
Abstract
We present a conservation genetics tool kit, which offers two ready-to-use workflows for the routine application of genetic methods in conservation management. The workflows were optimized for work load and costs and are accompanied by an easy-to-read and richly illustrated manual with guidelines regarding sampling design, sampling of genetic material, necessary permits, laboratory methods, statistical analyses and documentation of results in a practice-oriented way. The manual also provides a detailed interpretation help for the implementation of the results in conservation management. One workflow deals with the identification of pond-breeding amphibians based on metabarcoding and environmental DNA (eDNA) from water samples. This workflow also discriminates the morphologically similar water frogs (Pelophylax sp.) and other closely related species (e.g. Triturus cristatus and T. carnifex). The second workflow studies connectivity among populations using microsatellite markers. Its statistical analyses encompass the detection of genetic groups and historical, recent and current dispersal and gene flow. Using the two workflows does not involve academic research institutes; they can be applied by environmental consultancies, laboratories from the private sector, governmental agencies or non-governmental organisations. These and additional conservation genetic workflows will hopefully foster the routine use of genetic methods in conservation management.
Similar content being viewed by others


Introduction
Conservation genetics has an inherent practical goal, i.e., to use genetic theory and methods to conserve species and safeguard biodiversity (Frankham 1995). However, conservation genetics still is a mainly academic exercise. Applications in conservation management are mostly restricted to larger and emblematic vertebrates or wild species of economic or cultural value to human societies (Holderegger et al. 2019a, b). The myriads of other organisms like plants, insects, sponges, fungi or lichens are rarely dealt with in applied conservation genetics. In other words, there is a gap between conservation genetics science and practice (Sandström et al. 2019).
Many reasons for this science-practice gap have been suggested (Taylor et al. 2017; Grant et al. 2019). Amongst others, possible obstacles are the challenging understanding of conservation genetics and the latter’s prohibitive costs. In addition, conservation genetic results often do not fit the expectations of practitioners and are difficult to communicate to stakeholders (Taylor et al. 2017; Holderegger et al. 2019a, b). In our experience, however, conservation professionals acknowledge the potential of genetics in conservation management, but do not know how to initiate, apply or use genetic tools or how to interpret the output of conservation genetic analyses. Especially, laboratory capacities as well as knowledge to run statistical analyses are not accessible to practitioners. Therefore, help is needed to overcome these obstacles, allowing the use of genetic methods in every-day conservation management.
To understand the last point, a look at how conservation managers usually obtain the information they need is helpful. We illustrate how conservation managers get relevant data and results for their work with three examples. First, in order to describe the diversity of habitat types and landscape elements, conservation managers use, e.g., aerial photographs or existing GIS layers with topography or land cover (Turner et al. 2001). Conservation managers often do not do these analyses themselves, but contract private consultancies to carry them out. Second, if conservation agencies want to detect changes in the ecological conditions of nature reserves, they often apply vegetation analysis. Such vegetation analyses are generally mandated to environmental consultancies, which apply statistical analysis to analyse vegetation data (Wildi 2013). Third, if a wildlife organization wants to know the population size of an animal in a certain area, diverse methods can be used such as direct observation, transect surveys, traps or camera traps (Magurran 2003). Again, these surveys are often outsourced. In all these cases, governmental conservation agencies and non-governmental organizations (NGOs) give a mandate to an environmental consultancy to carry out investigations using routine tools and methods. Because of their long application in conservation management, professionals are used to the kinds of data and results of such studies and are able to interpret them. They are also sure that the results will be available in due time, because of deadlines fixed with the consultancies. Academic research institutions are not involved in the examples given above.
This is in marked contrast to how conservation genetics currently works. Most applied studies in conservation genetics are carried out by researchers or in close collaboration with academic institutions, zoological and botanical gardens or museums (Dufresnes et al. 2019). This fact prompts the question whether one needs to make conservation genetics a routine method—similar to the above examples—that can be readily applied by governmental authorities, NGOs, environmental consultancies and private-sector companies? If so, conservation genetic workflows are a way to move forward. Workflows define and describe the sequence of the steps involved in a process, from initiation to completion (www.merriam-webster.com). Such workflows may be a key component for a better implementation of conservation genetics in conservation management (Holderegger et al. 2019a, b).
