Modulation of Inflammasome and Pyroptosis by Olaparib, a PARP-1 Inhibitor, in the R6/2 Mouse Model of Huntington’s Disease

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Drug Administration
2.2. Survival and Weight
2.3. Animals Behavior
2.4. Tissue Processing and Immunohistochemical Studies
2.5. Western Blotting
2.6. Microglia Morphological Characterization
2.7. Statistical Analysis
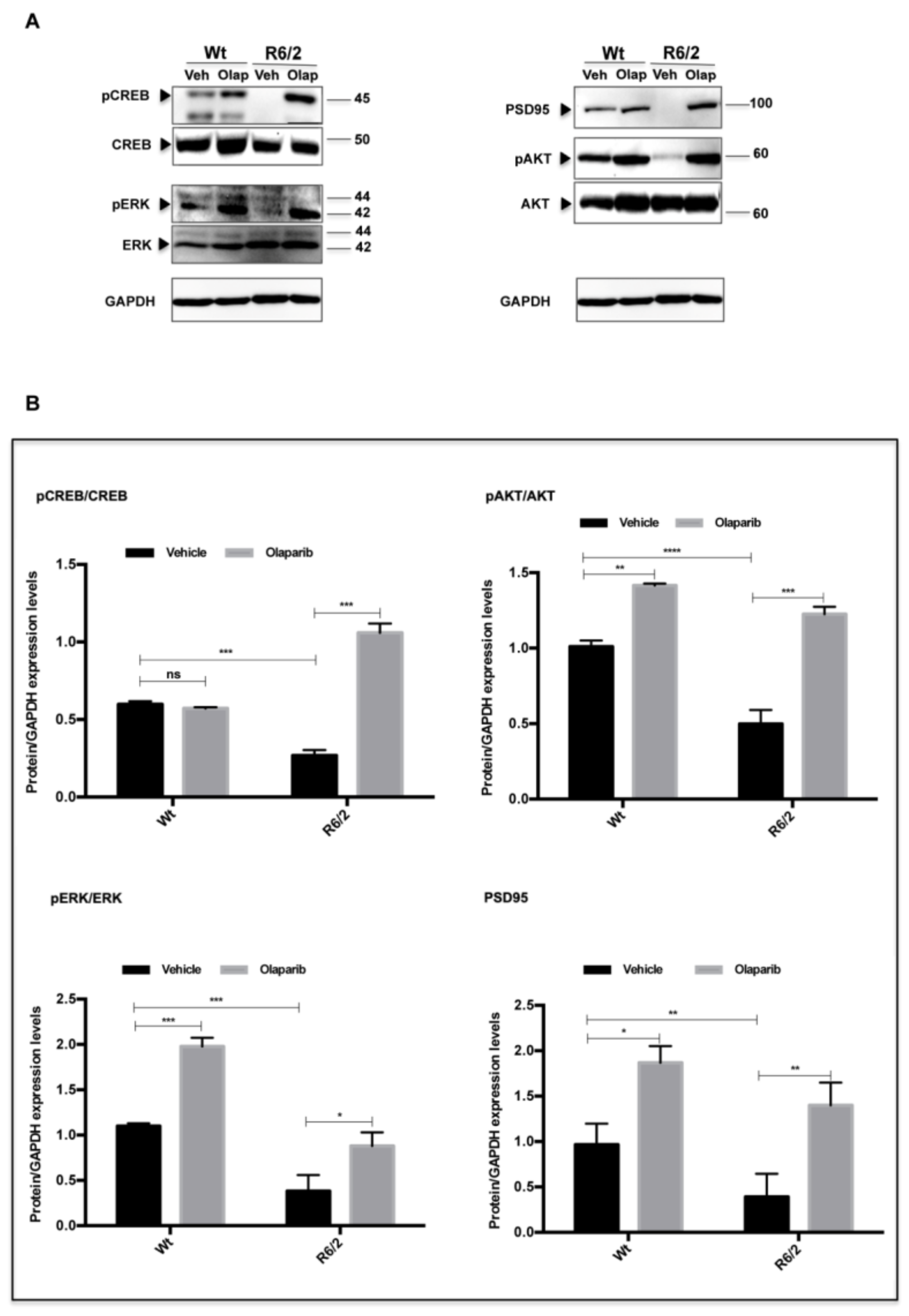
3. Results
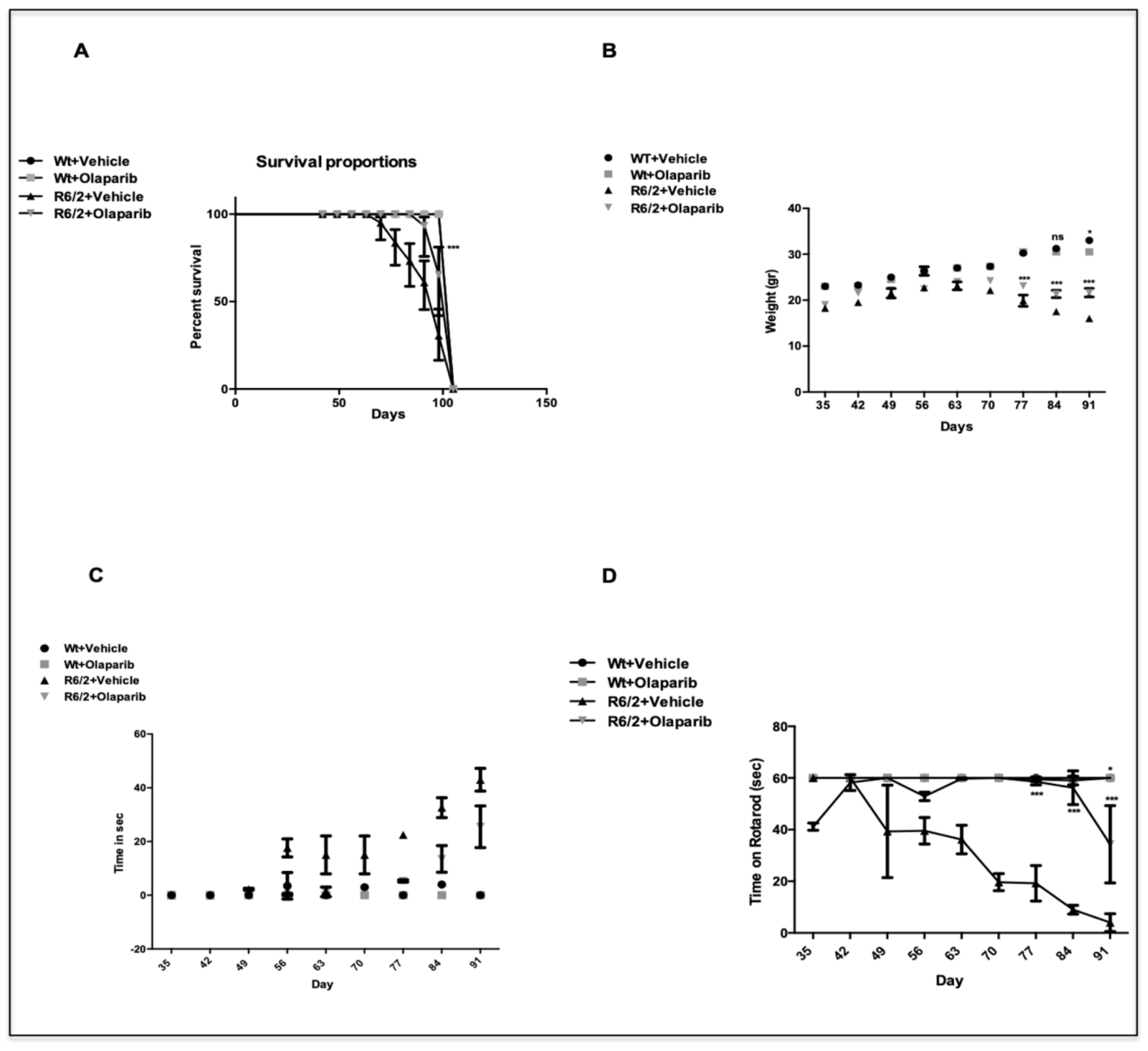
3.1. R6/2 Survival and Weight Changes
3.2. Olaparib Improves Neurological Deficits in R6/2 Mice
3.2.1. Clasping
3.2.2. Motor Behavior
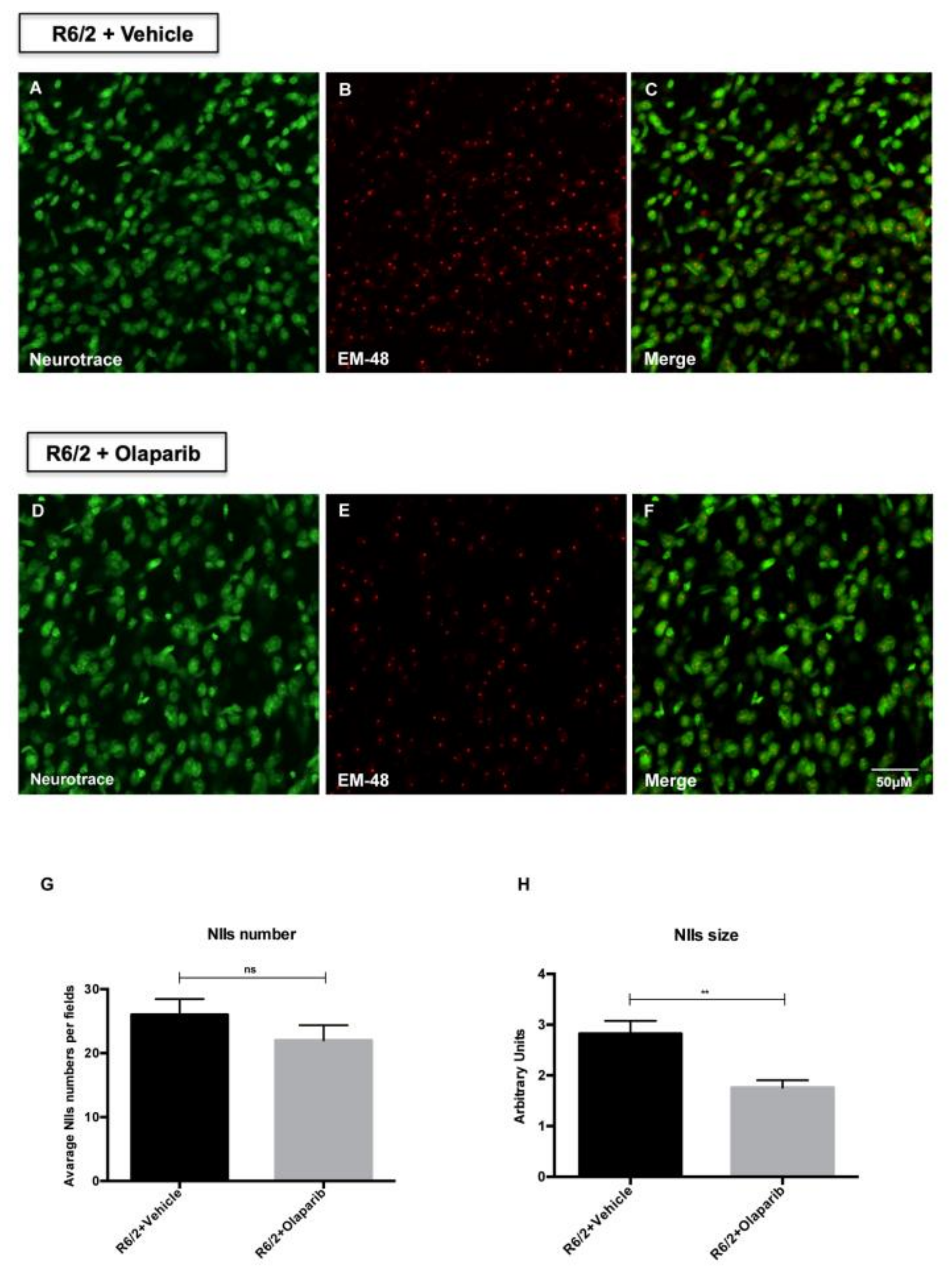
3.3. Evaluation of NIIs Number and Size
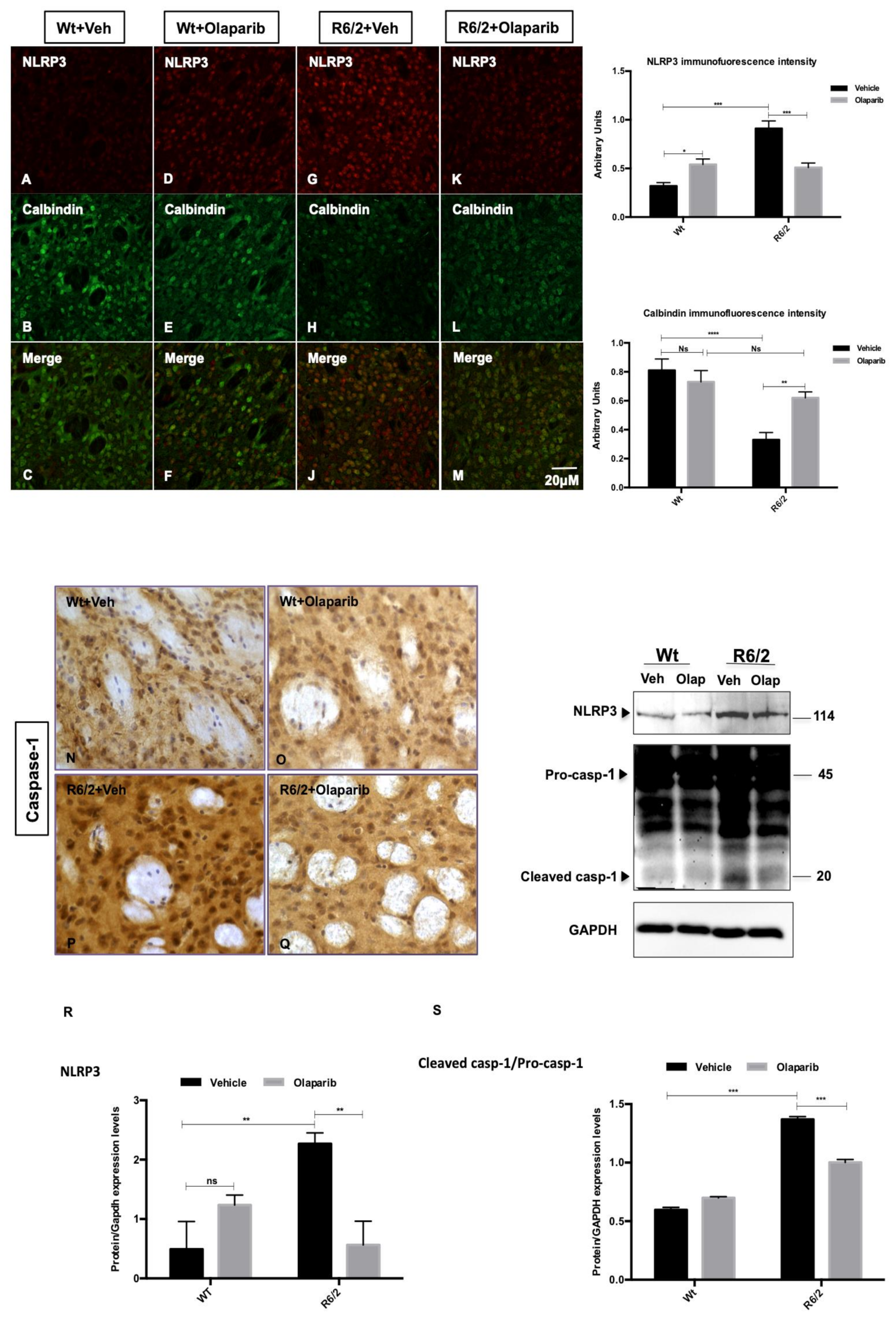
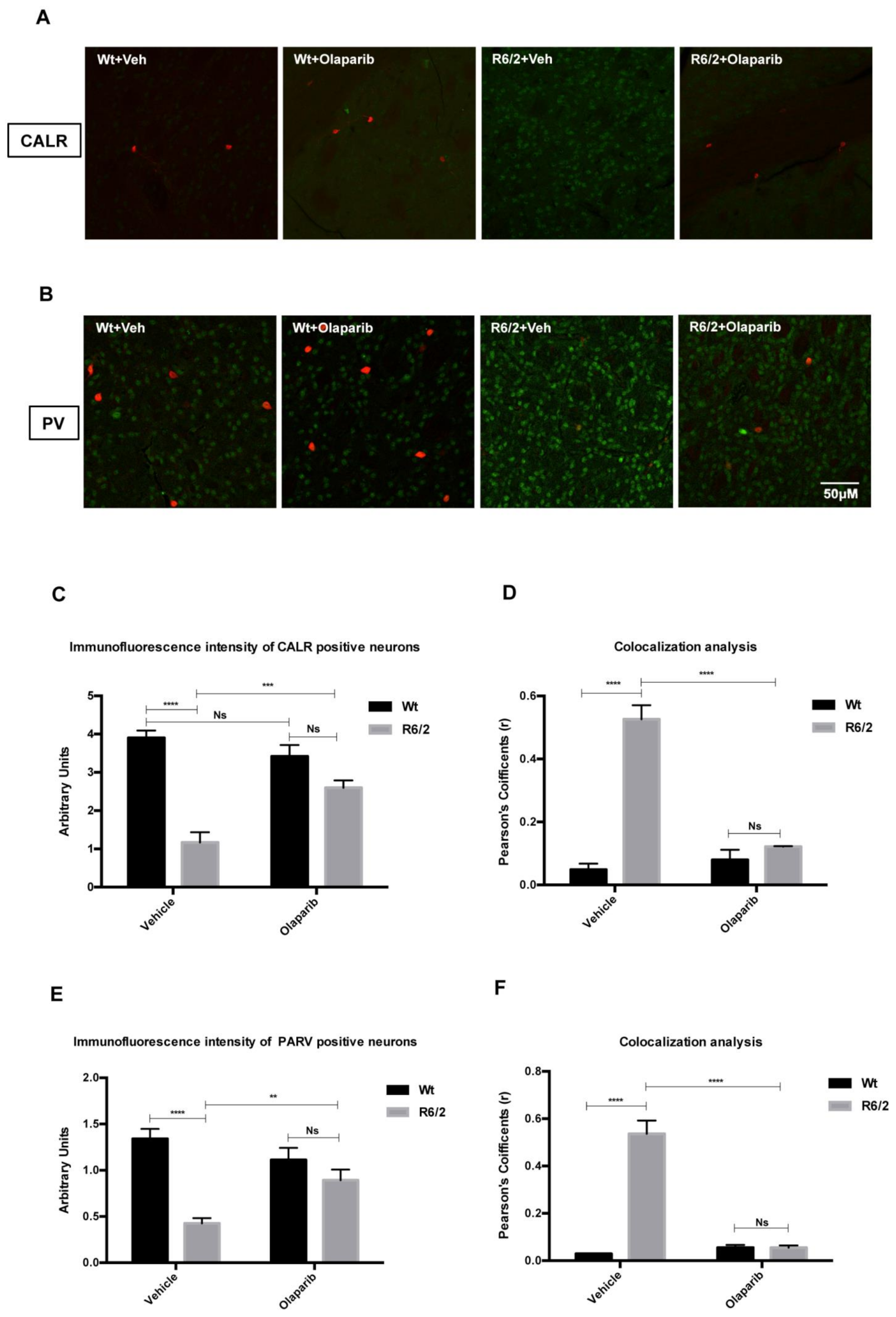
3.4. NLRP3 Expression in Striatal Projection Neurons
3.5. Analysis of NLRP3 Expression in the Striatal Interneurons
3.6. Microglial and Astrocytic Activation in R6/2 Mice
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Mangiarini, L.; Sathasivam, K.; Seller, M.; Cozens, B.; Harper, A.; Hetherington, C.; Lawton, M.; Trottier, Y.; Lehrach, H.; Davies, S.W.; et al. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 1996, 87, 493–506. [Google Scholar] [CrossRef] [Green Version]
- Wilson, R.S.; Como, P.G.; Garron, D.C.; Klawans, H.L.; Barr, A.; Klawans, D. Memory failure in Huntington’s disease. J. Clin. Exp. Neuropsychol. 1987, 9, 147–154. [Google Scholar] [CrossRef] [PubMed]
- The Huntington’s Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 1993, 72, 893–971. [Google Scholar] [CrossRef]
- Albin, R.L.; Tagle, D.A. Genetics and molecular biology of Huntington’s disease. Trends Neurosci. 1995, 18, 11–14. [Google Scholar] [CrossRef]
