General Aspects of Metal Ions as Signaling Agents in Health and Disease
by
 ,
,
Karolina Krzywoszyńska
1,*, 
Danuta Witkowska
1,*,
Jolanta Świątek-Kozłowska
1,
Agnieszka Szebesczyk
1 and
Henryk Kozłowski
1,2 1
Institute of Health Sciences, University of Opole, 68 Katowicka St., 45-060 Opole, Poland
2
Faculty of Chemistry, University of Wrocław, 14 F. Joliot-Curie St., 50-383 Wrocław, Poland
*
Authors to whom correspondence should be addressed.
Biomolecules 2020, 10(10), 1417; https://doi.org/10.3390/biom10101417
Submission received: 25 August 2020
/
Revised: 30 September 2020
/
Accepted: 2 October 2020
/
Published: 7 October 2020
(This article belongs to the Special Issue Toxic and Essential Metals in Human Health and Disease)
Abstract
:This review focuses on the current knowledge on the involvement of metal ions in signaling processes within the cell, in both physiological and pathological conditions. The first section is devoted to the recent discoveries on magnesium and calcium-dependent signal transduction—the most recognized signaling agents among metals. The following sections then describe signaling pathways where zinc, copper, and iron play a key role. There are many systems in which changes in intra- and extra-cellular zinc and copper concentrations have been linked to important downstream events, especially in nervous signal transduction. Iron signaling is mostly related with its homeostasis. However, it is also involved in a recently discovered type of programmed cell death, ferroptosis. The important differences in metal ion signaling, and its disease-leading alterations, are also discussed.
1. Introduction
Signal transduction and spreading is a key cellular process in maintaining life and its development. Typical chemical signaling comprises the release of a transmitter from one cell and its interaction with selected detectors on the surface of another. If the transmitter is taken up into the cell, it may bind to receptors on the inner part of cell membrane and, thus, stimulate it to react in a required manner to this signal. The molecular mechanisms of reaction to the signal are regulated by a strict spatiotemporal dynamic [1]. The signal effectiveness and transduction differ on the extra and intracellular side of the cell. In general, extracellular signaling depends on exposure to a sufficient carrier concentration. Simultaneously, the changes in the timing and frequency of a messenger are crucial for the intracellular signals [2]. Changes in the concentration of metal ions affect the signaling processes in both excitable and non-excitable cells on both sides of the cellular membrane [3,4,5,6].
The major groups of chemical neurotransmitters in excitable cells are amino acids, amines, or neuropeptides. Recent studies also indicate that metal ions, such as zinc and copper, may be released to the synaptic cleft [7,8,9]. This phenomenon is still not fully understood; however, it is a very interesting example of signaling involving metal ions. Many new studies and conclusions have appeared in this field recently and will be discussed in this review. Studies show that both the normal aging of the brain and the development of diseases, such as neurodegenerative and psychiatric disorders, are manifested by deregulation of the management of metal ions such as iron, zinc, and copper. That disproportion can have a direct impact on the neurotransmission coordinated by these ions and cellular processes necessary for proper functioning of nerve cells [10,11]. Furthermore, iron ions, along with reactive oxygen species (ROS), were connected with a recently discovered form of programmed cell death named ferroptosis [12]. While the pathological role of metal ions is still under investigation, it actually may arise from homeostasis disorders, which, in turn, may be related to disorders of metal ions sensing by different cells.
2. Magnesium
Magnesium (Mg) is an essential element that acts as a cofactor in many enzymes involved in the synthesis, folding, and stability of small and large biomolecules [13]. This most abundant free divalent cation in a cell, is one of the essential macronutrients in organism growth and development. Until now, more than 600 enzymatic reactions involving magnesium have been discovered [14]. About 99% of total body magnesium is distributed between different tissues in humans, amounting to approximately 25 g of magnesium in total for adults, with the largest proportion found in bones [15]; only 1–2% of the total magnesium is located in the blood. Mg2+ can act as an antagonist to reduce Ca2+ signaling in endothelium [16]. In spite of obvious chemical similarities between calcium and magnesium, major differences often prevail. For instance, the hydrated magnesium cation is hard to dehydrate, making it almost impossible for it to pass through narrow channels in biological membranes, which are no obstacles for calcium move [17]. Mg2+ binds water molecules more tightly than other cations, and for this reason the energy required for its transport is several times greater than that required for the transport of other cations [18]. Magnesium exhibits unique characteristics- the largest hydrated radius (0.428 nm) and the smallest ionic radius (0.072 nm) [19].
Mg2+ homeostasis is achieved through a balance of its uptake, intracellular storage, and efflux. Its deficiency can have destructive effects on the life of the cell [20]. Indeed, disorders of Mg2+ homeostasis are involved in neurodegenerative and cardiovascular diseases, bone disorders, asthma, cancer and diabetes [14,20,21,22]. It seems that magnesium deficiency can play an important role in the induction of inflammation in some of the mentioned pathologic conditions [21].
Its cellular homeostasis in vertebrates is regulated by the combined action of mitochondrial RNA splicing 2 (Mrs2), transient receptor potential melastatin 6/7(TRPM6/7), solute carrier family 41 (SLC41), membrane Mg2+ transporter 1 (MagT1), non-imprinted in Prader-Willi/Angelman syndrome protein (NIPA), membrane Mg2+ transporters (MMgTs), cyclin and cystathionine β-synthase domain magnesium transport mediators (CNNMs), and huntingtin-interacting protein 14 (HIP14) transporters [14,23,24]. The majority of proteins belonging to these families also transport other divalent cations across membranes (Figure 1B); only some of them (Figure 1A) are selective for Mg2+ ions [24]. Nonetheless, very recent data show that extracellular Mg2+ ions enter some tissues mainly through the TRPM7 channel and MagT1 transporter [16,25]. There are no evident uniform amino acid sequence similarities among the various magnesium transporters, even between MagT1 and NIPA2.
Mrs2 was the first mammalian magnesium transporter identified at the molecular level [26]. Using single channel patch clamping, Schindl et al. have shown that Mrs2 forms a Mg2+ selective channel of high conductance (155pS) [27]. Moreover, this channel is also permeable for Ni2+, with a lower conductance, and there was no permeability for Ca2+, Mn2+, and Co2+ ions [27].
MMgT1 and MMgT2 (membrane Mg2+ transporter 1 and 2) belong to a novel family of magnesium transporters with no known similarities to other transporters [24]. They were identified by differential gene expression using microarray analysis by Goytain and Quamme [28]. It was shown that MMgT1 and MMgT2 proteins reside in the Golgi and post-Golgi vesicles and both, as determined by two-electrode voltage-clamp analysis and fluorescence measurements, mediate magnesium ions [28].
Another protein responsible for magnesium homeostasis—magnesium transporter 1 (MagT1) is a plasma membrane Mg2+ transporter, highly-conserved across different eukaryotic species [29].
The full-length protein is composed of 367 amino acids with a large N-terminal segment, four transmembrane domains (TMs), and a small C-terminal tail (Figure 1A); it shows no structural similarity to any other magnesium transporters [30]. Transport of Mg2+ by this protein is rheogenic and voltage-dependent. It is highly selective for magnesium ions, as shown by fluorescence and voltage-clamp methods [24]. Recently, it has been demonstrated that humans lacking functional MagT1 have a selective deficiency in both immune and nonimmune glycoproteins [29]. Studies involving patients who had suffered from X-link immunodeficiency with magnesium defect and Epstein- Barr virus infection and neoplasia (XMEN) disease revealed that MagT1 serves as a kinetic regulator of signaling in lymphocyte T cells but not in B cells. It was suggested that Mg2+ influx may promote rapid spatial integration of antigen and costimulatory receptor signals critical for T cell activation. Moreover, MagT1 deficiency impairs T-cell receptor induced Mg2+ and Ca2+ influxes [31]. On the other hand, studies on mice with magT1 genetic deletion proved MagT1 is not required for lymphocyte homeostasis. MagT1-deficient cluster of differentiation 4+ (CD4+) proliferation was normal in multiple in vitro proliferation assays [32]. This discrepancy could arise due to differences in human and mouse physiology. It could be also that MagT1 has other functions that, when distorted, contribute to immunodeficiency [33].
The second highly Mg2+ selective transporter, non-imprinted in Prader-Willi/Angelman syndrome protein 2 (NIPA2) was identified using microarray analysis [34].
Other members of NIPA family belong to nonselective magnesium transporters. NIPA2 consists of 360 amino acids and has nine transmembrane protein domains (Figure 1A); it is located in many tissues, but particularly plentiful in renal cells [34]. It was suggested that NIPA2 mutations may contribute to childhood absence epilepsy, as mutant proteins were accumulated in the cytoplasm, which reduced intracellular Mg2+concentration in the neurons and affected neuronal excitability [35]. This hypothesis is supported by the results of a recent study, where the dysfunction of NIPA2 proved to reduce big potassium (BK) channel currents. Furthermore, it was shown that the decreased currents of BK channels enhanced neuronal excitability [36]. NIPA2 is also associated with type 2 diabetes [37]. This highly-selective magnesium ion transporter was shown to regulate osteoblast apoptosis by affecting the intracellular magnesium level and further affecting the osteogenic capacity of osteoblasts. These results suggest that NIPA2 could be a potential target for the treatment of type 2 diabetes osteoporosis [37].
CNNMs have been shown to be encoded by Acdp genes [24]. Some researchers suggest that CNNMs serve as direct transporters that extrude Mg2+ ions from the cell by exchanging it with Na+ ions [38,39]. On the other hand, there is an evidence suggesting that they can act either as intracellular Mg2+ sensors or as Mg2+ homeostatic mediators [40,41,42]. Nevertheless, several structural characteristics support their direct involvement in the Mg2+ extrusion [15].
Nowadays, TRPM7 and its homologue—TRPM6 seem to be the most investigated Mg2+ transporters. The significance of TRPM7 in cellular magnesium regulation has been analyzed in many cell types, including cardiomyocytes, osteoblasts, tumor cells and leukocytes, to name a few [43,44,45]. In vascular cells, that transporter occurs to be the central cation channel involved in controlling [Mg2+]. Montezano and coworkers have shown that magnesium prevents vascular calcification and osteogenic differentiation by restoring TRPM7 activity, counteracting calcium actions, and increasing expression of anticalcification proteins [46].
Both TRPM6 and TRPM7 proteins comprise 6 TMs and a channel pore, permeable also for Ca2+, Mn2+, Co2+ and Zn2+ ions [47], located between segments 5 and 6 (Figure 1B). In the plasma membrane, TRPM7 functions as a homodimer, although it can also heterodimerize with its analogue TRPM6. TRPM6 is mostly expressed in intestines and kidneys and TRPM7 is ubiquitously expressed [25,48]. The role of TRPM7 in Mg2+ homeostasis has been questioned by Jin et al., since the deletion of this transporter did not affect the maintenance of total cellular magnesium ions level [49]. Nonetheless, as stated above, Mg2+ is typically regulated by MagT1 in immune cells. In DT40 cells and colon carcinoma cells, the lack of TRPM7 was associated with increased expression of MagT1. This result implies that the discrepancies may come from different cell types studied [50,51].
Emerging evidence demonstrates a crucial role for TRPM6 and TRPM7 chanzymes (protein that shows fused channel and enzyme activities) in growth factor signaling through receptor tyrosine kinases (RTKs) [25]. RTKs, typically activated by growth factors, are membrane-associated receptors [52]. They induce phosphorylation and activation of intracellular non-receptor kinases which can trigger critical signaling pathways and cell functions, such as migration, contraction, proliferation, and differentiation [25].
Magnesium ions are important in the regulation of kinase activity. RTKs influence TRPM7, which in turn can influence tyrosine kinase signaling. Humans have around 60 known RTKs, which fall into 20 subfamilies [53]; however, the C-terminal kinase ofTRPM7 (alpha-kinase) displays little amino acid sequence similarity to other known kinases [45,54]. Recently, studies showing a significant role for TRPM7’s kinase in regulating proteasome-mediated turnover of the channel and controlling its cellular localization in polarized epithelial cells, have been conducted [55]. The phosphorylation of Ser-1360 has been shown to be critical for controlling protein stability and cellular localization of the channel [55]. The intrinsic kinase activity of TRPM7 constitutes a mechanism that allows the channel to respond rapidly to different cellular conditions and requirements.
Next magnesium transporter—HIP14 acts as a chanzyme as well. Goytain and Quamme demonstrated, by fluorescence and voltage-clamp techniques, that HIP14 mediates Mg2+ flux [30]. That work revealed that HIP14 trafficking from Golgi to post-Golgi vesicles increases when extracellular levels of magnesium ions are lowered [30]. Moreover, HIP14 contain 11 cysteine residues that might function as palmitoylation sites, what in turn can influence HIP14-mediated magnesium transport [24]. Palmitoylation (reversible, posttranslational covalent attachment of palmitic acid to cysteine residues) increases the hydrophobicity of proteins and thereby regulates their membrane association and subcellular localization [56]. It activates many cation transporters, such as Na+, K+, and Ca2+ channels [57,58]. Singaraja et al. revealed that altered palmitoylation of HIP14 substrates could contribute to the pathogenesis of Huntington disease (HD) [59]. Patients with HD often demonstrate abnormalities of iron homeostasis as well, what shows that huntingtin-HIP14 interactions are more complex.
Solute carrier family 41 member 1 (SLC41A1) protein was also shown to be differentially regulated by Mg2+ ions [60]. It is composed of 10 TMs (Figure 1B). SLC41A1 protein has been determined to transport, apart from magnesium ions, many other divalent cations. The transport of Mg2+by SLC41A1 had been previously shown to be rheogenic and voltage dependent, but not coupled to Na+ or Cl− ions [60]. More recent data suggest that SLC41A1 is an NME (Na+/Mg2+ exchanger) and the major cellular Mg2+ efflux system [61]. Under acute oxidative stress, SLC41A1 together with two other proteins might act as molecular mechanisms causing significant Mg2+ wasting. That situation can result in decreased cellular metabolism (mitochondrial dysfunction) and pro-apoptotic responses [20]. In 2017, an interesting study was carried out in humans regarding the efficacy of magnesium supplementation on the transcription of TRPM6/7 and SLC41A1 transporters [62]; the results showed a notable increase only in the expression of TRPM6. However, that study had several limitations, the most prominent of which being the fact that intracellular levels of magnesium were not measured [62].
A very interesting study on the effects of magnesium signaling on the structural and functional development of neuronal cells has been carried out by Yamanaka et al. [63]. It was shown that the activation of gamma-aminobutyric acid A (GABAA) receptors mediates the GABA-induced cytosolic [Mg]2+ increase in immature neurons independently of calcium signals. Moreover, cytosolic magnesium regulates the signaling activities of extracellular signal-regulated kinase (ERK) negatively, cyclic AMP response element binding protein (CREB) positively, and mammalian target of rapamycin (mTOR) sigmoidally [63]. As these intracellular signaling pathways regulate neuronal growth and differentiation, magnesium, by activating CREB and mTOR signaling, enhances the maturation of neural networks [64].
As a result of the contradictory observations regarding magnesium, its regulatory system and the roles of intracellular Mg2+ are controversial [64].
It is worth to mention that there are also unique prokaryotic Mg2+ transport systems with unusual mechanisms for mediating Mg2+ movement through the membrane [65]. However, they are beyond the scope of this review. For more information on magnesium transporters and signaling please see these excellent recent reviews [14,24,25,40].
3. Calcium
Calcium ions (Ca2+) are prevalent second messengers that regulate physiological cell functions in almost all living beings. The right level of calcium in the cytoplasm is maintained by calcium-permeable channels, transporters, and ATPases. Only 0.1% of calcium is actually present in extracellular fluid, where calcium exists in different fractions, such as protein-bound calcium (40%), free or ionized (48%), and complexed to other inorganic compounds (12%) [66]. When the cell is under resting conditions, the cytosolic free calcium concentration [Ca2+] is maintained at approximately 100 nM. More calcium is stored in some organelles, such as the Golgi apparatus and endoplasmic reticulum (ER), amounting to hundreds of microM [67]. A close relationship exists between the ER and endosomal system to initiate calcium signaling, or to store and buffer the Ca2+ after its release [68]. There are two pathways of increasing [Ca2+]i which coexist in cells: the release from intracellular stores, mainly ER, or the influx from the extracellular medium through the opening of calcium-permeable channels and Ca2+ transporters located in plasma membrane [69].