Developing workflows for conservation practice
Here, we present ready-to-use conservation genetic workflows developed by a Swiss consortium consisting of a university (University of Zürich), a national research institute (WSL), a university of applied sciences (HSR), a national centre for species distribution data (info fauna-karch), a private laboratory company (Microsynth Ecogenics GmbH) and an environmental consultancy (ARNAL). This consortium guaranteed that all the skills needed for the development of genetic workflows were covered, especially, knowledge on implementing science in practice, an understanding of the needs of conservation professionals and experience in outreach. It had support from national and regional authorities in Switzerland (and from Austria).
Based on a consultation with Swiss cantonal agencies, two topics for the development of conservation genetic workflows were identified: (1) identification of amphibian species based on environmental DNA (eDNA; Taberlet et al. 2018) from water samples; (2) analysis of connectivity among populations (Lowe and Allendorf 2010), eventually coupled with an evaluation of the success of connectivity measures such as over- and underpasses, stepping stones or corridors (Corlatti et al. 2009). These two topics are of special relevance to conservation professionals in Switzerland (Braunisch et al. 2012), because they are looking for alternative, potentially cheaper monitoring methods (Pesch et al. 2016) and because a nation-wide ecological infrastructure consisting of habitat nodes and links providing landscape-wide connectivity is currently implemented by the Swiss government (BAFU 2017).
In the following, we describe the two conservation genetic workflows. Detailed information is provided by Holderegger et al. (2019a, b). As conservation practitioners do hardly rely on information in foreign languages (Fabian et al. 2019), the two workflows were presented in a series of information events and outreach publications in the national languages of Switzerland specifically targeted to practitioners (e.g. Csencsics and Gugerli 2017; Stapfer et al. 2019).
Workflow: identification of amphibian species with eDNA and metabarcoding
The goal was to establish a simple ready-to-use genetic workflow for the identification of all pond-dwelling amphibians in Switzerland and adjacent regions based on eDNA from water samples using metabarcoding (Hawlitschek et al. 2016; Ficetola et al. 2019) rather than qPCR for the detection of single species (Thomsen et al. 2012).
The manual for this workflow (Holderegger et al. 2019b) first offers a detailed introduction to the pros and cons of eDNA from water samples for the detection of amphibians. Amongst others, some pros of eDNA in comparison to traditional methods are (Smart et al. 2015; Goldberg et al. 2016): clandestine species can be identified (e.g. Lissotriton vulgaris, Triturus cristatus); species impossible to morphologically discriminate can be identified to a degree useful for conservation practice (e.g. native and invasive Pelophylax sp.); field work can be done during the day and is independent of weather conditions; amphibians are not impaired by sampling. Some cons are: hybrids cannot be determined (e.g. T. cristatus ✕ T. carnifex); laboratory methods can create false positives (see below); DNA content in the water is highest during peak activity of amphibians and decreases rapidly thereafter. Finally, it is clearly stated that the abundance of an amphibian species cannot (yet) be determined based on metabarcoding of eDNA from water samples (but see Chambert et al. 2018).
Next, the manual gives a list of the pond-dwelling amphibian species of Switzerland and adjacent regions that can be identified with the workflow (Table 1). It allows the detection of different taxa or taxon groups of the hybridogenetic water frogs (Pelophylax sp.), namely the species P. bergeri und P. bedriagae and the two groups P. esculentus/P. lessonae and P. kurtmuelleri/P. ridibundus (Leuenberger et al. 2014). Closely related species such as T. cristatus und T. carnifex can also be determined.
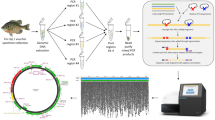
Subsequently, the manual describes the major steps of the workflow (Fig. 1). Detailed guidelines show at how many sites per pond water samples should be taken, mixed and then aliquoted to at least three sample replicates per pond for laboratory analysis (no filtering of water samples involved; Taberlet et al. 2018). Private laboratory companies and environmental consultancies provide advice and help with the sampling protocol. It is also stressed that permits, e.g., to enter nature reserves, must be requested. The workflow introduces the material necessary for field work and describes how water sampling and the labelling of samples (e.g. QR-codes provided by laboratory companies) are done. Special care is devoted to contamination issues, and it is stressed that thorough de-contamination of all (re-usable) equipment, shoes or wellingtons is mandatory after sampling a pond, in order to prevent the dispersal of pathogens such as the amphibian chytrid fungus (Batrachochytrium dendrobatidis; Schmidt et al. 2009).