- Figueredo-Cardenas, G.; Harris, C.L.; Anderson, K.D.; Reiner, A. Relative resistance of striatal neurons containing calbindin or parvalbumin to quinolinic acid-mediated excitotoxicity compared to other striatal neuron types. Exp. Neurol. 1998, 149, 356–372. [Google Scholar] [CrossRef] [PubMed]
- DiFiglia, M.; Sapp, E.; Chase, K.O.; Davies, S.W.; Bates, G.P.; Vonsattel, J.P.; Aronin, N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 1997, 277, 1990–1993. [Google Scholar] [CrossRef]
- Meade, C.A.; Deng, Y.-P.; Fusco, F.R.; Del Mar, N.; Hersch, S.; Goldowitz, D.; Reiner, A. Cellular localization and development of neuronal intranuclear inclusions in striatal and cortical neurons in R6/2 transgenic mice. J. Comp. Neurol. 2002, 449, 241–269. [Google Scholar] [CrossRef]
- Sugars, K.L.; Rubinsztein, D.C. Transcriptional abnormalities in Huntington disease. Trends Genet. 2003, 19, 233–238. [Google Scholar] [CrossRef]
- Zemskov, E.A.; Jana, N.R.; Kurosawa, M.; Miyazaki, H.; Sakamoto, N.; Nekooki, M.; Nukina, N. Pro-apoptotic protein kinase Cδ is associated with intranuclear inclusions in a transgenic model of Huntington’s disease. J. Neurochem. 2003, 87, 395–406. [Google Scholar] [CrossRef]
- Petersen, A.; Mani, K.; Brundin, P. Recent advances on the pathogenesis of Huntington’s disease. Exp. Neurol. 1999, 157, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Vis, J.C.; Schipper, E.; de Boer-van Huizen, R.T.; Verbeek, M.M.; de Waal, R.M.; Wesseling, P.; ten Donkelaar, H.J.; Kremer, B. Expression pattern of apoptosis-related markers in Huntington’s disease. Acta Neuropathol. 2005, 109, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Dragunow, M.; Faull, R.L.; Lawlor, P.; Beilharz, E.J.; Singleton, K.; Walker, E.B.; Mee, E. In situ evidence for DNA fragmentation in Huntington’s disease striatum and Alzheimer’s disease temporal lobes. Neuroreport 1995, 6, 1053–1057. [Google Scholar] [CrossRef] [PubMed]
