Clinical Genetics Can Solve the Pitfalls of Genome-Wide Investigations: Lesson from Mismapping a Loss-of-Function Variant in KANSL1

 , and
, and 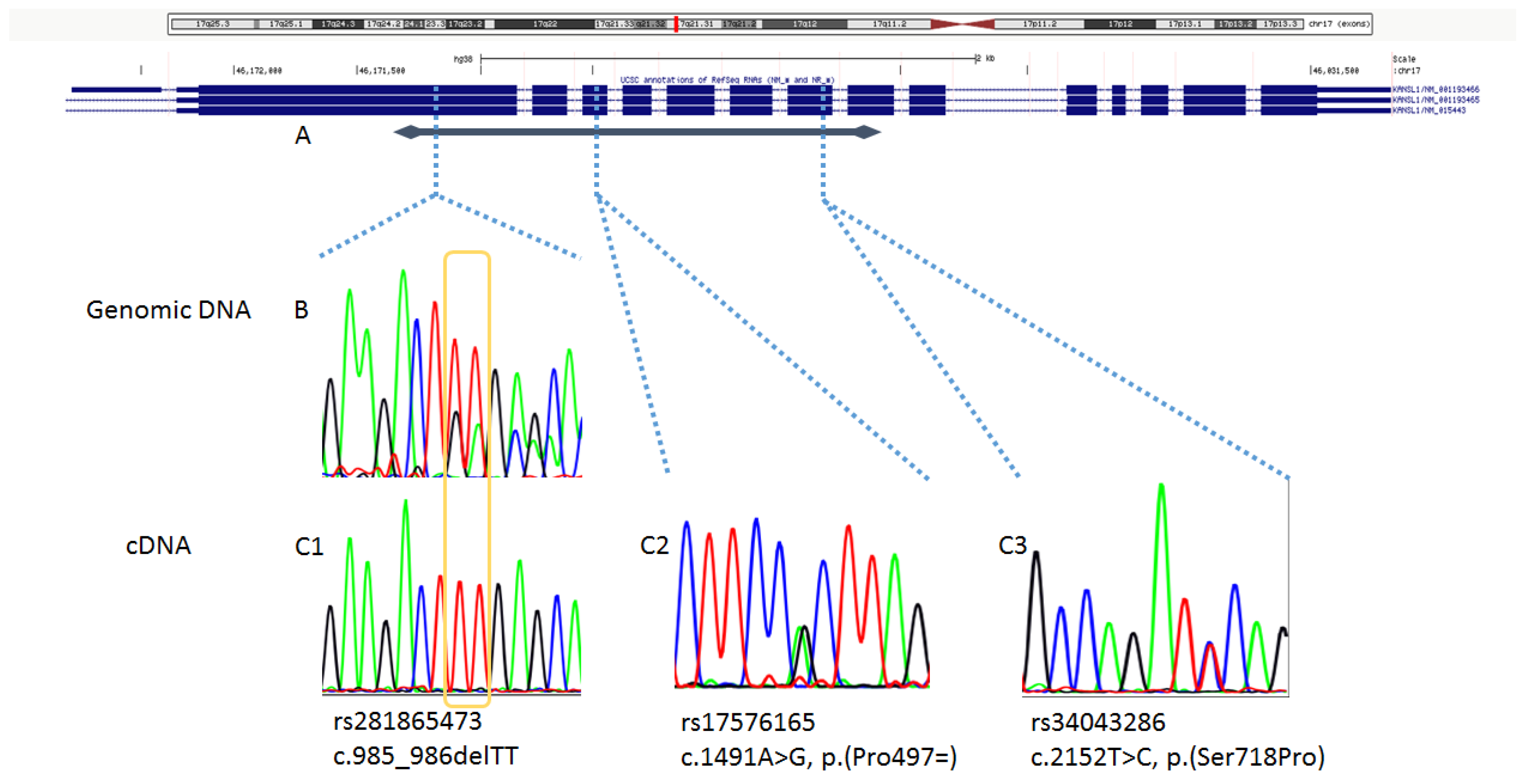{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Analyses on Genomic DNA
2.2. Analyses on mRNA
3. Results
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Koolen, D.A.; Vissers, L.E.; Pfundt, R.; de Leeuw, N.; Knight, S.J.; Regan, R.; Kooy, R.F.; Reyniers, E.; Romano, C.; Fichera, M.; et al. A new chromosome 17q21.31 microdeletion syndrome associated with a common inversion polymorphism. Nat. Genet. 2006, 38, 999–1001. [Google Scholar] [CrossRef] [PubMed]
- Shaw-Smith, C.; Pittman, A.M.; Willatt, L.; Martin, H.; Rickman, L.; Gribble, S.; Curley, R.; Cumming, S.; Dunn, C.; Kalaitzopoulos, D.; et al. Microdeletion encompassing MAPT at chromosome 17q21.3 is associated with developmental delay and learning disability. Nat. Genet. 2006, 38, 1032–1037. [Google Scholar] [CrossRef] [PubMed]
- Sharp, A.J.; Hansen, S.; Selzer, R.R.; Cheng, Z.; Regan, R.; Hurst, J.A.; Stewart, H.; Price, S.M.; Blair, E.; Hennekam, R.C.; et al. Discovery of previously unidentified genomic disorders from the duplication architecture of the human genome. Nat. Genet. 2006, 38, 1038–1042. [Google Scholar] [CrossRef] [PubMed]
- Koolen, D.A.; Sharp, A.J.; Hurst, J.A.; Firth, H.V.; Knight, S.J.; Goldenberg, A.; Saugier-Veber, P.; Pfundt, R.; Vissers, L.E.; Destrée, A.; et al. Clinical and molecular delineation of the 17q21.31 microdeletion syndrome. J. Med. Genet. 2008, 45, 710–720. [Google Scholar] [CrossRef] [PubMed]
- Zollino, M.; Orteschi, D.; Murdolo, M.; Lattante, S.; Battaglia, D.; Stefanini, C.; Mercuri, E.; Chiurazzi, P.; Neri, G.; Marangi, G. Mutations in KANSL1 cause the 17q21.31 microdeletion syndrome phenotype. Nat. Genet. 2012, 44, 636–638. [Google Scholar] [CrossRef] [PubMed]
- Koolen, D.A.; Kramer, J.M.; Neveling, K.; Nillesen, W.M.; Moore-Barton, H.L.; Elmslie, F.V.; Toutain, A.; Amiel, J.; Malan, V.; Tsai, A.C.; et al. Mutations in the chromatin modifier gene KANSL1 cause the 17q21.31 microdeletion syndrome. Nat. Genet. 2012, 44, 639–641. [Google Scholar] [CrossRef] [PubMed]
- Zollino, M.; Marangi, G.; Ponzi, E.; Orteschi, D.; Ricciardi, S.; Lattante, S.; Murdolo, M.; Battaglia, D.; Contaldo, I.; Mercuri, E.; et al. Intragenic KANSL1 mutations and chromosome 17q21.31 deletions: Broadening the clinical spectrum and genotype-phenotype correlations in a large cohort of patients. J. Med. Genet. 2015, 52, 804–814. [Google Scholar] [CrossRef] [PubMed]