Steps of the conservation genetic workflow for the identification of amphibians based on metabarcoding of eDNA from water samples. Steps given in white boxes are relevant to conservation professionals and must be understood by them. These steps are carried out with the help, advice and training provided by private laboratory companies and environmental consultancies. The steps given in grey boxes are the domain of private laboratory companies and environmental consultancies
We developed new primers in the 16 s rRNA of mtDNA for the metabarcoding of amphibians of Switzerland and adjacent regions (Microsynth Ecogenics 2018). These primers were optimised to avoid the amplification of human DNA. Special care was given to contamination issues in the laboratory as well as to negative and positive controls. The PCR reactions for metabarcoding also contained in process controls (short artificial DNA) as a quality check for amplification. Specimens of all Swiss amphibian species were Sanger sequenced, and a new, constantly up-dated sequence data base was created.
The manual then introduces the results per pond, which are sent to the purchaser. They show whether an amphibian species has been identified in a pond with high probability (presence), whether a species was only ambiguously detected (uncertain detection) or not detected (absence). The discrimination between presence and uncertain detection for each species was based on the number of Illumina reads in field as well as in control samples using a statistical framework accounting for false positives (Ficetola et al. 2016; Lahoz-Monfort et al. 2016). We also used different amounts of real amphibian and artificial DNAs to check for detectability thresholds.
Finally, the results are interpreted based on detailed guidelines given in the manual and illustrated by the results of water samples from real ponds to which the workflow had been applied. Special care is given to the fact that the results of the workflow on amphibian species identification do not always result in clear absence or presence information for a particular species, but that the above category of uncertain detection needs additional effort (e.g. field observation, independent repetition of the workflow with new water samples, targeted sequencing of the DNA of the species in question).
Workflow: assessing dispersal and gene flow among populations
Conservation professionals deal with issues of connectivity by planning structural connectivity measures such as underpasses, overpasses, corridors or stepping stones. However, it is difficult to measure functional connectivity among populations (Manel and Holderegger 2013) with traditional methods such as radio-tracking or mark-recapture. Therefore, conservation professionals are interested in using genetic tools to assess movement, dispersal and gene flow among populations and habitat patches (Lowe and Allendorf 2010; Holderegger et al. 2019a, b). Hence, our goal was to establish a ready-to-use genetic workflow for assessing dispersal (migration in genetic terms) and gene flow among populations using microsatellites. We decided to use microsatellites as they are relatively inexpensive once developed.
The manual (Holderegger et al. 2019b) shows all steps of this workflow (Fig. 2). It first offers guidelines to determine the appropriate sampling design in a given landscape. As the dispersal abilities, the number and size of populations and the compositions of landscape elements all differ among species and study areas, no general-purpose sampling design can be applied. Instead, five principles for the sampling design based on known species occurrences in the study landscape are given. (1) The appropriate spatial extent of the study area is defined by populations being within the maximum dispersal distance of the species (based on literature or expert knowledge). (2) Comparison of different situations of populations is fundamental, e.g., one needs to compare isolated and near-by populations or populations in fragmented and connected landscapes. (3) If barriers or connectivity measures are of interest, populations affected and unaffected (control) by these landscape elements need to be sampled. (4) Populations at different Euclidean distances should be sampled to detect the effect of geographic distance. (5) The number of populations sampled is defined by the number of comparisons needed to detect a certain effect. For instance, if one samples four populations on one side of a fenced motorway and four populations on the other side, one gets 16 comparisons of population pairs potentially fragmented by the motorway and 12 comparisons of potentially connected populations on either side of the motorway (Marsh et al. 2008). Generally, eight to ten sampled populations should be enough in an applied conservation genetic study at the landscape scale.

