
Abstract
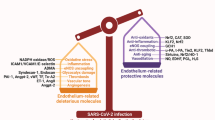
COVID-19 is caused by SARS-CoV-2 infection and characterized by diverse clinical symptoms. Type I interferon (IFN-I) production is impaired and severe cases lead to ARDS and widespread coagulopathy. We propose that COVID-19 pathophysiology is initiated by SARS-CoV-2 gene products, the NSP1 and ORF6 proteins, leading to a catastrophic cascade of failures. These viral components induce signal transducer and activator of transcription 1 (STAT1) dysfunction and compensatory hyperactivation of STAT3. In SARS-CoV-2-infected cells, a positive feedback loop established between STAT3 and plasminogen activator inhibitor-1 (PAI-1) may lead to an escalating cycle of activation in common with the interdependent signaling networks affected in COVID-19. Specifically, PAI-1 upregulation leads to coagulopathy characterized by intravascular thrombi. Overproduced PAI-1 binds to TLR4 on macrophages, inducing the secretion of proinflammatory cytokines and chemokines. The recruitment and subsequent activation of innate immune cells within an infected lung drives the destruction of lung architecture, which leads to the infection of regional endothelial cells and produces a hypoxic environment that further stimulates PAI-1 production. Acute lung injury also activates EGFR and leads to the phosphorylation of STAT3. COVID-19 patients’ autopsies frequently exhibit diffuse alveolar damage (DAD) and increased hyaluronan (HA) production which also leads to higher levels of PAI-1. COVID-19 risk factors are consistent with this scenario, as PAI-1 levels are increased in hypertension, obesity, diabetes, cardiovascular diseases, and old age. We discuss the possibility of using various approved drugs, or drugs currently in clinical development, to treat COVID-19. This perspective suggests to enhance STAT1 activity and/or inhibit STAT3 functions for COVID-19 treatment. This might derail the escalating STAT3/PAI-1 cycle central to COVID-19.
Similar content being viewed by others


Introduction
The etiology of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection and the progression of the resulting coronavirus disease of 2019 (COVID-19) disease are novel. A person may be contagious 48–72 h before clinical symptoms appear, and 44% of transmissions are presymptomatic [1]. Viral loads are high at symptom onset but decrease within days. In contrast, in the severe acute respiratory syndrome (SARS) disease caused by SARS-CoV-1, the highest viral shedding occurs 10 days after symptom onset [2]. SARS-CoV-2 also binds more tightly than SARS-CoV-1 to their common cell entry receptor, human angiotensin-converting enzyme 2 (ACE2) [3,4,5].
SARS-CoV-1 chiefly infects the lower airways, while SARS-CoV-2 is initially present in the upper respiratory tract and later moves to the lower airways. For SARS-CoV-2, the highest viral load in the upper respiratory tract occurred during the first 5 days, and viral load is higher and persists for longer in the lower respiratory tract of patients who are severely ill with COVID-19. For SARS, upper respiratory tract infection rarely occurred, and as a consequence, the transmission of SARS-CoV-1 was rare during the first 5 days of illness [6]. These differences accentuate the greater infectivity of SARS-CoV-2.
A strong correlation exists between SARS-CoV-2 viral load and disease severity and progression [7]. Pathological examinations have established that COVID-19 causes widespread thrombosis with microangiopathy in pulmonary vessels [8]. Among 184 COVID-19 patients in the intensive care unit (ICU) with pneumonia, the cumulative incidence of thrombotic complications was 31% [9]. Levels of D-dimer, a coagulopathy indicator, correlated well with COVID-19 illness severity [10]. Accordingly, coagulopathy and thrombosis are pervasive pathological features of COVID-19. Hospitalized patients also commonly exhibit lymphopenia [11].
Pronounced laboratory abnormalities are also present in severe COVID-19 disease. Immunopathologic phenomena include the temporal downregulation of the type I and type III interferon responses, with concomitant increases in proinflammatory cytokine and chemokine production [12]. In contrast to other viral infections [13], CRP levels are significantly elevated and positively correlated with disease severity in COVID-19 [14, 15]. These high CRP levels have never been observed in any other infectious viral disease [16]. Clinical studies of 5700 hospitalized patients revealed that the most common comorbidities were hypertension (56.6%), obesity (41.7%), and diabetes (33.8%) [17].
SARS-CoV-2 is a sarbecovirus with an overall structure similar to that of SARS-CoV-1. The SARS-CoV-2 genome contains the large 5′ open reading frame (ORF) 1ab encoding two polyproteins, including 16 nonstructural proteins (NSPs), namely NSP1–NSP16. The 3′ end of the genome encodes the structural proteins spike (S, composed of two subunits S1 and S2), envelope (E), membrane (M), and nucleocapsid (N). Interspersed among these genes are ORFs encoding the nonstructural accessory proteins ORF3a, ORF3b, ORF6, ORF7a, ORF7b, and ORF8 [18,19,20].
Several SARS-CoV-1 proteins antagonize the antiviral activities of IFNs and the downstream JAK (Janus kinase)-STAT signaling pathways they activate. JAK family kinases (JAK1, JAK2, JAK3, TYK2) display a wide range of functions during ontogeny, in immunity as well as in chronic inflammation, fibrosis, and cancer [21].
Comparative genetic structural studies have suggested that SARS-CoV-2 has similar IFN antagonist activity [18, 22]. After a careful review of the scientific literature, we realized that the SARS-CoV-2-mediated inhibition of IFN and STAT1, and the subsequent shift to a STAT3-dominant signaling network (see below), could result in almost all of the clinical features of COVID-19. Here, we discuss the pathophysiology of SARS-CoV-2, with a specific focus on SARS-CoV-2’s effects on IFN and JAK/STAT signaling. We propose that COVID-19 is a disease caused by a catastrophic cascade of failures stemming from the SARS-CoV-2-mediated dysregulation of STATs. Specifically, the dysfunctions of STAT1 and STAT3 induced by SARS-CoV-2 proteins may be the foundation of severe COVID-19 pathophysiology.
SARS-CoV-2 infection and effects on IFN and STATs
Target cell entry
SARS-CoV-2 cell entry depends on the binding of the Spike protein’s S1 subunit to ACE2 on the target cell surface [3,4,5]. Host proteases, furin [23], as well as TMPRSS2 (transmembrane serine protease 2) [24], then processes the S protein to facilitate membrane fusion. Cleavage at the S1/S2 junction and S2’ site mediates the fusion of the viral and cellular membranes, in a process driven by the S2 subunit [23, 24]. Furin is ubiquitously expressed and its cleavage site at the boundary between the S1/S2 subunits is not present in SARS-CoV-1 and SARS-related viruses [23, 24], allowing SARS-CoV-2 to have enhanced proteolytic activation in a wider range of tissues [23].
Potential host cells for SARS-CoV-2 express high levels of both ACE2 and TMPRSS2, and include type 2 alveolar cells, nasal goblet cells, nasal ciliated cells, corneal cells, and intestinal epithelial cells [25]. SARS-CoV-2 can also directly infect endothelial cells [8, 26] and engineered human blood vessels in kidney organoids in vitro [27]. Among immune cells, SARS-CoV-2 appears to infect mononuclear phagocytes, but not lymphocytes [28, 29].
Initiation of IFN production and signaling
Cells detect viral attack through membrane-bound or intracellular pattern recognition receptors (PRRs). Among these, coronaviruses activate the Toll-like receptors (TLRs), retinoic acid-inducible gene I (RIG-I), and melanoma differentiation-associated protein 5 (Fig. 1) [30]. Virus-induced oligomerization of PRRs leads to the activation of downstream interferon regulatory factors (IRFs) and nuclear factor-kappa B (NF-κB) transcription factors that induce the production of IFN-I, IFN-II, and IFN-III [30].

Molecular patterns derived from SARS-CoV-2-associated molecules, such as ssRNA, dsRNA, and viral proteins, bind to host PRRs and trigger the activation of signal transducers and transcription factors that drive the production of IFN-I and proinflammatory cytokines and chemokines. Soon after infection, the engagement of RIG-I and MDA-5 by these molecular patterns induces the activation of IRF3, or IRF7, through MAVS. In addition, viral ssRNA, dsRNA, and proteins can engage TLRs to trigger the MyD88- and TRIF-dependent pathways, primarily leading to the activation of the NF-κB (p50/p65) transcriptional complex. SARS-CoV-2 proteins that inhibit IFN-I production are indicated in black boxes, and the associated blocked pathways are indicated as dashed lines. Note that only the IFN-I production pathway, and not the secretion of proinflammatory cytokines/chemokines, is inhibited by the viral proteins. Proinflammatory cytokine/chemokine production is further activated by the engagement of TLRs by a high viral load.
The IFN released from infected cells binds to IFN receptors on neighboring cells, alerting them to a viral attack (Fig. 2). The IFN-I and IFN-II receptors are almost ubiquitously expressed, while IFN-III receptors are only expressed on cells lining the epithelial barrier [30]. The engagement of IFN-I and IFN-III receptors activates various members of the JAK (1, Fig. 3) and STAT families (2 and 3, Fig. 3), and specific transcription factor complexes are formed. For example, STAT1 interacts with STAT2 and IRF-9 to constitute the transcription factor complex “IFN-stimulated gene factor 3” (ISGF3) [30]. In contrast, IFN-II activates JAK1 and JAK2, producing a phosphorylated STAT1 homodimer known as “γ-interferon activation factor” (GAF) [31]. Interestingly, both ISGF3 and GAF can be evoked by all IFNs [32, 33]. In any case, before they can exert their transcription factor activity, the ISGF3 and GAF complexes must be transported to the nucleus and the subsequent upregulation of the interferon-stimulated gene products (ISGs) [31]. Karyopherin-α1 (KPNA1) is essential for the nuclear transport of STAT1 [34], and the interaction between STAT1 and KPNA1 (STAT1/KPNA1) involves a nonclassical nuclear localization signal (NLS). In addition to IFN signaling, STAT proteins are involved in signal transduction for other families of cytokines, including IL-6 [31, 35].

The IFN-I protein is secreted from infected cells and amplifies the IFN response by activating the ISGF3 complex (STAT1/STAT2/IRF9) to induce IFN-I-stimulated genes. The SARS-CoV-2 proteins that inhibit IFN-I signaling are indicated in black boxes, and the associated blocked pathways are indicated by dashed lines.

A IFN-I signaling before SARS-CoV-2 infection. JAK1 and TYK2 (1) are activated after IFN-I stimulation. STAT1 is normally activated in IFN-I signaling to induce ISGs (STAT1-ISGs) by ISGF3 (STAT1/STAT2/IRF9). STAT3 is also activated and becomes a homodimer, but the response is small. B IFN-I signaling with SARS-CoV-2 infection. After the infection, STAT1 activity is inhibited by the SARS-CoV-2 proteins, NSP1, and ORF6 (2). With STAT1 activity restricted, STAT3 (3) then becomes dominant and induces STAT3-ISGs. Both STAT1 and STAT3 induce SOCS1 and SOCS3 (4) that inhibit the kinase activity of JAKs for the negative feedback of IFN-I signaling. PIAS1 and PIAS3 (5) inhibit the binding of STAT1 and STAT3 to DNA, respectively, to regulate IFN-I signaling. The role of PIAS3 becomes critical when STAT3 is aberrantly activated and uncoupled from SOCSs regulation. Protein tyrosine phosphatases (PTPs, 6) have regulatory activities on activated JAKs and STATs, but their role in the viral infection needs further clarification. EGFR (7) is upregulated by acute lung injury or by reduced STAT1 activity in the SARS-CoV-2-infected lung. STAT3 is activated through directly binding to EGFR, through EGFR-activated SRC (8), or through JAK2 (data not shown). PIAS3 normally limits the activity of STAT3 but PAI-1 produced during infection blocks PIAS3 activity (9) and an escalating cascade in the STAT3/PAI-1 axis is established.
