
Abstract
Hydrogels that can mimic mechanical properties and functions of biological tissue have attracted great interest in tissue engineering and biofabrication. In these fields, new materials and approaches to prepare hydrogels without using toxic starting materials or materials that decompose into toxic compounds remain to be sought after. Here, we report the crosslinking of commercial, unfunctionalized hydrophilic poly(2-ethyl-2-oxazoline) using peroxide copolymers in their melt. The influence of temperature, peroxide copolymer concentration, and duration of the crosslinking process has been investigated. The method allows to create hydrogels from unfunctionalized polymers in their melt and to control the mechanical properties of the resulting materials. The design of hydrogels with a suitable mechanical performance is of crucial importance in many existing and potential applications of soft materials, including medical applications.
Similar content being viewed by others

Introduction
In the last decades, polymer hydrogels have drawn considerable attention as excellent soft materials with a wide variety of applications [1]. Hydrogels are a class of soft polymeric materials that have the ability to hold substantial amount of water, exhibit low interfacial tension, and can exhibit physical properties similar to those of living tissues [1,2,3,4]. Due to their permeability and biocompatibility (because of high water content), they have attracted much attention in the pharmaceutical and biomedical fields as key materials for various applications. Hydrogels are used, e.g., in drug delivery systems, tissue engineering materials, for contact lenses and protein separation, matrices for cell-encapsulation, cosmetic products or wound dressing, enzyme biosensors, chemical valves, metal particle preparation, and many more [5,6,7,8,9,10,11,12,13,14].
Hydrogels can be physically or chemically crosslinked. Inter alia, chemical crosslinking can be achieved by UV or reactive functional groups or enzymatic crosslinking [6, 8, 12, 15]. However, photo-polymerization requires a photo-sensitizer/initiator, suitable reactive groups on the polymer, and relatively high-energy irradiation. Crosslinking with various commercially available crosslinkers can lead to cytotoxic effects [15, 16]; alternatively specific biological triggers can be utilized [17]. Alternative crosslinking methods remain to be sought after.
It was previously reported that peroxides can be employed for crosslinking of polymers [18,19,20,21,22]. Their main advantages as crosslinking agents are the (1) ability to crosslink saturated and unsaturated polymers, (2) high-temperature resistance, (3) good elastic behavior at higher temperature, (4) low moisture uptake, and (5) no staining or discoloration of the resulting products. It was shown that the crosslinking was dependent on the reaction temperature, peroxide concentration, and crosslinking time [18, 23]. The effect of type and amount of peroxide and its decomposition rate on the crosslinking of several hydrophobic polymers was also investigated. Commercially available peroxides show relatively high hydrogen abstraction ability, which makes them good crosslinking agents for many organic polymers. Nevertheless, there are also a number of disadvantages. The decomposition products of peroxides can be volatile and exhibit a distinct smell and can lead to blooming effects. They may be extracted from the crosslinked compound by solvent, but this is clearly not a preferred approach [22]. Finally, the safety demands during the handling and storage of peroxides are considerable. As an alternative, polymer containing peroxide moieties in their side chains is known. For this, a plethora of different peroxide-containing monomers with different structures can be used for (co)polymerization. It has been suggested that 5-tert-butylperoxy-5-methyl-1-hexen-3-yne (PM) is a particularly interesting peroxide monomer due to its rather stable peroxide groups during storage and good reactivity during the polymerization [24]. In fact, the synthesis of copolymers containing PM along with other reactive groups has been reported previously [24]. These copolymers act as radical generators and have potential as compatibilization agents and surface modifiers. Therefore, such polymers have been studied for different applications, e.g., to interact with the interface of polymer colloidal systems, as coupling agents in polymer composites or as amphiphiles in aqueous solutions. In this, their ability to react simultaneously or sequentially by radical and condensation polymerization mechanism is of particular interest [24, 25].
The common crosslinking mechanism of polymers with peroxide has three main steps: (1) the primary radical formation from the thermal decomposition of peroxide, (2) hydrogen abstraction from the polymer by free radicals generated in step 1, and (3) the bimolecular radical recombination of polymer radicals from step 2 to generate covalent crosslinks [23].
Here, we report for the first time on the thermal crosslinking of a hydrophilic polymer in its melt and the swelling properties of the obtained hydrophilic covalent networks. A commercially available high molecular weight poly(2-ethyl-2-oxazoline)s (PEtOx) was chosen due to its low toxicity and potential in biomedical application [26].
At the moment the most commonly used hydrophilic polymers on the market for biomedical applications are poly(ethylene glycol) (PEG), poly(N-isopropyl acrylamide) (PNIPAAM), and other polymers which contain acrylic or methacrylic acid, partially hydrolysed poly(vinyl acetate), poly(vinyl alcohol), or poly(vinyl pyrrolidone). Poly(2-oxazoline)s (POx) are intensively investigated as alternative biomaterials, with one member of the family, poly(2-ethyl-2-oxazoline) (PEtOx) being FDA approved as indirect food additive. POx are a class of polymers obtained via (living) cationic ring opening polymerization, and because of structural similarities to poly(amino acids), they are sometimes also called pseudo-polypeptides or bioinspired polymers [27]. Furthermore they exhibit good thermal stability (decomposition temperature typically >300 °C) and are typically reported as safe biomaterials, which makes them promising materials in medical applications in general [26] and hydrogels in particular [26, 28,29,30,31]. Therefore, in recent years, they are intensively investigated for these purposes. Several strategies to obtain POx containing hydrogels can be found in the literature. Very early, Chujo et al. reported in a series of groundbreaking studies on chemical crosslinking of 2-methyl-2-oxazoline (MeOx) and 2-ethyl-2-oxazoline (EtOx) by Diels-Alder reaction, disulfide coupling, and other reversible reactions [32,33,34,35,36,37,38]. Wang and Hsiue reported the crosslinking of PEtOx dimethylacrylate with three-arm poly(D,L-lactide) trimethacrylate by photopolymerization [39]. Dargaville and co-workers reported the mild photo-crosslinking of copolymers of MeOx or EtOx, respectively, and 2-(dec-9-enyl)-2-oxazoline (DecEnOx) with dithiol crosslinkers and investigated the swelling properties of the resulting monolithic hydrogels [40]. Dargaville also reported on photo-crosslinking of water soluble copolymers of MeOx and 2-(3-butenyl)-2-oxazolin (PMeOx-co-ButenOx) as well as copolymers of MeOx and 2-undecenyl-2-oxazolin (PMeOx-DecenOx) copolymers using a low molecular dithiol [41]. Later, Hartlieb and co-workers reported on crosslinking copolymers of EtOx and 2-(5-amino valeric acid)-2-oxazoline and the resulting swelling properties of the hydrogels as well as their ability to absorb and release DNA [42]. Zahoranová et al. reported the synthesis of PEtOx-based hydrogels with different bis(2-oxazoline)s by “one pot” random copolymerization [43]. Haigh and coworkers reported on UV crosslinking of P(EtOx-co-ButenOx) for creation interconnected pores inside, resulting hydrogel’s meshes with potential application cell scaffolds or drug delivery systems [44]. All those reports dealt with chemical crosslinking. In contrast, Lorson et al. reported on thermogelling block copolymer based on the hydrophilic PMeOx and the thermoresponsive poly(2-n-propyl-2-oxazine) (PnPrOzi) which is an interesting material as bioink and biomaterial ink for three-dimensional (3D) cell culture and biofabrication [45,46,47]. Overall, POx-based hydrogels have seen a surge of interested in recent years [28, 29, 48,49,50,51], and recent reviews by Kelly and Wiesbrock as well as Dargaville and co-workers provide an excellent overview [29, 52].