- Portera-Cailliau, C.; Hedreen, J.C.; Price, D.L.; Koliatsos, V.E. Evidence for apoptotic cell death in Huntington disease and excitotoxic animal models. J. Neurosci. 1995, 15(Pt. 2), 3775–3787. [Google Scholar] [CrossRef] [Green Version]
- Thomas, L.B.; Gates, D.J.; Richfield, E.K.; O’Brien, T.F.; Schweitzer, J.B.; Steindler, D.A. DNA end labeling (TUNEL) in Huntington’s disease and other neuropathological conditions. Exp. Neurol. 1995, 133, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, Y.P.; Nicholson, D.W.; Rasper, D.M.; Kalchman, M.A.; Koide, H.B.; Graham, R.K.; Bromm, M.; Kazemi-Esfarjani, P.; Thornberry, N.A.; Vaillancourt, J.P.; et al. Cleavage of huntingtin by apopain, a proapoptotic cysteine protease, is modulated by the polyglutamine tract. Nat. Genet. 1996, 13, 442–449. [Google Scholar] [CrossRef]
- Hickey, M.A.; Chesselet, M.F. Apoptosis in Huntington’s disease. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2003, 27, 255–265. [Google Scholar] [CrossRef]
- Miao, E.A.; Rajan, J.V.; Aderem, A. Caspase-1-induced pyroptotic cell death. Immunol. Rev. 2011, 243, 206–214. [Google Scholar] [CrossRef]
- Yuan, B.; Zhou, X.M.; You, Z.Q.; Xu, W.-D.; Fan, J.-M.; Chen, S.-J.; Han, Y.-L.; Wu, Q.; Zhang, X. Inhibition of AIM2 inflammasome activation alleviates GSDMD-induced pyroptosis in early brain injury after subarachnoid haemorrhage. Cell Death Dis. 2020, 11, 76. [Google Scholar] [CrossRef] [Green Version]