Inhibition of IFN activity by SARS-CoV-2
The SARS coronaviruses use various mechanisms to hamper IFN production and response [30]. Consequently, target cells proximal to the site of the initial infection fail to receive critical and protective IFN signals, allowing the virus to spread and replicate without hindrance. A hallmark of SAR-CoV-2 infection is impaired IFN-I and III production and responses, which masks the IFN-related fever symptoms [36] and leads to naive spreading of the virus [12, 37, 38].
The SARS-CoV-1 NSP1 protein impedes STAT1 phosphorylation [39], and NSP3, NSP4, and NSP6 are involved in the establishment of double-membrane vesicles (DMVs) [40]. DMVs create a platform by assembling the replicase proteins, virus genomes, and host proteins required for replication, while physically separating the replication sites from the cytoplasmic sensors of the innate immune response [40, 41]. NSP16 and NSP10 prevent detection by the host’s RIG-I, which recognizes unmethylated RNAs as non-self [42]. SARS-CoV-1 structural proteins also quench the IFN response. The N protein binds to the E3 ubiquitin ligase TRIM25 (tripartite motif protein 25) and interferes with the association between TRIM25 and RIG-I [43]. The M protein impairs the formation of the TRAF3/TANK/TBK1/IKK\({\it{\epsilon }}\) complex, which is important in IRF3/IRF7 signaling [44]. The ORF3a protein induces serine phosphorylation within the degradation motif of IFN alpha-receptor subunit 1 (IFNAR1) and enhances IFNAR1 ubiquitination [45]. Finally, the SARS-CoV-1 ORF3b, ORF6, and N proteins block IRF3 phosphorylation [46].
SARS-CoV-2 proteins structurally resemble those of SARS-CoV-1 and thus are likely to have the similar effects on IFN production and response (Figs. 1 and 2), but there are some important differences. The SARS-CoV-2 ORF3b gene contains a premature stop codon, resulting in a truncated protein (22 aa) as compared to the SARS-CoV-1 ORF3b protein (154 aa) [18]. Although this SARS-CoV-2 ORF3b 22 aa peptide lacks the C-terminal NLS, it retains the ability to efficiently inhibit IFN-I signaling [47]. ORF3b proteins in bat coronaviruses (SL-CoVs) [48] also inhibit IFN-I and Konno et al. have described a larger, natural SARS-CoV-2 ORF3b variant (56 aa) with increased anti-IFN-I activity [47]. Yang et al. found that the SARS-CoV-2 inhibition of STAT1 phosphorylation leads to the attenuation of the interferon-stimulated genes transcription in monocyte-derived dendritic cells and macrophages [49]. Perhaps SARS-CoV-2 NSP1 blocks STAT1 phosphorylation in a similar manner as SARS-CoV-1 NSP1 [39], contributing to the inhibition of interferon response.
The SARS-CoV-1 ORF6 protein is a particularly vital viral factor that uses multiple measures to inhibit IFN production and responses. The SARS-CoV-2 ORF6 protein appears to have similar functions. Several lines of evidence demonstrate the effect of SARS-CoV-2 ORF6 on the IFN response.
-
(1)
Frieman et al. showed that SARS-CoV-1 ORF6 is an ER/Golgi membrane protein that binds KPNA2, together with KPNB1. This recruitment of KPNB1 into the membrane complex limits the bioavailability of KPNB1 needed for the nuclear import of STAT1/KPNA1 complexes. As a result, ORF6 blocks the nuclear import of STAT1 [50].
-
(2)
ORF6 is incorporated into mature SARS-CoV-1 virions [51], and thus the cell is exposed to ORF6 immediately upon viral infection. Moreover, when co-expressed with the SARS-CoV-1 S, M, and E proteins, ORF6 is incorporated into virus-like particles [51]. Therefore, the ORF6 protein may interfere with intracellular signaling prior to authentic viral replication.
-
(3)
SARS-CoV-1 ORF6 modifies membranes and produces perinuclear vesicles resembling DMVs [52]. ORF6 co-immunoprecipitated with viral RNAs and co-localized on cytoplasmic vesicles with replicating viral RNAs [53]. Thus, ORF6 is also involved in establishing DMVs for viral RNA replication.
-
(4)
The introduction of SARS-CoV-1 ORF6 into a sublethal mouse hepatitis virus (MHV) strain promoted viral proliferation and caused fatal encephalitis in infected mice [53, 54]. The expression of ORF6 before infection with wild-type MHV facilitated the production of significantly more progeny.
-
(5)
Yuen et al. analyzed the anti-IFN-I activities of 27 SARS-CoV-2-encoded proteins and found that the ORF6 protein was the most potent antagonist. Although SARS-CoV-2 ORF6 shares only 69% sequence homology with SARS-CoV-1 ORF6, the SARS-CoV-2 ORF6 protein reduced the IFN-beta promoter activity by 100-fold and suppressed IFN signaling to levels comparable to those imposed by SARS-CoV-1 ORF6. The SARS-CoV-1 and SARS-CoV-2 ORF6 proteins inhibit IFN-I and IFN-III secretion induced by Sendai virus infection, a method often used to elicit IFN-I production, and have profound effects on IRF3 phosphorylation, thus accounting for their potent inhibition of IFN-I production [20, 46].
STAT1 and STAT3 activation and compensation in IFN signaling
Once STAT1 function is impaired by the NSP1 or ORF6 protein of SARS-CoV-2 (2, Fig. 3), a concomitant and compensatory shift to signaling via STAT1-independent pathways may occur, and a STAT3-dependent transcriptional profile becomes dominant in many situations (3, Fig. 3) [55,56,57]. Notably, IFN-I signaling not only activates STAT1 but also STAT3, and STAT3 has a fine-tuning role in STAT1-mediated IFN-I responses [58].
Several potential mechanisms have been proposed as to how STAT3 inhibits the STAT1-mediated IFN-I response: (1) STAT3 prevents the formation of the STAT1 homodimer by making heterodimers with STAT1, (2) STAT3 inhibits the binding of ISGF3 (STAT1/STAT2/IRF9) to DNA in cooperation with repressors, (3) STAT3 directly and indirectly reduces the expression of ISGF3 components [58]. STAT3 may also compete with STAT1 for a nuclear translocation factor, KPNA1 [59]. If competition between STAT1 and STAT3 for these nuclear translocation factors occurs in SARS-CoV-2-infected cells, then STAT1 would have to outcompete STAT3 for binding to KPNA1 [59] and ORF6 for binding to KPNB2 [50] to achieve proper nuclear localization. These situations would create a higher functional STAT3:STAT1 ratio immediately after virus infection, shifting the dominant transcriptional network to that governed by STAT3, the STAT3-stimulated genes (STAT3-ISGs; 3, Fig. 3) [58].
Both STAT3 and STAT1 transcriptionally induce both suppressor of cytokine signaling1 (SOCS1) and SOCS3 that inhibit activity of JAKs (4, Fig. 3). In the nucleus, protein inhibitor of activated STAT3 (PIAS3) and PIAS1 can bind activated STAT3 and STAT1 dimers (5, Fig. 3), respectively, and block them from binding to DNA, thus inhibiting STAT mediated transcription. Tyrosine-phosphorylated STATs or JAKs are targets of tyrosine phosphatases, such as SHP1, SHP2, PTP1B, and TC-PTP [60] (6, Fig. 3). However, their roles in COVID-19 remains to be clarified.
Sometimes the negative regulation of STAT3 by SOCS1/SOCS3 becomes dysfunctional. Ramana et al. reported that the IFN-II induction of SOCS3 in STAT1-deficient mouse embryonic fibroblasts was derived from activated STAT3 [61]. In wild-type fibroblasts, STAT1 is normally dominant, but in STAT1-deficient fibroblasts STAT3 is activated strongly and in a sustained manner. The SRC kinase inhibitor SU6656 suppressed IFN-II activation of STAT3 significantly in STAT1-deficient fibroblasts but not in wild-type fibroblasts indicating that STAT3 is activated through SRC kinase activity in the absence of STAT1. STAT3 can be tyrosine-phosphorylated by the dimerized epidermal growth factor receptor (EGFR), EGFR-activated SRC (8, Fig. 3), or by EGFR-activated JAK2 [62], but only the latter pathway is regulated by SOCS3. Recently, EGFR signaling was shown to inhibit IFN-I through activated STAT3 [63]. EGFR is upregulated (7, Fig. 3) during acute lung injury [64], or when STAT1 is deficient [65]. This scenario is consistent with a study with an alpha coronavirus, porcine epidemic diarrhea virus. STAT1 was virologically suppressed by the infection [66] and IFN-I signaling was inhibited by upregulated EGFR and activated STAT3 [67]. Therefore, in COVID-19, EGFR signaling may become an alternative pathway that activates STAT3 specifically when the lung is damaged while the production of IFN-I is severely impaired by SARS-CoV-2 infection [12].
This aberrant transcriptional rewiring toward STAT3 may lead to the symptoms most commonly observed in hospitalized COVID-19 patients: rapid coagulopathy/thrombosis, proinflammatory conditions, profibrotic status, and T cell lymphopenia. The next sections describe how STAT3 hyperactivation can be linked to each of these abnormalities. A summary of these events, starting with the downregulation of STAT1 (1, Fig. 4) and compensatory upregulation of STAT3 (2, Fig. 4) induced by NSP1 and ORF6 of SARS-CoV-2, is presented in Fig. 4.

Infection by SARS-CoV-2 intracellularly delivers NSP1 and ORF6, which efficiently inhibit STAT1 function (1). Repression of STAT1 increases STAT3 (2) activity. STAT3 upregulates PAI-1 (3) by repressing miR-34a, a PAI-1 inhibitor (4). This increased PAI-1 reciprocally activates STAT3 by blocking PIAS3, a STAT3 inhibitor (5). STAT3 can activate HAS2, a hyaluronic acid synthase, which produces hyaluronan (HA) and leads to diffuse alveolar damage (DAD) characterized by hyaline membrane formation (6). Fragments of HA (LMW-HA) activate PAI-1 (3). An escalating cycle of stimulation between STAT3 and PAI-1 begins a catastrophic cascade of events, resulting in combinations of coagulopathy/thrombosis (7), macrophage production of cytokines and chemokines (8), and profibrotic changes (9). Hypoxia eventually results, which further induces PAI-1 transcription through HIF-1α (10). This elevated PAI-1 activity then drives IL-6 production via TLR4, which in turn stimulates even more STAT3 activity (No. 11). Elevated STAT3 also activates PD-L1 in endothelial cells, leading to T cell lymphopenia (No. 12). Details of these events are described in the main text. Italicized outside labels are cell types, and non-italicized outside labels are locations. Bolded italicized text indicates disease states. Bent arrows indicate the transcriptional induction of the indicated target proteins. Straight arrows indicate direct activation. Dashed lines indicate direct inhibition.
