Complete Chloroplast Genome of Argania spinosa: Structural Organization and Phylogenetic Relationships in Sapotaceae

 , ,
, ,
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Genome Assembly
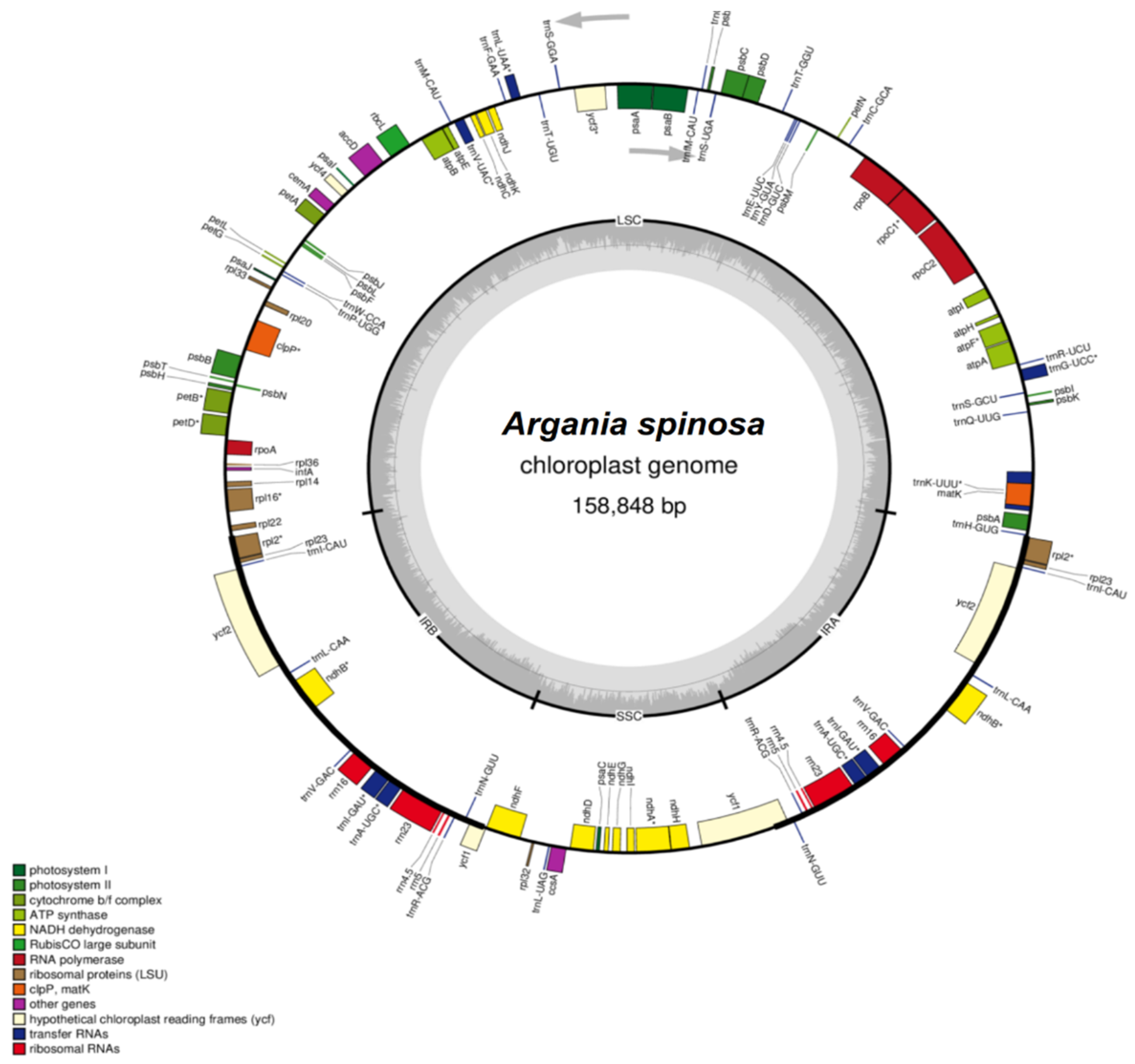
2.2. Features of the A. spinosa Chloroplast Genome
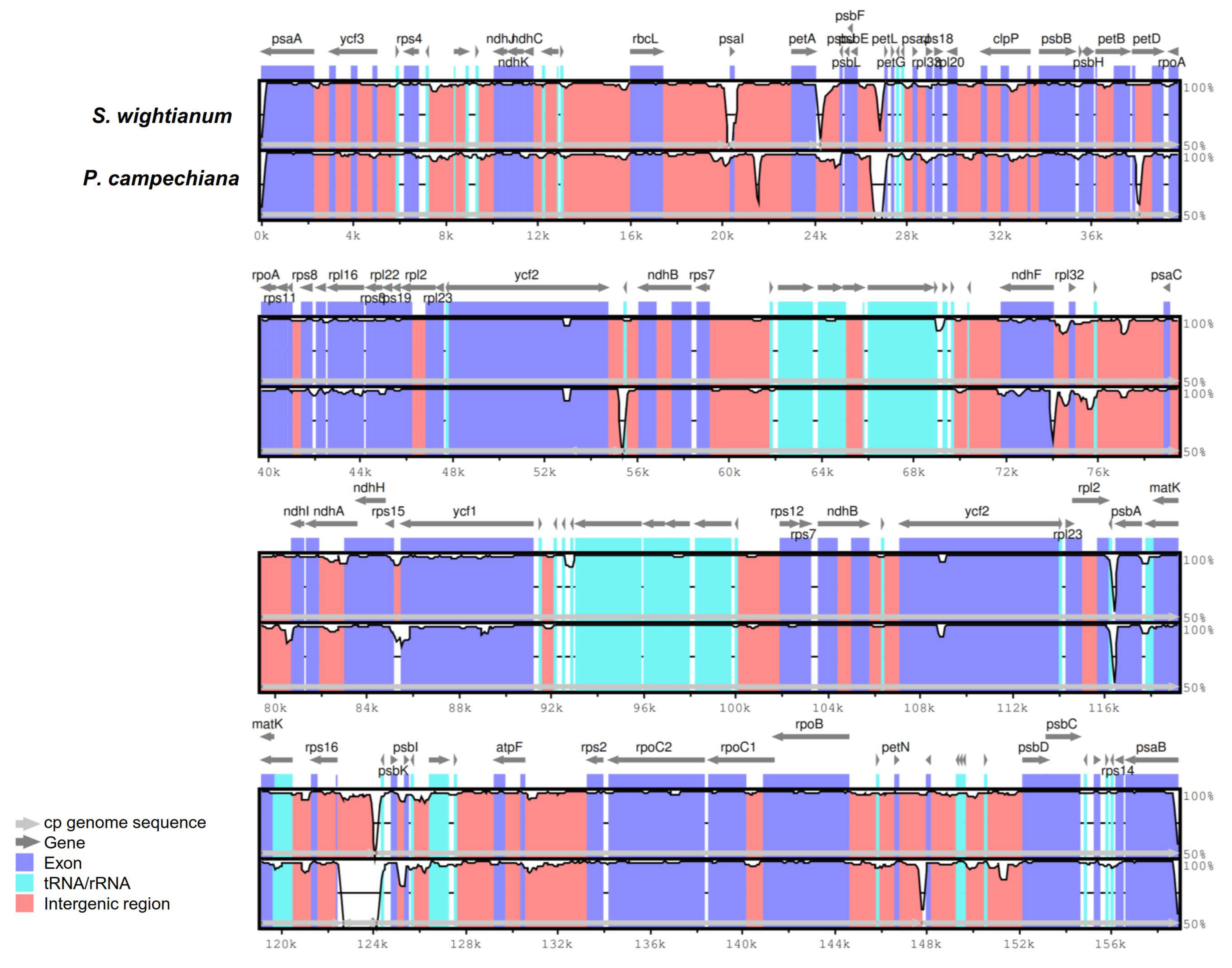
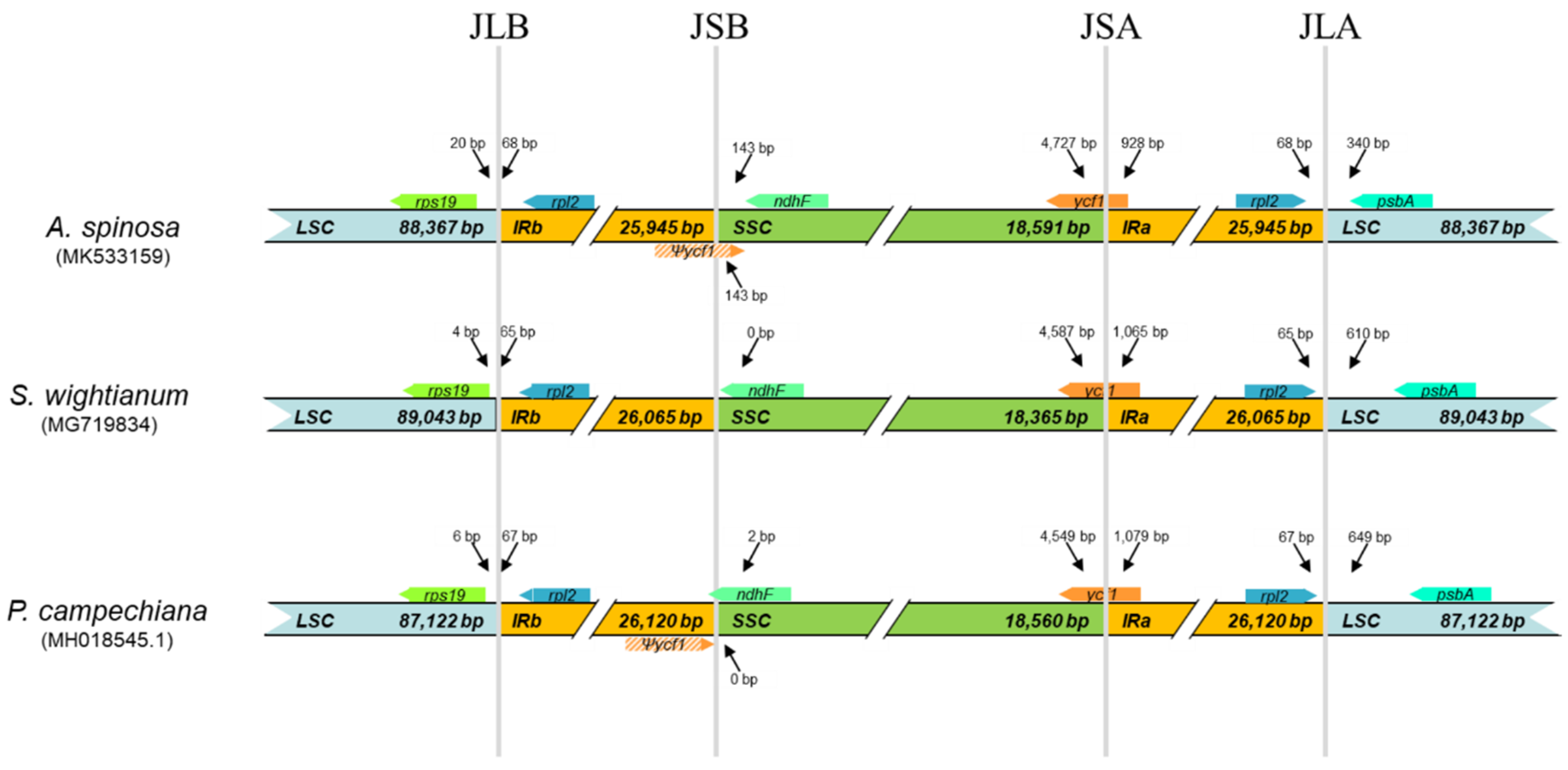
2.3. Comparative Analysis of cp Genome Structures
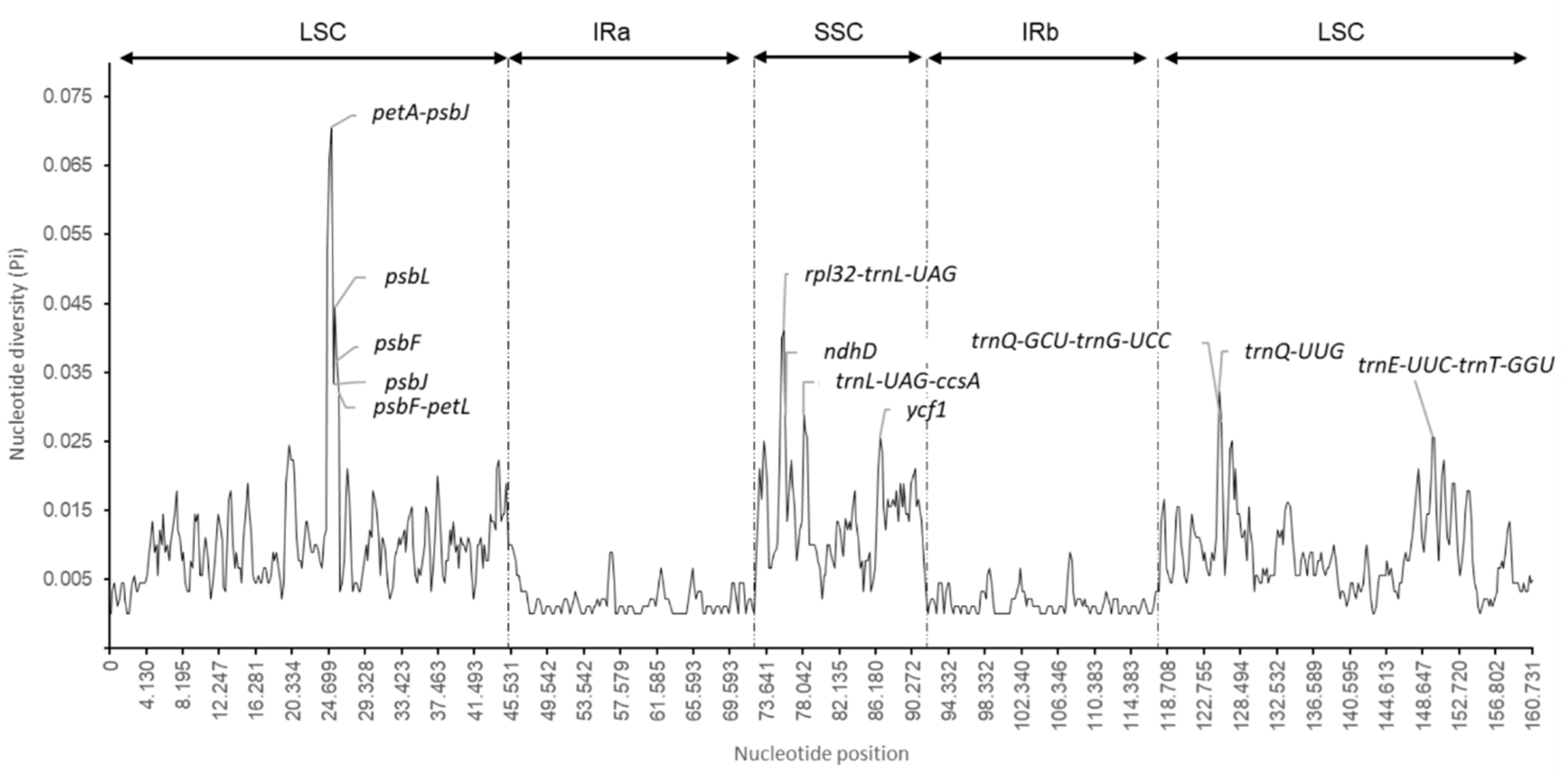
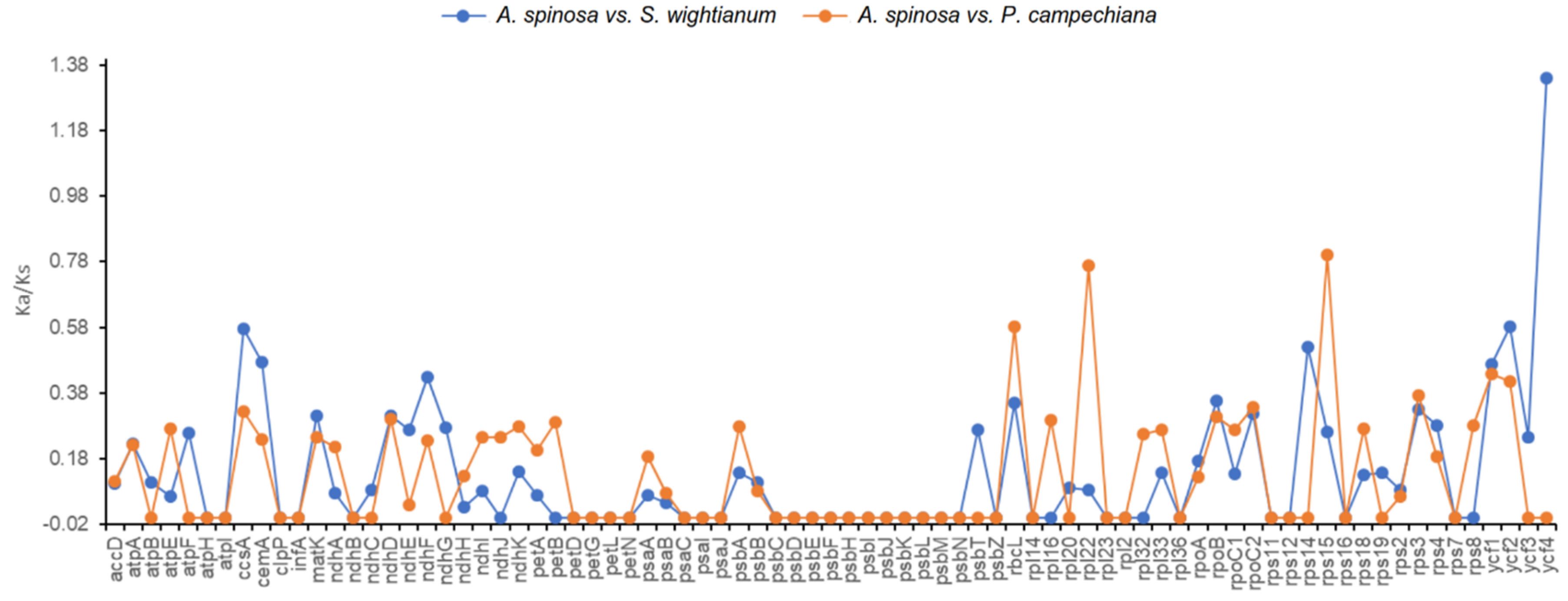
2.4. Nucleotide Diversity and Divergence of Coding Gene Sequences
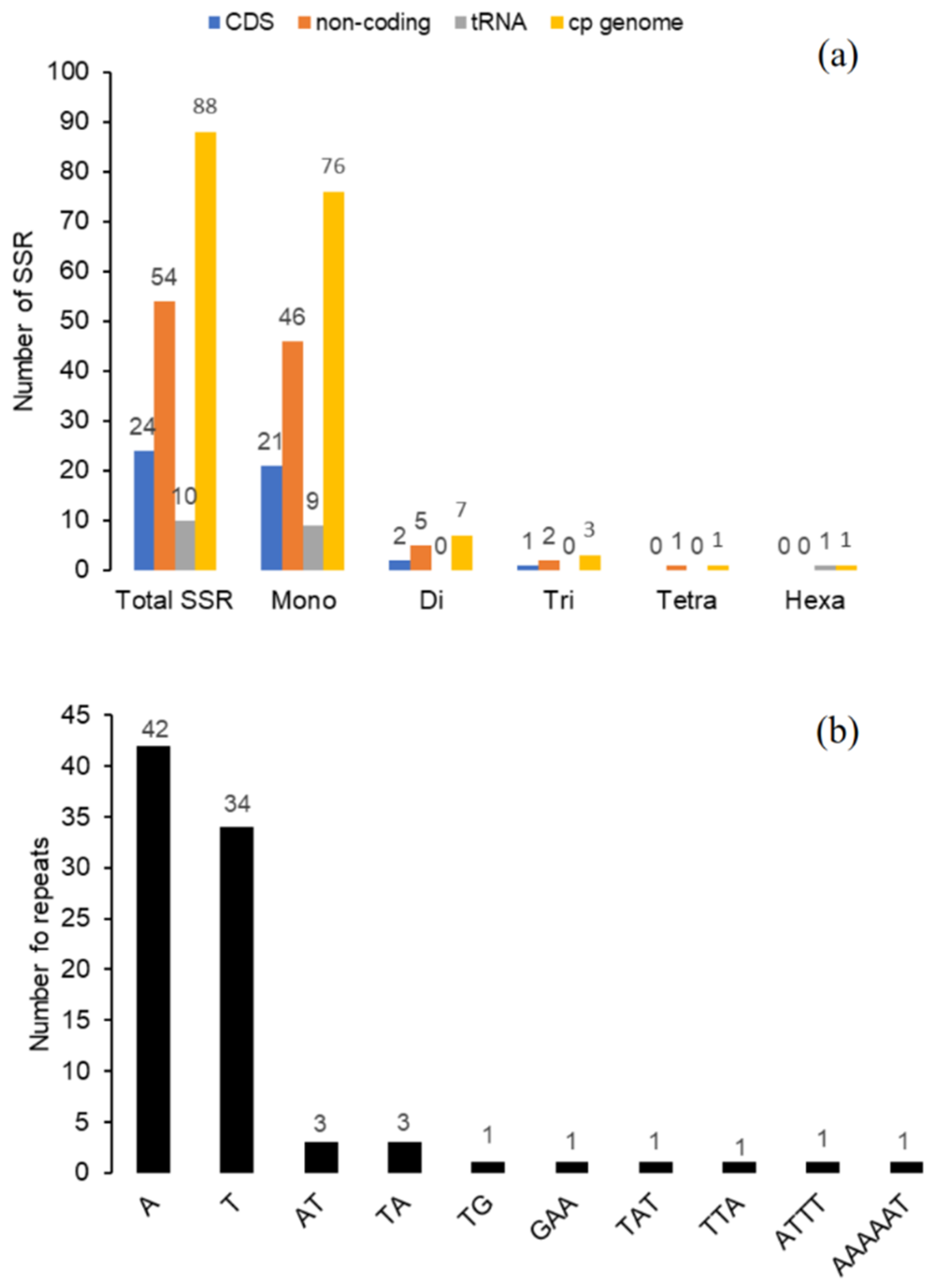
2.5. Long Repeat and Simple Sequence Repeats (SSR) Analysis
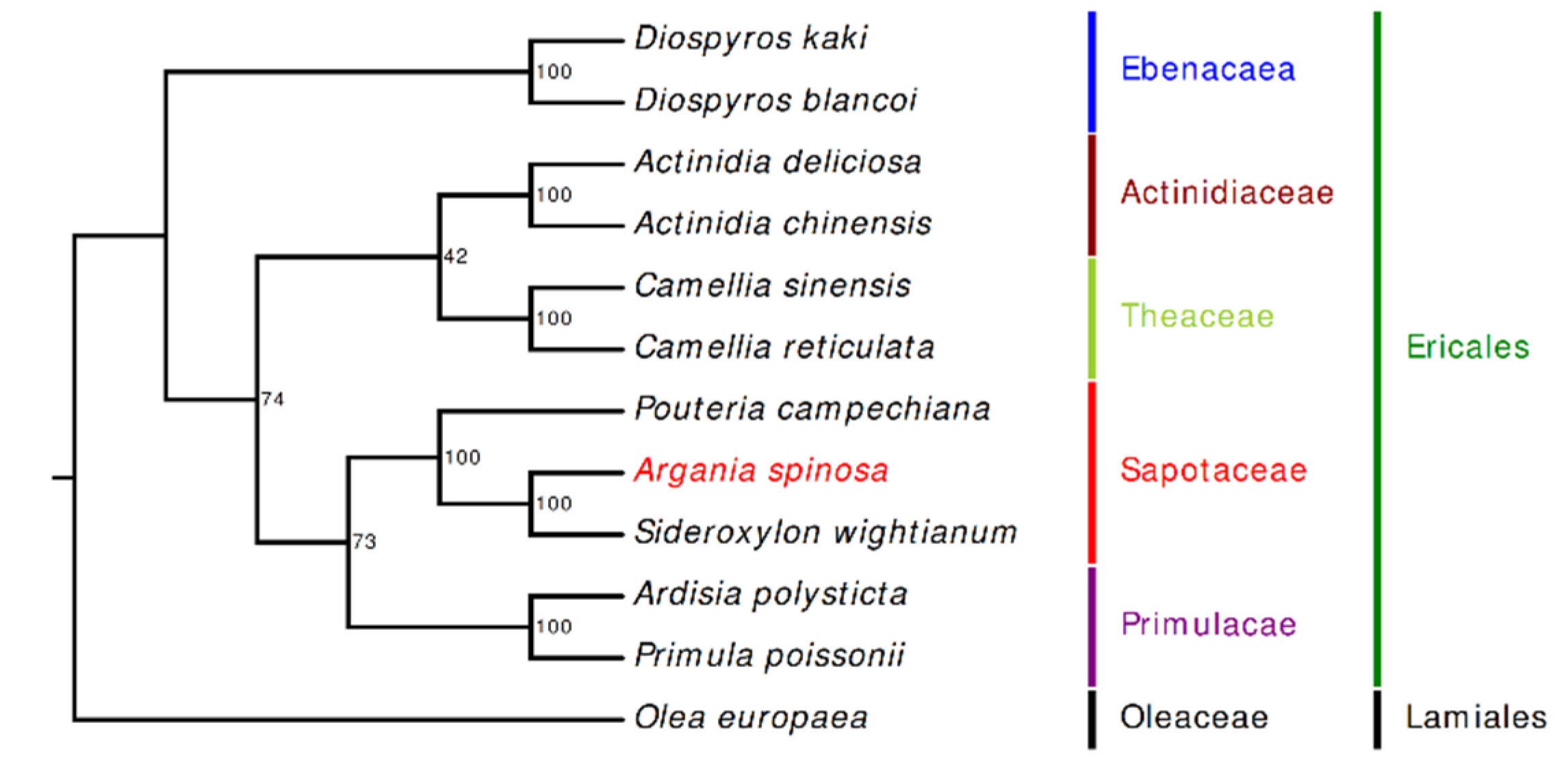
2.6. Phylogenetic Inference of A. spinosa
3. Materials and Methods
3.1. Plant Material, DNA Extraction, and Sequencing
3.2. Chloroplast Genome Assembly
3.3. Genome Annotation and Comparisons
3.4. Long Repetitive Sequences and Simple Sequence Repeat Analysis
3.5. Phylogenetic Analyses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kenny, L. Atlas De L’Arganier et de L’Arganeraie; Institut Agronomique et Vétérinaire Hassan II, Complexe Horticole d’Agadir; Agropolis International: Agadir, Montpellier, France, 2007; ISBN 978-2-909613-00-0. [Google Scholar]
- Charrouf, Z.; Guillaume, D. Argan oil: Occurrence, composition and impact on human health. Eur. J. Lipid Sci. Technol. 2008, 110, 632–636. [Google Scholar] [CrossRef]
- McGregor, H.V.; Dupont, L.; Stuut, J.-B.W.; Kuhlmann, H. Vegetation change, goats, and religion: A 2000-year history of land use in southern Morocco. Quat. Sci. Rev. 2009, 28, 1434–1448. [Google Scholar] [CrossRef]
- Le Polain de Waroux, Y.; Lambin, E.F. Monitoring degradation in arid and semi-arid forests and woodlands: The case of the argan woodlands (Morocco). Appl. Geogr. 2012, 32, 777–786. [Google Scholar] [CrossRef]