- Tschopp, J.; Schroder, K. NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nat. Rev. Immunol. 2010, 10, 210–215. [Google Scholar] [CrossRef]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [Green Version]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, J.; Zhao, F.; Chojnacki, J.E.; Fulp, J.; Klein, W.L.; Zhang, S.; Zhu, X. NLRP3 Inflammasome Inhibitor Ameliorates Amyloid Pathology in a Mouse Model of Alzheimer’s Disease. Mol. Neurobiol. 2018, 55, 1977–1987. [Google Scholar] [CrossRef] [PubMed]
- Paldino, E.; D’Angelo, V.; Sancesario, G.; Fusco, F.R. Pyroptotic cell death in the R6/2 mouse model of Huntington’s disease: New insight on the inflammasome. Cell Death Discov. 2020, 6, 69. [Google Scholar] [CrossRef] [PubMed]
- Beneke, S.; Diefenbach, J.; Burkle, A. Poly(ADP-ribosyl)ation inhibitors: Promising drug candidates for a wide variety of pathophysiologic conditions. Int. J. Cancer 2004, 111, 813–818. [Google Scholar] [CrossRef]
- Andrabi, S.A.; Kim, N.S.; Yu, S.W.; Wang, H.; Koh, D.W.; Sasaki, M.; Klaus, J.A.; Otsuka, T.; Zhang, Z.; Koehler, R.C.; et al. Poly(ADP-ribose) (PAR) polymer is a death signal. Proc. Natl. Acad. Sci. USA 2006, 103, 18308–18313. [Google Scholar] [CrossRef] [Green Version]
- Besson, V.C.; Croci, N.; Boulu, R.G.; Plotkine, M.; Marchand-Verrecchia, C. Deleterious poly(ADP-ribose)polymerase-1 pathway activation in traumatic brain injury in rat. Brain Res. 2003, 989, 58–66. [Google Scholar] [CrossRef]
- Koh, S.H.; Chang, D.I.; Kim, H.T.; Kim, J.; Kim, M.H.; Kim, K.S.; Bae, I.; Kim, H.; Kim, D.W.; Kim, S.H. Effect of 3-aminobenzamide, PARP inhibitor, on matrix metalloproteinase-9 level in plasma and brain of ischemic stroke model. Toxicology 2005, 214, 131–139. [Google Scholar] [CrossRef]
