
Abstract
Background
Clinicians have questioned whether any disorder involving seizures and neural antibodies should be called “(auto)immune epilepsy.” The concept of “acute symptomatic seizures” may be more applicable in cases with antibodies against neural cell surface antigens. We aimed at determining the probability of achieving seizure-freedom, the use of anti-seizure medication (ASM), and immunotherapy in patients with either constellation. As a potential pathophysiological correlate, we analyzed antibody titer courses.
Methods
Retrospective cohort study of 39 patients with seizures and neural antibodies, follow-up ≥ 3 years.
Results
Patients had surface antibodies against the N-methyl-d-aspartate receptor (NMDAR, n = 6), leucine-rich glioma inactivated protein 1 (LGI1, n = 11), contactin-associated protein-2 (CASPR2, n = 8), or antibodies against the intracellular antigens glutamic acid decarboxylase 65 kDa (GAD65, n = 13) or Ma2 (n = 1). Patients with surface antibodies reached first seizure-freedom (88% vs. 7%, P < 0.001) and terminal seizure-freedom (80% vs. 7%, P < 0.001) more frequently. The time to first and terminal seizure-freedom and the time to freedom from ASM were shorter in the surface antibody group (Kaplan–Meier curves: P < 0.0001 for first seizure-freedom; P < 0.0001 for terminal seizure-freedom; P = 0.0042 for terminal ASM-freedom). Maximum ASM defined daily doses were higher in the groups with intracellular antibodies. Seizure-freedom was achieved after additional immunotherapy, not always accompanied by increased ASM doses. Titers of surface antibodies but not intracellular antibodies decreased over time.
Conclusion
Seizures with surface antibodies should mostly be considered acute symptomatic and transient and not indicative of epilepsy. This has consequences for ASM prescription and social restrictions. Antibody titers correlate with clinical courses.
Similar content being viewed by others


Introduction
In its most recent classification paper, the International League Against Epilepsy (ILAE) introduced the new etiological category of “immune epilepsy.” The ILAE herewith referred to the emerging group of autoimmune encephalitides and explicitly mentioned antibodies against the N-methyl-d-aspartate receptor (NMDAR) and leucine-rich glioma inactivated protein 1 (LGI1) [40]. Autoimmune conditions with seizures have frequently been studied under the heading “autoimmune epilepsy” [3, 31, 39]. Recently, researchers [24] and the Autoimmunity/Inflammation Task Force [44] of the ILAE have suggested that patients with autoimmune encephalitides and pathogenic antibodies against cell surface antigens should be considered to have “seizures secondary to autoimmune encephalitis” in the sense of acute symptomatic seizures [1], should not regularly receive long-term anti-seizure medication (ASM), and be exempt from the social restrictions for epilepsy patients. In contrast, autoimmune-related seizures that recur in an unprovoked manner and are resistant to immunological therapy should be called “autoimmune-associated epilepsy.” These conditions are T cell-driven encephalitides [6], often with antibodies against intracellular antigens like glutamic acid decarboxylase 65 kDa (GAD65) [9], onconeural proteins [42] or Rasmussen encephalitis [50]. There are unfortunate cases with surface antibodies who also develop “autoimmune-associated epilepsy.” Geis and colleagues tentatively suggested one year as a cut-off between seizures secondary to autoimmune encephalitis and autoimmune epilepsy [24]; the ILAE task force felt that the database for such a border was still insufficient [44]. Here, we present long-term outcome data that support such a general distinction, but with a much longer time frame to become seizure-free for the group of patients with autoimmune encephalitides and surface antibodies.
Methods
Patients
We included patients of any age if they: (1) harbored neural antibodies in serum or CSF (CSF antibodies were needed for the diagnosis of NMDAR antibodies [25], serum titers ≥ 1:128 were required for contactin-associated protein-2 (CASPR2) antibodies [4], and ≥ 1:1500 for GAD65 antibodies [15]); (2) had > 1 seizure; (3) a follow-up of ≥ 36 months; and (4) at least three antibody titers (serum or CSF).
CGB identified the patients from the databases of the Bethel Antibody Laboratory and the Laboratory Krone (2011–2019). The treating physicians offered repeated consultations and examinations, including antibody tests for clinical reasons and the patients utilized these services. The responsible physicians retrospectively collected clinical information. AR and CGB extracted the following data: seizure frequency or occurrence of seizures (yes/no); ASM defined daily doses (DDD); time to antibody diagnosis and time to start of immunotherapy; types and numbers of immunotherapies and their duration. AR and CGB rated clinical performance by consensus according to the modified Rankin Scale (mRS) [26]. Values ≤ 2 indicate independent living of the patient and values > 2 increasing degrees of dependency. “Seizure-freedom” and “ASM-freedom” required this status for ≥ 12 months. We only counted clinical seizures.
Methods
Antibodies and their titers (multiples of 1:2) were determined in the Bethel Antibody Laboratory and (since 2016) in the Laboratory Krone, as described previously [2, 4]. Ma2 titers were determined with the tissue-based assay [2, 4].
Graphs and statistics
Demographic data are presented in Fig. 1. For group-wise comparisons, we used the Mann–Whitney U test or the two-tailed Fisher’s exact test (Prism, version 6, GraphPad Software Inc., San Diego, CA). We depicted the times to seizure and ASM-freedom using Kaplan–Meier curves (Fig. 2) and analyzed the differences with Mantel–Cox log-rank tests (Prism). For each patient, we prepared diagrams for the following parameters: seizures, ASM, mRS, immunotherapies, and antibody titers in serum and CSF; they are shown in the Supplementary Figure.