- Vaghani, S.N. Fruits of Tropical Climates | Fruits of the Sapotaceae. In Encyclopedia of Food Sciences and Nutrition; Elsevier: New York, NY, USA, 2003; pp. 2790–2800. ISBN 978-0-12-227055-0. [Google Scholar]
- Duangjai, S.; Wallnofer, B.; Samuel, R.; Munzinger, J.; Chase, M.W. Generic delimitation and relationships in Ebenaceae sensu lato: Evidence from six plastid DNA regions. Am. J. Bot. 2006, 93, 1808–1827. [Google Scholar] [CrossRef] [PubMed]
- Smedmark, J.E.E.; Anderberg, A.A. Boreotropical migration explains hybridization between geographically distant lineages in the pantropical clade Sideroxyleae (Sapotaceae). Am. J. Bot. 2007, 94, 1491–1505. [Google Scholar] [CrossRef]
- Smedmark, J.E.E.; Swenson, U.; Anderberg, A.A. Accounting for variation of substitution rates through time in Bayesian phylogeny reconstruction of Sapotoideae (Sapotaceae). Mol. Phylogenet. Evol. 2006, 39, 706–721. [Google Scholar] [CrossRef] [PubMed]
- Swenson, U.; Anderberg, A.A. Phylogeny, character evolution, and classification of Sapotaceae (Ericales). Cladistics 2005, 21, 101–130. [Google Scholar] [CrossRef]
- Stride, G.; Nylinder, S.; Swenson, U. Revisiting the biogeography of Sideroxylon (Sapotaceae) and an evaluation of the taxonomic status of Argania and Spiniluma. Aust. Syst. Bot. 2014, 27, 104. [Google Scholar] [CrossRef]
- Anderberg, A.A.; Rydin, C.; Kallersjo, M. Phylogenetic relationships in the order Ericales s.l.: Analyses of molecular data from five genes from the plastid and mitochondrial genomes. Am. J. Bot. 2002, 89, 677–687. [Google Scholar] [CrossRef]
- Jo, S.; Kim, H.-W.; Kim, Y.-K.; Cheon, S.-H.; Kim, K.-J. The first complete plastome sequence from the family Sapotaceae, Pouteria campechiana (Kunth) Baehni. Mitochondrial DNA Part B 2016, 1, 734–736. [Google Scholar] [CrossRef] [Green Version]