In this work, we introduce a new strategy to crosslink PEtOx (and potentially any other hydrophilic polymer) in the polymer melt without the need for any functionalization on POx. To this end, we employed polyperoxides (Fig. 1), which were obtained from PM, maleic anhydride (MA), and butyl acrylate (BA). The crosslinking reaction and the products were studied by Fourier transform infrared (FTIR) spectroscopy, thermogravimetric analysis (TGA), and rheology. It will be demonstrated that the crosslinking degree mainly depends on the curing temperature and crosslinker concentration. Overall, this study introduces a facile and new approach to access hydrogels potentially from almost any kind of hydrophilic polymer. Such solvent-free melt-crosslinking method could be interesting to prepare hydrogels with thermal powder-based printing techniques, such as selective laser sintering. Thermal treatment at high temperatures potentially allows concurrent crosslinking and sterilization of the chosen polymers. Common methods of sterilization of hydrogels for biomedical application are steam heating (121–124 °C at 1 bar for not less than 15 min) or dry heating (minimum of 180 °C for not less than 30 min) [53]. In addition we suppose that the proposed method also provides endotoxin removal from the hydrogel material [54].

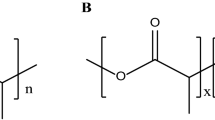
Schematic illustration of the chemical structure of the used polymers. P[PM-co-BA-co-MA] – poly(5-tert-butylperoxy-5-methyl-1-hexen-3-yne-co-butyl acrylate-co-maleic anhydride); P[PM-co-MA] – poly(5-tert-butylperoxy-5-methyl-1-hexen-3-yne-co-maleic anhydride); PEtOx – poly(2-ethyl-2-oxazoline)
Experimental section
Materials
Chloroform, maleic anhydride (MA), and butyl acrylate (BA) were purchased from Sigma-Aldrich and used without further purification, poly(2-ethyl-2-oxazoline) (PEtOx) was obtained from Kremer Pigmente (Munich, Germany). Dimethyl vinyl ethynyl carbinol (DVC, also known as Nazarov’s carbinol), was obtained from the Nairit plant CJSC (Yerevan, Armenia), was additionally purified via vacuum distillation, and was subsequently stabilized by adding of 0.1% of hydroquinone (HQ) before further use. Crude tert-butyl hydroperoxide (TBHP) with concentration of 65–70% has been synthesized in the laboratory of Organic Chemistry Chair at Lviv Polytechnic National University.
Synthesis of peroxide monomer (PM)
Crude TBHP (0.53 kg, 4 mol) and 0.46 kg (4 mol) of DVC were sequentially loaded into a glass reactor equipped with an overhead mechanical stirrer and water bath. The mixture was stirred and cooled to 0 °С by using ice water, followed by dropwise addition of 0.75 kg of 68% sulfuric acid to the reaction mixture over a period of 90 min, while keeping the temperature in the range of 0–5 °С. Subsequently, the mixture was stirred for an additional 90 min with a gradual raise of the temperature from 5 up to 10 °С and for another hour in which the mixture was allowed to warm to 10–12 °C. The reaction mixture was left to settle for 10 min; subsequently the upper organic layer was separated from the sulfuric acid layer, washed with water (160 mL) for 5 min under vigorous stirring, and again separated from the bottom water layer after 10 min’ settling. For the first alkaline washing, 100 mL of water and 40 mL of the 33 wt.% aqueous NaOH was added to the organic layer, and the mixture was stirred for 5 min, after which the phase were allowed to separate for 10 min. The bottom aqueous alkaline layer was removed. This was repeated with 80 mL of the 33 wt.% aqueous NaOH. Subsequently, the organic layer was washed twice with 160 mL of the 33 wt.% NaOH. Dry magnesium sulfate (76 g) and 0.6 g of hydroquinone were charged to the organic layer; this mixture was stirred for 30 min; afterwards the product obtained was filtered off from the solid residue sealed and stored in a refrigerator until further use. As a result, 0.66 kg (90% yield) of the final crude product 5-tert-butylperoxy-5-methyl-1-hexen-3-yne also cold peroxide monomer (PM) with a purity of 98% (determined by gas chromatography) was obtained.
Synthesis of polyperoxides
A typical procedure for synthesis of peroxide containing co- and terpolymers was as follows [55]. Copolymerization was performed in acetone at a temperature of 60 °C under argon (nitrogen) blanket at a total monomer concentration of 4 mol/L using benzoyl peroxide (BP) or lauryl peroxide (LP) as an initiator at concentration of 32 mmol/L. For the process control, the monomer conversion was determined by gas chromatography from aliquots of the reaction mixture taken every 2 h. When reaching the total monomer conversion of 85–90%, the process was stopped by cooling the reactive mixture down to room temperature, and the product was precipitated by dropwise addition of the reactive mixture into a mixture of hexane and petroleum ether (1/1; w/w). The products were further purified by double precipitation from acetone solution into a mixture of hexane and petroleum ether (1/1; w/w). Polymer composition was evaluated using elemental analysis, reverse potentiometric titration, and gas chromatography.
Gas chromatography
The content of the PM units in PC was determined using gas chromatography. Specifically, acetone and tert-butyl alcohol were quantified, formed by thermal decomposition of the peroxide groups at 180–200 °C in toluene in the presence of 2.6-di-tert-butyl-4-methyl phenol as a free-radical scavenger. The analysis was performed using gas chromatography “Selmichrom” (Selmi Co., Ukraine) supplied with 2 m metal columns filled by Inerton-super adsorbent, modified by 5% of Carbowax 40 M, and with a cathetometer as a detector. Helium was used as a carrier gas at a flow rate of 1.5 L/min.
Potentiometric titration
Potentiometric titration was carried out in a thermostatic cell using a I-130 pH meter (Belarus) supplied with the standard glass and reference Ag/AgCl electrodes. The analysis procedure was as follows: a sample of peroxide copolymer was dissolved in an excess amount of 0.1 N KOH aqueous solution for complete neutralization of the carboxylic group. Peroxide copolymer salt solution in the concentration range of 5–10 mmol/L (in moles of MA units contained in the copolymer composition) was thus obtained; afterwards an excess of KOH was titrated with 0.1 N aqueous hydrogen chloride.
Crosslinking via melt processing
In order to obtain homogeneous mixtures of PEtOx and polyperoxide (10, 15 and 20 wt.% of polyperoxide with respect to PEtOx, peroxide group content in the used polyperoxide is ≤ 10 wt.%); the polymers were dissolved in chloroform (5 wt.%). Chloroform was selected as a suitable solvent to dissolve both hydrogel components and obtain homogeneous polymer solution for the following powder preparation. The low boiling point of CHCl3 allows facile and fast removal with mild heating which leaves the peroxides intact. A homogeneous viscous solution was obtained after stirring continuously for 3–4 h at room temperature. Subsequently, the solvent was removed under reduced pressure to obtain a fine polymer blend which was ground into a powder. The obtained polymer powder (m = 0.2 g) was pressed into thin discs using a manual hydraulic press (Specac) applying 15 tons of pressure. Melting and crosslinking of polymer discs was performed at different temperatures and for predetermined times in a vacuum oven (BINDER GmbH, Germany).
Equilibrium water content
After temperature treatment, the polymer discs (0.2 g) were placed into deionized water (35 mL) for 48 h with constant shaking to remove non-crosslinked material. The water was exchanged every hour on the first day and every 3 h on the next day. The swollen polymer discs were removed from the water, were cut into small pieces, and were dried in a vacuum oven for 48 h at 45 °C until weight constancy. To obtain the equilibrium water content, samples of 1–5 mg were placed into deionized water (2.5 mL) for 1 h at ambient conditions. Subsequently, samples were removed, carefully wiped off with tissue paper to remove excess surface water, and weighed (ms).