- Czapski, G.A.; Cakala, M.; Gajkowska, B.; Strosznajder, J.B. Poly(ADP-ribose) polymerase-1 inhibition protects the brain against systemic inflammation. Neurochem. Int. 2006, 49, 751–755. [Google Scholar] [CrossRef]
- Eliasson, M.J.; Sampei, K.; Mandir, A.S.; Hurn, P.D.; Traystman, R.J.; Bao, J.; Pieper, A.; Wang, Z.-Q.; Dawson, T.M.; Snyder, S.H.; et al. Poly(ADP-ribose) polymerase gene disruption renders mice resistant to cerebral ischemia. Nat. Med. 1997, 3, 1089–1095. [Google Scholar] [CrossRef]
- Endres, M.; Wang, Z.Q.; Namura, S.; Waeber, C.; Moskowitz, M.A. Ischemic brain injury is mediated by the activation of poly(ADP-ribose) polymerase. J. Cereb. Blood Flow Metab. 1997, 17, 1143–1151. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.W.; Wang, H.; Poitras, M.F.; Coombs, C.; Bowers, W.J.; Federoff, H.J.; Poirier, G.G.; dawson, T.M.; Dawson, V.L. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science 2002, 297, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.W.; Andrabi, S.A.; Wang, H.; Kim, N.S.; Poirier, G.G.; Dawson, T.M.; Dawson, V.L. Apoptosis-inducing factor mediates poly(ADP-ribose) (PAR) polymer-induced cell death. Proc. Natl. Acad. Sci. USA 2006, 103, 18314–18319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Dawson, V.L.; Dawson, T.M. Poly(ADP-ribose) signals to mitochondrial AIF: A key event in parthanatos. Exp. Neurol. 2009, 218, 193–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tapodi, A.; Debreceni, B.; Hanto, K.; Bognar, Z.; Wittmann, I.; Gallyas, F., Jr.; Varbiro, G.; Sumegi, B. Pivotal role of Akt activation in mitochondrial protection and cell survival by poly(ADP-ribose)polymerase-1 inhibition in oxidative stress. J. Biol. Chem. 2005, 280, 35767–35775. [Google Scholar] [CrossRef] [Green Version]