Steps of the conservation genetic workflow on connectivity. For colour code see Fig. 1
The number of individuals sampled per population should be 15 to 20. These numbers are based on literature (e.g. Kalinowski 2005; Hoban et al. 2015) and a consultation with 20 conservation geneticists in Central Europe, in which most of them agreed that a sample size of 20 individuals per population is sufficient. We also performed a resampling analysis of published microsatellite datasets of two mammals and an herb (Biebach and Keller 2010; Buchalski et al. 2016; Fischer et al. 2017). These datasets contained 19 to 39 microsatellite markers and 20 to 36 individuals per population. In each dataset and for each possible number of markers and individuals per population (from 2 to the maximum − 1), we created 100 random subsets and calculated the average of several population genetic parameters, namely expected heterozygosity He, pairwise population differentiation FST, rarefied allelic richness Ar, and inbreeding coefficient FIS. For the herb, we also performed STRUCTURE analyses (Pritchard et al. 2000) and determined the putative number of genetic clusters using the classical (highest probability of the data, Pritchard et al. 2000), but also the delta K method (Evanno et al. 2005). We then compared the results of the random subsets with the observed results (i.e. the values with the complete set of markers and individuals) with a Pearson correlation and the ratio of resampled/observed values, indicating whether one under- or over-estimates the population genetic parameters with a smaller dataset.
This resampling exercise showed that population genetic parameters are clearly more sensitive to the number of markers than to the number of individuals per population. Acceptably stable results (Pearson’s r > 0.95) were obtained with about 15 individuals per population (in some cases even lower) and 20 microsatellite loci. There was a trade-off between the number of markers and the number of individuals; increasing the number of markers markedly decreased the required number of individuals to obtain accurate values. With high numbers of markers (> 30), around five individuals were usually enough for accurate population genetic assessments. There were some exceptions to this general pattern. For an accurate measurement of FIS, substantially more markers and individuals were required. Moreover, FIS and, naturally, Ar were clearly underestimated with low numbers of individuals. For identifying the number of genetic clusters in STRUCTURE, ten markers and ten individuals were enough using the likelihood method. Results were highly unstable with the delta K method. Finally, we found that in the species with the highest level of inbreeding, the above-mentioned rule of 15 individuals per population and 20 microsatellites was not sufficient for a sound estimation of all genetic parameters tested.
The next steps of the manual describe for many organismic groups (plants, arthropods, molluscs, fishes, amphibians, reptiles, birds and mammals) which genetic material is sampled in the field, how samples are labelled (e.g. QR codes provided by private laboratory companies), what kind of field material is needed and how samples are to be sent to the laboratory. It is stressed that individuals should be disturbed and handicapped as little as possible and that non-invasive or minimally invasive methods are preferable (Marschalek et al. 2013; Carroll et al. 2018; Zemanova 2019), e.g., buccal swabs (Broquet et al. 2007) or tissue punches in amphibians. Special emphasis is given to the necessary permits for entering protected or private land and regarding legal obligations for sampling protected, red-listed or rare species.
The manual then gives technical information on laboratory methods. We used microsatellites for genotyping because SNPs and bioinformatic pipelines are not yet ready for routine use in conservation practice (also see below; Puckett 2017; Holderegger et al. 2019a). Subsequently, the five standard statistical analyses of the workflow are presented. (1) As STRUCTURE is the most widely used method for genetic clustering (Janes et al. 2017), genetic groups are determined with STRUCTURE (admixture model, no LOCPRIOR; Pritchard et al. 2000) and CLUMPP (Jakobsson and Rosenberg 2007). STRUCTURE HARVESTER (Earl and von Holdt 2012) is used to define the optimal k using the log likelihood plot and the hierarchical development of genetic clusters k when moving from k = 1 to k = x (the Evanno method should not be used; Meirmans 2015; Janes et al. 2017; also see the resampling analysis above). It is also needed to create input files to CLUMPP. As an alternative to STRUCTURE, ordination methods such as multidimensional scaling MDS (Cox and Cox 2001) could be used for identifying genetic groups.