- Kuang, D.-Y.; Wu, H.; Wang, Y.-L.; Gao, L.-M.; Zhang, S.-Z.; Lu, L. Complete chloroplast genome sequence of Magnolia kwangsiensis (Magnoliaceae): Implication for DNA barcoding and population genetics. Genome 2011, 54, 663–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.-Y.; Yu, Y.; Deng, Y.-Q.; Li, J.; Huang, Z.-X.; Zhou, S.-D.; Liu, H.-Y.; Yu, Y.; Deng, Y.-Q.; Li, J.; et al. The Chloroplast Genome of Lilium henrici: Genome Structure and Comparative Analysis. Molecules 2018, 23, 1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niu, Y.-F.; Ni, S.-B.; Liu, Z.-Y.; Zheng, C.; Mao, C.-L.; Shi, C.; Liu, J. The complete chloroplast genome of tropical and sub-tropical fruit tree Lucuma nervosa (Sapotaceae). Mitochondrial DNA Part B 2018, 3, 440–441. [Google Scholar] [CrossRef] [Green Version]
- Ritland, K.; Clegg, M.T. Evolutionary Analysis of Plant DNA Sequences. Am. Nat. 1987, 130, S74–S100. [Google Scholar] [CrossRef]
- Pérez-Jiménez, M.; Besnard, G.; Dorado, G.; Hernandez, P. Varietal Tracing of Virgin Olive Oils Based on Plastid DNA Variation Profiling. PLoS ONE 2013, 8, e70507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, C.; Pereira, F. Identification of plant species using variable length chloroplast DNA sequences. Forensic Sci. Int. Genet. 2018, 36, 1–12. [Google Scholar] [CrossRef]
- Khayi, S.; Azza, N.E.; Gaboun, F.; Pirro, S.; Badad, O.; Claros, M.G.; Lightfoot, D.A.; Unver, T.; Chaouni, B.; Merrouch, R.; et al. First draft genome assembly of the Argane tree (Argania spinosa). F1000Research 2018, 7, 1310. [Google Scholar] [CrossRef]