- Cardinale, A.; Paldino, E.; Giampà, C.; Bernardi, G.; Fusco, F.R. PARP-1 Inhibition is neuroprotective in the R6/2 mouse model of Huntington’s disease. PLoS ONE 2015, 10, e0134482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hersch, S.M.; Ferrante, R.J. Translating therapies for Huntington’s disease from genetic animal models to clinical trials. NeuroRx 2004, 1, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Berger, N.A.; Besson, V.C.; Boulares, A.H.; Bürkle, A.; Chiarugi, A.; Clark, R.S.; Curtin, N.J.; Cuzzocrea, S.; Dawson, T.M.; Dawson, V.L.; et al. Opportunities for the repurposing of PARP inhibitors for the therapy of non-oncological diseases. Br. J. Pharmacol. 2018, 175, 192–222. [Google Scholar] [CrossRef]
- Rosado, M.; Bennici, E.; Novelli, F.; Pioli, C. Beyond DNA repair, the immunological role of PARP-1 and its siblings. Immunology 2013, 139, 428–437. [Google Scholar] [CrossRef]
- Jagtap, P.; Szabo, C. Poly(ADP-ribose) polymerase and the therapeutic effects of its inhibitors. Nat. Rev. Drug Discov. 2005, 4, 421–440. [Google Scholar] [CrossRef]
- Lupo, B.; Trusolino, L. Inhibition of poly(ADP-ribosyl)ation in cancer: Old and new paradigms revisited. Biochim. Biophys. Acta 2014, 1846, 201–215. [Google Scholar] [CrossRef] [Green Version]
- Sethi, G.S.; Sharma, S.; Naura, A.S. PARP inhibition by olaparib alleviates chronic asthma-associated remodeling features via modulating inflammasome signaling in mice. IUBMB Life 2019, 71, 1003–1013. [Google Scholar] [CrossRef] [PubMed]