Characteristics of the antibody-defined groups. ASM, anti-seizure medication; CASPR2, contactin-associated protein-2; DDD, defined daily dose; GAD65, glutamic acid decarboxylase 65 kDa; LGI1, leucine-rich glioma inactivated protein 1; NMDAR, N-methyl-d-aspartate receptor. Lines and whiskers indicate medians and interquartile ranges

Seizure and anti-seizure medication (ASM) freedom over time. Kaplan-Meier-curves. Lines: censored cases. CASPR2, contactin-associated protein-2; GAD65, glutamic acid decarboxylase 65 kDa; LGI1, leucine-rich glioma inactivated protein 1; NMDAR, N-methyl-d-aspartate receptor
We preliminarily set the significance level to P < 0.05 and used Bonferroni correction for 11 tests. Thus, the significance level was P < 0.0045. We averaged antibody titers (expressed as the percent of the individual’s highest recorded titer) using 15 neighboring points with a second order polynomial smoothing (Prism), see Fig. 3.

Courses of antibody titers in the cerebrospinal fluid (CSF, upper row, blue) and serum (lower row, red) during the first eight years after disease onset. The dots show all individual values, expressed as a percent of the individual’s highest titer. The blue dots in G and H are those of the patient with Ma2 antibodies. The lines are smoothed averages (see the Methods section)
Ethics
This study was approved by the Ethics Committee of the University of Münster, Germany (2018–436-f-S). Since this was a retrospective analysis of data from the authors’ own clinical practice, patients’ consent was not required.
Results
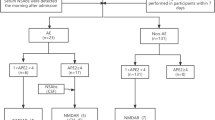
We included 39 patients. The median disease duration from disease manifestation to most recent follow-up was 7.5 years (range 3.0–35.1), and the median time from antibody detection to most recent follow-up was 6.4 years (3.0–13.9 years). The patients harbored antibodies directed against the following antigens: NMDAR, n = 6; LGI1, n = 11; CASPR2, n = 8 (cell surface antigens); GAD65, n = 13; Ma2, n = 1 (intracellular antigens). The patients’ characteristics are shown in Fig. 1 and Table 1; individual data and information, which patients had been included in previous studies [2, 4,5,6, 15, 36, 41] are given in Supplementary Table 1.
Demographics were similar to previous publications on surface [10], GAD65 [38], and Ma2 [11] antibodies. All patients with surface antibodies (apart from patient CASPR2-2) had definite autoimmune encephalitides according to recent diagnostic criteria [25], either in the form of definite limbic encephalitis [25] or faciobrachial dystonic seizures [46]. In contrast, only two patients with intracellular antibodies (GAD65-4, -9) started as definite autoimmune encephalitis, both in the form of a limbic encephalitis (P < 0.0001). The maximum ASM-DDD were higher in the patients with intracellular antibodies (median 3.0, range 0–5.0) compared to those with surface antibodies (median 1.3, range 0–6.8, P = 0.0003). The time from manifestation to antibody diagnosis was longer with intracellular antibodies (median 67, range 0–333 months) compared to surface antibodies (median 4, range 0–48 months, P = 0.0001). The following immunotherapies were administered (in brackets: number of treated patients): intravenous methylprednisolone (IVMP) or oral prednisolone (n = 37); intravenous immunoglobulins (IVIG, n = 15); immunoadsorption (n = 22); plasma exchange (n = 1); azathioprine (n = 17); mycophenolate mofetil (n = 11); rituximab (n = 7); cyclophosphamide (n = 6); natalizumab (n = 2); basiliximab (n = 1). The groups did not differ in the number of immunotherapies (surface antibodies: median 3, range 1–6; intracellular antibodies: median 2.5, range 1–7; P = 0.62) or duration of immunotherapies (surface antibodies: median 3.8, range 0–12.5 years; intracellular antibodies: median 2.9, range 0.4–13.8 years; P = 0.57).
Hippocampal sclerosis
Hippocampal sclerosis (HS), on magnetic resonance imaging (MRI), developed in patients with antibodies against LGI1 (LGI1-2, -4, -5, -6, -7, -9, -11; 64%), GAD65 (GAD65-4, -7, -9, -13; 31%), and CASPR2 (CASPR2-4; 20%), but not with NMDAR antibodies. The patient with Ma2 antibodies underwent an anteromedial temporal lobe resection ten years after disease onset; histologically, he had HS, ILAE type 3 with signs of chronic inflammation. HS had not been diagnosed preoperatively by MRI. A postoperative follow-up is not yet available. The following patients also underwent temporal lobe surgery, with histopathology congruent to MRI diagnoses: GAD65-4, HS (type 3); GAD65-5, no HS; GAD65-9, HS (type 3); GAD-13 HS (type 1). None of them became seizure-free.
Seizure-freedom, ASM-freedom
At most recent follow-up, the group with surface antibodies was superior regarding first and terminal seizure-freedom (Table 2). Median duration of seizure-freedom at most recent follow-up was 4.4 years (range 1.8–7.0 years). The time to first and terminal seizure-freedom and to ASM-freedom is depicted in Fig. 2 in the form of Kaplan–Meier curves. The patients with surface antibodies did better in all three parameters. Supplementary Table 2 shows the year-wise proportions of seizure-free patients.
While the patients with intracellular antibodies only exceptionally became seizure-free, seizure frequency decreased over the years by ≤ 50% compared to onset in 8/13 cases (in one case—GAD-7-no information about treatment during improvement was available). The seizure reduction was related to immunotherapy (GAD-12), ASM plus immunotherapy (GAD-4, -9 [plus epilepsy surgery], 10), ASM only (GAD-3, GAD-6) or no new intervention (GAD-1, -2).