STAT3 and coagulopathy
Pervasive hemostasis disorders are a life-threatening feature of COVID-19 [68]. Inflammation-induced coagulation is initiated by the expression of the transmembrane protein “Tissue Factor” (TF) [69]. In the lung, TF is expressed in alveolar epithelial cells [70], myeloid cells [71], and endothelial cells [72] in response to proinflammatory cytokines [73] or CRP [74,75,76,77,78]. TF promotes the transformation of prothrombin into thrombin, which converts circulating fibrinogen into fibrin, leading to fibrin-based blood clots [79]. Recombinant human CRP, administered in concentrations commonly seen in patients with inflammation, induced a 75-fold increase in the TF procoagulant activity of human PBMCs [74]. Because TF is induced by CRP, and CRP is a downstream target of STAT3 [80], aberrantly activated STAT3 may prime the initial phase of coagulation (7, Fig. 4). In addition, the full transcriptional induction of CRP requires the synergistic cooperation of STAT3, c-FOS, and T cell factor 7 (TCF-7, or HNF-1α) [81]. c-FOS is activated by SARS-CoV-1 ORF3b [82] and TCF-7 is expressed in lung cells [83]. Thus STAT3, c-FOS, and TCF-7 could participate in SARS-CoV-2-infected cells and act to raise plasma CRP levels. It then follows that the positive correlation between CRP level and disease severity in COVID-19 patients [14, 15] also reflects the number of virus-infected cells.
Another critical factor in coagulopathy and thrombosis is PAI-1 (SERPIN E1), a serine protease inhibitor secreted by vascular endothelial cells, hepatocytes, adipocytes, and epithelial cells [84, 85]. Several lines of evidence suggest that PAI-1 and STAT3 interact to promote coagulopathy and thrombosis in COVID-19 (7, Fig. 4).
-
(1)
PAI-1 is upregulated by STAT3. PAI-1 is expressed in damaged type 2 alveolar cells [86] and indirectly upregulated by STAT3 through five pathways. First, microRNA-34a (miR-34a) targets the PAI-1 gene in non-small cell lung carcinoma (NSCLC), and miR-34a transcription is suppressed by STAT3 [87]. This negative regulation of PAI-1 by miR-34a [88], and of miR-34a by STAT3 [89], has also been documented in cancer cell lines. Therefore, STAT3 indirectly upregulates PAI-1 through miR-34a (4, Fig. 4). Second, STAT3 (plus c-FOS and TCF-7) induces CRP [81], and CRP induces PAI-1 in human aortic endothelial cells [90]. Third, the tumor suppressor p53 activates PAI-1 production [91], and activated STAT3 induces p53 transcription [92]. Fourth, as detailed below, STAT3 indirectly activates TGF-β in the extracellular matrix (ECM), and TGF-β upregulates PAI-1 production [93]. Fifth, severe ARDS cases of COVID-19 result in diffuse alveolar damage (DAD) and the formation of hyaline membranes [94]. Hyaluronan (HA) is essential for many biological functions, but it is a causative agent in ARDS when it becomes a low-molecular-weight HA (LMW-HA) [95]. Both STAT3 and PAI-1 are associated with the production of HA. STAT3 activates the transcription of HAS2, the gene for HA synthase [96], and LMW-HA stimulate the production of PAI-1(6, Fig. 4) [97, 98]. Furthermore, variations in genes involved in HA synthesis control the levels of plasma PAI-1.(3, Fig. 4) [99] Indeed, the STAT3/PAI-1 axis is intimately connected to the production of HA.
-
(2)
PAI-1 may activate STAT3. PAI-1 can interact with PIAS3 (5, Fig. 4), an endogenous STAT3 inhibitor (9, Fig. 3), to regulate STAT3-dependent gene expression in NSCLC [87]. Other studies have found a more indirect effect. For example, PAI-1 engages TLR4 [100] and activates IL-6, which could then activate STAT3 [101]. In addition, PAI-1 can promote monocyte migration through its interaction with lipoprotein receptor protein-1 (LRP-1). PAI-1/LRP-1 binding influences monocyte/macrophage polarization toward the M2 type, which features transcriptional upregulation of IL-6 and activation of STAT3 [102].
-
(3)
PAI-1 is highly expressed in plasma and lungs in COVID-19 cases. SARS patients were characterized by increased plasma levels of PAI-1 [103] and high PAI-1 protein expression in alveolar cells [104] compared to controls. Consistently, PAI-1 is significantly elevated in the plasma of hospitalized COVID-19 patients [105]. Furthermore, PAI-1 mRNA levels are higher in the lungs of COVID-19 patients, as compared to those of uninfected or influenza patients [8].
-
(4)
PAI-1 is closely linked with COVID-19 risk factors. Network pathway analyses have identified PAI-1 as a key protein in obesity, diabetes [106], and cardiovascular disease [107]. Furthermore, higher levels of plasma PAI-1 are present in hypertensive patients [108, 109] and older adults [110, 111]. All of these conditions are high-risk factors for severe COVID-19 pathology.
The above studies suggest that the increase in PAI-1 levels due to STAT3 activation may efficiently inhibit both tissue-type plasminogen activator (tPA) and urokinase-type plasminogen activator, leading to coagulopathy/thrombosis (7, Fig. 4). In addition, IFN-I has been shown to upregulate tPA expression through STAT1 [112]. Therefore, the early repression of STAT1 and IFN activities imposed by viral proteins could synergistically inhibit tPA and promote coagulopathy/thrombosis.
In addition to the coagulopathic changes within the pulmonary vasculature, Copin et al. observed a peculiar pattern in the lungs of six COVID-19 patients that was different from the classical DAD pattern of ARDS [113]. The pattern, acute fibrinous and organizing pneumonia, consisted of a fibrin component and was characterized by extensive deposition of intra-alveolar fibrin (“fibrin balls”) in the lungs. McGonagle et al. suggested that the occurrence of enhanced thrombin generation and fibrin deposition within the bronchoalveolar system might be primarily driven by the upregulation of TF expression within the alveoli, coupled with a PAI-1-induced reduction in fibrinolysis [114]. Because TF is a downstream target of activated STAT3 [115] and produced in type 2 alveolar cells [70], the secretion of both TF and PAI-1 into the alveolar space may lead to the enhanced deposition of intra-alveolar fibrin.
STAT3 and induction of inflammatory cytokines and chemokines
Significant efforts are presently underway to define the proinflammatory cytokines and chemokines active in COVID-19. Huang et al. studied 41 in-patients (13 ICU patients and 28 non-ICU patients) and reported that the plasma levels of inflammatory cytokines, including IL-7, IL-10, G-CSF, IP-10, MCP-1, MIP1A, and TNF-α, were all elevated in critically ill patients [116]. In critically ill COVID-19 patients, serum IL-6 is significantly greater than those with moderate or severe disease [117]. The excessive production of these inflammatory cytokines in the lungs of COVID-19 patients appears to be due to increased macrophage activation [118,119,120]. Gupta et al. reported that PAI-1 activates macrophages and increases their proinflammatory cytokine production by binding to TLR4 [100] (8, Fig. 4). PAI-1 binding to TLR4 leads to the activation of NF-κB, which induces the production of chemokines and IL-6. IL-6 stimulates STAT3 (11, Fig. 4) to produce TGF-β [101] in a positively regulated reciprocal feedback loop involving PAI-1 [121], thereby amplifying the TLR4-mediated inflammatory response. PAI-1 thus appears to be partly responsible for the excessive production of inflammatory cytokines and chemokines by macrophages in the lungs of severely ill COVID-19 patients. In addition, accumulation of LMW-HA molecules in the small airways not only stimulates macrophages to release chemokines, cytokines, and growth factors, but also promotes fluid retention in the extracellular space, contributing to interstitial and alveolar edema [95].
Interstitial and alveolar edema severely impairs physiological gas exchange, causing hypoxia, which further stimulates PAI-1 expression through hypoxia-inducible factor 1 α (HIF-1α) [122, 123]. Thus, the central node involving STAT3 and PAI-1 may favor hypoxia through multiple routes (10, Fig. 4).
STAT3 and TGF-β-related profibrosis
TGF-β plays a key role in COVID-19. Xiong et al. performed transcriptome sequencing of RNAs isolated from bronchoalveolar lavage fluid and PBMC specimens of COVID-19 patients, and found that SARS-CoV-2 infection induced IL-10 and TGF-β production [124]. Many viral infections modulate TGF-β signaling to block cell apoptosis and promote fibroblast proliferation and myofibroblast differentiation. Thus, the increased expression of TGF-β in COVID-19 patients may drive the observed pulmonary fibrosis [124]. The possible sources of TGF-β in COVID-19 are the ECM, blood-circulating platelets, recruited neutrophils, macrophages, and infected type 2 alveolar cells [125, 126]. The latent form of TGF-β in the ECM requires activation by integrin αvβ6 and thrombospondin, which are induced by STAT3 [126,127,128,129,130]. Therefore, aberrantly activated STAT3 in type 2 alveolar cells may contribute to TGF-β activation and subsequent fibrosis (9, Fig. 4).
Experiments in a murine model of systemic sclerosis support the indirect upregulation of PAI-1 (3, Fig. 4) by STAT3 through TGF-β [93]. Activated TGF-β induces PAI-1 transcription [121], establishing another positive feedback loop involving STAT3 and PAI-1. The transcription of profibrotic genes encoding collagens, proteoglycans, integrins, connective tissue growth factor, and matrix metalloproteinases (MMPs) is also actuated by activated TGF-β [131], establishing a profibrotic environment. In fact, lessons from SARS-CoV-1 suggest that the SARS-CoV-2 N protein may enhance TGF-β-induced expression of PAI-1 and collagen I, and thus promote lung fibrosis [132].
STAT3 and T cell lymphopenia
Lymphopenia is a common characteristic of COVID-19 patients [11, 133, 134] and correlates with disease progression, but its etiology remains unclear. Diao et al. reported that, in 499 hospitalized cases, 76% of the patients had remarkably reduced total T cell counts, which were significantly lower in ICU patients than in non-ICU cases. They also observed that PD1 expression on T cells increased as patients progressed from the prodromal to the overtly symptomatic stage. Because PD1 is a marker of T cell exhaustion, this group proposed that the elevated proinflammatory cytokines in COVID-19 patients are responsible for their T cell lymphopenia [11]. Moreover, Chen et al. analyzed spleens and lymph nodes from six autopsy cases, and suggested that SARS-CoV-2 could directly infect tissue-resident CD169+ macrophages in secondary lymphoid organs and induce lymphocyte apoptosis [28].
Alternatively, T cell lymphopenia could be caused directly by SARS-CoV-2 entering T cells via CD147 [135]. Although SARS-CoV-2 cannot replicate in T cells, the apoptosis-inducing activity of the SARS-CoV-2 ORF3a protein [136] could effectuate T cell lymphopenia.
The effects of SARS-CoV-2 on PD1 and its binding partner PD-L1 differ by disease phase; i.e., the early/mild phase vs. the late/critical phase of COVID-19.