- Gerace, E.; Pellegrini-Giampietro, D.E.; Moroni, F.; Mannaioni, G. Poly(ADP-ribose) polymerase 1 (PARP-1) activation and Ca2+ permeable α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) channels in post-ischemic brain damage: New therapeutic opportunities? CNS Neurol. Disord. Drug Targets 2015, 14, 636–646. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, V.C.; White, J.K.; Gutekunst, C.A.; Vrbanac, V.; Weaver, M.; Li, Z.-J.; Li, S.-H.; Yi, H.; Vonsattel, J.-P.; Gusella, J.F.; et al. Long glutamine tracts cause nuclear localization of a novel form of huntingtin in medium spiny striatal neurons in HdhQ92 and HdhQ111 knock-in mice. Hum. Mol. Genet. 2000, 9, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Bjorkqvist, M.; Wild, E.J.; Thiele, J.; Silvestroni, A.; Andre, R.; Lahiri, N.; Raibon, E.; Lee, R.V.; Benn, C.L.; Soulet, D.; et al. A novel pathogenic pathway of immune activation detectable before clinical onset in Huntington’s disease. J. Exp. Med. 2008, 205, 1869–1877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGeer, P.L.; McGeer, E.G. Local neuroinflammation and the progression of Alzheimer’s disease. J. Neurovirol. 2002, 8, 529–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunningham, C.; Wilcockson, D.C.; Campion, S.; Lunnon, K.; Perry, V.H. Central and systemic endotoxin challenges exacerbate the local inflammatory response and increase neuronal death during chronic neurodegeneration. J. Neurosci. 2005, 25, 9275–9284. [Google Scholar] [CrossRef] [Green Version]
- Voet, S.; Srinivasan, S.; Lamkanfi, M.; van Loo, G. Inflammasomes in neuroinflammatory and neurodegenerative diseases. EMBO Mol. Med. 2019, 11, e10248. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, A.; Stockwell, B.R. Therapeutic approaches to preventing cell death in Huntington disease. Prog. Neurobiol. 2012, 99, 262–280. [Google Scholar] [CrossRef] [Green Version]