Of note, 13/39 patients achieved terminal seizure-freedom after > 1 year (Supplementary Table 1); the maximum lapse was ten years (patient LGI1-9). This was partly due to relapses in the surface group (there were no relapses with intracellular antibodies). During relapses with seizures, semiology was the same as before where sufficient data were available. In 7/25 patients in the surface group, even the time to first seizure-freedom lasted more than one year; five of them continuously had seizures before they achieved first seizure-freedom (median 21 months, range 15–87). When patients became seizure-free, they had received additional immunotherapy but not always more intense ASM (Supplementary Table 3).
Antibody titer courses
Titer courses started at different heights. The normalized and averaged serum and CSF antibody titers decreased over time in the patients with surface antibodies (exceptions on the individual level were only serum antibodies in patients CASPR2-5, -7,-8), but not in those with intracellular antibodies (Fig. 3).
In many individuals, the titer courses corresponded with fluctuations in seizure frequency and mRS (NMDAR-1, -4 in CSF; NMDAR-1, -4, -5, -6, LGI1-4, -6, -7, CASPR2-3, -7 in serum, partly in the absence of sufficient CSF studies). An elevation in serum LGI1 antibodies heralded the first relapse in patient LGI1-4. In other instances, titers increased again without clinical deterioration (LGI1-2, CASPR2-1, -4, -6, -8, all in serum). Occasionally, serum titers kept decreasing after seizures and elevated mRS had already remitted (LGI1-3, LGI1-11, CASPR2-2, all in serum). Elevated CSF antibodies became unmeasurable in only 2/15 cases with surface antibodies (both LGI1) and 3/13 with intracellular antibodies (all GAD65). Serum titers became undetectable in 7/11 cases with antibodies against LGI1, 2/5 against the NMDAR, 0/8 against CASPR2, 0/13 against GAD65, and 0/1 against Ma2. Apheresis had the strongest immediate effect on titers (NMDAR-2, CASPR2-3, -4 in CSF; LGI1-1, -8, -10, in serum). However, titers often rose again after such interventions, especially if there was insufficient steroid or immunosuppressive treatment after the apheresis (NMDAR-1, -3, -4, -5, LGI1-2, -4, -5, -11, CASPR2-1, -3, -4, -7, -8, all in serum, an exception being LGI1-10).
In most patients with intracellular antibodies, serum and CSF titers did not decrease in the long term. Periods of intense immunotherapy could lower the titers, but they eventually increased again (GAD65-1, -5, 6, -8, 9, 10 in serum; GAD65-2, -3, -10 in serum and CSF). The case of GAD65-12 was exceptional and spectacular. This patient, with recent manifestation of diabetes mellitus type I and Hashimoto thyroiditis, had her first seizure at age 15 years. There were no other features of limbic encephalitis. GAD65 antibodies were detected two weeks later, and she received her first monthly IVMP pulse with another 4 weeks later. Six pulses, five times of 1 g each, were administered. She never received ASM. Seizure frequency reduced. She had her last seizure 2.5 months after the first IVMP pulse. Her titers were strongly reduced.
Discussion
This retrospective study examined patients with neural autoantibodies and seizures and—as the most interesting novelty—analyzed long-term courses ≥ 3 years (median 7.5 years). Patients with surface antibodies achieved first and terminal seizure-freedom in 88% and 80% of cases. This is the same proportion as the 81% of patients with a seizure in close temporal association with a documented brain insult and without subsequent unprovoked seizures over ten years in a classical study [28]. These seizures were defined by the ILAE as “acute symptomatic” [1]. In contrast, only one patient with intracellular antibodies (7%) reached this favorable outcome (Table 2). The remaining rate of 93% of patients with subsequent unprovoked seizures lies in the range of the > 60% relapse risk that defines epilepsy according to the ILAE [22]. Patients with surface antibodies had lower maximum ASM-DDD (Fig. 1b), and ASM were discontinued earlier (Fig. 2a vs. 2E). Patients could become seizure-free after more than one year, always with additional immunotherapies, but not necessarily with ASM-DDD increases; in fact, the patients LGI1-2, -4, -11 became seizure-free while they were not taking any ASM (Supplementary Table 3). These observations correlate with the decreasing titers of surface but not of intracellular antibodies (Fig. 3). Thus, autoimmune encephalitides with NMDAR, LGI1, or CASPR2 antibodies are ictogenic but not usually epileptogenic. They force the brain to seize as long as the antibodies are present in a sufficiently high concentration but they rarely transform the central nervous system in a way that it generates recurrent unprovoked seizures, i.e., the observed seizures are but symptoms of a transient external disease process [1]. Intracellular antibodies, on the other hand, seem to be markers of a profound, enduring, and ASM-resistant propensity to generate recurrent unprovoked epileptic seizures, i.e., epilepsy [23]. These patients even respond poorly to epilepsy surgery, here and in the literature [8]. Out data thereby confirm previous hypotheses [24, 44]. They refine Geis’ and colleagues' preliminary suggestion that seizures for more than one year in patients with autoimmune encephalitides should lead to the diagnosis of epilepsy. According to our data, it can take up to seven years before continuously recurrent seizures secondary to an autoimmune encephalitis subside.
The term acute symptomatic seizures may appear stretched in such long-term courses. These are, however, exceptional in this sample. Whereas the demographic data of our cases were as expected from the antibody types, the long clinical follow-up was special. Delays to seizure-freedom were much longer in our patients than in a previous unselected series that reported on a median lag to seizure remission within a month after start of immunotherapy (interquartile range 0.3–2.4 months) [13]. Our long-term follow-up sample demonstrates the concept at its extremes. At the same time, it shows that terminal seizure-freedom in such cases was long-lasting and stable (≥ 21 months of terminal seizure-free follow-up).