The equilibrium water content (EWC, %) was calculated by:
where mdry is the weight of a dried crosslinked polymer sample after extraction of soluble components and ms is the weight of a swollen crosslinked polymer sample, respectively.
Sol-gel fraction
The prepared polymer discs (0.1 g) were heated in a vacuum oven at various temperatures for different times. Subsequently, the obtained samples were incubated in water (35 mL) for 48 h with constant shaking to remove the sol fraction. The water was exchanged twice. Thereafter, the samples were dried at 50 °C under vacuum for 48 h. The gel fraction (GF) was calculated gravimetrically as follows:
where m0 is the dry weight of the crude crosslinked polymer and m1 is the weight of the dried gel fraction.
NMR spectroscopy
NMR spectra were recorded on a Bruker Fourier 300 (1H: 300.12 MHz) at 298 K (Bruker BioSpin, Rheinstetten, Germany). The spectra were calibrated using the solvent signal (CD3OD 3.31 ppm).
Fourier transform infrared (FTIR) spectroscopy
Attenuated total reflectance infrared spectroscopy (ATR-IR) was recorded at room temperature on a FT/IR-4100 from 500 to 4000 cm −1 (Jasco, Groß-Umstadt, Germany) equipped with an ATR-unit.
Thermogravimetric analysis (TGA)
Thermal stability of prepolymers and crosslinked samples (3-5 mg) were characterized using a TG 209 F1 Iris® from NETZSCH (Selb, Germany) instrument (25–900 °C at 10 K/min under N2 flow).
Differential scanning calorimetry (DSC)
Temperature-modulated DSC measurements were conducted on crosslinked polymer samples using a DSC 204 F1 Phoenix® from NETZSCH (Selb, Germany) equipped with a CC200 F1 controller unit in an N2 atmosphere (20.0 mL/min). The crucibles used were aluminum crucibles from NETZSCH (Selb, Germany). All samples were at first heated to 200 °C (10 K/min) to remove thermal history followed by a cooling scan (10 K/min) to –20 °C. For each sample, three heating and two cooling cycles were performed.
Rheological measurements
Rheological experiments were performed using an Anton Paar (Ostfildern, Germany) Physica MCR 301 utilizing a plate-plate geometry (diameter 25 mm) in oscillating shear mode with a swollen polymer discs (1 mm × 25 mm). All experiments were performed in the linear viscoelastic region, which was determined by a prior amplitude sweep. All samples used for rheology were washed and swollen for 24 h. Oscillatory measurements were performed at a frequency range (ω) from 0.01 to 650 rad/s at an applied amplitude of 0.1%.
Scanning electron microscopy (SEM)
Crosslinked polymer discs were immersed into deionized water at room temperature to reach equilibrium state and then quickly frozen in liquid nitrogen and lyophilized in a freeze-drier at −40 °C. The freeze-dried samples were fixed on stubs and sputter coated with 4 nm platinum before observation. The morphology of the fractured sample surface was observed by SEM (‘Crossbeam 340’, Zeiss). The SEM analysis was done with an acceleration voltage (EHT) of 2 kV and by detecting secondary electrons (SE).
Cryo-SEM analysis
The morphology of hydrogels was examined through cryogenic-scanning electron microscopy (cryo-SEM). A sample was placed between two Al holders (d = 3 mm) both containing a notch with a diameter of 2 mm, inclosing the sample and creating a sandwich. Each sample was then flash-frozen in nitrogen slush at −210 °C, mounted on a (cryo-)transfer rod, and transferred with a cryo-shuttle to the cryo-chamber at –140 °C under high vacuum (< 1∙10−3 mbar) (Leica ACE 400 Sputter Coater; Leica EM VCT100 cryo-shuttle). Here the samples were freeze fractured by knocking of the upper half of the sample, freeze-etched by −85 °C for 15 min, and subsequently sputter coated with a 3-nm platinum layer. The sample was finally moved to the SEM chamber (with the cryo-shuttle) where either fracture or top surfaces were observed at −140 °C, using acceleration voltages between 2 and 8 kV.
Results and discussion
Prepolymer synthesis and characterization
The polyperoxides used in this study (Fig. 1) were obtained via free radical copolymerization of the corresponding monomers in acetone at 60 °C. As initiator, lauryl peroxide was used. The chemical composition of the obtained peroxide copolymers was characterized with 1H-NMR and FTIR spectroscopy. It was confirmed that under the chosen conditions, the polymer pending peroxides are stable and are incorporated into the copolymers.
The infrared spectra of the peroxide copolymers (Fig. 2a) shows absorption bands at 1860, 1782, and 1707 cm−1 which correspond to the antisymmetric and symmetric C=O stretching of maleic anhydride, respectively (Fig. 2a, violet band). The C=O of the ester groups of butyl acrylate yield an intense signal at 1730 cm−1 (Fig. 2a). Signals at 1245 and 1153 cm−1 are attributed to the C-O- stretching of MA and ester, respectively (Fig. 2a, orange band) [56, 57]. The signals at 1435 cm−1 and 753 cm−1 are attributed to the angular deformation of CH2 groups (Fig. 2a, blue). The signal at 1364 cm−1 is assigned to skeletal oscillation of −C(CH3)3 and − C(CH3)2 groups adjacent to the peroxide moieties (Fig. 2a, green). Other skeletal oscillations at 863 and 925 cm−1 correspond to (CH3)3C-O- groups (Fig. 2a, yellow). Finally, signals at 830–890 cm−1 can be attributed to peroxide groups in PM (Fig. 2a, red) [55, 58, 59], while the −C ≡ C− stretching is observed at 630 cm−1.

Characterization of the peroxide copolymers. (a) FTIR spectra of the P[PM-co-MA] and P[PM-co-BA-co-MA] respectively. (b) 1H NMR spectra of the P[PM-co-BA-co-MA] (red) and P[PM-co-MA] (black), respectively. (c) Thermogravimetric analysis of the used polymers of employed polyperoxides and commercial poly(2-ethyl-2-oxazoline) (Aquazol® 500)
The 1H-NMR spectra analysis of the product corroborates successful synthesis of the polyperoxides (Fig. 2b). The signals at 1.26 and 1.47 ppm are assigned to CH3 protons (signals 1, 2, Fig. 2b) of PM [60, 61]. The peak at 0.97 ppm is attributed to the methyl group of BA (signal 11, Fig. 2b, red). The signal at 4.09 ppm is attributed to the methylene group adjacent to the ester moiety in BA (signal 8, Fig. 2b, red). Weak and broad peaks that appear in the range 2.2–3.5 ppm are attributed to signals of MA and methine groups in the polymer backbone, respectively (signals 4, 5 and 7, Fig. 2b, both spectra). The peaks at 1.46 and 1.65 ppm are also attributed to signals of BA (signals 10, 9, Fig. 2b, red) and methylene group in the polymer backbone (signals 3, 6 Fig. 2b), respectively. At 6.31 ppm, a signal is present which belongs to C-H of residual MA monomer. The presence of this residual monomer (approx. 10%) must be kept in mind for the further discussion, as it must be expected to also react during the heat treatment.
Table 1 shows the respective values of weight average molar mass (Mw) and dispersity (Ð) of the used polymers, obtained from GPC analysis. Both copolymers were obtained with relatively similar molar mass (ca. 2 * 104 kg/mol) and molar mass distribution typical for free radical polymerization (Ð = 1.6 and 2.6, respectively).