Early/Mild phase
The early/mild phase of SARS-CoV-2 infection is limited to type 2 alveolar cells and alveolar macrophages. Once infected, these cells secrete chemokines, including IP-10 (CXCL10), in a process that requires STAT3 activation [137]. As a result, plasma IP-10 is increased in severely ill COVID-19 patients [138]. IP-10 recruits activated CXCR3+ T cells from the regional capillaries [138,139,140,141], and lymphocytes infiltrate the diseased lung tissues of these patients [8, 113, 114]. Because type 2 alveolar cells [142] and alveolar macrophages [143] express PD-L1 together with virus antigens, virus-specific T cells migrate to the area and then upregulate PD1 upon recognition of the antigens [144]. PD-L1 engagement with PD1 induces the apoptosis of the activated T cells [145, 146], resulting in moderate T cell lymphopenia (12, Fig. 4).
Late/Critical phase
In the severe phase of SARS-CoV-2 infection, activated STAT3 promotes the secretion of MMP-9 by type 2 alveolar cells [147] and alveolar macrophages [148]. MMP9 degrades collagen IV, the major component of the basement membrane, and compromises its integrity. The virus may then migrate into the interstitium and enter capillaries, where it could infect endothelial cells. The infection causes aberrant STAT3 regulation in these endothelial cells and may also induce PD-L1 activity, as demonstrated for some cancer cells with constitutively active STAT3 [149, 150]. In the critical stages of COVID-19, the PD1/PD-L1-induced death of T cells becomes systemic, resulting in severe T cell lymphopenia (12, Fig. 4).
Therapeutic considerations
We hypothesize that COVID-19 disease is due in large part to the actions of the SARS-CoV-2 NSP1 and ORF6 proteins, which cripple STAT1 function and predominantly promote STAT3 activation. STAT3 in turn upregulates PAI-1, and together these molecules serve as a central hub of reactions that perpetuate a catastrophic cascade. Our understanding of immune responses, coupled with lessons from SARS-CoV-1 and recent research on SARS-CoV-2, point to STAT1 and STAT3 as enticing drug targets because they function upstream of the cytokine storm and thrombosis. Developing vaccines will take some time, and attacking the downstream cytokine storm is difficult due to its many targets. Hence, in the short term, the manipulation of STAT1 and/or STAT3 may be the most practical strategy for treating COVID-19. Fortunately, many therapeutic regulators of these STATs have been, or are being, developed.
The first consideration in treating SARS-CoV-2 infection should be to preserve STAT1 function in its earliest stages. However, patients may be asymptomatic or presymptomatic for days while the virus replicates, spreads, and evades the IFN response. If the virus has already compromised STAT1 when symptoms arise, then treatment should focus on preventing the excessive activation of STAT3 that drives the release of proinflammatory cytokines and chemokines. Care must be taken to apply these therapeutic strategies in a stage-appropriate manner, because some approaches that may help in the earlier stages of the disease could be detrimental if used in its later stages. In the following sections, we discuss the potential use for COVID-19 treatment of existing drugs that enhance STAT1 or inhibit STAT3 functions.
Problems could arise from this STAT-approach to COVID-19 therapy. Chronic mucocutaneous candidiasis (CMC) was present in 98% of patients with gain of function (GOF) mutations in STAT1 in one study of 274 individuals, and the few that did not have CMC, had invasive fungal or bacterial infections. Generally, the immune profile was relatively normal, but 82% had decreased Th17 cells [151]. STAT1 and STAT3 reciprocally regulate each other, and consistently, STAT1 GOF patients have reduced levels of STAT3 [152]. STAT3 IgE syndrome (STAT3-HIES) is a rare autosomal dominant condition caused by loss of function mutations in the STAT3 gene. Patients have eczema, recurrent skin and respiratory tract infections, and usually high levels of IgE [153]. These examples of genetic defects are likely the most extreme consequences of increased STAT1 or decreased STAT3 activities. It is unknown if temporary treatment with STAT1 activators and STAT3 inhibitors would be detrimental, but it is promising that a high STAT1 to STAT3 ratio is advantageous in cancer treatment [154].
Enhancement of STAT1 function
IFN-I
At the very onset of infection, SARS-CoV-2 infects a minority of cells, making its invasion hard to detect. The virus acts to delay antiviral responses while it hijacks the cell’s functions for viral replication. If the infection is detected early, then an effective strategy might be to use IFN-I to activate STAT1 in neighboring uninfected cells. It is proposed that the timing of IFN treatment is critical [30]. Early induction of IFN-I signaling protects the patients, but delay in IFN administration not only fail to inhibit viral replication, but also increase proinflammatory cytokine production, leading to fatal pneumonia [30]. Since the upper respiratory tract is the primary entry site for SARS-CoV-2, mucosal treatments with IFN-I for prevention of COVID-19 is an ideal strategy. Meng et al. used IFN-I nasal drops prophylactically applied to more than 500 high-risk medical staff who were in direct contact with SARS-CoV-2-infected patients. Remarkably, the use of the IFN-I nasal drops, with thymosin-α1, an immune-stimulator, protected all of the high-risk staff from COVID-19 pneumonia [155].
IFN-I inducers
The use of agents that induce IFN production is another option for activating STAT1 in uninfected cells. Ampligen, poly(I:C(12)U), is a synthetic dsRNA polymer that stimulates IFN production [156]. Mice treated with Ampligen 16 h prior to lethal infection with SARS-CoV-1 were protected against death, showed reduced virus titers in the lungs, and exhibited significantly reduced lung disease scores and weight loss [157]. Notably, because IFN-I inducers can be administered prophylactically, the innate immune system can theoretically be primed to respond to a SARS-CoV-2 attack by immediately activating STAT1. However, such use of IFN inducers must be carefully monitored, because these agents can exacerbate the disease in the later stages of infection.
Histamine receptor-2 blocker (H-2 blocker)
Administration of the H-2 blocker famotidine to patients hospitalized with COVID-19, but not initially in the ICU, was associated with a twofold reduction in clinical deterioration leading to intubation or death [158]. Computational modeling indicated that famotidine directly binds and inhibits the SARS-CoV-2 processing enzyme NSP5 [158], but this drug lacked a direct anti-SARS-CoV-2 effect in vitro in Vero cells, and H-2-mediated antiviral effect is suggested [159]. In vitro, histamine pretreatment of C57BL/6 mouse splenocytes enhances STAT1 phosphorylation, and an H-2 antagonist (but not an H-1 antagonist) can augment STAT1 phosphorylation to a similar extent [160]. Famotidine may function as an H-2 receptor antagonist that promotes STAT1 activation and IFN responses. Therefore, early stage treatment with famotidine might significantly decrease the mortality rate of COVID-19 patients.
Ivermectin
Ivermectin displays broad-spectrum antiparasitic and antiviral activities. Caly et al. reported that a single dose of ivermectin reduced the amount of SARS-CoV-2 RNA by 5000-fold. They speculated that ivermectin inhibits the KPNA/KPNB1-mediated nuclear import of viral proteins [161]. Apart from its possible role in blocking nuclear transport, Ivermectin may promote a positive clinical outcome by inhibiting STAT3 and IL-6 production. Ivermectin inhibits p21 activated kinase 1 (PAK1), a serine/threonine kinase with oncogenic activity, which then compromises STAT3 activity. In this case, ivermectin suppresses Akt/mTOR signaling by promoting the ubiquitination-mediated degradation of PAK1 [162]. In addition, PAK1 physically binds to both JAK1 and STAT3, and the resultant PAK1/STAT3 complex activates IL-6 gene transcription [163]. When ivermectin inhibits JAK/STAT3 signaling by promoting PAK1 degradation, STAT3 activity is compromised and IL-6 production is decreased.
Inhibition of STAT3 function
There are a number of approved drugs, or drugs in cancer development, that inhibit STAT3 directly. STAT3 antagonists have been described comprehensively in recent reviews by Qin et al. [164] and Bharadwaj et al. [21]. A brief description of some prominent candidates follows.
Napabucasin
Napabucasin was initially identified by its ability to inhibit the properties of cancer cell stemness and STAT3 activity in gel retardation assays [21]. A Phase III clinical trial involving napabucasin in combination with other standard chemotherapeutic agents is currently underway for several advanced malignancies [21]. In rectal cancer, napabucasin reduced not only pSTAT3 levels but also angiogenesis through a ROS-mediated effect [165].
Danvatirsen
Another promising anti-STAT3 agent is the 16-mer antisense oligonucleotide AZD9150 (danvatirsen), which targets the 3′UTR of the STAT3 gene and inhibits its transcription [166]. A Phase II trial of danvatirsen plus anti-PD-L1 monoclonal antibody in patients with head-and-neck squamous cell carcinoma is currently underway, with encouraging results thus far [21].
Metformin
Metformin is a first-line oral antidiabetic drug that has been used to treat type 2 diabetic patients for over 60 years. Metformin inhibits STAT3 by specifically reducing its phosphorylation at Tyr705 and Ser727 [167]. In addition, metformin prevents both venous and arterial thrombosis by inhibiting platelet activation and extracellular mitochondrial DNA (mtDNA) release. mtDNA induces platelet activation through a DC-SIGN-dependent pathway [168]. Despite metformin’s apparent utility in reducing STAT3 activation and thrombosis, a major concern is that the dose that elicits these activities is very high (200–400 mg/kg/day).
PIAS3 activators
The regulation of endogenous inhibitors is another way to control aberrantly activated STAT3. PIAS3 is an ideal target because of its potency to inhibit the activated STAT3 [169]. Specifically, curcumin and resveratrol have been shown to suppress constitutive activation of STAT3, through upregulation of PIAS3 [170, 171] Although widely used for many indications, curcumin and resveratrol have not been shown to be conclusively effective in any randomized, placebo-controlled, clinical trial.
JAK inhibitors
The occurrence of a cytokine storm in COVID-19 (IL-2, IL-6, IL-7, IL-10, G-CSF, IFNγ, MIP1α, and TNF-α [116, 117], which triggers cytokine receptors coupled to the JAK-STAT pathway, suggests that inhibition of the JAK pathway may be an appropriate therapeutic strategy for the management of COVID-19. Although the JAK pathway affects many STATs, perhaps the inhibition of the JAK pathway may lead to the therapeutically favorable effect of quenching cytokine storm via STAT3 inhibition. The available JAK1/2 inhibitors [21] could be studied for their effects in COVID-19. However, as JAK inhibitors also inhibit STAT1 activation, the application of JAK inhibitors in COVID-19 cases needs careful consideration.
EGFR signaling inhibitors as an IFN-I potentiator
As discussed earlier and shown in Fig. 3, acute lung injury and/or the loss of functional STAT1 can lead to the upregulation of EGFR that may cause the constitutive activation of STAT3. Therefore, it is reasonable to assume that targeting EGFR signaling is an attractive strategy for COVID-19 treatment. A promising candidate is erlotinib. Lupberger et al. reported the combination of erlotinib and IFN-I resulted in a highly synergistic antiviral response against hepatitis C virus. Furthermore, erlotinib reduced IFN-I-induced STAT3 activity by induction of SOCS3 expression [63]. Very recently, the remarkable inhibition of SARS-CoV-2 replication by several growth factor signaling inhibitors was reported [172]. These include lonafarnib (RAS inhibitor), omipalisib (PI3K inhibitor), pictilisib (PI3K inhibitor), RO5126766 (RAF and MEK inhibitor), sorafenib (RAF inhibitor, STAT3 inhibitor [62]), because Ras/Raf/MAPK pathway [173] and PI3K pathway [174] are also involved in EGFR-mediated STAT3 activation, these inhibitors can possibly synergistically potentiate IFN-I’s anti-SARS-CoV-2 activity.