An epilepsy diagnosis results in marked driving restrictions and limited professional abilities. We suggest that patients with seizures secondary to autoimmune encephalitides and antibodies against NMDAR, LGI1, or CASPR2 should be individually evaluated for social restrictions, as suggested by a UK guideline that mentions “limbic encephalitis associated with seizures” together with acute encephalitides and meningitis, apart from chronic epilepsy [16]. Congruently, a review article reported a low long-term risk (< 15%) to develop epilepsy after encephalitides associated with surface antibodies [43].
The time to diagnosis of surface antibodies was shorter compared to intracellular antibodies (NMDAR < LGI1 < CASPR < < GAD65, Table 1 and Fig. 1d). This phenomenon is probably due to the subacute start in the autoimmune encephalitides with surface antibodies [13], which is only occasionally noted in patients with GAD65 antibodies and a limbic encephalitis at onset [37].
Individual titer courses of surface antibodies, to some extent, moved in parallel to the clinical courses. In some cases, antibodies increased again despite clinical recovery, or antibodies fell more slowly than patients recovered—especially CASPR2 antibodies. Antibody titers, thus, did not in general predict the immediately subsequent clinical course. Titers of intracellular antibodies remained high or increased again after transient reduction by intense immunotherapies. Titer courses confirmed chronicity but were otherwise not clinically informative.
HS occurred at similar frequencies in patients with surface and intracellular antibodies. It occurred most frequently with LGI1 antibodies (64%), despite the favorable seizure outcome in this group. This proportion is in line with existing figures of 41% [49] or 50% [21]. Other studies reported hippocampal abnormalities with NMDAR antibodies in < 10%, [36, 37] again congruent with our data (0%) [20, 30]. CASPR2 antibodies (20%) and GAD65 antibodies (31%) were between those values, again consistent with the literature reporting on 20–24% [32, 48] and 33–62% [17, 35] of cases, respectively, having hippocampal lesions or atrophy/sclerosis. Hence, in patients with neural antibodies, obvious hippocampal damage is neither necessary nor sufficient for the development of epilepsy. A caveat comes from the case with Ma2 antibodies, whose HS was only diagnosed under microscope. The true frequency of structural epileptogenic damage might be underestimated in this series.
Immunotherapy is the most relevant treatment in seizures secondary to autoimmune encephalitis. Recurrent seizures and cognitive decline may be prevented if immunotherapy is applied early in patients with surface antibodies [7, 13, 29, 46, 47] but not with intracellular antibodies [7, 36]. This hypothesis is confirmed by the present series. The first-line and second-line therapy concept derived from the retrospective analysis of treatment courses in anti-NMDAR encephalitis [12] has been widely adopted in autoimmune encephalitides with different surface antibodies [7, 33]. It can be recognized in cases of the present series, including those with anti-NMDAR encephalitis who only responded to second-line therapy with rituximab. Most patients had several immunological treatment approaches. Due to the retrospective and uncontrolled documentation of combination therapies, we did not attempt to disentangle the contribution of single interventions.
Patients with intracellular antigens did not benefit from immunotherapy, except for one patient (GAD65-12) who became seizure-free under IVMP pulses starting six weeks after disease onset. A similar case has recently been described [14]. In contrast, immunotherapy does not stop seizures in chronic patients with GAD65 antibodies [36].
Plasmapheresis or immunoadsorption had an immediate but no long-lasting effect on antibody courses. A special case was patient LGI1-10, who stopped having seizures and had reduced antibody titers after only one sequence of immunoadsorption.
ASMs are mostly considered as an add-on-therapy for patients with autoimmune encephalitides with surface antibodies [18], but usually not as a stand-alone or long-term treatment [13, 34]. They usually do not have a strong effect unless applied together with immunotherapy [19, 46]. This can also be observed in our sample: Only 1/11 cases, in which ASM was discontinued, had a seizure relapse (NMDAR-1, latency 2 days, 14 months after disease onset; immunotherapy had been stopped 3.5 months before, and there were still NMDAR antibodies in serum and CSF). Vice versa, only 1/3 cases with introduction of an ASM without a parallel intensification or introduction of immunotherapy experienced a reduction in seizure frequency (NMDAR-5, four years after onset); in the other two instances, mere introduction of ASM was without effect on seizure frequency (LGI1-7, 4.5 years after onset; CASPR2-4, 3.5 years after onset, both with HS).
This study has limitations. First, we documented and rated data retrospectively, a process that has inherent problems. Second, the study group is biased toward difficult-to-treat patients. Third, patients were treated by various ASM and immunotherapies in different orders and combinations. Fourth, the patients did not systematically undergo prolonged video-EEG monitoring to capture unnoticed or unaware seizures [45]. Fifth, the data do not directly determine ante hoc in an individual patient with surface antibodies when to diagnose an “epilepsy.” One may tentatively suggest to diagnose epilepsy if seizures go on for more than one year even though the antibodies in serum (LGI1 or CASPR2) or CSF (NMDAR) [27] have gone down by more than three or more than titer levels compared to onset, especially (but not necessarily), if potentially epileptogenic atrophic brain damage is evident. This phenomenon seems to be the case for these four patients: LGI1-7, CASPR2-4 (both with HS), and NMDAR-3, -5 (without structural damage). After symptom control, immunotherapies could be discontinued without early deteriorations, regardless of titers (NMDAR-6, LGI1-1, -3, -5, -7 [epilepsy persisted], -8, -9, 10, CASPR2-1, -4 [epilepsy persisted],-5, -6; two of these patients relapsed after > 1 year: NMDAR-6; LGI1-9). ASM contributed less than immunotherapies; they could be discontinued without provoking a seizure relapse (the only exception being NMDAR-1).