As was already mentioned before, thermal decomposition of peroxides group leads to the formation of free radicals followed by crosslinking via recombination reaction. The main low-molecular weight products obtained during this process are acetone and tert-butyl alcohol [62]. The thermal decomposition of the present polyperoxides was analyzed by TGA. The main mass loss was observed from ca. 110 to 200 °C, which is attributed to the decomposition of peroxide moieties (Fig. 2c). According to the suggested decomposition, the main products after full decomposition of present peroxide groups are poly(5-methyl-1-hexen-3-yne-5-ol) and poly(5-methyl-1-hexen-3-yne-5-one) (Fig. 3). Assuming that all peroxide groups decompose and are converted into poly(5-methylhept-3-yn-2-one), we can calculate the expected weight loss during peroxide decomposition. For P[PM-co-MA], we expect 74% residual mass, while 80% residual mass is expected for P[PM-co-BA-co-MA]. The experimental results give a residual for P[PM-co-MA] of 69% (137–215 °C) and ca. 71% (156–203 °C) for P[PM-co-BA-co-MA], respectively (Fig. 2c). Therefore, the mass loss is higher than based on the decomposition of the peroxides within the polymers alone. This may be attributed to other degradation pathways and/or incorrect determination of the peroxide content in the polymers.

Schematic illustration of the thermal decomposition of peroxide groups in peroxide containing copolymer
Following the established mechanism for crosslinking with peroxides, the crosslinking mechanism using polyperoxides should also comprise three main steps (Fig. 4). Taking into account the radical nature of the process, it is important to note that this scheme can only show a simplified picture of possible events, as radical reactions cannot be considered to be very specific and many side reactions will occur simultaneously, such as radical combination reactions of two small radicals or reaction of one macroradical and a small radical that do not lead to crosslinking.

Simplified schematic illustration of peroxide containing copolymer induced cross-linking: (a) thermal decomposition of peroxide copolymer into two radicals when exposed to heat followed by (b) hydrogen abstraction from polymer chain by primary radical and (c) recombination reaction of polymer radicals
Crosslinking of PEtOx in melt
From low molar mass peroxides, it is known that the factors which influence crosslinking are crosslinker concentration, temperature, and time. Accordingly, we suppose that variation of these parameters allows us to control the crosslinking density and resulting mechanical and swelling properties of the prepared hydrogels. If crosslinking of PEtOx by peroxide copolymers is successful, a covalent network should result, which swells in contact with water or other good solvents. The swelling behavior of hydrogels is one of their most important characteristics, and the equilibrium water content (EWC) correlates with the crosslinking density of the polymer network [63, 64]. In this exploratory study, we prepared homogeneous mixtures of PEtOx and polyperoxide in solution. However, for large-scale powder preparation, well-known mixing techniques, like ball-milling or cryo-milling [65,66,67,68], could be used.
FTIR spectroscopy of crosslinked polymers
FTIR spectroscopy was chosen as an easy and readily available method to investigate the chemical changes during crosslinking. The employed polyperoxides have an amphiphilic character bearing hydrophilic MA groups and hydrophobic peroxide/BA, respectively. According to the suggested crosslinking mechanism (Fig. 4), both reactive groups can partake in the crosslinking process. The IR spectrum of polyperoxide/PEtOx blends also show the absorption bands at 1860, 1782 and 1707 cm−1, which were attributed to the antisymmetric stretching of the carbonyl moieties in MA, while the band at 1730 cm−1 corresponds to the C=O stretching of the BA group (Fig. 5, blue).

FTIR spectra of the (crosslinked) PEtOx with 20 wt.% of P[PM-co-MA] (a) and P[PM-co-BA-co-MA] (b) crosslinked at different temperatures and 40 min of crosslinking time
Even though the polyperoxides containing blends show only weak signals in IR spectra due to the symmetric nature [58] and low concentration of the peroxides, we can expect that the corresponding signals should decrease during crosslinking [58, 69]. Similarly, the signals attributed to the anhydride motif in MA should also decrease [70,71,72]. IR spectra of PEtOx crosslinked with P[PM-co-MA] (Fig. 5a) and P[PM-co-BA-co-MA] (Fig. 5b), respectively, before and after crosslinking at 120–160 °C showed ambiguous results. An increase in temperature to 120 °C is accompanied by an increased signal attributed to ester groups with a corresponding decrease in the signal attributed to MA. An interesting observation was made in the region of 800–900 cm−1 (Fig. 5, red band). Before heating, the spectra exhibit a broad peak in this region. With increasing temperature, a splitting of the peak is observed concomitant with a minor increase of peak intensity at 880 cm−1 and decrease at 860 cm−1 (Fig. 5a). The increase at 880 cm−1 can be attributed to symmetric stretching of ether bonds that are formed during the crosslinking [73,74,75]. This observation was less noticeable when using P[PM-co-BA-co-MA] as a crosslinker (Fig. 5b). This may be due to the lower mol% of peroxide groups. Changes of a band at 1065 cm−1 (asymmetric stretching of the C-O-C group) can be explained with changes in the MA and ether contents [74, 75]. The signals at 1780 and 1725 cm−1, which correspond to C=O groups of MA and esters, respectively, initially increased with temperature (Fig. 5, blue). However, after treatment at 200 °C, the intensities at 1780 and 1725 cm−1 as well as at 800–900 cm−1 decreased. This corroborates that peroxide groups decomposed during heating. Decreasing intensity at 1780 and 1725 cm−1 could be explained due to decomposition of ester groups under these conditions.
Thermogravimetric analysis (TGA)
The thermal stability of polyperoxide-PEtOx blends were studied before and after crosslinking by TGA. The TG profiles of the polymer blends before (red) and after crosslinking (blue) show clear differences (Fig. 6a). The degradation process consists of various stages. For the crosslinked sample, a minor mass loss of about 3% below ca. 100 °C can be attributed to physically adsorbed water (Fig. 6a blue). In contrast, the non-crosslinked sample shows a more pronounced mass loss of 15% between 50 and 180 °C. As previously discussed, this is attributed to decomposition of the peroxide groups and evaporation of resulting tert-butanol (Fig. 6a). Further mass loss (from 180 °C to 410 °C) is observed and can be attributed in part to the decomposition of the MA groups [56, 57]. While the absence of decomposition < 200 °C in the crosslinked blend confirm complete decomposition of the peroxides, the thermal stability of the blend was otherwise not significantly improved by the crosslinking. While this excellent thermal stability of the crosslinked polymer is not relevant for use under physiological conditions, it is of course beneficial in the context of sterilization and endotoxin removal of the materials [53, 54].

TG (a) and DSC (b) analysis of the polymer mixture of commercial poly(2-ethyl-2-oxazoline) (Aquazol® 500) and P[PM-co-MA] before and after crosslinking. Crosslinking was carried out for 20 min at 200 °C using 20 wt.% peroxide copolymer. Measurement was done in the temperature range 25–900 °C (a) and − 50 to 200 °C (b) at heating rate is 10 °C min−1 and second heating cycle (b)
The DSC thermogram of pure PEtOx showed a Tg at 60.5 °C (Fig. 6b) while this value was shifted to higher temperature (69.5 °C) for the crosslinked PEtOx, corresponding to lower chain segment mobility as expected after crosslinking.
Morphology analysis
Scanning electron microscopy (SEM) was used to visualize the surface morphology of the crosslinked polymers. The surface morphology of pressed discs of PEtOx powder blends with 20 wt.% of P[PM-co-MA] was assessed after heat treatment. Furthermore, samples heated at 120–200 °C were studied. Before analysis heated samples were washed with water for several days to extract the sol fraction followed by lyophilization (Fig. 7).