- El Mousadik, A.; Petit, R.J. Chloroplast DNA phylogeography of the argan tree of Morocco. Mol. Ecol. 1996, 5, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Daniell, H.; Lin, C.-S.; Yu, M.; Chang, W.-J. Chloroplast genomes: Diversity, evolution, and applications in genetic engineering. Genome Biol. 2016, 17, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gitzendanner, M.A.; Soltis, P.S.; Yi, T.-S.; Li, D.-Z.; Soltis, D.E. Plastome Phylogenetics: 30 Years of Inferences Into Plant Evolution. In Advances in Botanical Research; Elsevier: New York, NY, USA, 2018; Volume 85, pp. 293–313. ISBN 978-0-12-813457-3. [Google Scholar]
- Raman, G.; Park, S. The Complete Chloroplast Genome Sequence of Ampelopsis: Gene Organization, Comparative Analysis, and Phylogenetic Relationships to Other Angiosperms. Front. Plant Sci. 2016, 7, 341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, I.; Kim, W.; Yeo, S.-M.; Choi, G.; Kang, Y.-M.; Piao, R.; Moon, B. The Complete Chloroplast Genome Sequences of Fritillaria ussuriensis Maxim. and Fritillaria cirrhosa D. Don, and Comparative Analysis with Other Fritillaria Species. Molecules 2017, 22, 982. [Google Scholar] [CrossRef] [Green Version]
- Asaf, S.; Waqas, M.; Khan, A.L.; Khan, M.A.; Kang, S.-M.; Imran, Q.M.; Shahzad, R.; Bilal, S.; Yun, B.-W.; Lee, I.-J. The Complete Chloroplast Genome of Wild Rice (Oryza minuta) and Its Comparison to Related Species. Front. Plant Sci. 2017, 8, 304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Hao, Z.; Xu, H.; Yang, L.; Liu, G.; Sheng, Y.; Zheng, C.; Zheng, W.; Cheng, T.; Shi, J. The complete chloroplast genome sequence of the relict woody plant Metasequoia glyptostroboides Hu et Cheng. Front. Plant Sci. 2015, 6, 447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, J.; Song, J.; Gao, H.; Zhu, Y.; Xu, J.; Pang, X.; Yao, H.; Sun, C.; Li, X.; Li, C.; et al. The complete chloroplast genome sequence of the medicinal plant Salvia miltiorrhiza. PloS ONE 2013, 8, e57607. [Google Scholar] [CrossRef] [PubMed]