In conclusion, this study shows, on a group level, that patients with autoimmune encephalitides and surface antibodies have acute symptomatic seizures that do usually not require long-term immunological or ASM therapy or social restrictions. In contrast, patients with intracellular antibodies and seizures—if not treated early on—develop epilepsy. Antibody titers can partially help to interpret the clinical courses, especially on the group level.
Availability of data material
Data will be shared upon request from any qualified investigator, while maintaining anonymization of the patients.
Abbreviations
- ASM:
-
Anti-seizure medication
- CASPR2:
-
Contactin-associated protein-2
- DDD:
-
Defined daily doses
- GAD65:
-
Glutamic acid decarboxylase 65 kDa
- HS:
-
Hippocampal sclerosis
- ILAE:
-
International league against epilepsy
- LGI1:
-
Leucine-rich glioma inactivated protein 1
- NMDAR:
-
N-Methyl-d-aspartate receptor
- IVMP:
-
Intravenous methylprednisolone
References
Beghi E, Carpio A, Forsgren L, Hesdorffer DC, Malmgren K, Sander JW, Tomson T, Hauser WA (2010) Recommendation for a definition of acute symptomatic seizure. Epilepsia 51:671–675. https://doi.org/10.1111/j.1528-1167.2009.02285.x
Bien CG, Bien CI, Dogan Onugoren M, De Simoni D, Eigler V, Haensch CA, Holtkamp M, Ismail FS, Kurthen M, Melzer N, Mayer K, von Podewils F, Rauschka H, Rossetti AO, Schabitz WR, Simova O, Witt K, Hoftberger R, May TW (2020) Routine diagnostics for neural antibodies, clinical correlates, treatment and functional outcome. J Neurol 267:2101–2114. https://doi.org/10.1007/s00415-020-09814-3
Bien CG, Holtkamp M (2017) "Autoimmune epilepsy": encephalitis with autoantibodies for epileptologists. Epilepsy Curr 17:134–141. https://doi.org/10.5698/1535-7511.17.3.134
Bien CG, Mirzadjanova Z, Baumgartner C, Onugoren MD, Grunwald T, Holtkamp M, Isenmann S, Kermer P, Melzer N, Naumann M, Riepe M, Schabitz WR, von Oertzen TJ, von Podewils F, Rauschka H, May TW (2017) Anti-contactin-associated protein-2 encephalitis: relevance of antibody titres, presentation and outcome. Eur J Neurol 24:175–186. https://doi.org/10.1111/ene.13180
Bien CG, Urbach H, Schramm J, Soeder BM, Becker AJ, Voltz R, Vincent A, Elger CE (2007) Limbic encephalitis as a precipitating event in adult-onset temporal lobe epilepsy. Neurology 69:1236–1244. https://doi.org/10.1212/01.wnl.0000276946.08412.ef
Bien CG, Vincent A, Barnett MH, Becker AJ, Blümcke I, Graus F, Jellinger KA, Reuss DE, Ribalta T, Schlegel J, Sutton I, Lassmann H, Bauer J (2012) Immunopathology of autoantibody-associated encephalitides: clues for pathogenesis. Brain 135:1622–1638. https://doi.org/10.1093/brain/aws082
Byun JI, Lee ST, Jung KH, Sunwoo JS, Moon J, Lim JA, Lee DY, Shin YW, Kim TJ, Lee KJ, Lee WJ, Lee HS, Jun J, Kim DY, Kim MY, Kim H, Kim HJ, Suh HI, Lee Y, Kim DW, Jeong JH, Choi WC, Bae DW, Shin JW, Jeon D, Park KI, Jung KY, Chu K, Lee SK (2016) Effect of immunotherapy on seizure outcome in patients with autoimmune encephalitis: a prospective observational registry study. PLoS ONE 11:e0146455. https://doi.org/10.1371/journal.pone.0146455
Carreño M, Bien CG, Asadi-Pooya AA, Sperling M, Marusic P, Elisak M, Pimentel J, Wehner T, Mohanraj R, Uranga J, Gomez-Ibanez A, Villanueva V, Gil F, Donaire A, Bargallo N, Rumia J, Roldan P, Setoain X, Pintor L, Boget T, Bailles E, Falip M, Aparicio J, Dalmau J, Graus F (2017) Epilepsy surgery in drug resistant temporal lobe epilepsy associated with neuronal antibodies. Epilepsy Res 129:101–105. https://doi.org/10.1016/j.eplepsyres.2016.12.010
Daif A, Lukas RV, Issa NP, Javed A, VanHaerents S, Reder AT, Tao JX, Warnke P, Rose S, Towle VL, Wu S (2018) Antiglutamic acid decarboxylase 65 (GAD65) antibody-associated epilepsy. Epilepsy Behav 80:331–336. https://doi.org/10.1016/j.yebeh.2018.01.021
Dalmau J, Graus F (2018) Antibody-mediated encephalitis. N Engl J Med 378:840–851. https://doi.org/10.1056/NEJMra1708712
Dalmau J, Graus F, Villarejo A, Posner JB, Blumenthal D, Thiessen B, Saiz A, Meneses P, Rosenfeld MR (2004) Clinical analysis of anti-Ma2-associated encephalitis. Brain 127:1831–1844. https://doi.org/10.1093/brain/awh203
Dalmau J, Lancaster E, Martinez-Hernandez E, Rosenfeld MR, Balice-Gordon R (2011) Clinical experience and laboratory investigations in patients with anti-NMDAR encephalitis. Lancet Neurol 10:63–74. https://doi.org/10.1016/S1474-4422(10)70253-2