The release from internal stores goes through a variety of messengers such as inositol-1,4,5-trisphosphate (IP3), cyclic ADP-ribose (cADPR), nicotinic acid adenine dinucleotide phosphate (NAADP) and others [67,70]. In many electrically non-excitable cells, both processes are coupled in a process that is known as the store-operated calcium entry (SOCE). This mechanism has two important functions in the cell: cellular signaling and store refilling [71]. The influx of Ca2+ ions is also regulated by the TRPM chanzymes, by their effects on other channels such as ligand- or voltage-gated Ca2+ channels, through modulation of the membrane potential [72]. There is some evidence showing a relationship between TRPM channels, SOCE and the ER store content [71,73]. Two kinds of Ca2+ entry channels can be found in cells: voltage-gated calcium channels (VGCCs), which are dominant in excitable cells, and non-voltage-gated channels, which are dominant in non-excitable cells [73]. Recent advances in the structural biology of voltage-gated sodium and calcium channels are reviewed by Catterall and coworkers [74]. Very recently, an interesting study on the influence of Ca2+ concentration on voltage-dependent L-type calcium channels’ in fish was published [75]. In another newly published paper, the authors review the interplay between three potassium channels and calcium ions [76]. The calcium ions are pumped out of the cytoplasm by the membrane Ca2+-ATPase and the sarcoplasmic/endoplasmic reticulum Ca2+-ATPase [77,78]. Schematic representations of calcium-permeable channels, transporters, and ATPases involved in calcium signaling in excitable and non-excitable cells are shown in Figure 2A,B, respectively.
Calcium ions are involved in controlling cell proliferation, differentiation, secretion, maturation, mobility, and contraction [53,69,79]. Environmental stimuli mobilize intrinsic calcium stores, and selectively regulate its function alone or employing primary cell function [80].
Functional abnormalities in proteins that mediate Ca2+ transport and homeostasis usually lead to a wide range of diseases and pathogenic states, including cancer, heart failure, diabetes, and neurodegenerative disease [73]. Moreover, Ca2+ plays numerous roles in the immune system; calcium signaling is essential for T cell activation, tolerance of self-antigens, differentiation, and development [81]. The key regulators of T cells are the nuclear factor of activated T cells (NFAT) proteins [82]. It has been shown recently that suppression of Ca2+-NFAT signaling weakens T cell activation, clonal expansion, and infection clearance in fish [83].
There are dendritic cells (DC) in the mammalian immune system, also known as accessory cells, whose role is to capture and process antigens, then present antigens on the cell surface to the T cells. An increase in the [Ca2+]i acts as a signal that influences a broad range of dendritic cell (DC) functions [84]. The initiation and maintenance of these functions is induced by antigen receptor engagement. It has been shown that Ca2+ ions activate nonselective cation channel TRPM4 in DC cells. Authors conclude that TRPM4-regulated calcium homeostasis has been important for DC mobility, but not its maturation [74,85].
Ca2+ signaling pathways have a crucial role in signaling in excitable cells [86]. The role of Ca2+ in neuronal cells is composed of synaptic transmission and modulation of many signaling cascades upon activation of calcium dependent proteins (mainly kinases) [73]. Release of intracellular Ca2+ is linked to apoptosis (programmed cell death) through the intrinsic pathway primarily involving processes that converge on caspase-3 signaling [87].
Moreover, Ca2+ signaling has a central role in triggering apoptosis in many kinds of cells [88]. Prolonged accumulation of mitochondrial Ca2+ may lead to a phenomenon known as the mitochondrial permeability transition (MPT), that is regarded by some researchers as a mechanism of pathological cell death, and by others as the regulation of apoptosis [88,89].
Describing calcium signaling, the role of calmodulin (CaM) must be mentioned. This ubiquitous 148-residue protein was shown to be a major Ca2+ sensor in non-muscle cells, that responds to and regulates intracellular calcium levels [90]. Upon binding of Ca2+ its conformation is altered and enabled to bind with target peptides or proteins [91]. CaM senses the changes in intracellular Ca2+, and tunes the activity of numerous protein kinases, such as CaMKs. One of them, CaMKIV, is expressed in distinct brain regions that regulate learning and memory, emotion, and motor function [92].
The complex of calmodulin and one of the calmodulin-dependent protein kinases (CaM-KK) activates calcium stimulation of insulin gene transcription, showing that Ca2+ ions can affect insulin synthesis in pancreatic beta-cells [93]. Furthermore, frequency- and intensity-modulated fluctuation in the cytosolic and organelles free Ca2+ concentration are transduced into signals able to control multiple molecular systems and cellular functions by the actions of other regulatory Ca2+-binding proteins. Nonetheless, CaM seems to be the most important calcium sensor in eukaryotic cells [94]. The role of CaM-dependent systems involved in cell migration, tumor cell invasiveness, and metastasis development has been discussed recently by Villalobo and Berchtold [94].
The total calcium balance is supported by the work of the calcium sensing receptor (CaSR), that modifies parathyroid hormone secretion or renal cation handling. In the parathyroid gland, the CaSR is located on the cell surface of chief cells and is composed of seven transmembrane domains. Calcium ions bind to CaSR, which allows this protein to monitor and regulate the amount of calcium in the blood. Vitamin D and calcium, both acting through negative regulation in the parathyroids, have been recognized for many years as key modifiers of parathyroid hormone (PTH) gene transcription, hormone synthesis, and parathyroid cell proliferation [95].
As briefly shown in this section, calcium represents a crucial signal for almost every aspect of cellular life, and because of that it is involved in complex signaling networks. However, even small disorders in calcium homeostasis can trigger destructive processes that contribute to a number of pathogenic states [73]. For this reason, calcium signaling remains an important component of many studies. When researchers develop the ability to trace calcium dynamics, novel targets and treatments of chronic human diseases can be developed [68,73].
4. Zinc
Zinc as a divalent metal ion plays an important role in numerous processes in the cells. Zn2+ is a multitasking tool required for the activity of many enzymes, regulating transcriptional processes, cell growth, and differentiation, as well as the immunological response. Such versatility of biological zinc functions suggests a huge cellular demand for this ion. After Iron, Zinc is the second most abundant transition metal within the human body [96]. The concentration of this metal differs among organs and tissues, and the brain is the most enriched location of this metal ion in the body [97]. The concentration of Zn2+ in this organ reaches levels 10 times higher than that observed in serum—this highlights the significant role of zinc signaling in the central nervous system (CNS) [98].
In physiological conditions, the majority of Zn2+ is bound by proteins and small chelators in both excitable and non-excitable cells. The remaining pool of free zinc is strictly controlled; this part of it is found to be released by the cell as a zinc signal. The zinc spark is a very interesting example of exocytosis of zinc ions observed in early development of activated mammalian egg cells [99,100]. The oocytes require zinc for proper maturation [101]. After fertilization, the cell releases accumulated zinc ions as a spark, and this phenomenon can be described by a set of parameters like amplitude and integrated intensity [102]. Previous studies performed on mouse cells indicate that a high amplitude zinc spark may be a useful biomarker that helps to select high quality embryos prepared for in vitro fertilization (IVF) procedure [102]. The zinc sparks are triggered by intracellular calcium fluxes [100]. The effect of such potent zinc releasing is correlated with the blocking of polyspermy soon after fertilization on the way of ovastacin (a zinc metalloendopeptidase) activity [103].
In nerve cells, zinc ions are co-released to the synaptic cleft with glutamic acid from zincergic neurons or zinc-enriched (ZEN) terminals located mostly in mossy fibers of the hippocampus [104,105]. Mossy fibers work as a specialized unit, the main task of which is to convert the signals it receives into the code necessary for memory formation in CA3 region of the hippocampus [106].
The release of zinc and its interaction with the receptors of the postsynaptic membrane plays a significant role in long-term potentiation (LTP) and thus contributes to synaptic plasticity, which has been largely studied using animal models [104,107,108,109]. Simultaneously, studies in rats have shown that chelatable zinc ion homeostasis, and their proportion outside and inside neurons, is disturbed during acute behavioral stress. This may lead to an increase in the influx of zinc ions into the hippocampal cells, which under these conditions surprisingly reduces the ability of mossy fibers to generate LTP [110].
The exact amount of free zinc ions in human presynaptic terminals is still intensively discussed, and the imaging of the true distribution of unbound Zn2+ in the brain and nervous system is still a major challenge for scientists. Some studies confirm that zinc is a locally acting neurotransmitter that passes through the synaptic cleft, interacting with receptors on the postsynaptic membrane [111]. However, the release of zinc ions into the synaptic cleft [96], or its neuromodulatory character, is still under discussion [112,113]. Nevertheless, there is experimental confirmation that zinc ions can be successfully released into the synaptic cleft as a result of electrical stimulation, for example [114]. Additionally, in some cases, enhancement of Zn2+-dependent potentiation is observed on the postsynaptic side, and may be regulated by changes in the concentration of other cations like K+ [108].
There are many indications that proper zinc management in zincergic neurons is related to the level of expression of various proteins, of which metallothionein 3 (MT-3) plays a significant role [115]. It is a specific isoform of metallothionein that is expressed in a significantly increased amount in nerve cells enriched in Zn2+ [115]. Therefore, this protein is recognized as a provider of zinc ions to the site of the formation of synaptic vesicles. Furthermore, due to the lack of saturation with metal ions under physiological conditions, MT-3 may be an element of the mechanism protecting the cell against the dangerous increase in the concentration of free zinc ions [116]. The transport of Zn2+ ions into the vesicle is possibly due to the activity of the ZnT-3 transporter [117]. It is worth mentioning that the same protein is associated with cadmium-dependent toxicity within the hippocampus [118].
In the membrane of postsynaptic neurons, zinc ions can cause a number of excitatory or inhibitory reactions by interacting with different receptors, and the best-known targets are N-methyl-D-aspartate glutamate receptor (NMDAR) [107,119], alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (APMAR) [120,121], or voltage-gated Ca2+ channels [122,123]. The concentration of zinc ions in the postsynaptic nerve terminals is controlled by the ZnT-1 transporter activity; the expression of this transporter depends on the number of free zinc ions within the cell [124]. The ZnT-1 placement in the postsynaptic membrane closely correlates with the location of the NMDAR, and the transporter itself can interact with this receptor through the GluN2A subunit of the NMDAR [125]. A illustration of the sequence of events related to the zinc release by ZEN neurons is shown in Figure 3.
The influx of Zn2+ into the cell awakens the Ca2+ related processes, which is observed not only in neurons [126], but also in other cells [127].
The changes in concentration of free or loosely bound zinc with the aging of the organism cause disturbances in the functioning of various centers of the nervous system in old age [128]. The increased concentration of zinc ions in many areas of the brain is associated with the occurrence of serious diseases, such as neurodegenerative diseases, and the level of this metal concentration can be an indicator of disease progression [129]. Furthermore, the age-related changes in serum copper to zinc ratio may be used as an efficient biomarker of health disturbances, and the effect of the change on cellular processes in mostly non-excitable cells was very interestingly described in a review by Malavolta et al. [130].
Higher levels of free zinc have been detected in Parkinson’s disease (PD) [131] and Alzheimer’s disease (AD) patients [132] in the olfactory bulb. Moreover, intranasal zinc exposition causes the death of nasal cells by necrosis [133]. The specimens prepared from the autopsied brains of people with AD show a close correlation between the increased concentration of calcium and zinc ions and indicate that zinc ions may be involved in the dysfunction of neurons at the early stages of the disease development [129]. The dyshomeostasis and increase of unbound Zn2+ are dangerous for neurons and may lead to their damage and death [134]. One of the most supported mechanisms of neuronal death induced by free zinc excess was proposed to be a result of zinc accumulation within mitochondria [135]. It was suggested that this phenomenon leads to direct zinc-induced inhibition of cellular energy production, however, recent studies reveal that a more plausible mechanism relies on the dependence between calcium and zinc concentration [136,137]. Zinc ions directly participate only in the inhibition of mitochondrial movement, and calcium ions are responsible for mitochondrial damage [136].
Studies on the permeability of the blood-brain barrier (BBB) place the higher availability of zinc ions in a different perspective, and may suggest positive aspects of this phenomenon. The increased concentration of extracellular zinc can affect the capacity of the BBB. Zinc loosens the tight junction between endothelial cells of brain capillaries in it. This phenomenon facilitates the cleansing of the central nervous system of toxins and waste in pathological conditions such as ischemia, but it was also postulated as a part of normal brain function under physiological conditions [138].
The opposite phenomenon, zinc deficiency, can be equally harmful. Physiologically, the amount of zinc in the human brain decreases with age [139]. A decrease in the intracellular concentration of zinc ions is correlated with an increase in glutamate release, the activity of which induces apoptosis in neurons [140]. Furthermore, there are studies showing that zinc dietary deficiency leads to disturbances in cognitive functions [141]. The lowering of zinc concentration in the brain, mostly by poor dietary intake, is related to many pathological conditions, among which mental disorders are included [142]. Recent studies correlate depression with a low zinc level, and some of them treat this metal ion supplementation as a potential therapy for this serious mental condition [143].
5. Copper
Copper belongs to the transition metals group. This metal can exist in two different stable forms of ions in biological systems—Cu+ and Cu2+. This characteristic allows it to participate in redox reactions, which can have a negative effect if copper homeostasis is disturbed [144]. For this reason, it is required to keep the copper ions bound to proteins and to strictly control the concentration of free copper ions, both outside and inside the cell. Similarly to zinc, the highest concentration of copper has been detected in the brain [145]. Copper is very important for cell development and functions. This ion is involved in the defense strategy against free radicals by its presence in Cu/Zn superoxide dismutase (Cu/Zn-SOD). Disturbances in the folding of this protein are related to the development of serious dysfunction of motor neurons [146].
In the CNS, copper signaling may influence synaptic transmission indirectly by modulating the synthesis of neurotransmitters, e.g., via the peptidylglycine α-amidating monooxygenase (PAM) [147] and dopamine β-hydroxylase (DBH) [148] pathways, or directly by being released into the synaptic cleft from the terminals of glutamatergic neurons [149,150]. However, the signal associated with copper ions has a different effect depending on the region where it is released; in the amygdala, copper acts towards enhancement of LTP [151], while in the hippocampus it shows a tendency to inhibit synaptic plasticity [145]. Interestingly, the LTP inhibition induced by copper is located on the presynaptic side of the transmission [152].
The management of the intracellular pool of copper is supervised by a set of different proteins. Copper enters the cell, e.g., via the Cu transporter 1 (CTR-1) [153,154], and it is immediately captured in the cytosol by a group of chaperones that deliver these ions to their target sites. Such a rapid response to the intracellular increase in copper concentration is important due to its high redox reactivity. The chaperones transport this metal to the target location, such as mitochondria [155] or the area of secretory vesicle formation, whilst further copper release from the neuronal cell is possible through ATP7A, the major copper transporter in the brain [156]. After diffusion through the synaptic cleft, copper ions interact with NMDA and AMPA receptors and may also modulate the activity of γ-aminobutyric acid (GABA) and other amino acid receptors [157,158].
An example of the cellular pathway related to the copper release by neurons is shown in Figure 4.
The natural aging of the brain is associated with copper accumulation [139]. The increased concentration of copper ions in the body is considered as a potential cause of cognitive decline observed in elderly people [159]. Interestingly, the results obtained after analyzing the brains of people suffering from AD showed the opposite phenomenon. In comparison to healthy patients, the concentration of copper was lower in the brain areas most affected by the action of this disease—hippocampus and amygdala [160]. Therefore, it has been suggested that such a significant reduction in the availability of copper ions may be an important factor in the pathogenesis of this disease [161]. This copper deficiency correlates with a simultaneous reduction of B12 vitamin availability, which altogether facilitates neurodegeneration [162].
Research shows that the action of copper ions may have a protective effect on nerve cells. On the molecular level, modulating the activity of the NMDA receptor through copper ions significantly reduces the influx of calcium ions, and thus reduces the risk of cell damage [163].
Disturbances in the expression of proteins related to the relocation of copper ions, such as ATP7A, lead to neuronal damage, mainly due to the hyperactivity of NMDA receptors. Therefore copper treatment of these neurons can reduce and reverse this phenomenon [163]. Additionally, the up-regulation of copper availability may be controlled by the iron regulatory protein 2 (IRP2), which can participate in the pathological redistribution of copper in neurodegenerative conditions [164].
The influence of copper ions on neurotransmission depends on their concentration and time of exposure, which can have an impact on normal brain function, and diseases such as AD and dementia. Studies on neuronal cell cultures revealed that acute exposure to Cu2+ ions increased the synaptic activity of neurons by the AMPA receptor pathway, but the changes provided by this phenomenon were reversed after 24 h of exposure, suggesting high activity of copper homeostatic mechanisms [165].
Simultaneously, animal studies have shown that chronic exposure to low doses of dietary copper led to an increase in its presence within the brain. This phenomenon directly induced greater neuronal degeneration and death following DNA damage and activation of apoptosis [166].
An important aspect of nerve cell function is access to glucose and its metabolism. It was shown that with aging, the amount of glucose in the brain increases, and its metabolism slows down [167]. Along with the fact that diabetes negatively affects copper ion transport mechanisms [168], researchers suggest that the highest level of glucose and reduced copper availability in the brain can serve as an indicator of neurodegeneration [167]. Such a relationship between the level of glucose metabolism and copper ion homeostasis can also be used in the adaptation of diagnostic techniques for the early detection of these dangerous changes by connecting positron emission tomography–computed tomography (PET/CT) diagnosis with 64Cu flux detection [169].
Apart from neurodegenerative diseases, mental disorders may also be caused by abnormal access to copper ions. In the case of socially isolated animals, a decrease in the concentration of copper ions in the brain has been detected and correlated with a reduction in cognitive functions and the development of depression [170].