- Asaf, S.; Khan, A.L.; Khan, M.A.; Shahzad, R.; Lubna; Kang, S.M.; Al-Harrasi, A.; Al-Rawahi, A.; Lee, I.-J. Complete chloroplast genome sequence and comparative analysis of loblolly pine (Pinus taeda L.) with related species. PLoS ONE 2018, 13, e0192966. [Google Scholar] [CrossRef] [Green Version]
- Palmer, J.D.; Thompson, W.F. Chloroplast DNA rearrangements are more frequent when a large inverted repeat sequence is lost. Cell 1982, 29, 537–550. [Google Scholar] [CrossRef]
- Nei, M.; Kumar, S. Molecular Evolution and Phylogenetics; Oxford University Press: Oxford, NY, USA, 2000; ISBN 978-0-19-513584-8. [Google Scholar]
- Wang, D.; Liu, F.; Wang, L.; Huang, S.; Yu, J. Nonsynonymous substitution rate (Ka) is a relatively consistent parameter for defining fast-evolving and slow-evolving protein-coding genes. Biol. Direct 2011, 6, 13. [Google Scholar] [CrossRef] [Green Version]
- Redwan, R.M.; Saidin, A.; Kumar, S.V. Complete chloroplast genome sequence of MD-2 pineapple and its comparative analysis among nine other plants from the subclass Commelinidae. BMC Plant Biol. 2015, 15. [Google Scholar] [CrossRef] [Green Version]
- Rousseau-Gueutin, M.; Bellot, S.; Martin, G.E.; Boutte, J.; Chelaifa, H.; Lima, O.; Michon-Coudouel, S.; Naquin, D.; Salmon, A.; Ainouche, K.; et al. The chloroplast genome of the hexaploid Spartina maritima (Poaceae, Chloridoideae): Comparative analyses and molecular dating. Mol. Phylogenet. Evol. 2015, 93, 5–16. [Google Scholar] [CrossRef]
- Xu, J.-H.; Liu, Q.; Hu, W.; Wang, T.; Xue, Q.; Messing, J. Dynamics of chloroplast genomes in green plants. Genomics 2015, 106, 221–231. [Google Scholar] [CrossRef]