de Bruijn MAAM, van Sonderen A, van Coevorden-Hameete MH, Bastiaansen AEM, Schreurs MWJ, Rouhl RPW, van Donselaar CA, Majoie M, Neuteboom RF, Sillevis Smitt PAE, Thijs RD, Titulaer MJ (2019) Evaluation of seizure treatment in anti-LGI1, anti-NMDAR, and anti-GABABR encephalitis. Neurology 92:e2185–e2196. https://doi.org/10.1212/WNL.0000000000007475
Di Giacomo R, Deleo F, Pastori C, Didato G, Andreetta F, Del Sole A, de Curtis M, Villani F (2019) Predictive value of high titer of GAD65 antibodies in a case of limbic encephalitis. J Neuroimmunol 337:577063. https://doi.org/10.1016/j.jneuroim.2019.577063
Dogan Onugoren M, Golombeck KS, Bien C, Abu-Tair M, Brand M, Bulla-Hellwig M, Lohmann H, Munstermann D, Pavenstadt H, Tholking G, Valentin R, Wiendl H, Melzer N, Bien CG (2016) Immunoadsorption therapy in autoimmune encephalitides. Neurol Neuroimmunol Neuroinflamm 3:e207. https://doi.org/10.1212/NXI.0000000000000207
Driver & Vehicle Licensing Agency (2020) Assessing fitness to drive—a guide for medical professionals. https://www.gov.uk/government/publications/assessing-fitness-to-drive-a-guide-for-medical-professionals. Accessed on 05.04.2020
Falip M, Rodriguez-Bel L, Castañer S, Sala-Padró J, Miro J, Jaraba S, Casasnovas C, Morandeira F, Berdejo J, Carreño M (2019) Hippocampus and insula are targets in epileptic patients with glutamic acid decarboxylase antibodies. Front Neurol 9:1143. https://doi.org/10.3389/fneur.2018.01143
Feyissa AM, Lamb C, Pittock SJ, Gadoth A, McKeon A, Klein CJ, Britton JW (2018) Antiepileptic drug therapy in autoimmune epilepsy associated with antibodies targeting the leucine-rich glioma-inactivated protein 1. Epilepsia Open 3:348–356. https://doi.10.1002/epi4.12226
Feyissa AM, Lopez Chiriboga AS, Britton JW (2017) Antiepileptic drug therapy in patients with autoimmune epilepsy. Neurol Neuroimmunol Neuroinflamm 4:e353. https://doi.org/10.1212/NXI.0000000000000353
Finke C, Kopp UA, Scheel M, Pech LM, Soemmer C, Schlichting J, Leypoldt F, Brandt AU, Wuerfel J, Probst C, Ploner CJ, Pruss H, Paul F (2013) Functional and structural brain changes in anti-N-methyl-d-aspartate receptor encephalitis. Ann Neurol 74:284–296. https://doi.org/10.1002/ana.23932
Finke C, Prüss H, Heine J, Reuter S, Kopp UA, Wegner F, Then Bergh F, Koch S, Jansen O, Münte T, Deuschl G, Ruprecht K, Stöcker W, Wandinger KP, Paul F, Bartsch T (2017) Evaluation of cognitive deficits and structural hippocampal damage in encephalitis with leucine-rich, glioma-inactivated 1 antibodies. JAMA Neurol 74:50–59. https://doi.org/10.1001/jamaneurol.2016.4226
Fisher RS, Acevedo C, Arzimanoglou A, Bogacz A, Cross JH, Elger CE, Engel J Jr, Forsgren L, French JA, Glynn M, Hesdorffer DC, Lee BI, Mathern GW, Moshe SL, Perucca E, Scheffer IE, Tomson T, Watanabe M, Wiebe S (2014) ILAE official report: a practical clinical definition of epilepsy. Epilepsia 55:475–482. https://doi.org/10.1111/epi.12550
Fisher RS, van Emde BW, Blume W, Elger C, Genton P, Lee P, Engel J Jr (2005) Epileptic seizures and epilepsy: definitions proposed by the International League Against Epilepsy (ILAE) and the International Bureau for Epilepsy (IBE). Epilepsia 46:470–472. https://doi.org/10.1111/j.0013-9580.2005.66104.x
Geis C, Planaguma J, Carreño M, Graus F, Dalmau J (2019) Autoimmune seizures and epilepsy. J Clin Invest 129:926–940. https://doi.org/10.1172/JCI125178
Graus F, Titulaer MJ, Balu R, Benseler S, Bien CG, Cellucci T, Cortese I, Dale RC, Gelfand JM, Geschwind M, Glaser CA, Honnorat J, Hoftberger R, Iizuka T, Irani SR, Lancaster E, Leypoldt F, Pruss H, Rae-Grant A, Reindl M, Rosenfeld MR, Rostasy K, Saiz A, Venkatesan A, Vincent A, Wandinger KP, Waters P, Dalmau J (2016) A clinical approach to diagnosis of autoimmune encephalitis. Lancet Neurol 15:391–404. https://doi.org/10.1016/S1474-4422(15)00401-9
Graus F, Vega F, Delattre JY, Bonaventura I, Rene R, Arbaiza D, Tolosa E (1992) Plasmapheresis and antineoplastic treatment in CNS paraneoplastic syndromes with antineuronal autoantibodies. Neurology 42:536–540. https://doi.org/10.1212/WNL.42.3.536
Gresa-Arribas N, Titulaer MJ, Torrents A, Aguilar E, McCracken L, Leypoldt F, Gleichman AJ, Balice-Gordon R, Rosenfeld MR, Lynch D, Graus F, Dalmau J (2014) Antibody titres at diagnosis and during follow-up of anti-NMDA receptor encephalitis: a retrospective study. Lancet Neurol 13:167–177. https://doi.org/10.1016/S1474-4422(13)70282-5