Subsequently, three measurements of dispersal or gene flow are introduced. (2) Historical gene flow (Whitlock and McCauley 1999) is estimated with pairwise FST-values with the R package strataG 2.0.2 (Archer et al. 2016), and a simple Mantel test is used to check for isolation by distance with the R package ecodist 2.0.1 (Goslee and Urban 2007). (3) BAYESASS 3.0 (Wilson and Rannala 2003) is applied to estimate bi-directional recent dispersal rates among populations. Bi-directional genetic exchange also allows detecting source and sink populations (Holderegger and Gugerli 2012). (4) Current dispersal is studied by detecting first generation migrants with GENECLASS 2.0 (Piry et al. 2004). However, it is made clear that a sample size of 15 to 20 individuals per population is generally not sufficient to detect first generation migrants (Kraaijeveld-Smit et al. 2005).
(5) Finally, and in case of an appropriate sampling scheme and the presence of potential barriers, a simple isolation by barrier (IBB) analysis is performed (Oyler-McCance et al. 2013). Here, the occurrence of a barrier between population pairs is coded in a 0/1 matrix. The statistical analysis incorporates a matrix of pairwise FST-values or averaged recent dispersal rates from BAYESASS 3.0 as dependent variables, a matrix of Euclidian distances and one or several barrier matrices and a covariance matrix, to account for the non-independence of genetic data, as independent variables. The data are then analysed with a generalized linear mixed effects model with Monte Carlo Markov Chain testing in the R package MCMCglmm 2.26 (Hadfield 2010). All results are generated and visualised in a geographical way. For instance, the genetic groups inferred from STRUCTURE are overlaid onto topographical maps, aerial photographs or Google maps. Such STRUCTURE applications are appealing to practitioners. The results are then sent to the purchaser.
The next step of the manual explains how to interpret the results on dispersal and gene flow. A first good overview is provided by STRUCTURE results. The manual offers richly illustrated guidelines and also provides the results of a real example to which the workflow had been applied. Special focus is given to the fact that one should watch out for the large patterns, e.g., comparing isolated with non-isolated sites or potential source with sink populations, and not interpret single details (“seeing the forest despite the trees”). Again, private laboratory companies and environmental consultancies provide help, advice and training with interpreting the genetic results of the workflow.
Challenges met during the development of the workflows
In the following, we discuss three main challenges that we encountered during this transdisciplinary project, which included both research institutions from the public domain as well as companies (i.e. a molecular laboratory company and an environmental consultancy) from the private sector.
First, eDNA and metabarcoding do not always result in a clear “yes or no”-result, and it is particularly difficult to define thresholds in order to keep the sensitivity of an assay as high as possible without increasing its specificity. This is a general problem in diagnostic tests (Altman and Bland 1994), but is even more pronounced if multiple species are detected in a single assay.
Many factors can influence the results of metabarcoding of eDNA from water samples. Some examples are low DNA quantity or quality, cross-contamination during field sampling, cross-contamination in the laboratory, primer competition among the DNA of different species during PCR or sequence mistakes introduced by TAQ-polymerase inconsistencies (Taberlet et al. 2018; Mathieu et al. 2020). All these phenomena can lead to the false negative or false positive identification of species in a sample or to some species having many sequencing reads, while other species show only a few reads. In the latter case, species identification in a particular sample remains questionable and uncertain. It is this uncertainty that can be difficult for conservation practitioners to accept. However, the situation is not really different from field observations of species; an approach to which practitioners are well used. Species can also be misidentified and overlooked in the field, leading to false negatives and false positives in observations as well (Cruickshank et al. 2019). In the workflow for the detection of amphibian species with eDNA from water samples by metabarcoding we thus decided to actively communicate uncertainties in species identification, by introducing a sample-specific threshold of the required number of sequencing reads for certain species detection (presence) or uncertain species detection (uncertain detection), respectively (see above). Scientists have long asked for the communication of uncertainties to practitioners, politics and the broader public (Fischhoff and Davis 2014; Papadopoulou et al. 2018).