- Zhou, T.; Chen, C.; Wei, Y.; Chang, Y.; Bai, G.; Li, Z.; Kanwal, N.; Zhao, G. Comparative Transcriptome and Chloroplast Genome Analyses of Two Related Dipteronia Species. Front. Plant Sci. 2016, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, R.I.; Sutherland, G.R. Simple repeat DNA is not replicated simply. Nat. Genet. 1994, 6, 114–116. [Google Scholar] [CrossRef] [PubMed]
- Echt, C.S.; DeVerno, L.L.; Anzidei, M.; Vendramin, G.G. Chloroplast microsatellites reveal population genetic diversity in red pine, Pinus resinosa Ait. Mol. Ecol. 1998, 7, 307–316. [Google Scholar] [CrossRef] [Green Version]
- Leclercq, S.; Rivals, E.; Jarne, P. Detecting microsatellites within genomes: Significant variation among algorithms. BMC Bioinform. 2007, 8, 125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, Z.; Cheng, T.; Zheng, R.; Xu, H.; Zhou, Y.; Li, M.; Lu, F.; Dong, Y.; Liu, X.; Chen, J.; et al. The Complete Chloroplast Genome Sequence of a Relict Conifer Glyptostrobus pensilis: Comparative Analysis and Insights into Dynamics of Chloroplast Genome Rearrangement in Cupressophytes and Pinaceae. PLoS ONE 2016, 11, e0161809. [Google Scholar] [CrossRef] [Green Version]
- Rose, J.P.; Kleist, T.J.; Löfstrand, S.D.; Drew, B.T.; Schönenberger, J.; Sytsma, K.J. Phylogeny, historical biogeography, and diversification of angiosperm order Ericales suggest ancient Neotropical and East Asian connections. Mol. Phylogenet. Evol. 2018, 122, 59–79. [Google Scholar] [CrossRef]
- Larson, D.A.; Walker, J.F.; Vargas, O.M.; Smith, S.A. A consensus phylogenomic approach highlights paleopolyploid and rapid radiation in the history of Ericales. Am. J. Bot. 2020, 107, 773–789. [Google Scholar] [CrossRef]
- Stull, G.W.; Soltis, P.S.; Soltis, D.E.; Gitzendanner, M.A.; Smith, S.A. Nuclear phylogenomic analyses of asterids conflict with plastome trees and support novel relationships among major lineages. Am. J. Bot. 2020, 107, 790–805. [Google Scholar] [CrossRef] [PubMed]