6. Iron
Iron is an essential metal, necessary for practically all living organisms, present in the form of ferric and ferrous ion, i.e., Fe3+ and Fe2+. The ability to form two oxidation states is responsible for iron’s role in redox reactions. Iron overload could be as dangerous as its deficiency. As there is no regulatory mechanism of the removal of iron excess from the body, its absorption has to provide coverage of the requirements for iron ions, while at the same time preventing iron overload. Therefore, perfect balance between iron absorption, distribution, accumulation, and excretion should be maintained. Iron sensing and signaling are mostly related with its homeostasis in the human body, in which several mechanisms are involved.
Dietary iron is absorbed in enterocytes in the small intestine, both as heme and non-heme iron. Heme-Fe is absorbed more efficiently than the non-heme one, likely due to the lack of essential interactions with dietary factors in the gastrointestinal tract [171]. Heme carrier protein (HCP1) was found to mediate the transport of heme-Fe to the enterocytes, but it was lately identified as folate transporter [172,173]. Nevertheless, the study of Le Blanc et al. showed, that HCP1 is involved in low-affinity heme-Fe transport [174]. Inside the enterocyte, ferrous ion from heme is released in the process mediated by heme oxygenase (HO) and enters the same pathway of utilization as non-heme iron [171]. Fe3+ must be reduced to Fe2+ before it can be transported by the divalent metal transporter 1 (DMT1) inside the enterocyte. Such reduction is made by duodenal cytochrome B (Dcytb), located in the apical membrane of enterocytes, other ferrireductases, or by non-enzymatic reductants such as ascorbate and/or superoxide, probably also amino acids, e.g., cysteine [175]. Inside the enterocyte, up to 4500 iron ions can be stored in the complex with ferritin, a spherical protein consisting of 24 subunits of light and heavy type subunits [176]. Heavy chains, responsible for the reuse of iron ions, present ferroxidase activity, and reduction to Fe2+ allows the mobilization of these ions, and their subsequent use or efflux via the basolateral membrane of the enterocyte.
The export of iron is provided by ferroportin-1 (FPN1, SLC40A1). FPN1 is associated with hephaestin, ferroxidase which oxidizes exported Fe2+ again to Fe3+, the Fe3+ is bound to transferrin (Tf) in the blood, and then transported to the cells [175]. Apo-Tf possess two iron-binding sites, and to be recognized by the transferrin receptor (TFRC, TFR1) both sites must be occupied. One TFRC is able to bind two holo-Tfs, and such complex is transported through the cell membrane and located in endosome. Inside the endosome the environment is acidified, Fe3+ is reduced to Fe2+ by STEAP3 protein, and then exported to the cytosol by DMT1. Occurring simultaneously with the iron ion reduction, the Tf-TFRC complex hydrolyzes, TFRC is recycled to the cell membrane, and apo-Tf is released outside the cell [177].
The regulation of proteins involved in iron metabolism takes place by the iron-sensing mechanism during post-transcriptional modification. Iron responsive elements (IREs) are hairpin structure fragments of mRNA located at 5′-untraslated region (5′-UTR) or 3′-UTR. One or two types of iron-responsive elements binding proteins (IRE-BP) can be bound to the IRE, namely iron regulatory protein 1 (IRP1) and IRP2. When iron is scarce, the IRE-BP binds to IRE with high affinity, which results in suppression of translation for mRNAs with IRE located on 5′-UTR (e.g., ferritin, both light and heavy chains, or FPN1). When IRE is located in the 3′-UTR, binding of IRE-BP enhances the mRNA stability, and encoded protein is synthesized (e.g., TFRC and DMT1). That allows the import of iron ions into the cell. When the desirable iron level is achieved, the labile iron pool (LIP) increases, and Fe2+ from LIP can bind to IRE-BP causing conformational changes, resulting in detachment of the Fe-IRE-BP complex from IRE. Translation of proteins encoded by mRNAs with IRE in 5′-UTR becomes possible (e.g., iron storage protein ferritin or exporting FPN1), while mRNA with IRE in 3′-UTR becomes sensitive for nuclease attack and undergoes degradation [175,176].
Besides the regulation of uptake by the cell, iron absorption needs to be controlled in the gastrointestinal tract. Iron-dependent regulation of this metal absorption from enterocytes is provided by the peptide hormone, hepcidin, which was shown to be the key regulator of iron homeostasis. This 27 kDa peptide is expressed in hepatocytes, in Kupffer cells, and also in small quantities in macrophages and adipocytes [178,179]. It is encoded by the hepcidin antimicrobial peptide (HAMP) gene, mutations of which are connected with severe iron overload diseases and hemochromatosis. Hepcidin is produced as pre-pro-peptide composed of 84 amino acids, which is processed to the 60 amino acid pro-hepcidin, and then to the 25 amino acid active form [179,180]. Hepcidin binds to FPN1 which triggers its degradation, resulting in iron sequestration inside cells (such as enterocytes, hepatocytes or macrophages). This leads to a decrease in the amount of iron available for erythropoiesis, what in turn causes reduction in the level of hepcidin. As a result, iron absorption in the gastrointestinal tract increases, along with ion release from iron stores [176].
The excretion of hepcidin is regulated by several factors, including iron-dependent proteins: human homeostatic iron regulator protein (HFE, hemochromatosis protein), Transferrin Receptor 2 (TFR2), Hemojuvelin (HJV), and Transmembrane Serine Protease 6 (matriptase-2, TMPRSS6). All of these proteins are expressed in the liver. Liver iron content and serum iron (bound to Tf) affect hepcidin expression in a different manner [181].
The main regulator of hepcidin expression is bone morphogenic protein (BMP)-SMAD signaling pathway. In general, BMP triggers the phosphorylation of SMAD1/5/8 proteins, which subsequently bind to SMAD4 and translocate to the nucleus, where the complex activates hepcidin expression via interaction with BMP responsive element (BMP-RE) in the HAMP gene (Figure 5) [182].
HFE interacts with TFRC, at the binding site for holo-Tf [183]. Therefore, when serum iron-Tf concentration is high, HFE detaches from TFRC and likely interacts with active receptor-like kinase 3 (ALK3), the BMP receptor. The formed complex prevents ALK3 degradation, increases its expression, which results in hepcidin expression [184].
The TFR2 protein also functions as an iron sensor. Under conditions of high iron concentration, it is stabilized by binding iron-Tf and is associated with HFE; however, it is unclear if TFR2 also interacts with ALK3. Tfr2 knockout mice exhibited decreased BMP-SMAD signaling and hepcidin expression. The effect is even more visible in double-knockout Hfe/Tfr2 mice [182,185].
HJV was found to be a co-receptor for BMP, as mutations in Hjv gene results in hemochromatosis, a disease manifested by iron overload. Interestingly, Hjv−/− mice exhibited both hepcidin expression and the BMP-SMAD signaling pathway, yet they were significantly attenuated. This results suggest that HJV may act not as a direct sensor, but as an enhancer of iron signaling in the hepcidin pathway [186].
TMPRSS6 protein cleaves HJV, therefore it acts as an inhibitor of HJV expression. The deficiency of TMPRSS6 causes unregulated BMP signaling, the hepcidin excess and anemia resulting from iron deficiency [187].
Studies on transgenic and wild type mice showed that higher amounts of iron in the diet result in higher hepatic iron levels, along with increased amounts of mRNA of enzymes responsible for ROS inactivation (Sod1 and Sod2) [188]. Moreover, hepcidin levels reach a plateau with a certain supply of iron. Further increases in the amount of iron in the diet results in the rise of iron concentration in the liver, what does not affect hepcidin regulation [188]. This result indicates that the knowledge of iron regulation via its concentration in the liver needs to be expanded.
The iron exporter, FPN1, is composed of 571 amino acids. Based on data from the model and the crystal structure of a putative bacterial homologue, it has 12 helical transmembrane domains, with the C- and N-terminus positioned intracellularly [189,190]. Mutations in the SLC40A1 gene lead to hemochromatosis type 4 (HC4), also called ferroportin disease [191,192].
Iron is essential for the development of all vital organs, including the brain. Iron deficiency in infancy results in the development of cognitive, motor, socio-emotional, and neurophysiological disorders [193]. Therefore, significant amounts of iron ions are absorbed in duodenal enterocytes, despite the fact that breast milk contains relatively little iron. During suckling more than 80% of iron is absorbed, while shortly after weaning the percentage decreases to 10–20%. The reason for such changes is not clear. It was shown that in the immature digestive system, the process of iron absorption is hypo-responsive to the inhibitory effect of hepcidin [194]. Nevertheless, ferroportin was shown to be pivotal for high iron absorption, as it was significantly decreased in ferroportin knockout mice.
Elevated iron levels were found in tumors, as they exhibit increased metabolism and rapid proliferation of the cells [195,196,197]. Moreover, increased iron dietary uptake and/or systemic iron levels correlate with an increased risk of developing certain types of cancer, including colorectal cancer (CRC) [198]. It was shown that iron demand is a key factor during CRC development, but its character varies in different CRC cell types [199]. Xue et al. presented results of investigations showing that elevated iron concentration in the cell leads to an activation of the signaling pathway involving Cyclin dependent kinase 1 (CDK1), Janus kinase 1 (JAK1) and Signal transducer and activator of transcription (STAT3), i.e. CDK1-JAK1-STAT3 signaling pathway [200]. This results in tumor cell proliferation, and may partially explain how high iron diet intake increases the risk of CRC development.
In 2012, a new form of non-apoptotic programmed cell death was identified. Morphological and biochemical characteristics of ferroptosis, induced by iron-dependent lipid peroxidation, include ROS accumulation (from iron metabolism), NADPH oxidase activity and lipid peroxidation products, cell volume shrinkage, and increased mitochondrial membrane density, with lack of typical apoptotic and necrotic features [12,201]. It was shown, that after the induction of ferroptosis by erastin, the cell death can be inhibited by iron chelators, such as deferoxamine, and antioxidant vitamin E, which provides evidence that ferroptosis depends on iron and ROS production [202]. The details of the dependence of ferroptosis on iron still need to be explained. In addition to iron chelators, ferroptosis can be inhibited by energy stress, while AMP-activated protein kinase (AMPK), the energy sensor in cell, is activated [203]. This promotes poly-unsaturated fatty acids (PUFAs) and other fatty acids biosynthesis, what results in ferroptosis inhibition.
Besides ferroptosis, iron overload and ROS generation can induce apoptotic cell death [204]. Recent results suggest that in myelodysplastic syndrome (MDS) patients, iron overload was related to the decrease of hypoxia inducible factor 1-a (HIF1-a). This process was shown to be iron concentration dependent as Fe2+ is a cofactor for prolyl hydroxylase domain 2, (PHD2), responsible for HIF1-a degradation. Triggering the HIF1-a/ROS signaling pathway led to mitochondrial-dependent apoptosis. On the other hand, apoptosis induced by H2O2 in colon-adenocarcinoma cell line (Caco-2) was abrogated by zinc or iron chelator, bathophenanthroline disulfonic acid (BPDS). Simultaneously with cell death induction, RNA binding activity of IRP1 increased. It resulted in increased DMT1 expression and iron uptake. Inhibition of this process by zinc means that zinc can act as an effective modulator of H2O2-induced iron signaling and cell death [205].
Additionally, superparamagnetic iron oxide nanoparticles (SPIONs), used as MRI contrast agents, were found to initiate autophagy processes in macrophages, in a manner suggesting that macrophages recognize SPIONs as microorganisms and their removal is carried out in a similar fashion [206]. Furthermore, SPIONs induced autophagy by increasing p62 mRNA expression, resulting from toll-like receptor 4 (TLR4) activation. Moreover, iron oxide nanoparticles (IONPs) have been found to reduce the expression of osteoclastogenesis-related genes. The way of action of IONPs is based on the inhibition of osteoclastogenesis through regulating the signaling complex involving the p62 protein (TRAF6-p62-CYLD) also [207].
7. Summary and Conclusions
Living cells simultaneously use various intracellular signaling systems to sense and interpret changes in their extracellular environment.
Many controversies still exist regarding the roles of magnesium in cell signaling. These controversies are caused mainly by the versatility and complexity of this metal ion. Studies show that it can be trophic or toxic, an activator or an inhibitor, and a cause of disease progression or regression. Magnesium has a crucial role in numerous cellular processes including enzymatic reactions, ion channel functions, metabolic cycles, and DNA/RNA stabilities. Disorders of Mg2+ homeostasis are involved in neurodegenerative and cardiovascular diseases, bone disorders, asthma, cancer, and diabetes. The hydrated magnesium cation is hard to dehydrate, which makes Mg2+ionsvery difficult to pass through narrow channels in biological membranes. Although a conserved magnesium-binding protein has not been reported for Mg2+ signaling pathways, phosphoryl transfer reactions in cells depend on this ion. Moreover, Mg2+ ions, because of their abundance and multivalence, are main players in neutralizing negatively charged biomolecules such as ROS or nucleic acids. Calcium cations should be better competitors for binding to negatively charged biomolecules, however, under normal and resting conditions the intracellular calcium ions are maintained at low levels. Calcium is a universal second messenger used to regulate versatile cellular processes ranging from contraction, through proliferation, secretion, fertilization, to learning and memory. Moreover, calcium signaling is essential for T cell activation, tolerance of self-antigens, differentiation, and development. Cells acquire signal Ca2+ ions from both internal and external sources. Calcium signals are detected and transmitted to downstream responses by a set of Ca2+ binding proteins that function as calcium sensors, with the prevailing role of the calmodulin protein. In normal conditions, free calcium concentration [Ca2+] is maintained at approximately 100 nM; much greater levels are stored in the ER and mitochondria. In electrically non-excitable cells, the Ca2+ ions are released from internal stores mainly in a process known as the store-operated calcium entry (SOCE). This mechanism has two important functions in the cell: cellular signaling and store refilling. Ca2+ flux through the excitable cell membrane is strictly limited in time due to biophysical properties of VGCCs and ligand-activated receptors. During calcium-mediated signal transduction these receptors and channels associate temporally and form the transient signaling complexes.
Zinc and transition metal ions, such as copper and iron, have been traditionally thought to be kinetically inert cofactors that are bound and buried within proteins and other molecules. However, recent data reveal that beyond their traditional functions as metabolic cofactors, they have also an important role in cell signaling. Copper, iron, and zinc are the three most abundant non-alkali metal ions in the brain, for this reason both deficiency and excess of these metal ions results in central nervous system disease.
Zinc might regulate the plasticity of synapses, however, how much zinc is released during synaptic activity remains highly elusive. Some research confirms that Zn2+ ions facilitate the transduction of a variety of signaling cascades in response to extracellular stimuli. Zinc pathways interact with calcium, redox, and magnesium signaling. The influx of Zn2+ into the cell triggers the Ca2+ related processes, what was observed not only in neurons but also in other cells. In physiological conditions, the majority of Zn2+ is bound by proteins and small chelators in both excitable and non-excitable cells. Zinc has an impact on the activity of many enzymes, transcriptional processes, cell growth and differentiation, and the immunological response. Very interesting events caused by zinc ions are called “zinc sparks”; functionally, the zinc sparks mediate a decrease in intracellular zinc content that is necessary for continued egg cell cycle progression. Recent studies reveal that this mechanism is dependent on intracellular calcium transients, which are tightly associated with embryonic development.
Copper ions may exist in two different forms in biological systems—Cu+ and Cu2+. Because of their potential toxicity to cause oxidative stress and free-radical damage, copper ions must be strictly controlled, e.g., through binding to specific proteins. The exchange of copper between a variety of target-specific cytosolic chaperones and their targets is driven presumably by an increase in the copper binding affinity. Copper containing enzymes and transcription factors are essential for cellular integrity, energy production, and proliferation. The intracellular level of copper is managed by a set of different proteins such as Cu-transporters and copper chaperones. Copper signaling may influence synaptic transmission indirectly by modulating the synthesis of neurotransmitters, or directly by being released into the synaptic cleft from the nerve terminals of glutamatergic neurons.
Iron is present in the living cells in the form of ferric and ferrous ion, i.e., Fe3+ and Fe2+. There is no regulatory mechanism for the removal of excess iron from the body. Therefore, the perfect balance between iron absorption, distribution, accumulation, and excretion should be maintained. Regulation of the proteins involved in iron metabolism is a form of iron-sensing mechanism during post-transcriptional modification. Elevated iron levels were found in tumors, as they exhibit increased metabolism and rapid proliferation of the cells. Iron ions, like copper ions, take part in the redox signaling process. The distinction between signaling and toxic redox processes is not always obvious. There is growing evidence that iron excess is a major risk for carcinogenesis, suggesting the importance of ferroptosis-resistance. Ferroptosis is a relatively recently described type of non-apoptotic cell death, induced by iron-dependent lipid peroxidation, that remains to be fully characterized.
There are suggestions that copper and iron redox mechanisms are interesting therapeutic targets for treating some diseases such as cancer and chronic lung inflammation.
Table 1 summarizes transporters and receptors engaged in the signaling of the metal ions characterized in this review, as well as their downstream signaling events.
This review shows the role of chosen metal ions in signaling processes. As we can see, variable thermodynamics and kinetics of metal actions blur the lines between metabolism and signaling, placing metals in a unique chemical and biological space.