Second, it is difficult for conservation professionals to understand that conservation genetics cannot provide a single parameter that shows whether there is “enough” gene flow or dispersal among populations. In fact, the exact value of many genetic parameters indicative of gene flow and dispersal such as FST have no particular meaning per se and vary, e.g., substantially among molecular marker types applied to the same samples (Fischer et al. 2017). While it is safe to conclude that two populations differentiated by a low FST value of 0.03 show more (historical) gene flow than two populations with a pairwise FST of 0.10, it cannot be inferred whether there is enough gene flow in one or the other case (unless one accepts the heavily criticised Nm = 1 rule; Whitlock and McCauley 1999). What is needed is a comparison of populations in different situations. For instance, one can compare populations that are separated by a motorway with populations that are not or populations in a dense setting with populations that are sparsely scattered across a landscape. This is why the manual for the workflow on dispersal and gene flow puts particular emphasis on major principles to set up an adequate sampling design: one such principle stresses the need for sampling and comparing populations in different situations (see above). This later point needs explanation and training to those conservation practitioners that will be involved in setting up sampling schemes for genetic analyses of dispersal and gene flow.
Third, researchers from the public sector and professionals from the private sector do not necessarily have the same goals in a transdisciplinary project (Enquist et al. 2017). Researchers—their salaries being paid by governmental agencies from tax money—want to make all approaches and methods as openly accessible as possible, so that everybody who wants to use them can do so. In contrast, private molecular laboratory companies and environmental consultancies must generate money to pay salaries, expenses and infrastructure. In other words, one of their main goals is to generate an economic benefit. Professionals from the private sector do thus not have an a priori interest in making approaches and methods openly available, which can lead to a conflict of interest. This is, e.g., illustrated by the fact that some primers for the metabarcoding of European amphibian species are patented and cannot be freely used for routine commercial use (e.g. Valentini et al. 2016). In the present project, such a conflict of interest was avoided by setting up a contract signed by all institutional participants of the transdisciplinary project right at the beginning. It clearly regulated the use and shared ownership of all methods and approaches developed during the project’s course.
Perspectives
The two workflows described above are intended to foster the use of genetic methods in conservation management by presenting genetic tools, which can readily be applied by environmental consultancies, governmental agencies or NGOS in collaboration with private companies and consultancies outside academia. Researchers play no role in the workflows, once the latter are set up. Researchers might act as external experts, e.g., for training, specific statistical analyses or special sampling designs, but in general, they are not involved in the application of the two conservation genetic workflows.
However, researchers play an important role in the enhancement of established workflows or in the development of additional workflows, in close collaboration with private laboratory companies and environmental consultancies. An obvious future development of the workflow on eDNA detection of amphibians is the extension to other water organisms, such as fishes (Valentini et al. 2016) or dragon- and damselflies (Thomsen et al. 2012). The latter is especially relevant as it allows the species identification of larvae of dragon- and damselflies in water bodies and thus indicates local reproduction. Another topic is the transition from microsatellites to SNPs in the workflow on dispersal and gene flow (Shafer et al. 2015). SNPs enable inference on genome-wide patterns of genetic diversity, and the results of studies using microsatellites or SNPs can differ substantially (Fischer et al. 2017; Bohling et al. 2019). For instance, large panels of SNPs allow for high precision when inferring the genetic structure of populations (Jeffries et al. 2016; Puckett and Eggert 2016). In conservation genetic research and in non-model species, SNPs from RADseq (Petersen et al. 2012) are often used. The laboratory and sequencing costs for SNPs from RADseq are already comparable to those of microsatellites (Puckett 2017), and the major hurdle is to set up bioinformatic pipelines that can be used in a routine way for many species without much adjustment (Shafer et al. 2017).
Additionally, new genetic workflows should be established. One example is a workflow on inbreeding. This topic is of relevance as many small and isolated populations are inbred with potentially strong fitness consequences caused by inbreeding depression (Frankham et al. 2017). Studies have shown that runs of homozygosity (ROHs) are the method of choice to measure inbreeding in wild populations (Purfield et al. 2012; Diez-del-Molino et al. 2018; Robinson et al. 2019). However, measuring inbreeding with ROHs requires high quality, contiguous genome data, which currently sets an obstacle to their routine use in conservation genetics, but also points to the need for a change from genetics to genomics in applied conservation genetics (Shafer et al. 2015). Corresponding efforts to set up genomic workflows for conservation management are currently undertaken, e.g., in the COST-actions DNAqua-Net (https://www.cost.eu/actions/CA15219/) and G-BIKE (https://www.cost.eu/actions/CA18134/).