- Krumsiek, J.; Arnold, R.; Rattei, T. Gepard: A rapid and sensitive tool for creating dotplots on genome scale. Bioinformatics 2007, 23, 1026–1028. [Google Scholar] [CrossRef] [Green Version]
- Wyman, S.K.; Jansen, R.K.; Boore, J.L. Automatic annotation of organellar genomes with DOGMA. Bioinformatics 2004, 20, 3252–3255. [Google Scholar] [CrossRef] [Green Version]
- Lohse, M.; Drechsel, O.; Kahlau, S.; Bock, R. OrganellarGenomeDRAW—A suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 2013, 41, W575–W581. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, W273–W279. [Google Scholar] [CrossRef] [PubMed]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Wang, D.; Zhang, Y.; Zhang, Z.; Zhu, J.; Yu, J. KaKs_Calculator 2.0: A Toolkit Incorporating Gamma-Series Methods and Sliding Window Strategies. Genom. Proteom. Bioinform. 2010, 8, 77–80. [Google Scholar] [CrossRef] [Green Version]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [Green Version]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Shen, W.; Le, S.; Li, Y.; Hu, F. SeqKit: A Cross-Platform and Ultrafast Toolkit for FASTA/Q File Manipulation. PLoS ONE 2016, 11, e0163962. [Google Scholar] [CrossRef]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinforma. Oxf. Engl. 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Group of Genes | Name of Genes |
|---|---|---|
| Self-replication | Ribosomal RNAs | rrn16, rrn 23, rrn4.5, rrn5 |
| Transfer RNAs | trnA-UGC, trnC-GCA, trnD-GUC, trnE-UUC, trnF-GAA, trnfM-CAU, trnG-UCC, trnH-GUG, trnI-CAU, trnI-GAU, trnK-UUU, trnL-CAA, trnL-UAA, trnL-UAG, trnM-CAU, trnN-GUU, trnS-GCU, trnP-UGG, trnQ-UUG, trnR-ACG, trnR-UCU trnW-CCA, trnY-GUA, trnS-GGA, trnS-UGA, trnT-GGU, trnT-UGU, trnV-GAC, trnV-UAC, trnG-GCC | |
| Small subunit of ribosome | rps2, rps3, rps4, rps7, rps8, rps11, rps12, rps14, rps15, rps16, rps18, rps19 | |