SEM micrographs at 2 kV EHT of the PEtOx/P[PM-co-MA] blends after heat treatment at different temperatures and time. All PEtOx/P[PM-co-MA] samples were obtained after incubation for 40 min with 20 wt.% of polyperoxide
Samples incubated at 120 °C exhibit the largest pores with a wide pore size distribution, the determined average pore areas range decreases with increasing incubation time from 205 to 51 μm2 (Fig. 8a). In addition, at higher incubation temperatures, smaller pore sizes were obtained, but no significant differences in pore size for samples crosslinked at 160 or 200 °C were observed. Interestingly, with increasing temperature and incubation time, an increasing number of agglomerates consisting of small, spherical particles were observed (Fig. 8b, c). The origin of this phenomenon is unclear at this moment but may be connected to an increased degradation of the polymer, as evidenced by the polymer discoloration.

(a) Impact of crosslinking temperature and incubation time on pore area. (b and c) Appearance of the agglomerates obtained during the heating process. EHT: 2 kV
It has become clear in recent years that freezing hydrogels at an insufficient cooling rate or increasing the temperature after freezing for, e.g., freeze-drying, may have a strong impact on the morphology due to the formation of cubic or hexagonal ice [76, 77]. Such experimental artifacts may be avoided using cryogenic SEM [78, 79]. By rapid freezing of small hydrogel samples in liquid nitrogen slush (LNS), liquid propane (LP), or ethane (LE), one can avoid water crystallization (vitrification). Accordingly, we analyzed vitrified swollen hydrogel samples crosslinked at different temperatures (Fig. 9). Clearly, the hydrogel morphology is indeed affected by the sample preparation. After vitrification, the observed pore size is much smaller than it was obtained from the SEM analysis of the lyophilized samples (Fig. 7). Also, the morphology is quite heterogeneous with the presence of rather large pores (a few hundred nanometers to a few micrometers) which are filled with another, finer network in the range of several tens of nanometers resulting in a double-pore structure. Interestingly, the hydrogel obtained at 160 °C shows the largest pores size compared with the hydrogels obtained at 120 and 200 °C, which clearly contradicts results obtained for lyophilized samples, showing once again the importance of sample preparation.

Cryo-SEM analysis (EHT: 8 kV) of the samples obtained after 40 min of treatment at different temperature. The concentration of P[PM-co-MA] is 20 wt.%
Equilibrium water content and sol-gel fraction
The EWC of the PEtOx/polyperoxide based hydrogels was determined after crosslinking with different concentrations of crosslinker (P[PM-co-MA]), crosslinking time, and temperature (Fig. 10). The investigation of swelling was performed in deionized water at room temperature. It appears that the EWC is affected by all three crosslinking parameters, but the crosslinking time shows only minor influence on EWC. In contrast, the crosslinker concentration and the crosslinking temperature appear to have a more pronounced effect. For P[PM-co-MA] crosslinker concentrations of 10 and 15 wt.% and at temperatures ≤ 160 °C, the influence on the EWC was not systematic (Fig. 10a). Interestingly, at 180 °C, there seems to be a small but systematic decrease in EWC with increasing incubation time. At 20 wt.% crosslinker, consistently the lowest EWC values were obtained (Fig. 10a). The most obvious correlation could be observed between the crosslinking temperature and EWC; a higher temperature leads to a lower EWC. For example, at 10 and 15 wt.% and 120–160 °C, EWC was found at 96.20 ± 0.31%, which decreases to 93.64 ± 0.37% at 180 °C and 91.89 ± 0.09*103% at 200 °C, respectively. At 20 wt.%, the overall EWC values are smaller but the trend remains clear. While at 120 °C, the EWC ranges from 94.36 ± 0.21%; it is reduced to 85.21 ± 0.40% at 200 °C.

Influence of the peroxide containing copolymer concentration (10–20 wt.%), temperature (120–200 °C), and crosslinking time 10–40 min, on the equilibrium water content (EWC) of PEtOx hydrogels. (a) P[PM-co-MA] and (b) P[PM-co-BA-co-MA]
Overall, a similar picture is observed with the other crosslinker (P[PM-co-BA-co-MA]). In this, the relative content of peroxide groups is lower at 20 mol%. It could be expected that the decreased PM content leads to an increase in EWC. However, no major difference in EWC was observed (Fig. 10b). We think that the presence of hydrophobic BA monomer units leads to reduced swelling of the hydrogels. Important to note, the obtained hydrogels appeared also much weaker during handling, which is in line with the lower crosslinking.
As the temperature may influence the swelling and therefore the mechanical properties of hydrogels [80], we investigated the influence of the incubation temperature (25, 35, and 60 °C) during the swelling (Fig. 11). It should be noted that all hydrogels were allowed to equilibrate for 60 min. In control experiments, we also incubated for 36 h, but no significant influence on EWC was observed.

Dependence of the swelling behavior on the temperature of swelling process. Hydrogels were obtained with the concentration of P[PM-co-MA] (a) 10 wt.% and (b) 15 wt.%
For this, we concentrated on samples crosslinked with 10 and 15 wt.% polyperoxides. For the samples crosslinked with 10 wt.%, several interesting observations were apparent. For the hydrogels crosslinked for the shortest period (10 min) and at the lowest temperature (120 °C), the EWC at room temperature reached 96.17 ± 0.15% while these samples completely dissolved when incubated at 35 °C (Fig. 11a). When crosslinked at higher temperature (140–160 °C), the resulting hydrogels show better stability. Interestingly, when we increase the incubation temperature further to 60 °C, all samples crosslinked at ≤ 160 °C dissolved completely (Fig. 11a). At 15 wt.% of crosslinker, this picture does not change much. Hydrogels obtained at 120–140 °C and relatively short crosslinking time dissolved at higher temperature, while samples crosslinked at 180 and 200 °C give stable hydrogels.
Overall, the crosslinking time does not have strong influence on EWC while the swelling temperature has a major influence. Surprisingly, hydrogels that were crosslinked at 120–160 °C were dissolved at higher temperatures (35–60 °C), but a better stability was noticed for crosslinking temperatures of 180–200 °C, even though slightly higher EWC values were observed. As it was already mentioned before, the crosslinking presumably takes place via radical mechanism as well as via the MA moieties. MA can form ester linkages with hydroxyl groups formed during peroxide degradation (Fig. 3), or, to a minor extend with OH moieties on PEtOx (from, e.g., terminal groups). Such esters can subsequently be hydrolyzed in aqueous environment, in particular at elevated temperature. Hydrogels crosslinked at lower temperature and shorter times have a lower degree of crosslinking and the ester linkages appear to be critical. When they are cleaved, the hydrogels disintegrate. To exclude any effects of the heat treatment on the solubility of PEtOx, the polymer was heated to 200 °C for 30 min, but no difference in solubility was observed.
Finally, the gel fractions of samples crosslinked at 120–200 °C were determined after extensive extraction of the sol fraction (Fig. 12). All samples showed a decrease of sol fraction with an increase of crosslinking temperature leveling off at 12% for samples obtained at 200 °C with 20 wt.% polyperoxide. At lower crosslinking temperature, significantly higher sol fractions were determined. Interestingly, samples crosslinked with P[PM-co-BA-co-MA] at 160 °C show a somewhat higher sol fraction for 20 wt.% crosslinker compared with 15 wt.% (Fig. 12b, green vs. black columns). Hydrogels obtained at 120 °C and 20 wt.% of P[PM-co-MA] show higher sol fractions than the samples prepared with P[PM-co-BA-co-MA]. For 15 wt.%, the situation was reversed. At this point, we cannot explain these trends. In most cases, heating time does not have significant influence on the sol fraction. Only at 120 °C, gel fraction increases notably with incubation time.