- Strosznajder, R.P.; Czubowicz, K.; Jesko, H.; Strosznajder, J.B. Poly (ADP-ribose) metabolism in brain and its role in ischemia pathology. Mol. Neurobiol. 2010, 41, 187–196. [Google Scholar] [CrossRef]
- Xu, J.C.; Fan, J.; Wang, X.; Eacker, S.M.; Kam, T.I.; Chen, L.; Yin, X.; Zhu, J.; Chi, Z.; Jiang, H.; et al. Cultured networks of excitatory projection neurons and inhibitory interneurons for studying human cortical neurotoxicity. Sci. Transl. Med. 2016. [Google Scholar] [CrossRef] [Green Version]
- Teng, F.; Zhu, L.; Su, J.; Zhang, X.; Li, N.; Nie, Z.; Jin, L. Neuroprotective effects of poly(ADP-ribose)polymerase inhibitor olaparib in transient cerebral ischemia. Neurochem. Res. 2016, 41, 1516–1526. [Google Scholar] [CrossRef] [PubMed]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paldino, E.; D’Angelo, V.; Laurenti, D.; Angeloni, C.; Sancesario, G.; Fusco, F.R. Modulation of Inflammasome and Pyroptosis by Olaparib, a PARP-1 Inhibitor, in the R6/2 Mouse Model of Huntington’s Disease. Cells 2020, 9, 2286. https://doi.org/10.3390/cells9102286
Paldino E, D’Angelo V, Laurenti D, Angeloni C, Sancesario G, Fusco FR. Modulation of Inflammasome and Pyroptosis by Olaparib, a PARP-1 Inhibitor, in the R6/2 Mouse Model of Huntington’s Disease. Cells. 2020; 9(10):2286. https://doi.org/10.3390/cells9102286
Chicago/Turabian StylePaldino, Emanuela, Vincenza D’Angelo, Daunia Laurenti, Cecilia Angeloni, Giuseppe Sancesario, and Francesca R. Fusco. 2020. "Modulation of Inflammasome and Pyroptosis by Olaparib, a PARP-1 Inhibitor, in the R6/2 Mouse Model of Huntington’s Disease" Cells 9, no. 10: 2286. https://doi.org/10.3390/cells9102286