Nonetheless, deficiency, as well as excess, of any essential metal ion, can lead to many disorders. Not only redox-inactive alkali metals, but also some transition metal ions, have important roles in signaling and other biological processes. Recent studies focus on the disruption of transition and non-transition metal ions homeostasis by such factors as the disturbances of their bioavailability, and changes in the chemical status of essential cations. This knowledge can help to prevent many disorders, and to design proper medication, such as inhibitors of the destructive signaling pathways, where these metal ions play an important role.
Many conclusions on the distribution of metal ions in excitable cells are based on conventional histochemical studies of nerve tissues. This technique is used primarily to show sites of only relatively high metal accumulation. Hence, the main far-reaching goal of novel research seems to be the improvement and development of techniques able to detect the chosen chemical form, concentration, and location of metal ions, especially zinc, iron, and copper, e.g., in the living brain. As described in this review, the proper cellular management of the copper and zinc levels is relevant for signal transduction and viability of neuronal cells. Additionally, the potent regulation of copper and zinc signals in excitable cells strongly influences calcium concentration.
Author Contributions
Literature search and review, writing—original draft preparation, K.K., D.W., A.S.; writing—review and editing, K.K., D.W., H.K.; participation in revision, J.Ś.-K. All authors have read and agreed to the published version of the manuscript.
Funding
This publication was supported by the University of Opole within the framework of its programs for the research grants of young scientists, WPBIN 3/19 to K.K. Research in our laboratories is also funded by the Polish National Science Centre (UMO-2018/31/D/ST4/02574 (to A.S.) and 2017/26/A/ST5/00363 (to H.K.)).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ross, B.; Mehtal, S.; Zhang, J. Molecular tools for acute spatiotemporal manipulation of signal transduction. Curr. Opin. Chem. Biol. 2016, 34, 135–142. [Google Scholar] [CrossRef] [Green Version]
- Lqbal, J.; Zaidi, M.; Avadhani, N.G. Cell signaling. Mol. Integr. Physiol. Musculoskelet. Syst. 2010, 1211, 3–8. [Google Scholar] [CrossRef]
- Penner, R.; Neher, E. The role of calcium in stimulus-secretion coupling in excitable and non-excitable cells. J. Exp. Biol. 1988, 139, 329–345. [Google Scholar] [PubMed]
- Hojyo, S.; Fukada, T. Roles of Zinc Signaling in the Immune System. J. Immunol. Res. 2016, 2016, 6762343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamano, H.; Koike, Y.; Nakada, H.; Shakushi, Y.; Takeda, A. Significance of synaptic Zn2+ signaling in zincergic and non-zincergic synapses in the hippocampus in cognition. J. Trace Elem. Med. Biol. 2016, 38, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Kardos, J.; Heja, L.; Simon, A.; Jablonkai, I.; Kovacs, R.; Jemnitz, K. Copper signalling: Causes and consequences. Cell Commun. Signal. 2018, 16, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, C.; Weth, A.; Walcher, S.; Lax, C.; Baumgartner, W. Modeling of Zinc Dynamics in the Synaptic Cleft: Implications for Cadherin Mediated Adhesion and Synaptic Plasticity. Front. Mol. Neurosci. 2018, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamano, H.; Morioka, H.; Nishio, R.; Takeuchi, A.; Takeda, A. Blockade of Rapid Influx of Extracellular Zn2+ into Nigral Dopaminergic Neurons Overcomes Paraquat-Induced Parkinson’s Disease in Rats. Mol. Neurobiol. 2019, 56, 4539–4548. [Google Scholar] [CrossRef]
- D’Ambrosi, N.; Rossi, L. Copper at synapse: Release, binding and modulation of neurotransmission. Neurochem. Int. 2015, 90, 36–45. [Google Scholar] [CrossRef]
- Ashraf, A.; Michaelides, C.; Walker, T.A.; Ekonomou, A.; Suessmilch, M.; Sriskanthanathan, A.; Abraha, S.; Parkes, A.; Parkes, H.G.; Geraki, K.; et al. Regional Distributions of Iron, Copper and Zinc and Their Relationships With Glia in a Normal Aging Mouse Model. Front. Aging Neurosci. 2019, 11. [Google Scholar] [CrossRef] [Green Version]
- Pal, A.; Prasad, R. Regional Distribution of Copper, Zinc and Iron in Brain of Wistar Rat Model for Non-Wilsonian Brain Copper Toxicosis. Indian J. Clin. Biochem. 2016, 31, 93–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixon, S.; Lemberg, K.; Lamprecht, M.; Skouta, R.; Zaitsev, E.; Gleason, C.; Patel, D.; Bauer, A.; Cantley, A.; Yang, W.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maguire, M.E.; Cowan, J.A. Magnesium chemistry and biochemistry. Biometals 2002, 15, 203–210. [Google Scholar] [CrossRef] [PubMed]
- de Baaij, J.H.F.; Hoenderop, J.G.J.; Bindels, R.J.M. Magnesium in man: Implications for health and disease. Physiol. Rev. 2015, 95, 1–46. [Google Scholar] [CrossRef]
- Gimenez-Mascarell, P.; Gonzalez-Recio, I.; Fernandez-Rodriguez, C.; Oyenarte, I.; Mueller, D.; Luz Martinez-Chantar, M.; Alfonso Martinez-Cruz, L. Current Structural Knowledge on the CNNM Family of Magnesium Transport Mediators. Int. J. Mol. Sci. 2019, 20, 1135. [Google Scholar] [CrossRef] [Green Version]
- Zhu, D.; You, J.; Zhao, N.; Xu, H. Magnesium Regulates Endothelial Barrier Functions through TRPM7, MagT1, and S1P1. Adv. Sci. 2019, 6. [Google Scholar] [CrossRef] [Green Version]
- Jahnen-Dechent, W.; Ketteler, M. Magnesium basics. Clin. Kidney J. 2012, 5, i3–i14. [Google Scholar] [CrossRef] [Green Version]
- Yatsimirsky, K.B. ELECTRONIC-STRUCTURE, HYDRATION ENERGY AND STABILITY OF METAL AQUAIONS. Teor. I Eksperimentalnaya Khimiya 1994, 30, 1–11. [Google Scholar] [CrossRef]
- Binding, Transport and Storage of Metal Ions in Biological Cells; Royal Society of Chemistry: London, UK, 2014; Volume 2, pp. 1–911. [CrossRef] [Green Version]
- Kolisek, M.; Montezano, A.C.; Sponder, G.; Anagnostopoulou, A.; Vormann, J.; Touyz, R.M.; Aschenbach, J.R. PARK7/DJ-1 dysregulation by oxidative stress leads to magnesium deficiency: Implications in degenerative and chronic diseases. Clin. Sci. 2015, 129, 1143–1150. [Google Scholar] [CrossRef]
- Shahi, A.; Aslani, S.; Ataollahi, M.; Mahmoudi, M. The role of magnesium in different inflammatory diseases. Inflammopharmacology 2019, 27, 649–661. [Google Scholar] [CrossRef]
- Rude, R.K.; Gruber, H.E.; Norton, H.J.; Wei, L.Y.; Frausto, A.; Kilburn, J. Reduction of dietary magnesium by only 50% in the rat disrupts bone and mineral metabolism. Osteoporos. Int. 2006, 17, 1022–1032. [Google Scholar] [CrossRef] [PubMed]
- Romani, A. Regulation of magnesium homeostasis and transport in mammalian cells. Arch. Biochem. Biophys. 2007, 458, 90–102. [Google Scholar] [CrossRef] [PubMed]
- Quamme, G.A. Molecular identification of ancient and modern mammalian magnesium transporters. Am. J. Physiol. Cell Physiol. 2010, 298, C407–C429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, Z.-G.; Rios, F.J.; Montezano, A.C.; Touyz, R.M. TRPM7, Magnesium, and Signaling. Int. J. Mol. Sci. 2019, 20, 1877. [Google Scholar] [CrossRef] [Green Version]
- Kolisek, M.; Zsurka, G.; Samaj, J.; Weghuber, J.; Schweyen, R.J.; Schweigel, M. Mrs2p is an essential component of the major electrophoretic Mg2+ influx system in mitochondria. Embo J. 2003, 22, 1235–1244. [Google Scholar] [CrossRef]
- Schindl, R.; Weghuber, J.; Romanin, C.; Schweyen, R.J. Mrs2p forms a high conductance Mg2+ selective channel in mitochondria. Biophys. J. 2007, 93, 3872–3883. [Google Scholar] [CrossRef] [Green Version]
- Goytain, A.; Quamme, G.A. Identification and characterization of a novel family of membrane magnesium transporters, MMgT1 and MMgT2. Am. J. Physiol. Cell Physiol. 2008, 294, C495–C502. [Google Scholar] [CrossRef] [Green Version]
- Matsuda-Lennikov, M.; Biancalana, M.; Zou, J.; Ravell, J.C.; Zheng, L.; Kanellopoulou, C.; Jiang, P.; Notarangelo, G.; Jing, H.; Masutani, E.; et al. Magnesium transporter 1 (MAGT1) deficiency causes selective defects in N-linked glycosylation and expression of immune-response genes. J. Biol. Chem. 2019, 294, 13638–13656. [Google Scholar] [CrossRef]
- Goytain, A.; Quamme, G.A. Identification and characterization of a novel mammalian Mg2+ transporter with channel-like properties. BMC Genom. 2005, 6. [Google Scholar] [CrossRef] [Green Version]
- Li, F.-Y.; Lenardo, M.J.; Chaigne-Delalande, B. Loss of MAGT1 abrogates the Mg2+ flux required for T cell signaling and leads to a novel human primary immunodeficiency. Magnes. Res. 2011, 24, S109–S114. [Google Scholar] [CrossRef]
- Holmes, D.; Carroll, K.; Brodeur, S.; Pashine, A. Characterizing the intracellular magnesium transporter MagT1 in murine lymphocyte function. J. Immunol. 2016, 196. [Google Scholar]
- Wu, N.; Veillette, A. IMMUNOLOGY Magnesium in a signalling role. Nature 2011, 475, 462–463. [Google Scholar] [CrossRef] [PubMed]
- Goytain, A.; Hines, R.M.; Quamme, G.A. Functional characterization of NIPA2, a selective Mg2+ transporter. Am. J. Physiol. Cell Physiol. 2008, 295, C944–C953. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Zhang, Y.; Zhang, P.; Wang, J.; Wu, Y.; Wu, X.; Netoff, T.; Jiang, Y. Functional Study of NIPA2 Mutations Identified from the Patients with Childhood Absence Epilepsy. PLoS ONE 2014, 9. [Google Scholar] [CrossRef]
- Liu, N.-N.; Xie, H.; Xiang-wei, W.-S.; Gao, K.; Wang, T.-S.; Jiang, Y.-W. The absence of NIPA2 enhances neural excitability through BK (big potassium) channels. CNS Neurosci. Ther. 2019, 25, 865–875. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.; Zhang, W.-L.; Yang, B.; Sun, J.; Yang, M.-W. NIPA2 regulates osteoblast function via its effect on apoptosis pathways in type 2 diabetes osteoporosis. Biochem. Biophys. Res. Commun. 2019, 513, 883–890. [Google Scholar] [CrossRef]
- Funato, Y.; Yamazaki, D.; Mizukami, S.; Du, L.; Kikuchi, K.; Miki, H. Membrane protein CNNM4-dependent Mg2+ efflux suppresses tumor progression. J. Clin. Investig. 2014, 124, 5398–5410. [Google Scholar] [CrossRef] [Green Version]
- Funato, Y.; Furutani, K.; Kurachi, Y.; Miki, H. CrossTalk proposal: CNNM proteins are Na+/Mg2+ exchangers playing a central role in transepithelial Mg2+ (re)absorption. J. Physiol. Lond. 2018, 596, 743–746. [Google Scholar] [CrossRef] [Green Version]
- Kolisek, M.; Sponder, G.; Pilchova, I.; Cibulka, M.; Tatarkova, Z.; Werner, T.; Racay, P. Magnesium Extravaganza: A Critical Compendium of Current Research into Cellular Mg2+ Transporters Other than TRPM6/7. Rev. Physiol. Biochem. Pharmacol. 2019, 176, 65–105. [Google Scholar] [CrossRef]
- Sponder, G.; Mastrototaro, L.; Kurth, K.; Merolle, L.; Zhang, Z.; Abdulhanan, N.; Smorodchenko, A.; Wolf, K.; Fleig, A.; Penner, R.; et al. Human CNNM2 is not a Mg2+ transporter per se. Pflug. Arch. Eur. J. Physiol. 2016, 468, 1223–1240. [Google Scholar] [CrossRef]
- Hardy, S.; Uetani, N.; Wong, N.; Kostantin, E.; Labbe, D.P.; Begin, L.R.; Mes-Masson, A.; Miranda-Saavedra, D.; Tremblay, M.L. The protein tyrosine phosphatase PRL-2 interacts with the magnesium transporter CNNM3 to promote oncogenesis. Oncogene 2015, 34, 986–995. [Google Scholar] [CrossRef] [PubMed]
- Nadler, M.J.S.; Hermosura, M.C.; Inabe, K.; Perraud, A.L.; Zhu, Q.Q.; Stokes, A.J.; Kurosaki, T.; Kinet, J.P.; Penner, R.; Scharenberg, A.M.; et al. LTRPC7 is a Mg center dot ATP-regulated divalent cation channel required for cell viability (vol 411, pg 590, 2001). Nature 2001, 412, 660. [Google Scholar] [CrossRef]
- Yu, Y.; Chen, S.R.; Xiao, C.Y.; Jia, Y.Y.; Guo, J.L.; Jiang, J.M.; Liu, P.Q. TRPM7 is involved in angiotensin II induced cardiac fibrosis development by mediating calcium and magnesium influx. Cell Calcium 2014, 55, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Stritt, S.; Nurden, P.; Favier, R.; Favier, M.; Ferioli, S.; Gotru, S.K.; van Eeuwijk, J.M.M.; Schulze, H.; Nurden, A.T.; Lambert, M.P.; et al. Defects in TRPM7 channel function deregulate thrombopoiesis through altered cellular Mg2+ homeostasis and cytoskeletal architecture. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef]
- Montezano, A.C.; Zimmerman, D.; Yusuf, H.; Burger, D.; Chignalia, A.Z.; Wadhera, V.; van Leeuwen, F.N.; Touyz, R.M. Vascular Smooth Muscle Cell Differentiation to an Osteogenic Phenotype Involves TRPM7 Modulation by Magnesium. Hypertension 2010, 56, 453–462. [Google Scholar] [CrossRef] [Green Version]
- Monteilh-Zoller, M.K.; Hermosura, M.C.; Nadler, M.J.S.; Scharenberg, A.M.; Penner, R.; Fleig, A. TRPM7 provides an ion channel mechanism for cellular entry of trace metal ions. J. Gen. Physiol. 2003, 121, 49–60. [Google Scholar] [CrossRef] [Green Version]
- Luongo, F.; Pietropaolo, G.; Gautier, M.; Dhennin-Duthille, I.; Ouadid-Ahidouch, H.; Wolf, F.I.; Trapani, V. TRPM6 is Essential for Magnesium Uptake and Epithelial Cell Function in the Colon. Nutrients 2018, 10, 784. [Google Scholar] [CrossRef] [Green Version]
- Jin, J.; Desai, B.N.; Navarro, B.; Donovan, A.; Andrews, N.C.; Clapham, D.E. Deletion of Trpm7 Disrupts Embryonic Development and Thymopoiesis Without Altering Mg(2+) Homeostasis. Science 2008, 322, 756–760. [Google Scholar] [CrossRef] [Green Version]
- Cazzaniga, A.; Moscheni, C.; Trapani, V.; Wolf, F.I.; Farruggia, G.; Sargenti, A.; Iotti, S.; Maier, J.A.M.; Castiglioni, S. The different expression of TRPM7 and MagT1 impacts on the proliferation of colon carcinoma cells sensitive or resistant to doxorubicin. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [Green Version]
- Deason-Towne, F.; Perraud, A.-L.; Schmitz, C. The Mg2+ transporter MagT1 partially rescues cell growth and Mg2+ uptake in cells lacking the channel-kinase TRPM7. FEBS Lett. 2011, 585, 2275–2278. [Google Scholar] [CrossRef] [Green Version]
- Butti, R.; Das, S.; Gunasekaran, V.P.; Yadav, A.S.; Kumar, D.; Kundu, G.C. Receptor tyrosine kinases (RTKs) in breast cancer: Signaling, therapeutic implications and challenges. Mol. Cancer 2018, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemmon, M.A.; Schlessinger, J. Cell Signaling by Receptor Tyrosine Kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Middelbeek, J.; Clark, K.; Venselaar, H.; Huynen, M.A.; van Leeuwen, F.N. The alpha-kinase family: An exceptional branch on the protein kinase tree. Cell. Mol. Life Sci. 2010, 67, 875–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, N.; Lou, L.; Al-Saadi, N.; Tetteh, S.; Runnels, L.W. The kinase activity of the channel-kinase protein TRPM7 regulates stability and localization of the TRPM7 channel in polarized epithelial cells. J. Biol. Chem. 2018, 293, 11491–11504. [Google Scholar] [CrossRef] [Green Version]