Hesdorffer DC, Benn EK, Cascino GD, Hauser WA (2009) Is a first acute symptomatic seizure epilepsy? Mortality and risk for recurrent seizure. Epilepsia 50:1102–1108. https://doi.org/10.1111/j.1528-1167.2008.01945.x
Iorio R, Assenza G, Tombini M, Colicchio G, Della Marca G, Benvenga A, Damato V, Rossini PM, Vollono C, Plantone D (2015) The detection of neural autoantibodies in patients with antiepileptic-drug-resistant epilepsy predicts response to immunotherapy. Eur J Neurol 22:70–78. https://doi.org/10.1111/ene.12529
Irani SR, Bera K, Waters P, Zuliani L, Maxwell S, Zandi MS, Friese MA, Galea I, Kullmann DM, Beeson D, Lang B, Bien CG, Vincent A (2010) N-methyl-d-aspartate antibody encephalitis: temporal progression of clinical and paraclinical observations in a predominantly non-paraneoplastic disorder of both sexes. Brain 133:1655–1667. https://doi.org/10.1093/brain/awq113
Irani SR, Bien CG, Lang B (2011) Autoimmune epilepsies. Curr Opin Neurol 24:146–153. https://doi.org/10.1097/WCO.0b013e3283446f05
Joubert B, Saint-Martin M, Noraz N, Picard G, Rogemond V, Ducray F, Desestret V, Psimaras D, Delattre JY, Antoine JC, Honnorat J (2016) Characterization of a subtype of autoimmune encephalitis with anti-contactin-associated protein-like 2 antibodies in the cerebrospinal fluid, prominent limbic symptoms, and seizures. JAMA Neurol 73:1115–1124. https://doi.org/10.1001/jamaneurol.2016.1585
Lee WJ, Lee ST, Byun JI, Sunwoo JS, Kim TJ, Lim JA, Moon J, Lee HS, Shin YW, Lee KJ, Kim S, Jung KH, Jung KY, Chu K, Lee SK (2016) Rituximab treatment for autoimmune limbic encephalitis in an institutional cohort. Neurology 86:1683–1691. https://doi.org/10.1212/WNL.0000000000002635
Liu X, Yan B, Wang R, Li C, Chen C, Zhou D, Hong Z (2017) Seizure outcomes in patients with anti-NMDAR encephalitis: a follow-up study. Epilepsia 58:2104–2111. https://doi.org/10.1111/epi.13929
Mäkelä K-M, Hietaharju A, Brander A, Peltola J (2018) Clinical management of epilepsy with glutamic acid decarboxylase antibody positivity: the interplay between immunotherapy and anti-epileptic drugs. Front Neurol 9:579. https://doi.org/10.3389/fneur.2018.00579
Malter MP, Frisch C, Zeitler H, Surges R, Urbach H, Helmstaedter C, Elger CE, Bien CG (2015) Treatment of immune-mediated temporal lobe epilepsy with GAD antibodies. Seizure 30:57–63. https://doi.org/10.1016/j.seizure.2015.05.017
Malter MP, Helmstaedter C, Urbach H, Vincent A, Bien CG (2010) Antibodies to glutamic acid decarboxylase define a form of limbic encephalitis. Ann Neurol 67:470–478. https://doi.org/10.1002/ana.21917
Muñoz-Lopetegi A, de Bruijn MA, Boukhrissi S, Bastiaansen AE, Nagtzaam MM, Hulsenboom ES, Boon AJ, Neuteboom RF, de Vries JM, Sillevis Smitt PA, Schreurs MW, Titulaer MJ (2020) Neurologic syndromes related to anti-GAD65: Clinical and serologic response to treatment. Neurol Neuroimmunol Neuroinflamm 7:e696. https://doi.org/10.1212/NXI.0000000000000696
Quek AM, Britton JW, McKeon A, So E, Lennon VA, Shin C, Klein C, Watson RE Jr, Kotsenas AL, Lagerlund TD, Cascino GD, Worrell GA, Wirrell EC, Nickels KC, Aksamit AJ, Noe KH, Pittock SJ (2012) Autoimmune epilepsy: clinical characteristics and response to immunotherapy. Arch Neurol 69:582–593. https://doi.org/10.1001/archneurol.2011.2985
Scheffer IE, Berkovic S, Capovilla G, Connolly MB, French J, Guilhoto L, Hirsch E, Jain S, Mathern GW, Moshé SL (2017) ILAE classification of the epilepsies: position paper of the ILAE commission for classification and terminology. Epilepsia 58:512–521. https://doi.org/10.1111/epi.13709
Schimmel M, Frühwald MC, Bien CG (2018) Limbic encephalitis with LGI1 antibodies in a 14-year-old boy. Eur J Paediatr Neurol 22:190–193. https://doi.org/10.1016/j.ejpn.2017.08.004
Serafini A, Lukas RV, VanHaerents S, Warnke P, Tao JX, Rose S, Wu S (2016) Paraneoplastic epilepsy. Epilepsy Behav 61:51–58. https://doi.org/10.1016/j.yebeh.2016.04.046
Spatola M, Dalmau J (2017) Seizures and risk of epilepsy in autoimmune and other inflammatory encephalitis. Curr Opin Neurol 30:345–353. https://doi.org/10.1097/WCO.0000000000000449
Steriade C, Britton J, Dale RC, Gadoth A, Irani SR, Linnoila J, McKeon A, Shao XQ, Venegas V, Bien CG (2020) Acute symptomatic seizures secondary to autoimmune encephalitis and autoimmune-associated epilepsy: conceptual definitions. Epilepsia 61:1341–1351. https://doi.org/10.1111/epi.16571
Steriade C, Moosa ANV, Hantus S, Prayson RA, Alexopoulos A, Rae-Grant A (2018) Electroclinical features of seizures associated with autoimmune encephalitis. Seizure 60:198–204. https://doi.org/10.1016/j.seizure.2018.06.021