Sol investigations of PEtOx networks obtained with different concentrations of (a) P[PM-co-MA] and (b) P[PM-co-BA-co-MA] obtained at different crosslinking temperatures and time
Rheological properties
To investigate the viscoelastic properties of hydrogels, rheological measurements of the hydrated samples were performed (Fig. 13). Since samples crosslinked at 120 °C were too weak to handle properly, only hydrogels crosslinked at 160–200 °C and crosslinker concentrations of 15 and 20 wt.% were studied. As G’˃˃G” for all samples and all measurement parameters, all samples can be characterized as hydrogels [81]. The G’ and G” values were relatively independent of the frequency behavior with deviations only becoming apparent at or above 100 rad/s. As can be expected, an increase in crosslinker concentration and temperature (and therefore degree of crosslinking) resulted in an increase of G’. Despite the fact that the concentration of polyperoxide crosslinker is relatively high, the amount/content of peroxide groups that are involved in crosslinking reaction is much lower (≤ 2 wt.%). Therefore, it is not surprising that rather weak hydrogels are obtained. Liu et al. also reported on the peroxide crosslinking of the low-density polyethylene (LDPE) with the concentration of low molecular weight peroxide (dicumyl peroxide) (DCP) from 0.3 wt.% up to 2.5 wt.% [20]. Gel fraction of the resulting hydrogel was in the range of 70–85%, which is in good agreement with values obtained in our material. Wei and McDonald studied peroxide crosslinking of poly(3-hydroxybutyrate) and poly(L-lactic acid) with DCP (the concentration of peroxide was 0.25–1 wt.%) and also obtained material with similar values [23]. The G’/G” values of the hydrogels obtained by Wei and McDonald were higher compared with values for our material, but the measurements were done in polymer melts and not in swollen hydrogels.

Frequency sweep of PEtOx/polyperoxide-based hydrogels obtained after incubation for 40 min at different crosslinking temperatures (160–200 °C) and different concentrations of peroxide containing copolymer crosslinker: (a) P[PM-co-MA] 15 wt.%, (b) P[PM-co-MA] 20 wt.%, and (c) P[PM-co-BA-co-MA] 15 wt.%. (d) Visual appearance of hydrogel, obtained at 160 °C after 40 min using P[PM-co-MA] at 20 wt.%
The chemical composition of the polyperoxide does not have significant influence on the viscoelastic properties of the obtained hydrogels (Fig. 13c). The visual appearance of a sample obtained after 40 min incubation at 160 °C shows a rough surface and air bubbles inside the investigated samples (Fig. 13d). Increase in incubation time and temperature leads to an increased amount of bubbles. We posit that these bubbles in the structure occur due to evaporation of resulting tert-butanol and acetone after decomposition of the peroxide groups. Therefore, the obtained hydrogels have a sponge-like structure. Such a highly porous and irregular structure was typical for all investigated hydrogels. It must be noted that such heterogeneities will have a significant influence on the rheological measurements and its reproducibility and the numerical values should be considered with great care, as they will not reflect the actual values for the crosslinked material but that of the foam. However, the rheological analysis clearly corroborates the chemically crosslinking of the polymers.
Conclusions
In summary, we report on a novel approach to obtain hydrogels by chemical crosslinking of unfunctionalized hydrophilic PEtOx using polyperoxides of different chemical compositions by simple heating of the mixed powders. The influence of temperature, time, and concentration as well as chemical composition of the polyperoxide on the crosslinking process and the properties of the resulting hydrogels has been investigated. Furthermore, we found that the swelling temperature has an impact on the degree of swelling and the stability of ChemRxiv the obtained hydrogels, presumably due to hydrolysis of ester crosslinks. The crosslinking degree increases with temperature and concentration of polyperoxide. Rheological measurements confirm that the PEtOx/polyperoxide-based hydrogels are rather soft materials with storage moduli of a few kPa. We also demonstrated that the chemical composition of polyperoxide crosslinker does have a minor effect on mechanical properties of the resulting hydrogels. Comparison of SEM on freeze-dried samples and samples observed by cryo-SEM confirmed the significant impact of sample preparation on the sample morphology. The latter suggests double-pore structure, which should be investigated in more detail. The present approach of using polyperoxide crosslinker is particularly simple and should be applicable to a wide range of hydrophilic polymers without the need of any functionalization or modification and may be useful for different applications.
References
Na YH, Oh HY, Ahn YJ, Han Y (2015) Suturing property of tough double network hydrogels for bio-repair materials. Korea Aust Rheol J 27(1):25–31
Demirgöz D, Navarro R, Pérez M, Reinecke H, Gallardo A (2010) Asymmetric bihomologous crosslinkers for bicomponent gels—the way to strongly increased elastic moduli. J Appl Polym Sci 115(2):896–900
Zhao Y, Nakajima T, Yang JJ, Kurokawa T, Liu J, Lu J, Mizumoto S, Sugahara K, Kitamura N, Yasuda K, Daniels AUD, Gong PJ (2014) Proteoglycans and Glycosaminoglycans improve toughness of biocompatible double network hydrogels. Adv Mater 26(3):436–442
Zheng WJ, Gao Y, Fan XX, Cui XF, Zou W, Zheng D, Yan J, Li B (2018) Physically linked PVA/P(AAm-co-SMA) hydrogels: tough, fast recovery, Antifatigue, and self-healing soft materials. Macromol Mater Eng 303(7):1800104
Park KR, Nho YC (2003) Synthesis of PVA/PVP hydrogels having two-layer by radiation and their physical properties. Radiat Phys Chem 67(3–4):361–365
Lanouar S, Aid-Launais R, Oliveira A, Bidault L, Closs B, Labour M-N, Letourneur D (2018) Effect of cross-linking on the physicochemical and in vitro properties of pullulan/dextran microbeads. J Mater Sci Mater Med 29(6):77
Balakrishnan B, Mohanty M, Umashankar P, Jayakrishnan A (2005) Evaluation of an in situ forming hydrogel wound dressing based on oxidized alginate and gelatin. Biomaterials 26(32):6335–6342
Hennink WE, van Nostrum CF (2012) Novel crosslinking methods to design hydrogels. Adv Drug Deliv Rev 64:223–236
Karadaǧ E, Üzüm ÖB, Saraydin D (2002) Swelling equilibria and dye adsorption studies of chemically crosslinked superabsorbent acrylamide/maleic acid hydrogels. Eur Polym J 38(11):2133–2141
Wu S, Li H, Chen JP, Lam K (2004) Modeling investigation of hydrogel volume transition. Macromol Theory Simul 13(1):13–29