PAI-1 inhibitors and TLR4 inhibitors
As noted above, PAI-1 is intimately involved in the pathogenesis of COVID-19, and its inhibition may be a key point at which to treat the disease once the escalating cycle between PAI-1 and STAT3 has been established. Unfortunately, no FDA-approved PAI-1 inhibitor is currently available. PAI-1’s labile structure appears to make it a difficult target for the development of small-molecule inhibitors [175]. Alternatively, therapeutically targeting TLR4, PAI-1’s binding partner, may be equally effective. TLR4 has already attracted keen interest as a therapeutic target for sepsis cases. Although the molecular mechanisms have yet to be clarified, it is worth investigating whether PAI-1/TLR4 binding can be inhibited by the several TLR4 antagonists in development, or by approved drugs with anti-TLR4 activity. One TLR4 inhibitor, Eritoran, is now in clinical trials to treat ARDS in COVID-19 [176].
Concluding remarks
The SARS-CoV-2 virus has evolved multiple tools to escape immune detection and destruction, and thus has become the most formidable virus in over 100 years. Our survey of the current literature reveals that severe cases of COVID-19 are commonly dependent on the over-stimulation of the STAT3/PAI-1 signaling network. We believe that this shared node may be the Achilles’ Heel of COVID-19 and a vulnerable point of the disease. We therefore urge the immediate investigation and application of STAT therapy as a treatment for this perplexing disorder, and hope that our article provides a sound foundation for doing so.
A final note: during our analysis of the literature on COVID-19 and related topics, we noticed many similarities in the pathogenesis of COVID-19 and cancers. In fact, both STAT3 and PAI-1 have been separately implicated in cancer development and are the subjects of extensive clinical investigations. A shared node of STAT3 and PAI-1 activities may also function in some pSTAT3-positive cancers, creating a cascade of harmful responses comparable to those of COVID-19. Any STAT-related agents developed to treat COVID-19 may therefore eventually enjoy much wider clinical use.
Facts
-
Specific SARS-CoV proteins inhibit the functions of STAT1 and IFNs.
-
In the absence of STAT1, STAT3 is activated in a compensatory manner.
-
STAT3 upregulates PAI-1 through five signaling pathways mediated by miR-34a, CRP, p53, transforming growth factor-β (TGF-), or hyaluronan fragments.
-
PAI-1 is upregulated in aged individuals and in those suffering from hypertension, obesity, or diabetes, which are risk factors for COVID-19.
-
In severe cases of COVID-19, there is a common escalating cycle of STAT3 and PAI-1 activation that is shared among diverse disease manifestations and leads to catastrophic consequences.
Open questions
-
Is IFN production different between asymptomatic carriers, and patients with less or more severe cases of COVID-19?
-
Is IFN production different between people infected with SARS-CoV-2, SARS-CoV-1, MERS coronavirus, or other coronaviruses?
-
Is STAT3 activated in SARS-CoV-2-infected tissues?
-
Will prophylactic activation of STAT1 protect against the severe symptoms of COVID-19?
-
Will inhibition of the STAT3/PAI-1 axis decrease the severities of the cytokine storm and thrombosis/coagulopathy in COVID-19?
-
Will inhibition of EGFR signaling potentiate the antiviral activity of IFN-I against SARS-CoV-2?
-
Will inhibition of EGFR signaling ameliorate the disease severity?
-
Are genetic polymorphisms of STAT1, STAT3, PAI-1, HA, or EGFR related to the severity of COVID-19?
-
How does the STAT3/PAI-1 axis contribute to other disease conditions, including cancer?
References
He X, Lau EHY, Wu P, Deng X, Wang J, Hao X et al. Temporal dynamics in viral shedding and transmissibility of COVID-19. Nat Med. 2020. https://doi.org/10.1038/s41591-020-0869-5.
Petrosillo N, Viceconte G, Ergonul O, Ippolito G, Petersen E. COVID-19, SARS and MERS: are they closely related? Clin Microbiol Infect. 2020. https://doi.org/10.1016/j.cmi.2020.03.026.
Shang J, Ye G, Shi K, Wan Y, Luo C, Aihara H, et al. Structural basis of receptor recognition by SARS-CoV-2. Nature. 2020;581:221–4.
Wang Q, Zhang Y, Wu L, Niu S, Song C, Zhang Z et al. Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell. 2020. https://doi.org/10.1016/j.cell.2020.03.045.
Wan Y, Shang J, Graham R, Baric RS, Li F. Receptor recognition by the novel coronavirus from Wuhan: an analysis based on decade-long structural studies of SARS coronavirus. J Virol. 2020;94. https://doi.org/10.1128/JVI.00127-20.
Petersen E, Koopmans M, Go U, Hamer DH, Petrosillo N, Castelli F et al. Comparing SARS-CoV-2 with SARS-CoV and influenza pandemics. Lancet Infect Dis. 2020. https://doi.org/10.1016/S1473-3099(20)30484-9.
Zheng S, Fan J, Yu F, Feng B, Lou B, Zou Q, et al. Viral load dynamics and disease severity in patients infected with SARS-CoV-2 in Zhejiang province, China, January-March 2020: retrospective cohort study. BMJ. 2020;369:m1443.
Ackermann M, Verleden SE, Kuehnel M, Haverich A, Welte T, Laenger F et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid-19. N Engl J Med. 2020. https://doi.org/10.1056/NEJMoa2015432.
Klok FA, Kruip MJHA, van der Meer NJM, Arbous MS, Gommers DAMPJ, Kant KM et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb Res. 2020. https://doi.org/10.1016/j.thromres.2020.04.013.
Zheng Z, Peng F, Xu B, Zhao J, Liu H, Peng J et al. Risk factors of critical & mortal COVID-19 cases: a systematic literature review and meta-analysis. J Infect. 2020. https://doi.org/10.1016/j.jinf.2020.04.021.
Diao B, Wang C, Tan Y, Chen X, Liu Y, Ning L, et al. Reduction and functional exhaustion of T cells in patients with coronavirus disease 2019 (COVID-19). Front Immunol. 2020;11:827.
Hadjadj J, Yatim N, Barnabei L, Corneau A, Boussier J, Smith N, et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science. 2020;369:718–24.
Haran JP, Beaudoin FL, Suner S, Lu S. C-reactive protein as predictor of bacterial infection among patients with an influenza-like illness. Am J Emerg Med. 2013;31:137–44.
Wang L. C-reactive protein levels in the early stage of COVID-19. Med Mal Infect. 2020;50:332–4.
Chen W, Zheng KI, Liu S, Yan Z, Xu C, Qiao Z. Plasma CRP level is positively associated with the severity of COVID-19. Ann Clin Microbiol Antimicrob. 2020;19:18.
Kayser S, Kunze R, Sheriff A Selective C-reactive protein apheresis for Covid-19 patients suffering from organ damage. Ther Apher Dial. 2020. https://doi.org/10.1111/1744-9987.13532.
Richardson S, Hirsch JS, Narasimhan M, Crawford JM, McGinn T, Davidson KW et al. Presenting characteristics, comorbidities, and outcomes among 5700 patients hospitalized with COVID-19 in the New York City Area. JAMA. 2020. https://doi.org/10.1001/jama.2020.6775.
Lokugamage KG, Hage A, Schindewolf C, Rajsbaum R, Menachery VD. SARS-CoV-2 is sensitive to type I interferon pretreatment. bioRxiv. 2020. https://doi.org/10.1101/2020.03.07.982264.
Wang C, Liu Z, Chen Z, Huang X, Xu M, He T et al. The establishment of reference sequence for SARS-CoV-2 and variation analysis. J Med Virol. 2020. https://doi.org/10.1002/jmv.25762.
Yuen C-K, Lam J-Y, Wong W-M, Mak L-F, Wang X, Chu H et al. SARS-CoV-2 nsp13, nsp14, nsp15 and orf6 function as potent interferon antagonists. Emerg Microbes Infect. 2020;9:1–29.
Bharadwaj U, Kasembeli MM, Robinson P, Tweardy DJ. Targeting janus kinases and signal transducer and activator of transcription 3 to treat inflammation, fibrosis, and cancer: rationale, progress, and caution. Pharm Rev. 2020;72:486–526.
Sardar R, Satish D, Birla S, Gupta D. Comparative analyses of SAR-CoV2 genomes from different geographical locations and other coronavirus family genomes reveals unique features potentially consequential to host-virus interaction and pathogenesis. bioRxiv. 2020. https://doi.org/10.1101/2020.03.21.001586.
Walls AC, Park Y-J, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell. 2020;181:281–92.e6.
Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181:271–80.
Sungnak W, Huang N, Bécavin C, Berg M, Queen R, Litvinukova M, et al. SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat Med. 2020;26:681–7.
Varga Z, Flammer AJ, Steiger P, Haberecker M, Andermatt R, Zinkernagel AS, et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet. 2020;395:1417–8.
Monteil V, Kwon H, Prado P, Hagelkrüys A, Wimmer RA, Stahl M et al. Inhibition of SARS-CoV-2 infections in engineered human tissues using clinical-grade soluble human ACE2. Cell. 2020. https://doi.org/10.1016/j.cell.2020.04.004.
Chen Y, Feng Z, Diao B, Wang R, Wang G, Wang C et al. The novel severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) directly decimates human spleens and lymph nodes. Infectious Diseases (except HIV/AIDS). 2020. https://doi.org/10.1101/2020.03.27.20045427.
Corley MJ, Sugai C, Schotsaert M, Schwartz RE, Ndhlovu LC. Comparative in vitro transcriptomic analyses of COVID-19 candidate therapy hydroxychloroquine suggest limited immunomodulatory evidence of SARS-CoV-2 host response genes. bioRxiv. 2020. https://doi.org/10.1101/2020.04.13.039263.
Park A, Iwasaki A. Type I and type III interferons—induction, signaling, evasion, and application to combat COVID-19. Cell Host Microbe. 2020;27:870–8.
Stockinger S, Decker T. STATs and Infection. Landes Biosci. 2013. https://www.ncbi.nlm.nih.gov/books/NBK6266/.
Takaoka A, Mitani Y, Suemori H, Sato M, Yokochi T, Noguchi S, et al. Cross talk between interferon-γ and -α/β signaling components in caveolar membrane domains. Science. 2000;288:2357–60.
Gough DJ, Messina NL, Hii L, Gould JA, Sabapathy K, Robertson APS, et al. Functional crosstalk between type I and II interferon through the regulated expression of STAT1. PLoS Biol. 2010;8:e1000361.
Sekimoto T, Imamoto N, Nakajima K, Hirano T, Yoneda Y. Extracellular signal-dependent nuclear import of Stat1 is mediated by nuclear pore-targeting complex formation with NPI-1, but not Rch1. EMBO J. 1997;16:7067–77.
Velazquez-Salinas L, Verdugo-Rodriguez A, Rodriguez LL, Borca MV. The role of interleukin 6 during viral infections. Front Microbiol. 2019;10:1057.
Takahashi K. Current clinical applications of interferon. JMAJ. 2004;47:60–3.
Gandhi M, Yokoe DS, Havlir DV. Asymptomatic Transmission, the Achilles’ Heel of Current Strategies to Control Covid-19. N. Engl. J. Med. 2020;382:2158–60.