We hope that the conservation genetic workflows presented here for Switzerland could serve as a model for establishing similar workflows in other countries and that they help to implement genetic methods as routine tools in practical conservation management and to safeguard threatened populations and species. The methods described in the workflow identification of amphibians with eDNA and metabarcoding are already in routine use in the monitoring of the nationally important habitats in Switzerland, which encompass more than 250 amphibian breeding sites (BAFU 2020). First results already inform the current update of the new Swiss Red List of amphibians (Benedikt Schmidt, unpubl. data). This example certifies that the developed workflows find their way into practical conservation management.
References
Altman DG, Bland JM (1994) Diagnostic tests 1: sensitivity and specificity. BMJ 308:1552
Archer FI, Adams PE, Schneiders BB (2016) STRATAG: an R package for manipulating, summarizing and analysing population genetic data. Mol Ecol Res 17:5–11
BAFU (2017) Aktionsplan Strategie Biodiversität Schweiz. BAFU, Berne
BAFU (2020) Monitoring und Wirkungskontrolle Biodiversität. Übersicht zu nationalen Programmen und Anknüpfungspunkten. BAFU, Berne
Biebach I, Keller LF (2010) Inbreeding in reintroduced populations: the effects of early reintroduction history and contemporary processes. Conserv Genet 11:527–538
Bohling J, Small M, Van Bargen J, Louden A, DeHaan P (2019) Comparing inferences derived from microsatellite and RADseq datasets: a case study involving threatened bull trout. Conserv Genet 20:329–342
Braunisch V, Home R, Pellet J, Arlettaz R (2012) Conservation science relevant to action: a research agenda identified and prioritized by practitioners. Biol Conserv 153:201–210
Broquet T, Berset-Braendli L, Emaresi G, Fumagalli L (2007) Buccal swabs allow efficient and reliable microsatellite genotyping in amphibians. Conserv Genet 8:509–511
Buchalski MR, Sacks BN, Gille DA, Penedo MCT, Ernest HB, Morrison SA, Boyce WM (2016) Phylogeographic and population genetic structure of bighorn sheep (Ovis canadensis) in North American deserts. J Mammal 97:823–838
Carroll E, Bruford MW, DeWoody JA, Leroy G, Strand A, Wairs L, Wang J (2018) Genetic and genomic monitoring with minimally invasive sampling methods. Evol Appl 11:094–1119
Chambert T, Pilliod DS, Goldberg CS, Doi H, Takahara T (2018) An analytical framework for estimating aquatic species density from environmental DNA. Ecol Evol 8:3468–3477
Csencsics D, Gugerli F (2017) Naturschutzgenetik. WSL Ber 60:1–82
Corlatti L, Hackländer K, Frey-Roos F (2009) Ability of wildlife-overpasses to provide connectivity and prevent genetic isolation. Conserv Biol 23:548–556
Cox TF, Cox MAA (2001) Multidimensional scaling. Chapman and Hall, London
Cruickshank SS, Bühler C, Schmidt BR (2019) Quantifying data quality in a citizen science monitoring program: false negatives, false positives and occupancy trends. Conserv Sci Pract 1:e54
Diez-del-Molino D, Sanchez-Barreiro F, Barnes I, Gilbert MTP, Dalen L (2018) Quantifying temporal genomic erosion in endangered species. Trends Ecol Evol 33:176–185
Dufresnes C, Dejean T, Zumbach S, Schmidt BR, Fumagalli L, Ramseier P, Dubey S (2019) Early detection and spatial monitoring of an emerging biological invasion by population genetics and environmental DNA metabarcoding. Conserv Sci Pract 1:e86
Earl DA, von Holdt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Res 4:359–361
Enquist AF, Garfin GM, Davis FW, Gerber LR, Littell JA, Tank JL, Terando AJ, Wall TU, Halpern B, Hiers JK, Morelli TL, McNie E, Stephenson NL, Williamson MA, Woodhouse CA, Yung L, Brunson MW, Hall KR, Hallett LM, Lawson DM, Moritz MA, Nydick K, Pairis A, Ray AJ, Regan C, Safford HD, Schwartz MW, Shaw MR (2017) Foundations of translational ecology. Frontiers Ecol Environ 15:541–550