Li S, Wang L, Yu X, Wang C, Wang Z (2018) Synthesis and characterization of a novel double cross-linked hydrogel based on Diels-Alder click reaction and coordination bonding. Mater Sci Eng C 82:299–309
Maitra J, Shukla VK (2014) Cross-linking in Hydrogels-A Review. Am J Pol Sci 4(2):25–31
Wen Q, Dong Y (2016) Gels Handbook Fundamentals, Properties and Applications Volume 1: Fundamentals of Hydrogels, edited by Demirci U and Khademhosseini A, Singapure, pp.1172
Kabiri K, Omidian H, Zohuriaan-Mehr M, Doroudiani S (2011) Superabsorbent hydrogel composites and nanocomposites: a review. Polym Compos 32(2):277–289
Balakrishnan B, Jayakrishnan A (2005) Self-cross-linking biopolymers as injectable in situ forming biodegradable scaffolds. Biomaterials 26(18):3941–3951
Kirchhof S, Brandl FP, Hammer N, Goepferich AM (2013) Investigation of the Diels–Alder reaction as a cross-linking mechanism for degradable poly (ethylene glycol) based hydrogels. J Mater Chem B 1(37):4855–4864
Belka J, Nickel J, Kurth DG (2019) Growth on metallo-supramolecular coordination polyelectrolyte (MEPE) stimulates osteogenic differentiation of human osteosarcoma cells (MG63) and human bone marrow derived mesenchymal stem cells. Polymers 11(7):1090
Loan L (1972) Peroxide crosslinking reactions of polymers pure. Appl Chem 30(1–2):173–180
Takamura M, Nakamura T, Takahashi T, Koyama K (2008) Effect of type of peroxide on cross-linking of poly(L-lactide). Polym Degrad Stab 93(10):1909–1916
Liu S-Q, Gong W-G, Zheng B-C (2014) The effect of peroxide cross-linking on the properties of low-density polyethylene. J Macromol Sci B 53(1):67–77
Jiang Q, Li J, Huang W, Zhang D, Chen J, Yang H, Xue X, Jiang B (2017) Radical polymerization in the presence of a peroxide monomer: an approach to branched vinyl polymers. Polym Chem 8(30):4428–4439
Naskar K, Noordermeer JW (2004) Dynamically vulcanized PP/EPDM blends: multifunctional peroxides as crosslinking agents - part I rubber. Chem Technol. 77(5):955–971
Wei L, McDonald AG (2015) Peroxide induced cross-linking by reactive melt processing of two biopolyesters: Poly(3-hydroxybutyrate) and poly(L-lactic acid) to improve their melting processability. J Appl Polym Sci 132(13)
Voronov S, Tokarev V, Oduola K, Lastukhin Y (2000) Polyperoxide surfactants for interface modification and compatibilization of polymer colloidal systems. I. synthesis and properties of polyperoxide surfactants. J Appl Polym Sci 76(8):1217–1227
Voronov S, Tokarev V, Datsyuk V, Seredyuk V, Bednarska O, Oduola K, Adler H, Puschke C, Pich A, Wagenknecht U (2000) Polyperoxidic surfactants for interface modification and compatibilization of polymer colloidal systems. II. Design of compatibilizing layers. J Appl Polym Sci 76(8):1228–1239
Lorson T, Lübtow MM, Wegener E, Haider MS, Borova S, Nahm D, Jordan R, Sokolski-Papkov M, Kabanov AV, Luxenhofer R (2018) Poly(2-oxazoline)s based biomaterials: a comprehensive and critical update. Biomaterials 178:204–280
Zahoranová A, Kronek J (2016) Hydrogels based on poly(2-oxazoline)s for pharmaceutical applications, Handbook of Polymers for Pharmaceutical Technologies, Volume 4, edited by Thakur VK and Thakur MK, Scrivener Publishing LLC, pp. 231–258
Hartlieb M, Kempe K, Schubert US (2015) Covalently cross-linked poly(2-oxazoline) materials for biomedical applications–from hydrogels to self-assembled and templated structures. J Mater Chem B 3(4):526–538
Kelly AM, Wiesbrock F (2012) Strategies for the synthesis of poly(2-oxazoline)-based hydrogels. Macromol Rapid Commun 33(19):1632–1647
Kelly AM, Hecke A, Wirnsberger B, Wiesbrock F (2011) Synthesis of poly(2-oxazoline)-based hydrogels with tailor-made swelling degrees capable of stimuli-triggered compound release. Macromol Rapid Commun 32(22):1815–1819
Hartlieb M, Pretzel D, Englert C, Hentschel M, Kempe K, Gottschaldt M, Schubert US (2014) Matrix supported poly(2-oxazoline)-based hydrogels for DNA catch and release. Biomacromolecules 15(6):1970–1978
Chujo Y, Sada K, Saegusa T (1990) Reversible gelation of polyoxazoline by means of Diels-Alder reaction. Macromolecules 23(10):2636–2641
Chujo Y, Sada K, Saegusa T (1993) Cobalt (III) bipyridyl-branched polyoxazoline complex as a thermally and redox reversible hydrogel. Macromolecules 26(24):6320–6323
Chujo Y, Sada K, Saegusa T (1993) Iron (II) bipyridyl-branched polyoxazoline complex as a thermally reversible hydrogel. Macromolecules 26(24):6315–6319
Chujo Y, Sada K, Nomura R, Naka A, Saegusa T (1993) Photogelation and redox properties of anthracene-disulfide-modified polyoxazolines. Macromolecules 26(21):5611–5614
Miyamoto M, Naka K, Shiozaki M, Chujo Y, Saegusa T (1990) Preparation and enzymatic activity of poly[( N-acy1imino)ethylenel] - modified catalase. Macromolecules 23(13):3201–3205
Chujo Y, Sada K, Matsumoto K, Saegusa T (1990) Synthesis of nonionic hydrogel, lipogel, and amphigel by copolymerization of 2-oxazolines and a bisoxazoline. Macromolecules 23(5):1234–1237
Imai Y, Itoh H, Naka K, Chujo Y (2000) Thermally reversible IPN organic-inorganic polymer hybrids utilizing the Diels-Alder reaction. Macromolecules 33(12):4343–4346
Wang CH, Hsiue GH (2002) Synthesis and characterization of temperature- and pH-sensitive hydrogels based on poly(2-ethyl-2-oxazoline) and poly(D,L-lactide). J Polym Sci A Pol Chem 40(8):1112–1121
Dargaville TR, Forster R, Farrugia BL, Kempe K, Voorhaar L, Schubert US, Hoogenboom R (2012) Poly(2-oxazoline) hydrogel monoliths via thiol-ene coupling. Macromol Rapid Commun 33(19):1695–1700
Dargaville TR, Lava K, Verbraeken B, Hoogenboom R (2016) Unexpected switching of the photogelation chemistry when cross-linking poly(2-oxazoline) copolymers. Macromolecules 49(13):4774–4783
Hartlieb M, Pretzel D, Kempe K, Fritzsche C, Paulus RM, Gottschaldt M, Schubert US (2013) Cationic poly(2-oxazoline) hydrogels for reversible DNA binding. Soft Matter 9(18):4693–4704
Zahoranová A, Kroneková Z, Zahoran M, Chorvát D, Janigová I, Kronek J (2016) Poly(2-oxazoline) hydrogels crosslinked with aliphatic bis(2-oxazoline)s: properties, cytotoxicity, and cell cultivation. J Polym Sci A Polym Chem 54(11):1548–1559
Haigh JN, Chuang YM, Farrugia B, Hoogenboom R, Dalton PD, Dargaville TR (2016) Hierarchically structured porous poly(2-oxazoline) hydrogels. Macromol Rapid Commun 37(1):93–99
Lorson T, Jaksch S, Lübtow MM, Jüngst T, Groll J, Lühmann T, Luxenhofer R (2017) A thermogelling supramolecular hydrogel with sponge-like morphology as a cytocompatible bioink. Biomacromolecules 18(7):2161–2171
Lübtow MM, Mrlik M, Hahn L, Altmann A, Beudert M, Lühmann T, Luxenhofer R (2019) Temperature-dependent rheological and viscoelastic investigation of a poly(2-methyl-2-oxazoline)-b-poly(2-iso-butyl-2-oxazoline)-b-poly(2-methyl-2-oxazoline)-based thermogelling hydrogel. J Funct Biomater 10(3):36