Blanco-Melo D, Nilsson-Payant BE, Liu W-C, Uhl S, Hoagland D, Møller R, et al. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell. 2020;181:1036–45.
Wathelet MG, Orr M, Frieman MB, Baric RS. Severe acute respiratory syndrome coronavirus evades antiviral signaling: role of nsp1 and rational design of an attenuated strain. J Virol. 2007;81:11620–33.
Angelini MM, Akhlaghpour M, Neuman BW, Buchmeier MJ. Severe acute respiratory syndrome coronavirus nonstructural proteins 3, 4, and 6 induce double-membrane vesicles. MBio. 2013;4. https://doi.org/10.1128/mBio.00524-13.
Emmanuelle Blanchard PR. Virus-induced double-membrane vesicles. Cell Microbiol. 2015;17:45–50.
Decroly E, Debarnot C, Ferron F, Bouvet M, Coutard B, Imbert I, et al. Crystal structure and functional analysis of the SARS-coronavirus RNA cap 2’-O-methyltransferase nsp10/nsp16 complex. PLoS Pathog. 2011;7:e1002059.
Hu Y, Li W, Gao T, Cui Y, Jin Y, Li P et al. The severe acute respiratory syndrome coronavirus nucleocapsid inhibits type I interferon production by interfering with TRIM25-mediated RIG-I ubiquitination. J Virol. 2017;91:e02143-16.
Siu K-L, Kok K-H, Ng M-HJ, Poon VKM, Yuen K-Y, Zheng B-J, et al. Severe acute respiratory syndrome coronavirus M protein inhibits type I interferon production by impeding the formation of TRAF3.TANK.TBK1/IKKepsilon complex. J Biol Chem. 2009;284:16202–9.
Minakshi R, Padhan K, Rani M, Khan N, Ahmad F, Jameel S. The SARS coronavirus 3a protein causes endoplasmic reticulum stress and induces ligand-independent downregulation of the type 1 interferon receptor. PLoS ONE. 2009;4:e8342.
Kopecky-Bromberg SA, Martínez-Sobrido L, Frieman M, Baric RA, Palese P. Severe acute respiratory syndrome coronavirus open reading frame (ORF) 3b, ORF 6, and nucleocapsid proteins function as interferon antagonists. J Virol. 2007;81:548–57.
Konno Y, Kimura I, Uriu K, Fukushi M, Irie T, Koyanagi Y et al. SARS-CoV-2 ORF3b is a potent interferon antagonist whose activity is further increased by a naturally occurring elongation variant. bioRxiv. 2020. https://doi.org/10.1016/j.celrep.2020.108185.
Zhou P, Li H, Wang H, Wang L-F, Shi Z. Bat severe acute respiratory syndrome-like coronavirus ORF3b homologues display different interferon antagonist activities. J Gen Virol. 2012;93:275–81.
Yang D, Chu H, Hou Y, Chai Y, Shuai H, Lee AC-Y et al. Attenuated interferon and pro-inflammatory response in SARS-CoV-2-infected human dendritic cells is associated with viral antagonism of STAT1 phosphorylation. J Infect Dis. 2020. https://doi.org/10.1093/infdis/jiaa356.
Frieman M, Yount B, Heise M, Kopecky-Bromberg SA, Palese P, Baric RS. Severe acute respiratory syndrome coronavirus ORF6 antagonizes STAT1 function by sequestering nuclear import factors on the rough endoplasmic reticulum/Golgi membrane. J Virol. 2007;81:9812–24.
Huang C, Peters CJ, Makino S. Severe acute respiratory syndrome coronavirus accessory protein 6 is a virion-associated protein and is released from 6 protein-expressing cells. J Virol. 2007;81:5423–6.
Zhou H, Ferraro D, Zhao J, Hussain S, Shao J, Trujillo J, et al. The N-terminal region of severe acute respiratory syndrome coronavirus protein 6 induces membrane rearrangement and enhances virus replication. J Virol. 2010;84:3542–51.
Tangudu C, Olivares H, Netland J, Perlman S, Gallagher T. Severe acute respiratory syndrome coronavirus protein 6 accelerates murine coronavirus infections. J Virol. 2007;81:1220–9.
Pewe L, Zhou H, Netland J, Tangudu C, Olivares H, Shi L, et al. A severe acute respiratory syndrome-associated coronavirus-specific protein enhances virulence of an attenuated murine coronavirus. J Virol. 2005;79:11335–42.
Wang J, Schreiber RD, Campbell IL. STAT1 deficiency unexpectedly and markedly exacerbates the pathophysiological actions of IFN-alpha in the central nervous system. Proc Natl Acad Sci USA. 2002;99:16209–14.
Shao W-H, Gamero AM, Zhen Y, Lobue MJ, Priest SO, Albandar HJ, et al. Stat1 regulates lupus-like chronic graft-versus-host disease severity via interactions with Stat3. J Immunol. 2015;195:4136–43.
Avalle L, Pensa S, Regis G, Novelli F, Poli V. STAT1 and STAT3 in tumorigenesis: a matter of balance. JAKSTAT. 2012;1:65–72.
Tsai M-H, Pai L-M, Lee C-K. Fine-tuning of type I interferon response by STAT3. Front Immunol. 2019;10:1448.
Wang H, Yuan M, Wang S, Zhang L, Zhang R, Zou X et al. STAT3 regulates the type I IFN-mediated antiviral response by interfering with the nuclear entry of STAT1. Int J Mol Sci. 2019;20:4870.
Arimoto K-I, Miyauchi S, Stoner SA, Fan J-B, Zhang D-E Negative regulation of type I IFN signaling. J Leukoc Biol. 2018;103:1099–116.
Ramana CV, Kumar A, Enelow R. Stat1-independent induction of SOCS-3 by interferon-gamma is mediated by sustained activation of Stat3 in mouse embryonic fibroblasts. Biochem Biophys Res Commun. 2005;327:727–33.
Rosmorduc O, Desbois-Mouthon C. Targeting STAT3 in hepatocellular carcinoma: sorafenib again. J Hepatol. 2011;55:957–9.
Lupberger J, Duong FHT, Fofana I, Zona L, Xiao F, Thumann C, et al. Epidermal growth factor receptor signaling impairs the antiviral activity of interferon-alpha. Hepatology. 2013;58:1225–35.
Finigan JH, Downey GP, Kern JA. Human epidermal growth factor receptor signaling in acute lung injury. Am J Respir Cell Mol Biol. 2012;47:395–404.
Venkataraman T, Coleman CM, Frieman MB. Overactive epidermal growth factor receptor signaling leads to increased fibrosis after severe acute respiratory syndrome coronavirus infection. J Virol. 2017;91:e00182-17.
Guo L, Luo X, Li R, Xu Y, Zhang J, Ge J, et al. Porcine epidemic diarrhea virus infection inhibits interferon signaling by targeted degradation of STAT1. J Virol. 2016;90:8281–92.
Yang L, Xu J, Guo L, Guo T, Zhang L, Feng L et al. Porcine epidemic diarrhea virus-induced epidermal growth factor receptor activation impairs the antiviral activity of type I interferon. J Virol. 2018;92:e02095-17.
Joly BS, Siguret V, Veyradier A. Understanding pathophysiology of hemostasis disorders in critically ill patients with COVID-19. Intens Care Med. 2020. https://doi.org/10.1007/s00134-020-06088-1.
Grover SP, Mackman N. Tissue factor: an essential mediator of hemostasis and trigger of thrombosis. Arterioscler Thromb Vasc Biol. 2018;38:709–25.
Bastarache JA, Wang L, Geiser T, Wang Z, Albertine KH, Matthay MA, et al. The alveolar epithelium can initiate the extrinsic coagulation cascade through expression of tissue factor. Thorax. 2007;62:608–16.
Shaver CM, Grove BS, Clune JK, Mackman N, Ware LB, Bastarache JA. Myeloid tissue factor does not modulate lung inflammation or permeability during experimental acute lung injury. Sci Rep. 2016;6:22249.
Kirchhofer D, Tschopp TB, Hadváry P, Baumgartner HR. Endothelial cells stimulated with tumor necrosis factor-alpha express varying amounts of tissue factor resulting in inhomogenous fibrin deposition in a native blood flow system. Effects of thrombin inhibitors. J Clin Investig. 1994;93:2073–83.
Del Prete G, De Carli M, Lammel RM, d’Elios MM. Th1 and Th2 T-helper cells exert opposite regulatory effects on procoagulant activity and tissue factor production by human monocytes. 1995;86:250–57.
Cermak J, Key NS, Bach RR, Balla J, Jacob HS, Vercellotti GM. C-reactive protein induces human peripheral blood monocytes to synthesize tissue factor. Blood. 1993;82:513–20.
Ji Y, Fish PM, Strawn TL, Lohman AW, Wu J, Szalai AJ, et al. C-reactive protein induces expression of tissue factor and plasminogen activator inhibitor-1 and promotes fibrin accumulation in vein grafts. J Thromb Haemost. 2014;12:1667–77.
Cirillo P, Golino P, Calabrò P, Calì G, Ragni M, De Rosa S, et al. C-reactive protein induces tissue factor expression and promotes smooth muscle and endothelial cell proliferation. Cardiovasc Res. 2005;68:47–55.
Wu J, Stevenson Meredith J, Brown Jordan M, Grunz Elizabeth A, Strawn Tammy L, Fay William P. C-reactive protein enhances tissue factor expression by vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2008;28:698–704.
Devaraj S, Dasu MR, Singh U, Rao LVM, Jialal I. C-reactive protein stimulates superoxide anion release and tissue factor activity in vivo. Atherosclerosis. 2009;203:67–74.
Cimmino G, Cirillo P. Tissue factor: newer concepts in thrombosis and its role beyond thrombosis and hemostasis. Cardiovasc Diagn Ther. 2018;8:581–93.
Zhang D, Sun M, Samols D, Kushner I. STAT3 participates in transcriptional activation of the C-reactive protein gene by interleukin-6. J Biol Chem. 1996;271:9503–9.
Nishikawa T, Hagihara K, Serada S, Isobe T, Matsumura A, Song J, et al. Transcriptional complex formation of c-Fos, STAT3, and hepatocyte NF-1 alpha is essential for cytokine-driven C-reactive protein gene expression. J Immunol. 2008;180:3492–501.
Varshney B, Lal SK. SARS-CoV accessory protein 3b induces AP-1 transcriptional activity through activation of JNK and ERK pathways. Biochemistry. 2011;50:5419–25.
Douglas IS, Diaz del Valle F, Winn RA, Voelkel NF. Beta-catenin in the fibroproliferative response to acute lung injury. Am J Respir Cell Mol Biol. 2006;34:274–85.
Yamamoto K, Takeshita K, Kojima T, Takamatsu J, Saito H. Aging and plasminogen activator inhibitor-1 (PAI-1) regulation: implication in the pathogenesis of thrombotic disorders in the elderly. Cardiovasc Res. 2005;66:276–85.
Katz J, Tadi P. Physiology, Plasminogen Activation. In: StatPearls. Treasure Island (FL): StatPearls Publishing; 2019.
Osterholzer JJ, Christensen PJ, Lama V, Horowitz JC, Hattori N, Subbotina N, et al. PAI-1 promotes the accumulation of exudate macrophages and worsens pulmonary fibrosis following type II alveolar epithelial cell injury. J Pathol. 2012;228:170–80.