| Large subunit of ribosome | rpl14, rpl16, rpl2, rpl20, rpl22, rpl23, rpl32, rpl33, rpl36 | |
| Translational initiation factor | infA | |
| DNA-dependent RNA polymerase | rpoA, rpoB, rpoC1, rpoC2 | |
| Genes for photosynthesis | NADH dehydrogenase | ndhA, ndhB, ndhC |
| ndhD, ndhE, ndhF, ndhG, ndhH, ndhI, ndhJ, ndhK | ||
| PS1 | psaA, psaB, psaC, psaI, psaJ | |
| PS2 | psaA, psaB, psaC, psaI, psaJ, psbA, psbB, psbC, psbD, psbE, psbF, psbH, psbI, psbJ, psbK, psbL, psbM, psbN, psbT, psbZ | |
| Cytochromeb/f complex | petA, petB, petD, petG, petL, petN | |
| ATP synthase | atpA, atpB, atpE, atpF, atpH, atpI | |
| rubisco large | rbcL | |
| Other genes | maturase | matK |
| protease | clpP | |
| Envelope membrane protein | cemA | |
| Subunit acetyl-co carboxylase | accD | |
| c-type cytochrome synthesis gene | ccsA | |
| Genes with unknown function | ORF ycf | Ycf1, ycf2, ycf3, ycf4 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khayi, S.; Gaboun, F.; Pirro, S.; Tatusova, T.; El Mousadik, A.; Ghazal, H.; Mentag, R. Complete Chloroplast Genome of Argania spinosa: Structural Organization and Phylogenetic Relationships in Sapotaceae. Plants 2020, 9, 1354. https://doi.org/10.3390/plants9101354
Khayi S, Gaboun F, Pirro S, Tatusova T, El Mousadik A, Ghazal H, Mentag R. Complete Chloroplast Genome of Argania spinosa: Structural Organization and Phylogenetic Relationships in Sapotaceae. Plants. 2020; 9(10):1354. https://doi.org/10.3390/plants9101354
Chicago/Turabian StyleKhayi, Slimane, Fatima Gaboun, Stacy Pirro, Tatiana Tatusova, Abdelhamid El Mousadik, Hassan Ghazal, and Rachid Mentag. 2020. "Complete Chloroplast Genome of Argania spinosa: Structural Organization and Phylogenetic Relationships in Sapotaceae" Plants 9, no. 10: 1354. https://doi.org/10.3390/plants9101354