- Huang, K.; El-Husseini, A. Modulation of neuronal protein trafficking and function by palmitoylation. Curr. Opin. Neurobiol. 2005, 15, 527–535. [Google Scholar] [CrossRef]
- Mies, F.; Spriet, C.; Heliot, L.; Sariban-Sohraby, S. Epithelial Na+ channel stimulation by n-3 fatty acids requires proximity to a membrane-bound A-kinase-anchoring protein complexed with protein kinase A and phosphodiesterase. J. Biol. Chem. 2007, 282, 18339–18347. [Google Scholar] [CrossRef] [Green Version]
- Qin, N.; Platano, D.; Olcese, R.; Costantin, J.L.; Stefani, E.; Birnbaumer, L. Unique regulatory properties of the type 2a Ca2+ channel beta subunit caused by palmitoylation. Proc. Natl. Acad. Sci. USA 1998, 95, 4690–4695. [Google Scholar] [CrossRef] [Green Version]
- Singaraja, R.R.; Huang, K.; Sanders, S.S.; Milnerwood, A.J.; Hines, R.; Lerch, J.P.; Franciosi, S.; Drisdel, R.C.; Vaid, K.; Young, F.B.; et al. Altered palmitoylation and neuropathological deficits in mice lacking HIP14. Hum. Mol. Genet. 2011, 20, 3899–3909. [Google Scholar] [CrossRef] [Green Version]
- Goytain, A.; Quamme, G.A. Functional characterization of human SLC41A1, a Mg2+ transporter with similarity to prokaryotic MgtE Mg2+ transporters. Physiol. Genom. 2005, 21, 337–342. [Google Scholar] [CrossRef]
- Kolisek, M.; Nestler, A.; Vormann, J.; Schweigel-Roentgen, M. Human gene SLC41A1 encodes for the Na+/Mg2+ exchanger. Am. J. Physiol. Cell Physiol. 2012, 302, C318–C326. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Ramirez, M.; Rodriguez-Moran, M.; Reyes-Romero, M.A.; Guerrero-Romero, F. Effect of oral magnesium supplementation on the transcription of TRPM6, TRPM7, and SLC41A1 in individuals newly diagnosed of pre-hypertension. A randomized, double-blind, placebo-controlled trial. Magnes. Res. 2017, 30, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, R.; Shindo, Y.; Hotta, K.; Suzuki, K.; Oka, K. GABA-Induced Intracellular Mg2+ Mobilization Integrates and Coordinates Cellular Information Processing for the Maturation of Neural Networks. Curr. Biol. 2018, 28, 3984–3991.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamanaka, R.; Shindo, Y.; Oka, K. Magnesium Is a Key Player in Neuronal Maturation and Neuropathology. Int. J. Mol. Sci. 2019, 20, 3439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moncrief, M.B.C.; Maguire, M.E. Magnesium transport in prokaryotes. J. Biol. Inorg. Chem. 1999, 4, 523–527. [Google Scholar] [CrossRef]
- Moore, E.W. Ionized calcium in normal serum, ultrafiltrates, and whole blood determined by ion-exchange electrodes. J. Clin. Investig. 1970, 49, 318–334. [Google Scholar] [CrossRef]
- Berridge, M.J. Calcium microdomains: Organization and function. Cell Calcium 2006, 40, 405–412. [Google Scholar] [CrossRef]
- Rimessi, A.; Pedriali, G.; Vezzani, B.; Tarocco, A.; Marchi, S.; Wieckowski, M.R.; Giorgi, C.; Pinton, P. Interorganellar calcium signaling in the regulation of cell metabolism: A cancer perspective. Semin. Cell Dev. Biol. 2020, 98, 167–180. [Google Scholar] [CrossRef]
- Munaron, L. Calcium signalling and control of cell proliferation by tyrosine kinase receptors (review). Int. J. Mol. Med. 2002, 10, 671–676. [Google Scholar] [CrossRef]
- Krebs, J.; Agellon, L.B.; Michalak, M. Ca2+ homeostasis and endoplasmic reticulum (ER) stress: An integrated view of calcium signaling. Biochem. Biophys. Res. Commun. 2015, 460, 114–121. [Google Scholar] [CrossRef]
- Faouzi, M.; Kilch, T.; Horgen, F.D.; Fleig, A.; Penner, R. The TRPM7 channel kinase regulates store-operated calcium entry. J. Physiol. Lond. 2017, 595, 3165–3180. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Fliegert, R.; Guse, A.H.; Lu, W.; Du, J. A structural overview of the ion channels of the TRPM family. Cell Calcium 2020, 85. [Google Scholar] [CrossRef] [PubMed]
- Giorgi, C.; Danese, A.; Missiroli, S.; Patergnani, S.; Pinton, P. Calcium Dynamics as a Machine for Decoding Signals. Trends Cell Biol. 2018, 28, 258–273. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A.; Lenaeus, M.J.; Gamal El-Din, T.M. Structure and Pharmacology of Voltage-Gated Sodium and Calcium Channels. Annu. Rev. Pharmacol. Toxicol. 2020, 60, 133–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Q.; Chu, P.; Gu, J.; Zhang, H.; Feng, R.; Wen, X.; Wang, D.; Xiong, W.; Wang, T.; Yin, S. The influence of Ca2+ concentration on voltage-dependent L-type calcium channels’ expression in the marbled eel (Anguilla marmorata). Gene 2020, 722. [Google Scholar] [CrossRef]
- Brown, B.M.; Shim, H.; Christophersen, P.; Wulff, H. Pharmacology of Small- and Intermediate-Conductance Calcium-Activated Potassium Channels. Annu. Rev. Pharmacol. Toxicol. 2020, 60, 219–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choong, G.; Liu, Y.; Templeton, D.M. Interplay of calcium and cadmium in mediating cadmium toxicity. Chem. Biol. Interact. 2014, 211, 54–65. [Google Scholar] [CrossRef]
- Guerini, D. The Ca2+ pumps and the Na+/Ca2+ exchangers. Biometals 1998, 11, 319–330. [Google Scholar] [CrossRef]
- Mateos-Aparicio, P.; Rodriguez-Moreno, A. Calcium Dynamics and Synaptic Plasticity. Adv. Exp. Med. Biol. 2020, 1131, 965–984. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.X.; Ma, J.; Parrington, J.; Calcraft, P.J.; Galione, A.; Evans, A.M. Calcium signaling via two-pore channels: Local or global, that is the question. Am. J. Physiol. Cell Physiol. 2010, 298, C430–C441. [Google Scholar] [CrossRef] [Green Version]
- Fracchia, K.M.; Pai, C.Y.; Walsh, C.M. Modulation of T cell metabolism and function through calcium signaling. Front. Immunol. 2013, 4. [Google Scholar] [CrossRef] [Green Version]
- Macian, F. NFAT proteins: Key regulators of T-cell development and function. Nat. Rev. Immunol. 2005, 5, 472–484. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.M.; Li, H.Y.; Zhang, Y.; Li, C.; Li, K.; Ai, K.T.; Yang, J.L. Ca2+-Calcineurin Axis-Controlled NFAT Nuclear Translocation Is Crucial for Optimal T Cell Immunity in an Early Vertebrate. J. Immunol. 2020, 204, 569–585. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.S. Calcium signaling mechanisms in T lymphocytes. Annu. Rev. Immunol. 2001, 19, 497–521. [Google Scholar] [CrossRef] [PubMed]
- Barbet, G.; Demion, M.; Moura, I.C.; Serafini, N.; Leger, T.; Vrtovsnik, F.; Monteiro, R.C.; Guinamard, R.; Kinet, J.-P.; Launay, P. The calcium-activated nonselective cation channel TRPM4 is essential for the migration but not the maturation of dendritic cells. Nat. Immunol. 2008, 9, 1148–1156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santulli, G.; Marks, A.R. Essential Roles of Intracellular Calcium Release Channels in Muscle, Brain, Metabolism, and Aging. Curr. Mol. Pharmacol. 2015, 8, 206–222. [Google Scholar] [CrossRef] [Green Version]
- Olofsson, M.H.; Havelka, A.M.; Brnjic, S.; Shoshan, M.C.; Linder, S. Charting calcium-regulated apoptosis pathways using chemical biology: Role of calmodulin kinase II. Bmc Chem. Biol. 2008, 8, 2. [Google Scholar] [CrossRef] [Green Version]
- Danese, A.; Patergnani, S.; Bonora, M.; Wieckowski, M.R.; Previati, M.; Giorgi, C.; Pinton, P. Calcium regulates cell death in cancer: Roles of the mitochondria and mitochondria-associated membranes (MAMs). Biochim. Biophys. Acta Bioenergy 2017, 1858, 615–627. [Google Scholar] [CrossRef]
- Li, Y.J.; Wang, C.; Lian, Y.J.; Zhang, H.F.; Meng, X.H.; Yu, M.Y.; Xie, N.C. Role of the mitochondrial calcium uniporter in Mg2+-free-induced epileptic hippocampal neuronal apoptosis. Int. J. Neurosci. 2020, 9. [Google Scholar] [CrossRef]
- Lukas, T.J.; Haiech, J.; Lau, W.; Craig, T.A.; Zimmer, W.E.; Shattuck, R.L.; Shoemaker, M.O.; Watterson, D.M. CALMODULIN AND CALMODULIN-REGULATED PROTEIN-KINASES AS TRANSDUCERS OF INTRACELLULAR CALCIUM SIGNALS. Cold Spring Harb. Symp. Quant. Biol. 1988, 53, 185–193. [Google Scholar] [CrossRef]
- Chowdhury, S.R.; Jaiswal, S.; Lu, H.P. Compressive-force induced activation of apo-calmodulin in protein signalling. Phys. Chem. Chem. Phys. PCCP 2020, 22, 1092–1096. [Google Scholar] [CrossRef]
- Song, Z.X.; Chen, Q.; Ding, Q.; Zheng, F.; Li, C.W.; Xu, L.P.; Wang, H.B. Function of Ca2+-/calmodulin-dependent protein kinase IV in Ca2+-stimulated neuronal signaling and behavior. Sci. China Life Sci. 2015, 58, 6–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.; Murao, K.; Sayo, Y.; Imachi, H.; Cao, W.M.; Ohtsuka, S.; Niimi, M.; Tokumitsu, H.; Inuzuka, H.; Wong, N.C.W.; et al. The role of calcium/calmodulin-dependent protein kinase cascade in glucose upregulation of insulin gene expression. Diabetes 2004, 53, 1475–1481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villalobo, A.; Berchtold, M.W. The Role of Calmodulin in Tumor Cell Migration, Invasiveness, and Metastasis. Int. J. Mol. Sci. 2020, 21, 765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoorn, E.J.; Zietse, R. Disorders of calcium and magnesium balance: A physiology-based approach. Pediatr. Nephrol. 2013, 28, 1195–1206. [Google Scholar] [CrossRef] [PubMed]
- Paoletti, P.; Vergnano, A.M.; Barbour, B.; Casado, M. Zinc at glutamatergic synapses. Neuroscience 2009, 158, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Mocchegiani, E.; Bertoni-Freddari, C.; Marcellini, F.; Malavolta, M. Brain, aging and neurodegeneration: Role of zinc ion availability. Prog. Neurobiol. 2005, 75, 367–390. [Google Scholar] [CrossRef] [PubMed]
- Frederickson, C.J.; Klitenick, M.A.; Manton, W.I.; Kirkpatrick, J.B. CYTOARCHITECTONIC DISTRIBUTION OF ZINC IN THE HIPPOCAMPUS OF MAN AND THE RAT. Brain Res. 1983, 273, 335–339. [Google Scholar] [CrossRef]
- Lee, H.C.; Edmonds, M.E.; Duncan, F.E.; O’Halloran, T.V.; Woodruff, T.K. Zinc exocytosis is sensitive to myosin light chain kinase inhibition in mouse and human eggs. Mol. Hum. Reprod. 2020, 26, 228–239. [Google Scholar] [CrossRef]
- Kim, A.M.; Bernhardt, M.L.; Kong, B.Y.; Ahn, R.W.; Vogt, S.; Woodruff, T.K.; O’Halloran, T.V. Zinc Sparks Are Triggered by Fertilization and Facilitate Cell Cycle Resumption in Mammalian Eggs. ACS Chem. Biol. 2011, 6, 716–723. [Google Scholar] [CrossRef]
- Kim, A.M.; Vogt, S.; O’Halloran, T.V.; Woodruff, T.K. Zinc availability regulates exit from meiosis in maturing mammalian oocytes. Nat. Chem. Biol. 2010, 6, 674–681. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N.; Duncan, F.E.; Que, E.L.; O’Halloran, T.V.; Woodruff, T.K. The fertilization-induced zinc spark is a novel biomarker of mouse embryo quality and early development. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tokuhiro, K.; Dean, J. Glycan-Independent Gamete Recognition Triggers Egg Zinc Sparks and ZP2 Cleavage to Prevent Polyspermy. Dev. Cell 2018, 46, 627–640.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slomianka, L. Neurons of origin of zinc-containing pathways and the distribution of zinc-containing boutons in the hippocampal region of the rat. Neuroscience 1992, 48, 325–352. [Google Scholar] [CrossRef]
- Haug, F.M.S. Electron microscopical localization of zinc in hippocampal mossy fibre synapses by a modified sulfide silver procedure. Histochemie 1967, 8, 355–368. [Google Scholar] [CrossRef]
- Evstratova, A.; Toth, K. Information processing and synaptic plasticity at hippocampal mossy fiber terminals. Front. Cell. Neurosci. 2014, 8. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, J.A.; Zhang, X.-L.; Sullivan, A.P.; Vose, L.R.; Moghadam, A.A.; Fried, V.A.; Stanton, P.K. Zinc enhances hippocampal long-term potentiation at CA1 synapses through NR2B containing NMDA receptors. PLoS ONE 2018, 13. [Google Scholar] [CrossRef]
- Bastos, F.M.C.; Lopes, S.A.; Corceiro, V.N.; Matias, C.M.; Dionisio, J.C.; Sampaio dos Aidos, F.D.S.; Mendes, P.J.; Santos, R.M.; Quinta-Ferreira, R.M.; Emilia Quinta-Ferreira, M. Postsynaptic zinc potentiation elicited by KCl depolarization at hippocampal mossy fiber synapses. Gen. Physiol. Biophys. 2017, 36, 289–296. [Google Scholar] [CrossRef]
- Takeda, A.; Fuke, S.; Ando, M.; Oku, N. Positive modulation of long-term potentiation at hippocampal ca1 synapses by low micromolar concentrations of zinc. Neuroscience 2009, 158, 585–591. [Google Scholar] [CrossRef]
- Takeda, A.; Ando, M.; Kanno, S.; Oku, N. Unique response of zinc in the hippocampus to behavioral stress and attenuation of subsequent mossy fiber long-term potentiation. Neurotoxicology 2009, 30, 712–717. [Google Scholar] [CrossRef]
- Vogt, K.; Mellor, J.; Tong, G.; Nicoll, R. The actions of synaptically released zinc at hippocampal mossy fiber synapses. Neuron 2000, 26, 187–196. [Google Scholar] [CrossRef] [Green Version]
- Kay, A.R. Evidence for chelatable zinc in the extracellular space of the hippocampus, but little evidence for synaptic release of Zn. J. Neurosci. 2003, 23, 6847–6855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kay, A.R.; Toth, K. Is Zinc a Neuromodulator? Sci. Signal. 2008, 1. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Hough, C.J.; Frederickson, C.J.; Sarvey, J.M. Induction of mossy fiber -> CA3 long-term potentiation requires translocation of synaptically released Zn2+. J. Neurosci. 2001, 21, 8015–8025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erickson, J.C.; Hollopeter, G.; Thomas, S.A.; Froelick, G.J.; Palmiter, R.D. Disruption of the metallothionein-III gene in mice: Analysis of brain zinc, behavior, and neuron vulnerability to metals, aging, and seizures. J. Neurosci. 1997, 17, 1271–1281. [Google Scholar] [CrossRef] [Green Version]
- Palumaa, P.; Eriste, E.; Njunkova, O.; Pokras, L.; Jornvall, H.; Sillard, R. Brain-specific metallothionein-3 has higher metal-binding capacity than ubiquitous metallothioneins and binds metals noncooperatively. Biochemistry 2002, 41, 6158–6163. [Google Scholar] [CrossRef]
- Palmiter, R.D.; Cole, T.B.; Quaife, C.J.; Findley, S.D. ZnT-3, a putative transporter of zinc into synaptic vesicles. Proc. Natl. Acad. Sci. USA 1996, 93, 14934–14939. [Google Scholar] [CrossRef] [Green Version]
- Ben Mimouna, S.; Le Charpentier, T.; Lebon, S.; Van Steenwinckel, J.; Messaoudi, I.; Gressens, P. Involvement of the synapse-specific zinc transporter ZnT3 in cadmium-induced hippocampal neurotoxicity. J. Cell. Physiol. 2019, 234, 15872–15884. [Google Scholar] [CrossRef]