Lin X, Lin B-W, Chen X-L, Zhang B-L, Xiao X-J, Shi J-S, et al. PAI-1/PIAS3/Stat3/miR-34a forms a positive feedback loop to promote EMT-mediated metastasis through Stat3 signaling in Non-small cell lung cancer. Biochem Biophys Res Commun. 2017;493:1464–70.
Zhang Y, Pan Y, Xie C, Zhang Y. miR-34a exerts as a key regulator in the dedifferentiation of osteosarcoma via PAI-1-Sox2 axis. Cell Death Dis. 2018;9:777.
Rokavec M, Öner MG, Li H, Jackstadt R, Jiang L, Lodygin D, et al. IL-6R/STAT3/miR-34a feedback loop promotes EMT-mediated colorectal cancer invasion and metastasis. J Clin Investig. 2014;124:1853–67.
Devaraj S, Xu DY, Jialal I. C-reactive protein increases plasminogen activator inhibitor-1 expression and activity in human aortic endothelial cells: implications for the metabolic syndrome and atherothrombosis. Circulation. 2003;107:398–404.
Shetty SK, Bhandary YP, Marudamuthu AS, Abernathy D, Velusamy T, Starcher B, et al. Regulation of airway and alveolar epithelial cell apoptosis by p53-Induced plasminogen activator inhibitor-1 during cigarette smoke exposure injury. Am J Respir Cell Mol Biol. 2012;47:474–83.
Liu Y, Lv J, Liu J, Liang X, Jin X, Xie J, et al. STAT3/p53 pathway activation disrupts IFN-β-induced dormancy in tumor-repopulating cells. J Clin Investig. 2018;128:1057–73.
Chakraborty D, Šumová B, Mallano T, Chen C-W, Distler A, Bergmann C, et al. Activation of STAT3 integrates common profibrotic pathways to promote fibroblast activation and tissue fibrosis. Nat Commun. 2017;8:1130.
Mong MA, Awkal JA, Marik PE. Accelerated hyaluronan concentration as the primary driver of morbidity and mortality in high-risk COVID-19 patients: with therapeutic introduction of an oral hyaluronan inhibitor in the prevention of Induced Hyaluronan Storm Syndrome. Public and Global Health. 2020. https://doi.org/10.1101/2020.04.19.20071647.
Lonati C, Fumagalli J, Zanella A, Spinelli E, Mauri T. Hyaluronan in acute respiratory distress syndrome (ARDS): simply a biomarker or a deeper insight into ARDS mechanisms? J Lab Precis Med. 2018;3:49.
Jokela TA, Kärnä R, Makkonen KM, Laitinen JT, Tammi RH, Tammi MI. Extracellular UDP-glucose activates P2Y14 Receptor and Induces Signal Transducer and Activator of Transcription 3 (STAT3) Tyr705 phosphorylation and binding to hyaluronan synthase 2 (HAS2) promoter, stimulating hyaluronan synthesis of keratinocytes. J Biol Chem. 2014;289:18569–81.
Horton MR, Olman MA, Bao C, White KE, Choi AM, Chin BY et al. Regulation of plasminogen activator inhibitor-1 and urokinase by hyaluronan fragments in mouse macrophages. Am J Physiol Lung Cell Mol Physiol 2000;279:L707–15.
Hsieh Y-S, Yang S-F, Lue K-H, Chu S-C, Lu K-H. Effects of different molecular weight hyaluronan products on the expression of urokinase plasminogen activator and inhibitor and gelatinases during the early stage of osteoarthritis. J Orthop Res. 2008;26:475–84.
Lanktree MB, Johansen CT, Anand SS, Davis AD, Miller R, Yusuf S, et al. Genetic variation in hyaluronan metabolism loci is associated with plasma plasminogen activator inhibitor-1 concentration. Blood. 2010;116:2160–3.
Gupta KK, Xu Z, Castellino FJ, Ploplis VA. Plasminogen activator inhibitor-1 stimulates macrophage activation through Toll-like Receptor-4. Biochem Biophys Res Commun. 2016;477:503–8.
Zhu C, Shen H, Zhu L, Zhao F, Shu Y. Plasminogen activator inhibitor 1 promotes immunosuppression in human non-small cell lung cancers by enhancing TGF-β1 expression in macrophage. Cell Physiol Biochem. 2017;44:2201–11.
Kubala MH, Punj V, Placencio-Hickok VR, Fang H, Fernandez GE, Sposto R, et al. Plasminogen activator inhibitor-1 promotes the recruitment and polarization of macrophages in cancer. Cell Rep. 2018;25:2177–91.
Wu Y, Wei R, Liu Z, Chen B, Lisman T, Ren D, et al. Analysis of thrombotic factors in severe acute respiratory syndrome (SARS) patients. Thromb Haemost. 2006;96:100–1.
Zhao X, Nicholls JM, Chen Y-G. Severe acute respiratory syndrome-associated coronavirus nucleocapsid protein interacts with Smad3 and modulates transforming growth factor-beta signaling. J Biol Chem. 2008;283:3272–80.
Goshua G, Pine AB, Meizlish ML, Chang C-H, Zhang H, Bahel P, et al. Endotheliopathy in COVID-19-associated coagulopathy: evidence from a single-centre, cross-sectional study. Lancet Haematol. 2020. https://www.thelancet.com/action/showPdf?pii=S2352-3026%2820%2930216-7.
Kaur P, Reis MD, Couchman GR, Forjuoh SN, Greene JF, Asea A. SERPINE 1 links obesity and diabetes: a pilot study. J Proteom Bioinform. 2010;3:191–9.
Tofler GH, Massaro J, O’Donnell CJ, Wilson PWF, Vasan RS, Sutherland PA, et al. Plasminogen activator inhibitor and the risk of cardiovascular disease: the Framingham Heart Study. Thromb Res. 2016;140:30–35.
Peng H, Yeh F, de Simone G, Best LG, Lee ET, Howard BV, et al. Relationship between plasma plasminogen activator inhibitor-1 and hypertension in American Indians: findings from the Strong Heart Study. J Hypertens. 2017;35:1787–93.
Jacobs A, Schutte AE, Ricci C, Pieters M. Plasminogen activator inhibitor-1 activity and the 4 G/5 G polymorphism are prospectively associated with blood pressure and hypertension status. J Hypertens. 2019;37:2361–70.
Lasierra-Cirujeda J, Coronel P, Pascual-Salcedo MJA, Gimeno M, Mm AP-S, Lasierra-Ibañez A et al. Stress/inflammation and pai-1 as stellar processes in the aging and associated pathologies. Integr Mol Med. 2018;5:1–2
Cesari M, Pahor M, Incalzi RA. Plasminogen activator inhibitor-1 (PAI-1): a key factor linking fibrinolysis and age-related subclinical and clinical conditions. Cardiovasc Ther. 2010;28:e72–91.
Strandin T, Hepojoki J, Laine O, Mäkelä S, Klingström J, Lundkvist Å, et al. Interferons induce STAT1–dependent expression of tissue plasminogen activator, a pathogenicity factor in puumala hantavirus disease. J Infect Dis. 2016;213:1632–41.
Copin M-C, Parmentier E, Duburcq T, Poissy J, Mathieu D, Lille COVID-19 ICU and anatomopathology group. Time to consider histologic pattern of lung injury to treat critically ill patients with COVID-19 infection. Intens Care Med. 2020. https://doi.org/10.1007/s00134-020-06057-8.
McGonagle D, O’Donnell JS, Sharif K, Emery P, Bridgewood C. Immune mechanisms of pulmonary intravascular coagulopathy in COVID-19 pneumonia. Lancet Rheumatol. 2020. https://doi.org/10.1016/S2665-9913(20)30121-1.
Yeh H-H, Chang W-T, Lu K-C, Lai W-W, Liu H-S, Su W-C. Upregulation of tissue factor by activated Stat3 contributes to malignant pleural effusion generation via enhancing tumor metastasis and vascular permeability in lung adenocarcinoma. PLoS ONE. 2013;8:e75287.
Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497–506.
Han H, Ma Q, Li C, Liu R, Zhao L, Wang W, et al. Profiling serum cytokines in COVID-19 patients reveals IL-6 and IL-10 are disease severity predictors. Emerg Microbes Infect. 2020;9:1123–30.
Beck-Schimmer B, Schwendener R, Pasch T, Reyes L, Booy C, Schimmer RC. Alveolar macrophages regulate neutrophil recruitment in endotoxin-induced lung injury. Respir Res. 2005;6:61.
McQuattie-Pimentel AC, Budinger GRS, Ballinger MN. Monocyte-derived alveolar macrophages: the dark side of lung repair? Am J Respir Cell Mol Biol. 2018;58:5–6.
Johnston LK, Rims CR, Gill SE, McGuire JK, Manicone AM. Pulmonary macrophage subpopulations in the induction and resolution of acute lung injury. Am J Respir Cell Mol Biol. 2012;47:417–26.
Kutz SM, Hordines J, McKeown-Longo PJ, Higgins PJ. TGF-beta1-induced PAI-1 gene expression requires MEK activity and cell-to-substrate adhesion. J Cell Sci. 2001;114:3905–14.
Kietzmann T, Roth U, Jungermann K. Induction of the plasminogen activator inhibitor-1 gene expression by mild hypoxia via a hypoxia response element binding the hypoxia-inducible factor-1 in rat hepatocytes. Blood. 1999;94:4177–85.
Ueno M, Maeno T, Nomura M, Aoyagi-Ikeda K, Matsui H, Hara K, et al. Hypoxia-inducible factor-1α mediates TGF-β-induced PAI-1 production in alveolar macrophages in pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2011;300:L740–52.
Xiong Y, Liu Y, Cao L, Wang D, Guo M, Jiang A, et al. Transcriptomic characteristics of bronchoalveolar lavage fluid and peripheral blood mononuclear cells in COVID-19 patients. Emerg Microbes Infect. 2020;9:761–70.
Chen W. A potential treatment of COVID-19 with TGF-β blockade. Int J Biol Sci. 2020;16:1954–5.
Lodyga M, Hinz B. TGF-β1—A truly transforming growth factor in fibrosis and immunity. Semin Cell Dev Biol. 2020;101:123–39.
Xu M, Chen X, Yin H, Yin L, Liu F, Fu Y, et al. Cloning and characterization of the human integrin β6 gene promoter. PLoS ONE. 2015;10:e0121439.
Azare J, Leslie K, Al-Ahmadie H, Gerald W, Weinreb PH, Violette SM, et al. Constitutively activated Stat3 induces tumorigenesis and enhances cell motility of prostate epithelial cells through integrin beta 6. Mol Cell Biol. 2007;27:4444–53.
Bao Q, Zhang B, Suo Y, Liu C, Yang Q, Zhang K et al. Intermittent hypoxia mediated by TSP1 dependent on STAT3 induces cardiac fibroblast activation and cardiac fibrosis. Elife. 2020;9. https://doi.org/10.7554/eLife.49923.
Aluwihare P, Munger JS. What the lung has taught us about latent TGF-beta activation. Am J Respir Cell Mol Biol. 2008;39:499–502.
Walton KL, Johnson KE, Harrison CA. Targeting TGF-β mediated SMAD signaling for the prevention of fibrosis. Front Pharm. 2017;8:461.
Zuo W, Zhao X, Chen Y-G. SARS coronavirus and lung fibrosis. In: Lal SK, editor. Molecular Biology of the SARS Coronavirus, Berlin Heidelberg: Springer-Verlag; 2010. pp247-258.
