Assembly. Int. J. Thermophys. 2013, 34, 1229–1238. [Google Scholar] [CrossRef]
- The author does not exclude the possibility that the reported systems form a dissipative structure, because there may be additional symmetry-breaking phenomena that are not described in the papers.
- Ragazzon, G.; Baroncini, M.; Silvi, S.; Venturi, M.; Credi, A. Light-powered autonomous and directional molecular motion of a dissipative self-assembling system. Nat. Nanotechnol. 2014, 10, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.R.; Solà, J.; Carlone, A.; Goldup, S.M.; Lebrasseur, N.; Leigh, D.A. An autonomous chemically fuelled small-molecule motor. Nature 2016, 534, 235–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fletcher, S.P.; Dumur, F.; Pollard, M.M.; Feringa, B.L. A Reversible, Unidirectional Molecular Rotary Motor Driven by Chemical Energy. Science 2005, 310, 80–82. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
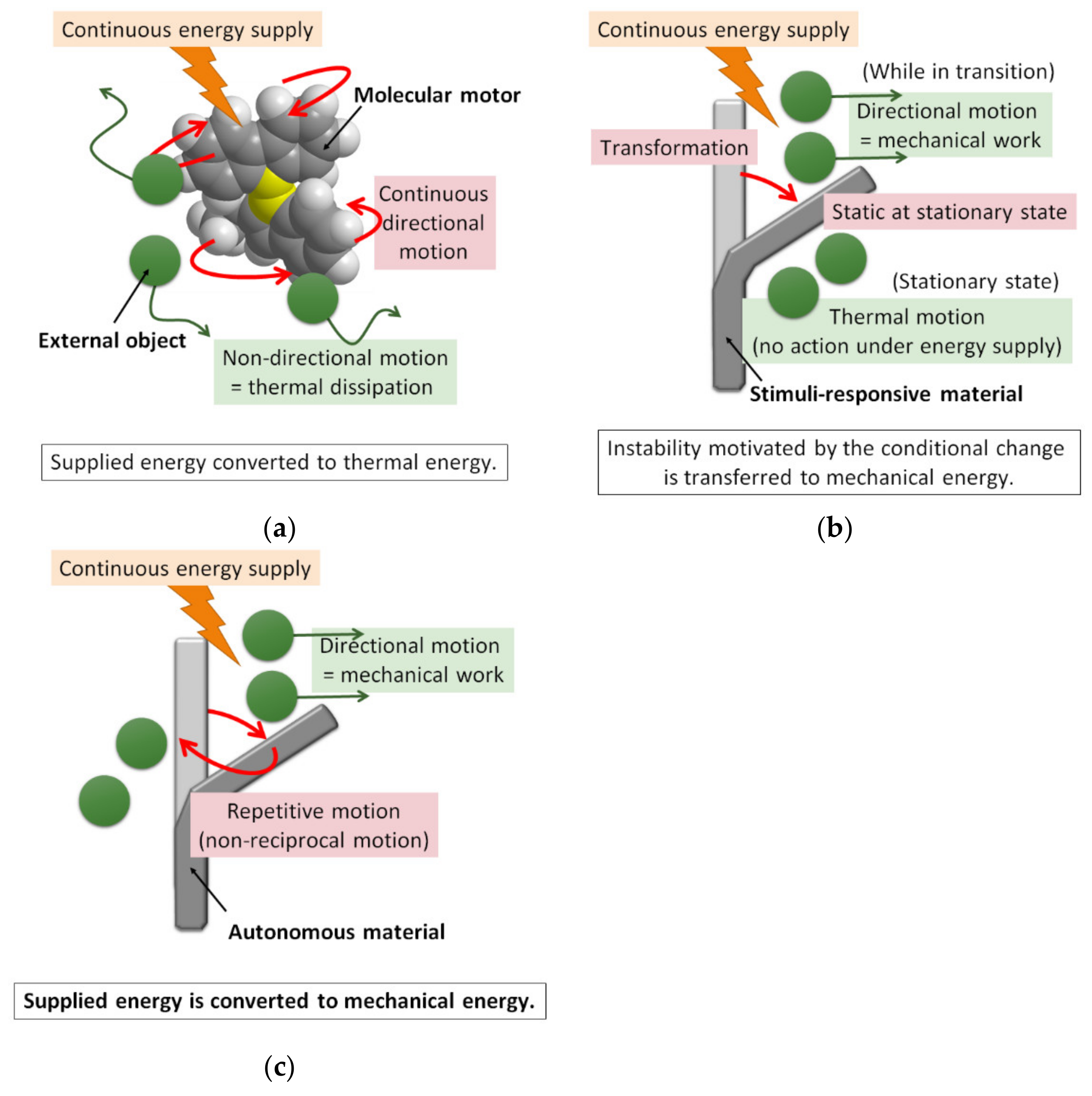
Figure 1.
How to compel molecular materials to work. (a) A small molecular motor cannot carry out mechanical work for several reasons, one of which is that the transferred energy is converted to thermal energy immediately. (b) A stimuli-responsive material can perform work only while relaxing the instability motivated by a shift in an external condition. (c) Autonomous transformation of a molecular system can perform work via energy transformation. The green circles denote objects subjected to force in the surroundings.
Figure 1.
How to compel molecular materials to work. (a) A small molecular motor cannot carry out mechanical work for several reasons, one of which is that the transferred energy is converted to thermal energy immediately. (b) A stimuli-responsive material can perform work only while relaxing the instability motivated by a shift in an external condition. (c) Autonomous transformation of a molecular system can perform work via energy transformation. The green circles denote objects subjected to force in the surroundings.