- Quinta-Ferreira, M.E.; Sampaio dos Aidos, F.D.S.; Matias, C.M.; Mendes, P.J.; Dionisio, J.C.; Santos, R.M.; Rosario, L.M.; Quinta-Ferreira, R.M. Modelling zinc changes at the hippocampal mossy fiber synaptic cleft. J. Comput. Neurosci. 2016, 41, 323–337. [Google Scholar] [CrossRef]
- Blakemore, L.J.; Trombley, P.Q. Mechanisms of zinc modulation of olfactory bulb AMPA receptors. Neuroscience 2019, 410, 160–175. [Google Scholar] [CrossRef]
- Ha, H.T.T.; Leal-Ortiz, S.; Lalwani, K.; Kiyonaka, S.; Hamachi, I.; Mysore, S.P.; Montgomery, J.M.; Garner, C.C.; Huguenard, J.R.; Kim, S.A. Shank and Zinc Mediate an AMPA Receptor Subunit Switch in Developing Neurons. Front. Mol. Neurosci. 2018, 11. [Google Scholar] [CrossRef] [Green Version]
- Noh, J.; Chung, J.-M. Modulation of Dopaminergic Neuronal Excitability by Zinc through the Regulation of Calcium-related Channels. Exp. Neurobiol. 2019, 28, 578–592. [Google Scholar] [CrossRef] [PubMed]
- Ekstein, D.; Benninger, F.; Daninos, M.; Pitsch, J.; van Loo, K.M.J.; Becker, A.J.; Yaari, Y. Zinc induces long-term upregulation of T-type calcium current in hippocampal neurons in vivo. J. Physiol. Lond. 2012, 590, 5895–5905. [Google Scholar] [CrossRef] [PubMed]
- Sindreu, C.; Bayes, A.; Altafaj, X.; Perez-Clausell, J. Zinc transporter-1 concentrates at the postsynaptic density of hippocampal synapses. Mol. Brain 2014, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mellone, M.; Pelucchi, S.; Alberti, L.; Geneazzani, A.A.; Di Luca, M.; Gardoni, F. Zinc transporter-1: A novel NMDA receptor-binding protein at the postsynaptic density. J. Neurochem. 2015, 132, 159–168. [Google Scholar] [CrossRef] [Green Version]
- Schulien, A.J.; Justice, J.A.; Di Maio, R.; Wills, Z.P.; Shah, N.H.; Aizenman, E. Zn2+-induced Ca2+ release via ryanodine receptors triggers calcineurin-dependent redistribution of cortical neuronal Kv2.1 K+ channels. J. Physiol. Lond. 2016, 594, 2647–2659. [Google Scholar] [CrossRef] [Green Version]
- Hershfinkel, M.; Moran, A.; Grossman, N.; Sekler, I. A zinc-sensing receptor triggers the release of intracellular Ca2+ and regulates ion transport. Proc. Natl. Acad. Sci. USA 2001, 98, 11749–11754. [Google Scholar] [CrossRef] [Green Version]
- Bredesen, D.E. Metabolic profiling distinguishes three subtypes of Alzheimer’s disease. Aging-Us 2015, 7, 595–600. [Google Scholar] [CrossRef] [Green Version]
- Ishihara, R.; Ide-Ektessabi, A.; Ikeda, K.; Mizuno, Y.; Fujisawa, S.; Takeuchi, T.; Ohta, T. Investigation of cellular metallic elements in single neurons of human brain tissues. Neuroreport 2002, 13, 1817–1820. [Google Scholar] [CrossRef]
- Malavolta, M.; Piacenza, F.; Basso, A.; Giacconi, R.; Costarelli, L.; Mocchegiani, E. Serum copper to zinc ratio: Relationship with aging and health status. Mech. Ageing Dev. 2015, 151, 93–100. [Google Scholar] [CrossRef]
- Gardner, B.; Dieriks, B.V.; Cameron, S.; Mendis, L.H.S.; Turner, C.; Faull, R.L.M.; Curtis, M.A. Metal concentrations and distributions in the human olfactory bulb in Parkinson’s disease. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- Samudralwar, D.L.; Diprete, C.C.; Ni, B.F.; Ehmann, W.D.; Markesbery, W.R. Elemental imbalances in the olfactory pathway in Alzheimers-diseasE. J. Neurol. Sci. 1995, 130, 139–145. [Google Scholar] [CrossRef]
- Lim, J.H.; Davis, G.E.; Wang, Z.; Li, V.; Wu, Y.; Rue, T.C.; Storm, D.R. Zicam-Induced Damage to Mouse and Human Nasal Tissue. PLoS ONE 2009, 4. [Google Scholar] [CrossRef] [Green Version]
- Eom, J.-W.; Lee, J.-M.; Koh, J.-Y.; Kim, Y.-H. AMP-activated protein kinase contributes to zinc-induced neuronal death via activation by LKB1 and induction of Bim in mouse cortical cultures. Mol. Brain 2016, 9. [Google Scholar] [CrossRef] [Green Version]
- Qi, Z.; Shi, W.; Zhao, Y.; Ji, X.; Liu, K.J. Zinc accumulation in mitochondria promotes ischemia-induced BBB disruption through Drp1-dependent mitochondria fission. Toxicol. Appl. Pharmacol. 2019, 377. [Google Scholar] [CrossRef] [PubMed]
- Pivovarova, N.B.; Stanika, R.I.; Kazanina, G.; Villanueva, I.; Andrews, S.B. The interactive role of zinc and calcium in mitochondrial dysfunction and neurodegeneration. J. Neurochem. 2014, 128, 592–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharaf, M.S.; Van den Heuvel, M.R.; Stevens, D.; Kamunde, C. Zinc and calcium modulate mitochondrial redox state and morphofunctional integrity. Free Radic. Biol. Med. 2015, 84, 142–153. [Google Scholar] [CrossRef] [PubMed]
- Xiao, R.; Yuan, L.; He, W.; Yang, X. Zinc ions regulate opening of tight junction favouring efflux of macromolecules via the GSK3 beta/snail-mediated pathway. Metallomics 2018, 10, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Vasudevaraju, P.; Bharathi, T.J.; Shamasundar, N.M.; Subba Rao, K.; Balaraj, B.M.; Ksj, R.; Ts, S.R. New evidence on iron, copper accumulation and zinc depletion and its correlation with DNA integrity in aging human brain regions. Indian J. Psychiatry 2010, 52, 140–144. [Google Scholar] [CrossRef]
- Tian, K.; Wang, Y.-x.; Li, L.-x.; Liu, Y.-q. Neuronal death/apoptosis induced by intracellular zinc deficiency associated with changes in amino-acid neurotransmitters and glutamate receptor subtypes. J. Inorg. Biochem. 2018, 179, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Takeda, A.; Tamano, H. Significance of the degree of synaptic Zn2+ signaling in cognition. Biometals 2016, 29, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Sowa-Kucma, M.; Szewczyk, B.; Sadlik, K.; Piekoszewski, W.; Trela, F.; Opoka, W.; Poleszak, E.; Pilc, A.; Nowak, G. Zinc, magnesium and NMDA receptor alterations in the hippocampus of suicide victims. J. Affect. Disord. 2013, 151, 924–931. [Google Scholar] [CrossRef] [PubMed]
- Szewczyk, B.; Kotarska, K.; Siwek, A.; Olech, L.; Kuter, K. Antidepressant activity of zinc: Further evidence for the involvement of the serotonergic system. Pharmacol. Rep. 2017, 69, 456–461. [Google Scholar] [CrossRef] [PubMed]
- Kozlowski, H.; Kolkowska, P.; Watly, J.; Krzywoszynska, K.; Potocki, S. General Aspects of Metal Toxicity. Curr. Med. Chem. 2014, 21, 3721–3740. [Google Scholar] [CrossRef] [PubMed]
- Doreulee, N.; Yanovsky, Y.; Haas, H.L. Suppression of long-term potentiation in hippocampal slices by copper. Hippocampus 1997, 7, 666–669. [Google Scholar] [CrossRef]
- Silverman, J.M.; Christy, D.; Shyu, C.C.; Moon, K.-M.; Fernando, S.; Gidden, Z.; Cowan, C.M.; Ban, Y.; Stacey, R.G.; Grad, L.I.; et al. CNS-derived extracellular vesicles from superoxide dismutase 1 (SOD1)(G93A) ALS mice originate from astrocytes and neurons and carry misfolded SOD1. J. Biol. Chem. 2019, 294, 3744–3759. [Google Scholar] [CrossRef] [Green Version]
- Gaier, E.D.; Miller, M.B.; Ralle, M.; Aryal, D.; Wetsel, W.C.; Mains, R.E.; Eipper, B.A. Peptidylglycine alpha-amidating monooxygenase heterozygosity alters brain copper handling with region specificity. J. Neurochem. 2013, 127, 605–619. [Google Scholar] [CrossRef]
- Schmidt, K.; Ralle, M.; Schaffer, T.; Jayakanthan, S.; Bari, B.; Muchenditsi, A.; Lutsenko, S. ATP7A and ATP7B copper transporters have distinct functions in the regulation of neuronal dopamine--hydroxylase. J. Biol. Chem. 2018, 293, 20085–20098. [Google Scholar] [CrossRef] [Green Version]
- Kapkaeva, M.R.; Popova, O.V.; Kondratenko, R.V.; Rogozin, P.D.; Genrikhs, E.E.; Stelmashook, E.V.; Skrebitsky, V.G.; Khaspekov, L.G.; Isaev, N.K. Effects of copper on viability and functional properties of hippocampal neurons in vitro. Exp. Toxicol. Pathol. 2017, 69, 259–264. [Google Scholar] [CrossRef]
- Kardos, J.; Kovacs, I.; Hajos, F.; Kalman, M.; Simonyi, M. Nerve-endings from rat-brain tissue release copper upon depolarization—A possible role in regulating neuronal excitability. Neurosci. Lett. 1989, 103, 139–144. [Google Scholar] [CrossRef]
- Gaier, E.D.; Rodriguiz, R.M.; Zhou, J.; Ralle, M.; Wetsel, W.C.; Eipper, B.A.; Mains, R.E. In vivo and in vitro analyses of amygdalar function reveal a role for copper. J. Neurophysiol. 2014, 111, 1927–1939. [Google Scholar] [CrossRef] [Green Version]
- Salazar-Weber, N.L.; Smith, J.P. Copper Inhibits NMDA Receptor-Independent LTP and Modulates the Paired-Pulse Ratio after LTP in Mouse Hippocampal Slices. Int. J. Alzheimer’s Dis. 2011, 2011, 864753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maryon, E.B.; Molloy, S.A.; Yu, K.I.H.; Kaplan, J.H. Rate and Regulation of Copper Transport by Human Copper Transporter 1 (hCTR1). J. Biol. Chem. 2013, 288, 18035–18046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheiber, I.F.; Mercer, J.F.B.; Dringen, R. Copper accumulation by cultured astrocytes. Neurochem. Int. 2010, 56, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Dong, D.; Kang, Y.J. Copper chaperone for superoxide dismutase-1 transfers copper to mitochondria but does not affect cytochrome c oxidase activity. Exp. Biol. Med. 2013, 238, 1017–1023. [Google Scholar] [CrossRef]
- Vest, K.E.; Paskavitz, A.L.; Lee, J.B.; Padilla-Benavides, T. Dynamic changes in copper homeostasis and post-transcriptional regulation of Atp7a during myogenic differentiation. Metallomics 2018, 10, 309–322. [Google Scholar] [CrossRef] [Green Version]
- Horning, M.S.; Trombley, P.Q. Zinc and copper influence excitability of rat olfactory bulb neurons by multiple mechanisms. J. Neurophysiol. 2001, 86, 1652–1660. [Google Scholar] [CrossRef] [Green Version]
- Marchetti, C.; Baranowska-Bosiacka, I.; Gavazzo, P. Multiple effects of copper on NMDA receptor currents. Brain Res. 2014, 1542, 20–31. [Google Scholar] [CrossRef]
- Meramat, A.; Rajab, N.F.; Shahar, S.; Sharif, R.A. DNA damage, copper and lead associates with cognitive function among older adults. J. Nutr. Health Aging 2017, 21, 539–545. [Google Scholar] [CrossRef]
- Deibel, M.A.; Ehmann, W.D.; Markesbery, W.R. Copper, iron, and zinc imbalances in severely degenerated brain regions in Alzheimer’s disease: Possible relation to oxidative stress. J. Neurol. Sci. 1996, 143, 137–142. [Google Scholar] [CrossRef]
- Xu, J.; Church, S.J.; Patassini, S.; Begley, P.; Waldvogel, H.J.; Curtis, M.A.; Faull, R.L.M.; Unwin, R.D.; Cooper, G.J.S. Evidence for widespread, severe brain copper deficiency in Alzheimer’s dementia. Metallomics 2017, 9, 1106–1119. [Google Scholar] [CrossRef] [Green Version]
- Kumar, N. Copper deficiency myelopathy (human swayback). Mayo Clin. Proc. 2006, 81, 1371–1384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlief, M.L.; West, T.; Craig, A.M.; Holtzman, D.M.; Gitlin, J.D. Role of the Menkes copper-transporting ATPase in NMDA receptor-mediated neuronal toxicity. Proc. Natl. Acad. Sci. USA 2006, 103, 14919–14924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mueller, C.; Magaki, S.; Schrag, M.; Ghosh, M.C.; Kirsch, W.M. Iron Regulatory Protein 2 is Involved in Brain Copper Homeostasis. J. Alzheimers Dis. 2009, 18, 201–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, C.; Munoz, B.; Sepulveda, F.J.; Urrutia, J.; Quiroz, M.; Luza, S.; De Ferrari, G.V.; Aguayo, L.G.; Opazo, C. Biphasic effects of copper on neurotransmission in rat hippocampal neurons. J. Neurochem. 2011, 119, 78–88. [Google Scholar] [CrossRef]
- Yu, H.; Wang, D.; Zou, L.; Zhang, Z.; Xu, H.; Zhu, F.; Ren, X.; Xu, B.; Yuan, J.; Liu, J.; et al. Proteomic alterations of brain subcellular organelles caused by low-dose copper exposure: Implication for Alzheimer’s disease. Arch. Toxicol. 2018, 92, 1363–1382. [Google Scholar] [CrossRef]
- Xu, J.; Begley, P.; Church, S.J.; Patassini, S.; McHarg, S.; Kureishy, N.; Hollywood, K.A.; Waldvogel, H.J.; Liu, H.; Zhang, S.; et al. Elevation of brain glucose and polyol-pathway intermediates with accompanying brain-copper deficiency in patients with Alzheimer’s disease: Metabolic basis for dementia. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, H.; Amarsingh, G.V.; Cheung, C.C.H.; Hogl, S.; Narayanan, U.; Zhang, L.; McHarg, S.; Xu, J.; Gong, D.; et al. Diabetic cardiomyopathy is associated with defective myocellular copper regulation and both defects are rectified by divalent copper chelation. Cardiovasc. Diabetol. 2014, 13. [Google Scholar] [CrossRef] [Green Version]
- Peng, F.; Xie, F.; Muzik, O. Alteration of Copper Fluxes in Brain Aging: A Longitudinal Study in Rodent Using (CuCl2)-Cu-64-PET/CT. Aging Dis. 2018, 9, 109–118. [Google Scholar] [CrossRef]
- Famitafreshi, H.; Karimian, M. Modulation of catalase, copper and zinc in the hippocampus and the prefrontal cortex in social isolation-induced depression in male rats. Acta Neurobiol. Exp. 2019, 79, 184–192. [Google Scholar] [CrossRef] [Green Version]
- Han, O. Molecular mechanism of intestinal iron absorption. Metallomics 2011, 3, 103–109. [Google Scholar] [CrossRef]
- Inoue, K.; Nakai, Y.; Ueda, S.; Kamigaso, S.; Ohta, K.; Hatakeyama, M.; Hayashi, Y.; Otagiri, M.; Yuasa, H. Functional characterization of PCFT/HCP1 as the molecular entity of the carrier-mediated intestinal folate transport system in the rat model. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G660–G668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laftah, A.; Latunde-Dada, G.; Fakih, S.; Hider, R.; Simpson, R.; Mckie, A. Haem and folate transport by proton-coupled folate transporter/haem carrier protein 1 (SLC46A1). Br. J. Nutr. 2009, 101, 1150–1156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Blanc, S.; Garrick, M.; Arredondo, M. Heme carrier protein 1 transports heme and is involved in heme-Fe metabolism. Am. J. Physiol. Cell Physiol. 2012, 302, C1780–C1785. [Google Scholar] [CrossRef] [PubMed]
- Lawen, A.; Lane, D. Mammalian Iron Homeostasis in Health and Disease: Uptake, Storage, Transport, and Molecular Mechanisms of Action. Antioxid. Redox Signal. 2013, 18, 2473–2507. [Google Scholar] [CrossRef] [PubMed]
- Gumienna-Kontecka, E.; Pyrkosz-Bulska, M.; Szebesczyk, A.; Ostrowska, M. Iron Chelating Strategies in Systemic Metal Overload, Neurodegeneration and Cancer. Curr. Med. Chem. 2014, 21, 3741–3767. [Google Scholar] [CrossRef] [PubMed]
- Gkouvatsos, K.; Papanikolaou, G.; Pantopoulos, K. Regulation of iron transport and the role of transferrin. Biochim. Et Biophys. Acta-Gen. Subj. 2012, 1820, 188–202. [Google Scholar] [CrossRef] [PubMed]