Zhao Q, Meng M, Kumar R, Wu Y, Huang J, Deng Y, et al. Lymphopenia is associated with severe coronavirus disease 2019 (COVID-19) infections: a systemic review and meta-analysis. Int J Infect Dis. 2020;96:131–5.
Liu Y, Yang Y, Zhang C, Huang F, Wang F, Yuan J, et al. Clinical and biochemical indexes from 2019-nCoV infected patients linked to viral loads and lung injury. Sci China Life Sci. 2020;63:364–74.
Ulrich H, Pillat MM. CD147 as a target for COVID-19 treatment: suggested effects of azithromycin and stem cell engagement. Stem Cell Rev Rep. 2020;16:434–40.
Ren Y, Shu T, Wu D, Mu J, Wang C, Huang M, et al. The ORF3a protein of SARS-CoV-2 induces apoptosis in cells. Cell Mol Immunol. 2020;17:881–3.
Xu W, Joo H, Clayton S, Dullaers M, Herve M-C, Blankenship D, et al. Macrophages induce differentiation of plasma cells through CXCL10/IP-10. J Exp Med. 2012;209:1813–23.
Yang Y, Shen C, Li J, Yuan J, Wei J, Huang F et al. Plasma IP-10 and MCP-3 levels are highly associated with disease severity and predict the progression of COVID-19. J Allergy Clin Immunol. 2020. https://doi.org/10.1016/j.jaci.2020.04.027.
Wang J, Oberley-Deegan R, Wang S, Nikrad M, Funk CJ, Hartshorn KL, et al. Differentiated human alveolar type II cells secrete antiviral IL-29 (IFN-lambda 1) in response to influenza A infection. J Immunol. 2009;182:1296–304.
Khan IA, MacLean JA, Lee FS, Casciotti L, DeHaan E, Schwartzman JD, et al. IP-10 is critical for effector T cell trafficking and host survival in Toxoplasma gondii infection. Immunity. 2000;12:483–94.
Dufour JH, Dziejman M, Liu MT, Leung JH, Lane TE, Luster AD. IFN-gamma-inducible protein 10 (IP-10; CXCL10)-deficient mice reveal a role for IP-10 in effector T cell generation and trafficking. J Immunol. 2002;168:3195–204.
Ahmadvand N, Khosravi F, Lingampally A, Wasnick R, Vazquez-Armendariz I, Heiner M et al. Identification a novel subset of alveolar type 2 cells expanding following pneumonectomy and enriched in PD-L1. bioRxiv. https://doi.org/10.1101/2020.05.20.106005.
Igarashi T, Teramoto K, Ishida M, Hanaoka J, Daigo Y. Scoring of PD-L1 expression intensity on pulmonary adenocarcinomas and the correlations with clinicopathological factors. ESMO Open. 2016;1:e000083.
Schönrich G, Raftery MJ. The PD-1/PD-L1 axis and virus infections: a delicate balance. Front Cell Infect Microbiol. 2019;9:207.
Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8:793–800.
Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008;26:677–704.
Pardo A, Barrios R, Maldonado V, Meléndez J, Pérez J, Ruiz V, et al. Gelatinases A and B are up-regulated in rat lungs by subacute hyperoxia: pathogenetic implications. Am J Pathol. 1998;153:833–44.
Lemjabbar H, Gosset P, Lechapt-Zalcman E, Franco-Montoya ML, Wallaert B, Harf A, et al. Overexpression of alveolar macrophage gelatinase B (MMP-9) in patients with idiopathic pulmonary fibrosis: effects of steroid and immunosuppressive treatment. Am J Respir Cell Mol Biol. 1999;20:903–13.
Jahangiri A, Dadmanesh M, Ghorban K. STAT3 inhibition reduced PD-L1 expression and enhanced antitumor immune responses. J Cell Physiol. 2020. https://doi.org/10.1002/jcp.29750.
Zerdes I, Wallerius M, Sifakis EG, Wallmann T, Betts S, Bartish M et al. STAT3 activity promotes programmed-death ligand 1 expression and suppresses immune responses in breast cancer. Cancers. 2019;11:1479.
Toubiana J, Okada S, Hiller J, Oleastro M, Lagos Gomez M, Aldave, et al. Heterozygous STAT1 gain-of-function mutations underlie an unexpectedly broad clinical phenotype. Blood. 2016;127:3154–64.
Zheng J, van de Veerdonk FL, Crossland KL, Smeekens SP, Chan CM, Al Shehri T, et al. Gain-of-function STAT1 mutations impair STAT3 activity in patients with chronic mucocutaneous candidiasis (CMC). Eur J Immunol. 2015;45:2834–46.
Deng Y, Li T, Xie X, Xia D, Ding L, Xiang H, et al. Hyper IgE syndrome associated with novel and recurrent STAT3 mutations: Two case reports. Medicine. 2019;98:e14003.
Friedrich K, Dolznig H, Han X, Moriggl R. Steering of carcinoma progression by the YIN/YANG interaction of STAT1/STAT3. Biosci Trends. 2017;11:1–8.
Meng Z, Wang T, Li C, Chen X, Li L, Qin X et al. An experimental trial of recombinant human interferon alpha nasal drops to prevent coronavirus disease 2019 in medical staff in an epidemic area. Infectious Diseases (except HIV/AIDS). 2020. https://doi.org/10.1101/2020.04.11.20061473.
De Clercq E. Interferon and its inducers—a never-ending story: ‘old’ and ‘new’ data in a new perspective. J Infect Dis. 2006;194:S19–S26.
Day CW, Baric R, Cai SX, Frieman M, Kumaki Y, Morrey JD, et al. A new mouse-adapted strain of SARS-CoV as a lethal model for evaluating antiviral agents in vitro and in vivo. Virology. 2009;395:210–22.
Freedberg DE, Conigliaro J, Wang TC, Tracey KJ, Callahan MV, Abrams JA et al. Famotidine use is associated with improved clinical outcomes in hospitalized COVID-19 patients: a propensity score matched retrospective cohort study. Gastroenterology. 2020. https://doi.org/10.1053/j.gastro.2020.05.053.
Malone RW, Tisdall P, Fremont-Smith P, Liu Y, Huang X-P, White KM et al. COVID-19: Famotidine, histamine, mast cells, and mechanisms. In review. 2020. https://doi.org/10.21203/rs.3.rs-30934/v1.
Sakhalkar SP, Patterson EB, Khan MM. Involvement of histamine H1 and H2 receptors in the regulation of STAT-1 phosphorylation: inverse agonism exhibited by the receptor antagonists. Int Immunopharmacol. 2005;5:1299–309.
Caly L, Druce JD, Catton MG, Jans DA, Wagstaff KM. The FDA-approved drug ivermectin inhibits the replication of SARS-CoV-2 in vitro. Antivir Res. 2020;178:104787.
Dou Q, Chen H-N, Wang K, Yuan K, Lei Y, Li K, et al. Ivermectin induces cytostatic autophagy by blocking the PAK1/Akt axis in breast cancer. Cancer Res. 2016;76:4457–69.
Kim J-H, Choi HS, Kim S-L, Lee D-S. The PAK1-Stat3 signaling pathway activates IL-6 gene transcription and human breast cancer stem cell formation. Cancers. 2019;11:1527.
Qin J-J, Yan L, Zhang J, Zhang W-D. STAT3 as a potential therapeutic target in triple negative breast cancer: a systematic review. J Exp Clin Cancer Res. 2019;38:195.
Nagaraju GP, Farran B, Farren M, Chalikonda G, Wu C, Lesinski GB et al. Napabucasin (BBI 608), a potent chemoradiosensitizer in rectal cancer. Cancer. 2020. https://doi.org/10.1002/cncr.32954.
Reilley MJ, McCoon P, Cook C, Lyne P, Kurzrock R, Kim Y, et al. STAT3 antisense oligonucleotide AZD9150 in a subset of patients with heavily pretreated lymphoma: results of a phase 1b trial. J Immunother Cancer. 2018;6:119.
Deng X-S, Wang S, Deng A, Liu B, Edgerton SM, Lind SE, et al. Metformin targets Stat3 to inhibit cell growth and induce apoptosis in triple-negative breast cancers. Cell Cycle. 2012;11:367–76.
Xin G, Wei Z, Ji C, Zheng H, Gu J, Ma L, et al. Metformin uniquely prevents thrombosis by inhibiting platelet activation and mtDNA release. Sci Rep. 2016;6:36222.
Chung CD, Liao J, Liu B, Rao X, Jay P, Berta P, et al. Specific inhibition of Stat3 signal transduction by PIAS3. Science. 1997;278:1803–5.
Saydmohammed M, Joseph D, Syed V. Curcumin suppresses constitutive activation of STAT-3 by up-regulating protein inhibitor of activated STAT-3 (PIAS-3) in ovarian and endometrial cancer cells. J Cell Biochem. 2010;110:447–56.
Zhang P, Yang B, Yao Y-Y, Zhong L-X, Chen X-Y, Kong Q-Y, et al. PIAS3, SHP2 and SOCS3 expression patterns in cervical cancers: relevance with activation and resveratrol-caused inactivation of STAT3 signaling. Gynecol Oncol. 2015;139:529–35.
Klann K, Bojkova D, Tascher G, Ciesek S, Münch C, Cinatl J. Growth factor receptor signaling inhibition prevents SARS-CoV-2 replication. 2020. https://doi.org/10.1101/2020.05.14.095661.
Chung J, Uchida E, Grammer TC, Blenis J. STAT3 serine phosphorylation by ERK-dependent and -independent pathways negatively modulates its tyrosine phosphorylation. Mol Cell Biol. 1997;17:6508–16.
Abdelhamed S, Ogura K, Yokoyama S, Saiki I, Hayakawa Y. AKT-STAT3 pathway as a downstream target of EGFR signaling to regulate PD-L1 expression on NSCLC cells. J Cancer. 2016;7:1579–86.
Li S-H, Reinke AA, Sanders KL, Emal CD, Whisstock JC, Stuckey JA, et al. Mechanistic characterization and crystal structure of a small molecule inactivator bound to plasminogen activator inhibitor-1. Proc Natl Acad Sci USA. 2013;110:E4941–9.
Acknowledgements
The authors apologize to the scientists that are not cited because of space limitation. We would like to thank Brenda Guthrie Yoshinaga (Skybay Scientific Editing), Masaru Taniguchi (Riken Institute), Ryuichi Amakawa (Japan Baptist Hospital), David B. Eisner (Fellow of the American College of Emergency Physicians), Mary E. Saunders (Princess Margaret Cancer Centre), and Thierry J. Molina (Paris and Descartes University) for their critical reading of the manuscript. We thank Hiroshi Noguchi (Kitasato University) and Satoshi Omura (Kitasato University) for valuable comments. We are grateful to Tarek Alam (Bangladesh Medical College) for providing clinical information, and to Rufus Watson and Nobue Sakanishi for providing updated information on COVID-19.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Edited by G. Melino
Rights and permissions
About this article

Cite this article
Matsuyama, T., Kubli, S.P., Yoshinaga, S.K. et al. An aberrant STAT pathway is central to COVID-19. Cell Death Differ 27, 3209–3225 (2020). https://doi.org/10.1038/s41418-020-00633-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-020-00633-7