Hahn L, Maier M, Stahlhut P, Beudert M, Flegler V, Forster S, Altmann A, Töppke F, Fischer K, Seiffert S (2019) Inverse thermogelation of aqueous triblock copolymer solutions into macroporous shear-thinning 3D printable inks. ACS Appl Mater Interfaces 12(11):12445–12456
Rueda JC, Komber H, Cedrón JC, Voit B, Shevtsova G (2003) Synthesis of new hydrogels by copolymerization of poly(2-methyl-2-oxazoline) bis(macromonomers) and N-vinylpyrrolidone. Macromol Chem Phys 204(7):947–953
Zhu Z, Li X (2014) Silicone hydrogels based on a novel amphiphilic poly(2-methyl-2-oxazoline)-b-poly(dimethyl siloxane) copolymer. J Appl Polym Sci 131(3):39867 (1–7)
Farrugia BL, Kempe K, Schubert US, Hoogenboom R, Dargaville TR (2013) Poly(2-oxazoline) hydrogels for controlled fibroblast attachment. Biomacromolecules 14(8):2724–2732
Wilson P, Ke PC, Davis TP, Kempe K (2017) Poly(2-oxazoline)-based micro-and nanoparticles: a review. Eur Polym J 88:486–515
Dargaville TR, Park JR, Hoogenboom R (2018) Poly(2-oxazoline) hydrogels: state-of-the-art and emerging applications. Macromol Biosci 1800070
Galante R, Pinto TJ, Colaço R, Serro AP (2018) Sterilization of hydrogels for biomedical applications: a review. J Biomed Mater Res B Appl Biomater 106(6):2472–2492
Miyamoto T, Okano S, Kasai N (2009) Inactivation of Escherichia coli endotoxin by soft hydrothermal processing. Appl Environ Microbiol 75(15):5058–5063
Kurganskii V, Puchin V, Voronov S, Tokarev V (1983) The synthesis of heterofunctional polymers with peroxide and anhydride groups. Polym Sci USSR 25(5):1152–1161
Murillo EA, López BL (2015) Effect of the maleic anhydride content on the structural, thermal, rheological and film properties of the n-butyl methacrylate–maleic anhydride copolymers. Prog Org Coat 78:96–102
Boztuğ A, Zengin H, Basan S (2004) Thermomechanical and thermogravimetric analysis of blends of poly(vinyl chloride) (PVC) with maleic anhydride – allyl propionate copolymer. J Mol Struct 697(1–3):61–64
Vacque V, Sombret B, Huvenne J, Legrand P, Suc S (1997) Characterisation of the O-O peroxide bond by vibrational spectroscopy. Spectrochim Acta A 53(1):55–66
Kazitsina L, Kupletskaya N, Primenenie Uf-,Ik-, Yamr-Spektroskopii v organicheskoj Chimii (1971) Russian Edition ed., Ripol Class
Popadyuk A, Tarnavchyk I, Popadyuk N, Kohut A, Samaryk V, Voronov S, Voronov A (2013) A novel copolymer of N-[(tert-butylperoxy) methyl] acrylamide and maleic anhydride for use as a reactive surfactant in emulsion polymerization. React Funct Polym 73(9):1290–1298
Samaryk V, Voronov A, Tarnavchyk I, Kohut A, Nosova N, Varvarenko S, Voronov S (2009) A versatile approach to develop porous hydrogels with a regular pore distribution and investigation of their physicomechanical properties. J Appl Polym Sci 114(4):2204–2212
Voronov S, Kohut A, Tarnavchyk I, Voronov A (2014) Advances in reactive polymeric surfactants for interface modification. Curr Opin Colloid Interface Sci 19(2):95–121
Gupta P, Vermani K, Garg S (2002) Hydrogels: from controlled release to pH-responsive drug delivery. Drug Discov Today 7(10):569–579
Berger J, Reist M, Mayer JM, Felt O, Peppas N, Gurny R (2004) Structure and interactions in covalently and ionically crosslinked chitosan hydrogels for biomedical applications. Eur J Pharm Biopharm 57(1):19–34
Delogu F, Gorrasi G, Sorrentino A (2017) Fabrication of polymer nanocomposites via ball milling: present status and future perspectives. Prog Mater Sci 86:75–126
Kong L, Zhu W, Tan O (2000) Preparation and characterization of Pb (Zr0. 52Ti0. 48) O3 ceramics from high-energy ball milling powders. Mater Lett 42(4):232–239
Uemoto Y, Kondo K, Niwa T (2019) Cryo-milling using a spherical sugar: contamination-free media milling technology. Eur J Pharm Sci 136:104934
Uemoto Y, Kondo K, Niwa T (2020) Cryo-milling with spherical crystalline cellulose beads: a contamination-free and safety conscious technology. Eur J Pharm Sci 143:105175
Kurganskii V, Puchin V, Voronov S, Tokarev V (1983) The synthsis of heterofunctional polymers with peroxide and ahnydride groups. Polymer Science USSR 25(5):1152–1161
De Roover B, Sclavons M, Carlier V, Devaux J, Legras R, Momtaz A (1995) Molecular characterization of maleic anhydride-functionalized polypropylene. J Polym Sci A Polym Chem 33(5):829–842
Nakason C, Kaesaman A, Supasanthitikul P (2004) The grafting of maleic anhydride onto natural rubber. Polym Test 23(1):35–41
Mirone P, Chiorboli P (1962) Infrared and Raman spectra and vibrational assignment of maleic anhydride. Spectrochim Acta 18(11):1425–1432
Wang H, Liu Y, Tian J, Sun B, Huang S (2013) FT-IR analysis of molecular structure evolvement of poly(ether urethanes) in ozone atmosphere, Advanced Materials Research, Trans Tech Publ, pp. 215–219
Smith BC (2017) The C-O Bond III: Ethers By a Knockout. http://www.spectroscopyonline.com/c-o-bond-iii-ethers-knockout?pageID=1:. (accessed 8.04.2019.)
Lu Y, Zhang W, Li X, Xu S (2017) Synthesis of new polyether titanate coupling agents with different polyethyleneglycol segment lengths and their compatibilization in calcium sulfate whisker/poly(vinyl chloride) composites. RSC Adv 7(50):31628–31640
Wowk B (2010) Thermodynamic aspects of vitrification. Cryobiology 60(1):11–22
Aston R, Sewell K, Klein T, Lawrie G, Grøndahl L (2016) Evaluation of the impact of freezing preparation techniques on the characterisation of alginate hydrogels by cryo-SEM. Eur Polym J 82:1–15
Harraß K (2013) Approaches for improving the mechanical strength of micro heterogeneous NCO-sP (EO-stat-PO) hydrogels. Rheinisch-Westfälische Technische Hochschule Aachen, Aachen
Harrass K, Krüger R, Möller M, Albrecht K, Groll J (2013) Mechanically strong hydrogels with reversible behaviour under cyclic compression with MPa loading. Soft Matter 9(10):2869–2877
Bae YH, Okano T, Kim SW (1990) Temperature dependence of swelling of crosslinked poly(N, N′-alkyl substituted acrylamides) in water. J Polym Sci B Polym Phys 28(6):923–936
Mezger TG, Das Rheologie Handbuch, edited by Vincent Network, Hannover, 2010, pp. 426
Acknowledgments
S.B. would like to thank the Bavarian Academic Center for Central, Eastern, and Southeastern Europe (BAYHOST) for scholarship support and the University Würzburg. We also thank the Deutsche Forschungsgemeinschaft for funding the crossbeam scanning electron microscope Zeiss CB 340 (INST 105022/58-1 FUGG) within the DFG State Major Instrumentation Programme.
Funding
Open Access funding provided by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Borova, S., Tokarev, V., Stahlhut, P. et al. Crosslinking of hydrophilic polymers using polyperoxides. Colloid Polym Sci 298, 1699–1713 (2020). https://doi.org/10.1007/s00396-020-04738-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00396-020-04738-w