- Ganz, T.; Nemeth, E. Hepcidin and iron homeostasis. Biochim. Biophys. Acta Mol. Cell Res. 2012, 1823, 1434–1443. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, A.; Yee, J. Hepcidin. Adv. Chronic Kidney Dis. 2019, 26, 298–305. [Google Scholar] [CrossRef]
- Rochette, L.; Gudjoncik, A.; Guenancia, C.; Zeller, M.; Cottin, Y.; Vergely, C. The iron-regulatory hormone hepcidin: A possible therapeutic target? Pharmacol. Ther. 2015, 146, 35–52. [Google Scholar] [CrossRef]
- Corradini, E.; Meynard, D.; Wu, Q.; Chen, S.; Ventura, P.; Pietrangelo, A.; Babitt, J. Serum and Liver Iron Differently Regulate the Bone Morphogenetic Protein 6 (BMP6)-SMAD Signaling Pathway in Mice. Hepatology 2011, 54, 273–284. [Google Scholar] [CrossRef] [Green Version]
- Corradini, E.; Rozier, M.; Meynard, D.; Odhiambo, A.; Lin, H.; Feng, Q.; Migas, M.; Britton, R.; Babitt, J.; Fleming, R. Iron Regulation of Hepcidin Despite Attenuated Smad1,5,8 Signaling in Mice Without Transferrin Receptor 2 or Hfe. Gastroenterology 2011, 141, 1907–1914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barton, J.; Edwards, C.; Acton, R. HFE gene: Structure, function, mutations, and associated iron abnormalities. Gene 2015, 574, 179–192. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Wang, Y.; Wu, Q.; Cheng, W.; Liu, W.; Zhao, Y.; Mayeur, C.; Schmidt, P.; Yu, P.; Wang, F.; et al. HFE interacts with the BMP type I receptor ALK3 to regulate hepcidin expression. Blood 2014, 124, 1335–1343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sangkhae, V.; Nemeth, E. Regulation of the Iron Homeostatic Hormone Hepcidin. Adv. Nutr. 2017, 8, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Gkouvatsos, K.; Fillebeen, C.; Daba, A.; Wagner, J.; Sebastiani, G.; Pantopoulos, K. Iron-Dependent Regulation of Hepcidin in Hjv-/- Mice: Evidence That Hemojuvelin Is Dispensable for Sensing Body Iron Levels. PLoS ONE 2014, 9. [Google Scholar] [CrossRef] [PubMed]
- Nai, A.; Rubio, A.; Campanella, A.; Gourbeyre, O.; Artuso, I.; Bordini, J.; Gineste, A.; Latour, C.; Besson-Fournier, C.; Lin, H.; et al. Limiting hepatic Bmp-Smad signaling by matriptase-2 is required for erythropoietin-mediated hepcidin suppression in mice. Blood 2016, 127, 2327–2336. [Google Scholar] [CrossRef] [Green Version]
- Rishi, G.; Secondes, E.; Subramaniam, V. Hemochromatosis: Evaluation of the dietary iron model and regulation of hepcidin. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 2550–2556. [Google Scholar] [CrossRef]
- Taniguchi, R.; Kato, H.; Font, J.; Deshpande, C.; Wada, M.; Ito, K.; Ishitani, R.; Jormakka, M.; Nureki, O. Outward- and inward-facing structures of a putative bacterial transition-metal transporter with homology to ferroportin. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef]
- di Patti, M.; Polticelli, F.; Cece, G.; Cutone, A.; Felici, F.; Persichini, T.; Musci, G. A structural model of human ferroportin and of its iron binding site. FEBS J. 2014, 281, 2851–2860. [Google Scholar] [CrossRef]
- Ward, D.; Kaplan, J. Ferroportin-mediated iron transport: Expression and regulation. Biochim. Biophys. Acta Mol. Cell Res. 2012, 1823, 1426–1433. [Google Scholar] [CrossRef] [Green Version]
- Neves, J.; Leitz, D.; Kraut, S.; Brandenberger, C.; Agrawal, R.; Weissmann, N.; Muhlfeld, C.; Mall, M.; Altamura, S.; Muckenthaler, M. Disruption of the Hepcidin/Ferroportin Regulatory System Causes Pulmonary Iron Overload and Restrictive Lung Disease. Ebiomedicine 2017, 20, 230–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lozoff, B.; Beard, J.; Connor, J.; Felt, B.; Georgieff, M.; Schallert, T. Long-lasting neural and behavioral effects of iron deficiency in infancy. Nutr. Rev. 2006, 64, S34–S43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frazer, D.; Wilkins, S.; Darshan, D.; Mirciov, C.; Dunn, L.; Anderson, G. Ferroportin Is Essential for Iron Absorption During Suckling, But Is Hyporesponsive to the Regulatory Hormone Hepcidin. Cell. Mol. Gastroenterol. Hepatol. 2017, 3, 410–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torti, S.; Torti, F. Iron and cancer: More ore to be mined. Nat. Rev. Cancer 2013, 13, 342–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bebber, C.; Muller, F.; Clemente, L.; Weber, J.; von Karstedt, S. Ferroptosis in Cancer Cell Biology. Cancers 2020, 12, 164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Yu, L.; Ding, J.; Chen, Y. Iron Metabolism in Cancer. Int. J. Mol. Sci. 2019, 20, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bastide, N.; Pierre, F.; Corpet, D. Heme Iron from Meat and Risk of Colorectal Cancer: A Meta-analysis and a Review of the Mechanisms Involved. Cancer Prev. Res. 2011, 4, 177–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sornjai, W.; Van Long, F.; Pion, N.; Pasquer, A.; Saurin, J.; Marcel, V.; Diaz, J.; Mertani, H.; Smith, D. Iron and hepcidin mediate human colorectal cancer cell growth. Chem. Biol. Interact. 2020, 319. [Google Scholar] [CrossRef]
- Xue, X.; Ramakrishnan, S.; Weisz, K.; Triner, D.; Xie, L.; Attili, D.; Pant, A.; Gyorffy, B.; Zhan, M.; Carter-Su, C.; et al. Iron Uptake via DMT1 Integrates Cell Cycle with JAK-STAT3 Signaling to Promote Colorectal Tumorigenesis. Cell Metab. 2016, 24, 447–461. [Google Scholar] [CrossRef] [Green Version]
- Latunde-Dada, G. Ferroptosis: Role of lipid peroxidation, iron and ferritinophagy. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 1893–1900. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Stockwell, B. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem. Biol. 2008, 15, 234–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.; Zandkarimi, F.; Zhang, Y.; Meena, J.; Kim, J.; Zhuang, L.; Tyagi, S.; Ma, L.; Westbrook, T.; Steinberg, G.; et al. Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat. Cell Biol. 2020, 22. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Zhao, Y.; Guo, J.; Zhao, S.; Song, L.; Fei, C.; Zhang, Z.; Li, X.; Chang, C. Iron overload promotes erythroid apoptosis through regulating HIF-1a/ROS signaling pathway in patients with myelodysplastic syndrome. Leuk. Res. 2017, 58, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Kilari, S.; Pullakhandam, R.; Nair, K. Zinc inhibits oxidative stress-induced iron signaling and apoptosis in Caco-2 cells. Free Radic. Biol. Med. 2010, 48, 961–968. [Google Scholar] [CrossRef]
- Jin, R.; Liu, L.; Zhu, W.; Li, D.; Yang, L.; Duan, J.; Cai, Z.; Nie, Y.; Zhang, Y.; Gong, Q.; et al. Iron oxide nanoparticles promote macrophage autophagy and inflammatory response through activation of toll-like Receptor-4 signaling. Biomaterials 2019, 203, 23–30. [Google Scholar] [CrossRef]
- Liu, L.; Jin, R.; Duan, J.; Yang, L.; Cai, Z.; Zhu, W.; Nie, Y.; He, J.; Xia, C.; Gong, Q.; et al. Bioactive iron oxide nanoparticles suppress osteoclastogenesis and ovariectomy-induced bone loss through regulating the TRAF6-p62-CYLD signaling complex. Acta Biomater. 2020, 103, 281–292. [Google Scholar] [CrossRef]
Figure 1.
Schematic diagrams of the representative members of mammalian (A) selective and (B) non-selective Mg2+ transporters.
Figure 1.
Schematic diagrams of the representative members of mammalian (A) selective and (B) non-selective Mg2+ transporters.

Figure 2.
(A) Schematic representation of calcium signaling in excitable cells. Voltage-gated calcium channels (VGCC) are found in the membrane of excitable cells. Other sources of calcium influx are calcium-permeable α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and N-methyl-D-aspartate (NMDA) glutamate-type receptors, and transient receptor potential type C (TRPC) channels. cAMP-dependent protein kinases and the Ca2+calmodulin-dependent proteins play crucial roles in local signaling and synaptic plasticity. Calcium release from internal stores is mediated by inositol trisphosphate receptors (IP3R), ryanodine receptors (RyR), and mitochondrial permeability transition pore (PTP). Ca2+ efflux is mediated by the plasma membrane calcium ATPase, the Na2+/Ca2+ exchanger, and the sarco-/endoplasmic reticulum calcium ATPase (SERCA) and the mitochondrial uniporter (UP). (B) Schematic representation of calcium signaling in non-excitable cells. Following the stimulation of G protein-coupled receptors, IP3 produced by phospholipase C (PLC) activates the IP3 receptor (IP3R) and triggers the depletion of ER calcium stores. The stromal interaction molecule 1(STIM1), representing Ca2+ sensor, and Orai, belonging to the group of store-operated calcium channels (SOCC) builds the ion-conducting transmembrane protein complex. STIM1 interacts with Orai1 to trigger SOCC opening. Then, SERCA pumps Ca2+ back into the ER to refill the stores with Ca2+. Transient receptor potential channels (TRPC) can be also activated by phospholipase C stimulation.
Figure 2.
(A) Schematic representation of calcium signaling in excitable cells. Voltage-gated calcium channels (VGCC) are found in the membrane of excitable cells. Other sources of calcium influx are calcium-permeable α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and N-methyl-D-aspartate (NMDA) glutamate-type receptors, and transient receptor potential type C (TRPC) channels. cAMP-dependent protein kinases and the Ca2+calmodulin-dependent proteins play crucial roles in local signaling and synaptic plasticity. Calcium release from internal stores is mediated by inositol trisphosphate receptors (IP3R), ryanodine receptors (RyR), and mitochondrial permeability transition pore (PTP). Ca2+ efflux is mediated by the plasma membrane calcium ATPase, the Na2+/Ca2+ exchanger, and the sarco-/endoplasmic reticulum calcium ATPase (SERCA) and the mitochondrial uniporter (UP). (B) Schematic representation of calcium signaling in non-excitable cells. Following the stimulation of G protein-coupled receptors, IP3 produced by phospholipase C (PLC) activates the IP3 receptor (IP3R) and triggers the depletion of ER calcium stores. The stromal interaction molecule 1(STIM1), representing Ca2+ sensor, and Orai, belonging to the group of store-operated calcium channels (SOCC) builds the ion-conducting transmembrane protein complex. STIM1 interacts with Orai1 to trigger SOCC opening. Then, SERCA pumps Ca2+ back into the ER to refill the stores with Ca2+. Transient receptor potential channels (TRPC) can be also activated by phospholipase C stimulation.

Figure 3.
Schematic representation of the events related to the zinc signal release from zincergic synapses. After zinc influx, Zn2+ ions are bound by metallothionein 3 (MT-3). This protein delivers zinc to the zone where vesicles are formed. The influx of Zn2+ into the vesicle is catalyzed by ZnT-1 transporter. After release, Zn2+ appears in a synaptic cleft and diffuses through it. This phenomenon leads to the interaction of Zn2+ with postsynaptic N-methyl-D-aspartate glutamate receptor (NMDAR) and the influx of these ions into the postsynaptic nerve terminal. The level of Zn2+ in a postsynaptic neuron is regulated by the ZnT-1 transporter responsible for efflux of Zn2+ excess. The location of ZnT-1 in the membrane allows this transporter to interact with the GluN2A subunit of NMDAR (blue sphere).
Figure 3.
Schematic representation of the events related to the zinc signal release from zincergic synapses. After zinc influx, Zn2+ ions are bound by metallothionein 3 (MT-3). This protein delivers zinc to the zone where vesicles are formed. The influx of Zn2+ into the vesicle is catalyzed by ZnT-1 transporter. After release, Zn2+ appears in a synaptic cleft and diffuses through it. This phenomenon leads to the interaction of Zn2+ with postsynaptic N-methyl-D-aspartate glutamate receptor (NMDAR) and the influx of these ions into the postsynaptic nerve terminal. The level of Zn2+ in a postsynaptic neuron is regulated by the ZnT-1 transporter responsible for efflux of Zn2+ excess. The location of ZnT-1 in the membrane allows this transporter to interact with the GluN2A subunit of NMDAR (blue sphere).

Figure 4.
The schematic representation of an example sequence of events related to copper release possible in nerve cells. Copper ions are taken up to the cell by a Cu transporter 1 (CTR-1). The presence of free Cu+ ions immediately activates a set of copper chaperones. These proteins transport copper within the cell and deliver it to its destination. In neurons, that can release copper into the synaptic cleft, chaperones introduce copper ions to the ATP7A protein, which transport it to the lumen of a vesicle. After Cu release, it can interact with e.g., NMDAR.
Figure 4.
The schematic representation of an example sequence of events related to copper release possible in nerve cells. Copper ions are taken up to the cell by a Cu transporter 1 (CTR-1). The presence of free Cu+ ions immediately activates a set of copper chaperones. These proteins transport copper within the cell and deliver it to its destination. In neurons, that can release copper into the synaptic cleft, chaperones introduce copper ions to the ATP7A protein, which transport it to the lumen of a vesicle. After Cu release, it can interact with e.g., NMDAR.

Figure 5.
Hepcidin expression via bone morphogenic protein (BMP)-SMAD signaling pathway in hepatocytes. High iron concentration in plasma increases transferrin saturation, which results in homeostatic iron regulator protein (HFE) detachment from the transferrin receptor (TFRC)/HFE complex, then its interaction with ALK3 (BMP receptor), which leads to increased expression of ALK3 and activation of the BMP-SMAD signaling pathway. High plasma iron also causes stabilization of TFR2, which is associated with HFE, and may also interact with ALK3 (but this is not confirmed). Hemojuvelin (HJV) is a co-receptor for BMP, and TMPRSS6 protein cleaves HJV acting as an inhibitor of HJV expression.
Figure 5.
Hepcidin expression via bone morphogenic protein (BMP)-SMAD signaling pathway in hepatocytes. High iron concentration in plasma increases transferrin saturation, which results in homeostatic iron regulator protein (HFE) detachment from the transferrin receptor (TFRC)/HFE complex, then its interaction with ALK3 (BMP receptor), which leads to increased expression of ALK3 and activation of the BMP-SMAD signaling pathway. High plasma iron also causes stabilization of TFR2, which is associated with HFE, and may also interact with ALK3 (but this is not confirmed). Hemojuvelin (HJV) is a co-receptor for BMP, and TMPRSS6 protein cleaves HJV acting as an inhibitor of HJV expression.