- Koolen, D.A.; Pfundt, R.; Linda, K.; Beunders, G.; Veenstra-Knol, H.E.; Conta, J.H.; Fortuna, A.M.; Gillessen-Kaesbach, G.; Dugan, S.; Halbach, S.; et al. The Koolen-de Vries syndrome: A phenotypic comparison of patients with a 17q21.31 microdeletion versus a KANSL1 sequence variant. Eur. J. Hum. Genet. 2016, 24, 652–659. [Google Scholar] [CrossRef] [PubMed]
- Boettger, L.M.; Handsaker, R.E.; Zody, M.C.; McCarroll, S.A. Structural haplotypes and recent evolution of the human 17q21.31 region. Nat. Genet. 2012, 44, 881–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinberg, K.M.; Antonacci, F.; Sudmant, P.H.; Kidd, J.M.; Campbell, C.D.; Vives, L.; Malig, M.; Scheinfeldt, L.; Beggs, W.; Ibrahim, M.; et al. Structural diversity and African origin of the 17q21.31 inversion polymorphism. Nat. Genet. 2012, 44, 872–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Squeo, G.M.; Augello, B.; Massa, V.; Milani, D.; Colombo, E.A.; Mazza, T.; Castellana, S.; Piccione, M.; Maitz, S.; Petracca, A.; et al. Customised next-generation sequencing multigene panel to screen a large cohort of individuals with chromatin-related disorder. J. Med. Genet. 2020. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Bjornsson, H.T. The Mendelian disorders of the epigenetic machinery. Genome Res. 2015, 25, 1473–1481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larizza, L.; Finelli, P. Developmental disorders with intellectual disability driven by chromatin dysregulation: Clinical overlaps and molecular mechanisms. Clin. Genet. 2019, 95, 231–240. [Google Scholar] [CrossRef] [PubMed]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bigoni, S.; Marangi, G.; Frangella, S.; Panfili, A.; Ognibene, D.; Squeo, G.M.; Merla, G.; Zollino, M. Clinical Genetics Can Solve the Pitfalls of Genome-Wide Investigations: Lesson from Mismapping a Loss-of-Function Variant in KANSL1. Genes 2020, 11, 1177. https://doi.org/10.3390/genes11101177
Bigoni S, Marangi G, Frangella S, Panfili A, Ognibene D, Squeo GM, Merla G, Zollino M. Clinical Genetics Can Solve the Pitfalls of Genome-Wide Investigations: Lesson from Mismapping a Loss-of-Function Variant in KANSL1. Genes. 2020; 11(10):1177. https://doi.org/10.3390/genes11101177
Chicago/Turabian StyleBigoni, Stefania, Giuseppe Marangi, Silvia Frangella, Arianna Panfili, Davide Ognibene, Gabriella Maria Squeo, Giuseppe Merla, and Marcella Zollino. 2020. "Clinical Genetics Can Solve the Pitfalls of Genome-Wide Investigations: Lesson from Mismapping a Loss-of-Function Variant in KANSL1" Genes 11, no. 10: 1177. https://doi.org/10.3390/genes11101177