Thompson J, Bi M, Murchison AG, Makuch M, Bien CG, Chu K, Farooque P, Gelfand JM, Geschwind MD, Hirsch LJ, Somerville E, Lang B, Vincent A, Leite MI, Waters P, Irani SR (2018) The importance of early immunotherapy in patients with faciobrachial dystonic seizures. Brain 141:348–356. https://doi.org/10.1093/brain/awx323
Toledano M, Britton JW, McKeon A, Shin C, Lennon VA, Quek AM, So E, Worrell GA, Cascino GD, Klein CJ, Lagerlund TD, Wirrell EC, Nickels KC, Pittock SJ (2014) Utility of an immunotherapy trial in evaluating patients with presumed autoimmune epilepsy. Neurology 82:1578–1586. https://doi.org/10.1212/WNL.0000000000000383
van Sonderen A, Arino H, Petit-Pedrol M, Leypoldt F, Körtvélyessy P, Wandinger KP, Lancaster E, Wirtz PW, Schreurs MW, Sillevis Smitt PA, Graus F, Dalmau J, Titulaer MJ (2016) The clinical spectrum of Caspr2 antibody-associated disease. Neurology 87:521–528. https://doi.org/10.1212/WNL.0000000000002917
van Sonderen A, Thijs RD, Coenders EC, Jiskoot LC, Sanchez E, de Bruijn MA, van Coevorden-Hameete MH, Wirtz PW, Schreurs MW, Sillevis Smitt PA, Titulaer MJ (2016) Anti-LGI1 encephalitis: clinical syndrome and long-term follow-up. Neurology 87:1449–1456. https://doi.org/10.1212/WNL.0000000000003173
Varadkar S, Bien CG, Kruse CA, Jensen FE, Bauer J, Pardo CA, Vincent A, Mathern GW, Cross JH (2014) Rasmussen's encephalitis: clinical features, pathobiology, and treatment advances. Lancet Neurol 13:195–205. https://doi.org/10.1016/S1474-4422(13)70260-6
Funding
Open Access funding enabled and organized by Projekt DEAL. None.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
Dr Bien obtained honoraria for speaking engagements from UCB (Monheim, Germany) and Desitin (Hamburg, Germany). He receives research support from the Deutsche Forschungsgemeinschaft (German Research Council, Bonn, Germany) and Gerd-Altenhof-Stiftung (Deutsches Stiftungs-Zentrum, Essen, Germany). Dr Gobbi reports that the Employer of the Department of Neurology, Neurocenter of Southern Switzerland (NSI), 6900 Lugano, Switzerland receives financial support from Biogen Idec, Celgene, Sanofi, Merck Serono, Novartis Roche, and Teva. The submitted work is not related to these agreements. Dr von Oertzen reports grants, personal fees, and non-financial support from Novartis Pharma, personal fees from Roche Pharma, personal fees from Biogen Idec Austria, personal fees from Liva Nova, grants from Grossegger & Drbal GmbH, grants from Merck, personal fees from Indivior Austria GmbH, personal fees and non-financial support from gtec GmbH Austria, personal fees and non-financial support from Boehringer-Ingelheim, personal fees from Philips, personal fees and non-financial support from UCB Pharma, personal fees from Almirall, personal fees from Eisai, outside the submitted work; and he is web editor in chief of the European Academy of Neurology (EAN), co-chair of the EAN scientific panel for epilepsy, and vice-president of the Österreichische Gesellschaft für Epileptologie (Austrian ILAE chapter). Dr Surges has received fees as a consultant from Bial, Desitin, Eisai, Liva Nova, Novartis, and UCB Pharma. He currently receives research support from the Federal Ministry of Health. Dr. Bien obtained honoraria for speaking engagements from UCB (Monheim, Germany), Desitin (Hamburg, Germany), and Euroimmun (Lübeck, Germany). He receives research support from Deutsche Forschungsgemeinschaft (German Research Council, Bonn, Germany) and Gerd-Altenhof-Stiftung (Deutsches Stiftungs-Zentrum, Essen, Germany). The other authors did not declare any conflict.
Ethics approval
This study was approved by the Ethics Committee of the University of Münster, Germany (2018-436-f-S).
Consent to participate
Since this was a retrospective analysis of data from the authors’ own clinical practice, patients’ consent was not required.
Consent for publication
Since this was a retrospective analysis of data from the authors’ own clinical practice, patients’ consent was not required.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rada, A., Birnbacher, R., Gobbi, C. et al. Seizures associated with antibodies against cell surface antigens are acute symptomatic and not indicative of epilepsy: insights from long-term data. J Neurol 268, 1059–1069 (2021). https://doi.org/10.1007/s00415-020-10250-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-020-10250-6