{kind=link}
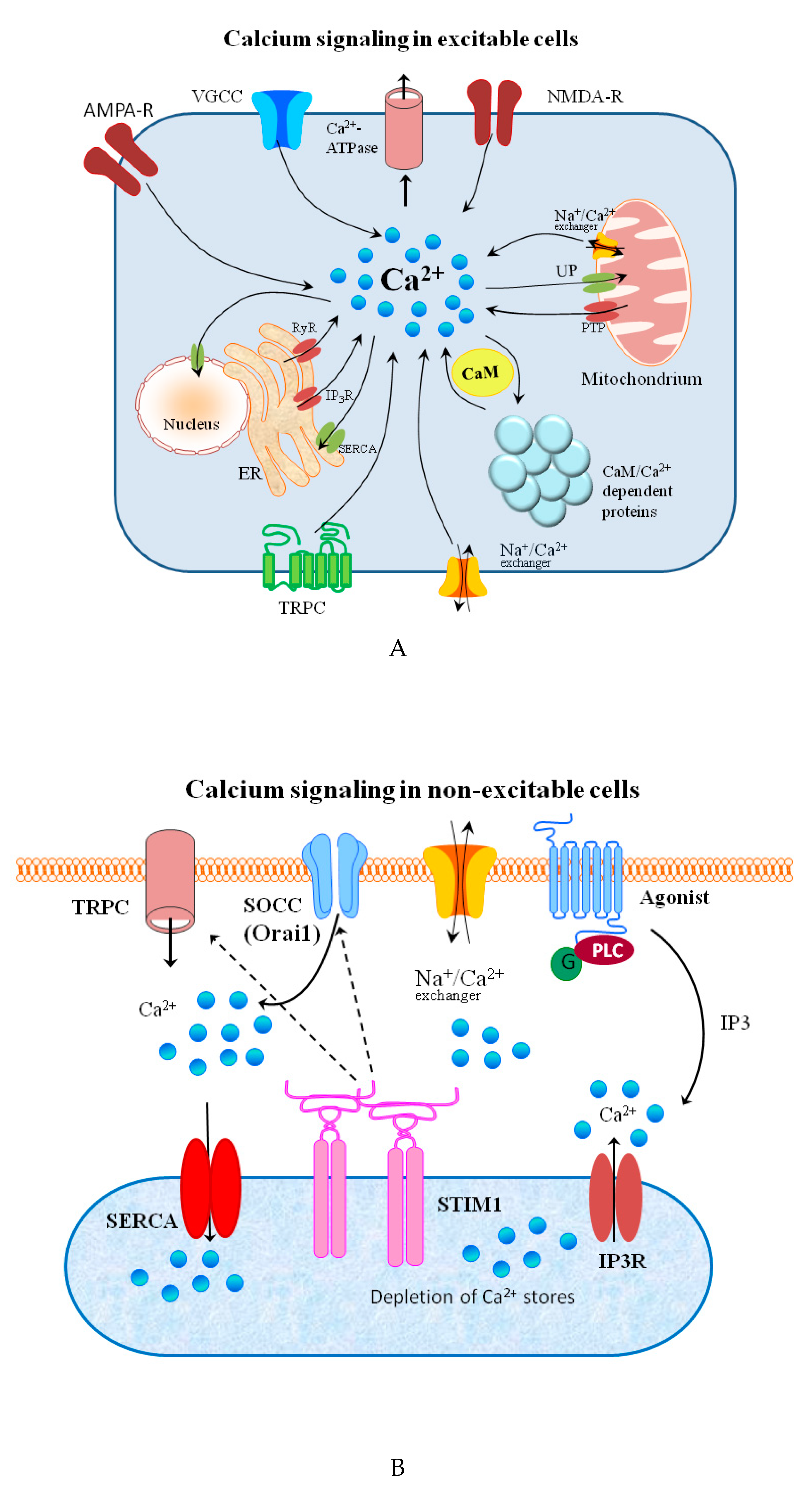{kind=link}
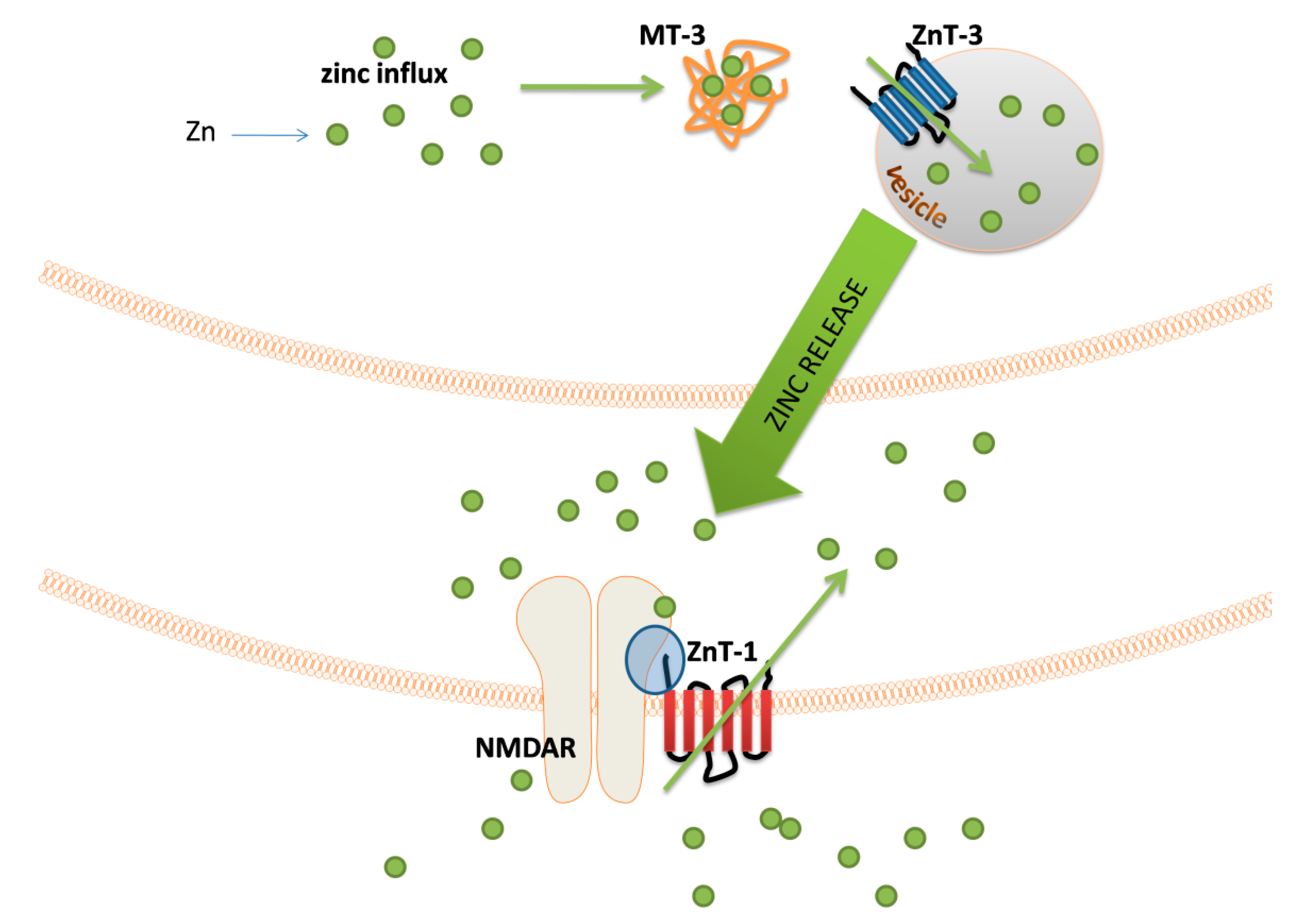{kind=link}
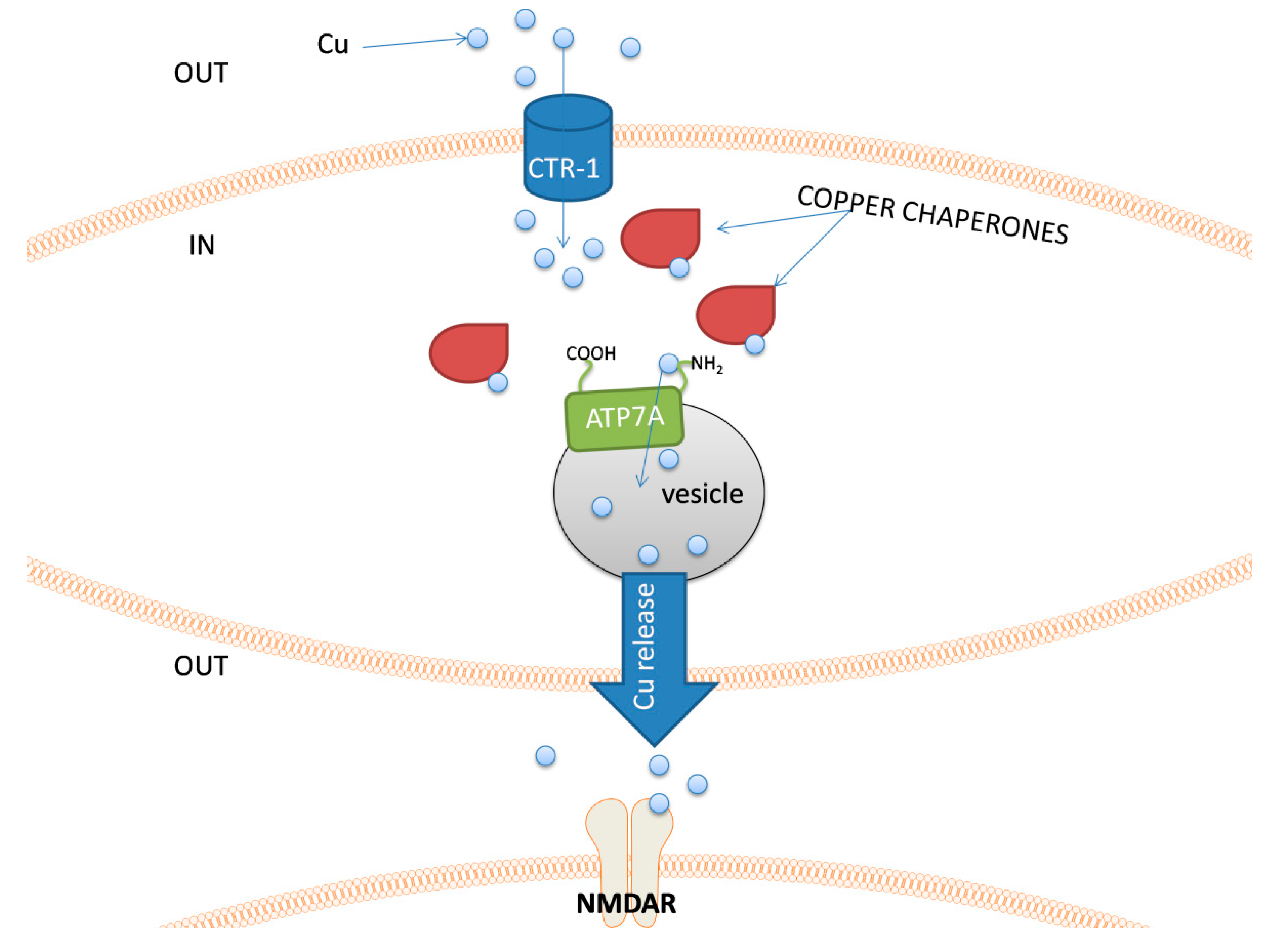{kind=link}
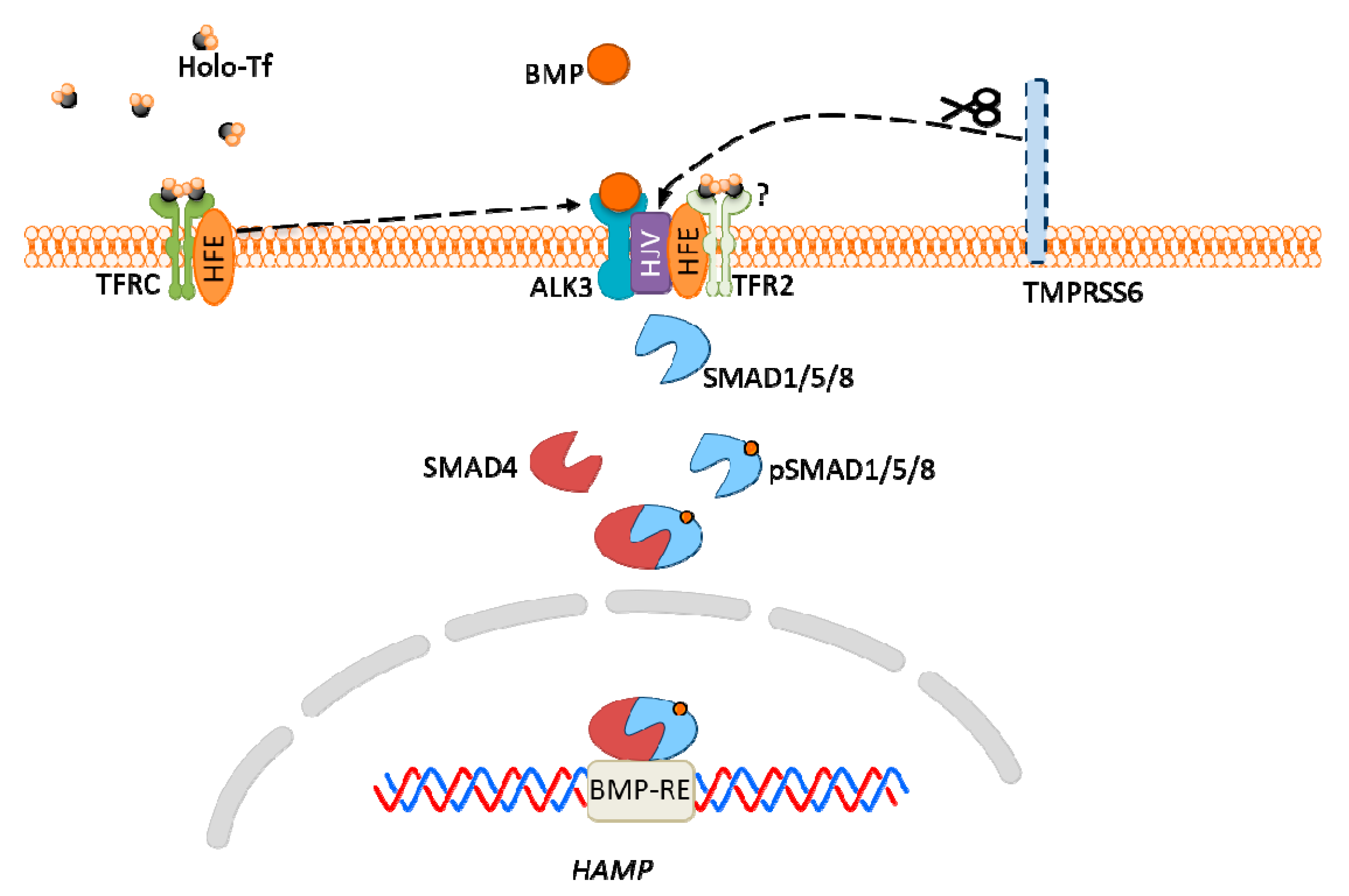{kind=link}
Table 1.
Summary of described signaling functions of chosen metal ions.
| Metal | Metal Ion Location during Signal Induction | Transporters and Receptors Involved in Sensing | Downstream Results |
|---|---|---|---|
| Mg | Intracellular | HIP14 MRS2 MMgT1/2 | - suppression of ROS toxicity - dynamics of cytoskeleton - ribosomal biosynthesis, regulation of translation via mTOR pathway, - antagonizing the Ca2+ signal - PTP inhibition - repair of DNA damage - inhibition of protein aggregation |
| Extracellular | TRPM6 and TRPM7 MagT1 NIPA SLC41A1(controversial: Na+/Mg2+ transporter or Mg2+ sensor) CNNM (controversial: Na+/Mg2+ transporter or Mg2+ sensor) | - growth factor signaling - membrane stabilization - channel regulation - osteoblast apoptosis | |
| Ca | Intracellular | IP3Rs RYRs NFAT NAADP cADPR Calmodulin STIM1 | - gene transcription - T-cell activation and development - CaMKs activation - insulin synthesis - fertilization - learning and memory |
| Extracellular | TRPMs TRPCs VGCCs Kinases Caspase-3 CaSR Orai1 (Calcium Release-Activated Calcium Modulator 1) | - membrane potential modulation - response to many kinds of stresses - signal transduction - neuronal synaptic transmission - apoptosis - regulation of PTH - cell proliferation, mobility | |
| Zn | Intracellular | MT-3 ZnT-3 ZnT-1 | - modulation of mitochondrial activity - vesicle formation - regulation of postsynaptic signal transduction |
| Extracellular | NMDAR APMAR VGCCs | - synaptic signal transduction and/or neuromodulation | |
| Cu | Intracellular | Chaperones ATP7A | - proper functioning of mitochondria - vesicle formation - regulation of cell redox processes |
| Extracellular | PAM DBH NMDAR AMPAR | - neurotransmitter synthesis modulation - reduced influx of Ca2+ - modulation of GABA and other amino-acids receptors | |
| Fe | Intracellular (LIP) | RE-BP bound to IRE at mRNA CDK1-JAK1-STAT3 pathway PHD2 | - ferritin and FPN1 translation; suppression of translation of TFRC and DMT1 - tumor cell proliferation - apoptosis - ferroptosis |
| Extracellular (iron-Tf) | HFE/TFRC TFR2 | - hepcidin expression |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Krzywoszyńska, K.; Witkowska, D.; Świątek-Kozłowska, J.; Szebesczyk, A.; Kozłowski, H. General Aspects of Metal Ions as Signaling Agents in Health and Disease. Biomolecules 2020, 10, 1417. https://doi.org/10.3390/biom10101417
AMA Style
Krzywoszyńska K, Witkowska D, Świątek-Kozłowska J, Szebesczyk A, Kozłowski H. General Aspects of Metal Ions as Signaling Agents in Health and Disease. Biomolecules. 2020; 10(10):1417. https://doi.org/10.3390/biom10101417
Chicago/Turabian StyleKrzywoszyńska, Karolina, Danuta Witkowska, Jolanta Świątek-Kozłowska, Agnieszka Szebesczyk, and Henryk Kozłowski. 2020. "General Aspects of Metal Ions as Signaling Agents in Health and Disease" Biomolecules 10, no. 10: 1417. https://doi.org/10.3390/biom10101417
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.