Figure 2.
Numerical analyses for composition curves of reactions involving autocatalysis. (a) Logistic-type composition curve. (b) Lotka–Volterra-type composition oscillation. (c) Composition curve for a triangle reaction including one autocatalytic process. (d,e) Composition curves for triangle reactions, including two autocatalytic processes with and without considering reverse processes, respectively. (f,g) Oscillatory composition curves in triangle reactions, including three autocatalytic processes with and without considering reverse processes, respectively. (h) The variation in total heat of formations of A, B, and C while the change is shown in (g). Because the exteriorization of energy from the system occurs when the sum of the heat of formations decreases, an external energy supply is required to maintain the oscillation. Similarly, all calculations assumed consistent kinetic constants, meaning that an adequate energy supply and relevant emissions exist, although the fact is not indicated in the schemes. The red, orange, and blue lines denote compounds A, B, and C, respectively. The equations for the numerical analysis are described in Appendix A.
Figure 2.
Numerical analyses for composition curves of reactions involving autocatalysis. (a) Logistic-type composition curve. (b) Lotka–Volterra-type composition oscillation. (c) Composition curve for a triangle reaction including one autocatalytic process. (d,e) Composition curves for triangle reactions, including two autocatalytic processes with and without considering reverse processes, respectively. (f,g) Oscillatory composition curves in triangle reactions, including three autocatalytic processes with and without considering reverse processes, respectively. (h) The variation in total heat of formations of A, B, and C while the change is shown in (g). Because the exteriorization of energy from the system occurs when the sum of the heat of formations decreases, an external energy supply is required to maintain the oscillation. Similarly, all calculations assumed consistent kinetic constants, meaning that an adequate energy supply and relevant emissions exist, although the fact is not indicated in the schemes. The red, orange, and blue lines denote compounds A, B, and C, respectively. The equations for the numerical analysis are described in Appendix A.

Scheme 1.
Conceptual scheme for an autonomous system.

Figure 3.
Self-sustaining mechanical motion of azobenzene assembly under continuous blue-light irradiation [24,25]. (a) Molecular-level repetitive transformation of the component molecule (an achiral azobenzene derivative, AZ). (b) Micrographs of the macroscopic autonomous flipping motion under light irradiation. (c) Schematic energy diagram for the autonomous cycle; mechanical work was performed at the steps of exergonic morphological change. All figures were reproduced from the original data with permission from Wiley-VCH, 2016.
Figure 3.
Self-sustaining mechanical motion of azobenzene assembly under continuous blue-light irradiation [24,25]. (a) Molecular-level repetitive transformation of the component molecule (an achiral azobenzene derivative, AZ). (b) Micrographs of the macroscopic autonomous flipping motion under light irradiation. (c) Schematic energy diagram for the autonomous cycle; mechanical work was performed at the steps of exergonic morphological change. All figures were reproduced from the original data with permission from Wiley-VCH, 2016.

Figure 4.
The difference between (a) non-autocatalytic and (b) autocatalytic vesicular self-reproduction.
Figure 4.
The difference between (a) non-autocatalytic and (b) autocatalytic vesicular self-reproduction.

Figure 5.
Conceptual illustration for a hierarchical system composed of autocatalytic assembling processes. Spatially and temporally larger oscillation generating a molecular assembly that appears and disappears repetitively.
Figure 5.
Conceptual illustration for a hierarchical system composed of autocatalytic assembling processes. Spatially and temporally larger oscillation generating a molecular assembly that appears and disappears repetitively.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kageyama, Y. Robust Dynamics of Synthetic Molecular Systems as a Consequence of Broken Symmetry. Symmetry 2020, 12, 1688. https://doi.org/10.3390/sym12101688
AMA Style
Kageyama Y. Robust Dynamics of Synthetic Molecular Systems as a Consequence of Broken Symmetry. Symmetry. 2020; 12(10):1688. https://doi.org/10.3390/sym12101688
Chicago/Turabian StyleKageyama, Yoshiyuki. 2020. "Robust Dynamics of Synthetic Molecular Systems as a Consequence of Broken Symmetry" Symmetry 12, no. 10: 1688. https://doi.org/10.3390/sym12101688
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.