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software structure: a simulation study. Mol Ecol 14(8):2611–2620
Fabian Y, Bollmann K, Brang P, Heiri C, Olschewski R, Rigling A, Stofer S, Holderegger R (2019) How to close the science-practice gap in nature conservation? Information sources used by practitioners. Biol Conserv 235:93–101
Ficetola GF, Taberlet P, Coissac E (2016) How to limit false positives in environmental DNA and metabarcoding? Mol Ecol Res 16:604–607
Ficetola GF, Manenti R, Taberlet P (2019) Environmental DNA and metabarcoding for the study of amphibians and reptiles: species distribution, the microbiome, and much more. Amphib-Reptil 40:129–148
Fischer MC, Rellstab C, Leuzinger M, Roumet M, Gugerli F, Shimizu K, Holderegger R, Widmer A (2017) Estimating genomic diversity and population differentiation - an empirical comparison of microsatellite and SNP variation in Arabidopsis halleri. BMC Genom 18:69
Fischhoff B, Davis AL (2014) Communicating scientific uncertainty. Proc Natl Acad Sci USA 111:13664–13671
Frankham R (1995) Conservation genetics. Ann Rev Genet 29:305–327
Frankham R, Ballou JD, Ralls K, Eldridge MDB, Dudash MR, Fenster CB, Lacy RC, Sunnucks P (2017) Genetic management of fragmented animal and plant populations. Oxford University Press, Oxford
Goldberg CS, Turner CR, Deiner K, Klymus KE, Thomsen PF, Murphy MA, Spear FS, McKnee A, Oyler-McCance SJ, Cornman RS, Laramie MB, Mahon AR, Lance RF, Pilliod DS, Strickler KM, Waits LP, Fremier AK, Takahara T, Herder JE, Taberlet P (2016) Critical considerations for the application of environmental DNA methods to detect aquatic species. Methods Ecol Evol 7:1299–1307
Goslee SC, Urban DL (2007) The ecodist package for dissimilarity-based analysis of ecological data. J Stat Software 22:1–19
Grant EHC, Muths E, Schmidt BR, Petrovan SO (2019) Amphibian conservation in the Anthropocene. Biol Conserv 236:543–547
Hawlitschek O, Moriniere J, Dunz A, Franzen M, Rödder D, Glaw F, Haszprunar G (2016) Comprehensive DNA barcoding of the herpetofauna of Germany. Mol Ecol Res 16:242–253
Hadfield JD (2010) MCMC methods for multi-response generalized linear mixed models: the MCMCglmm R package. J Stat Software 33:1–22
Hoban S, Arntzen JW, Bruford MW, Godoy JA, Hoelzel R, Segelbacher G, Vila C, Bertorelle G (2015) Comparative evaluation of potential indicators and temporal sampling protocols for monitoring genetic erosion. Evol Appl 7:984–998
Holderegger R, Gugerli F (2012) Where do you come from, where do you go? Directional migration rates in landscape genetics. Mol Ecol 21:5640–5642
Holderegger R, Balkenhol N, Bolliger J, Engler JO, Gugerli F, Hochkirch A, Nowak C, Segelbacher G, Widmer A, Zachos FE (2019a) Conservation genetics: linking science with practice. Mol Ecol 28:3848–3856
Holderegger R, Stapfer A, Schmidt B, Grünig C, Meier R, Csencsics D, Gassner M (2019b) Werkzeugkasten Naturschutzgenetik: eDNA Amphibien und Verbund. WSL Ber 81:1–56
Jakobsson M, Rosenberg NA (2007) CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinform 23:1801–1806
Janes JK, Miller JM, Dupuis JR, Malenfant RM, Gorrell JC, Cullingham CI, Andrew RL (2017) The k = 2 conundrum. Mol Ecol 26:3594–3602
Jeffries DL, Copp GH, Handley LL, Olsen KH, Sayer CD, Hänfling B (2016) Comparing RADseq and microsatellites to infer complex phylogeographic patterns, an empirical perspective in the Crucian carp, Carassius carassius, L. Mol Ecol 25:2997–3018
Kalinowski ST (2005) Do polymorphic loci require large sample sizes to estimate genetic distances? Heredity 94:33–36
Kraaijeveld-Smit FJL, Beebee TJC, Griffiths RA, Moore RD, Schley L (2005) Low gene flow but high genetic diversity in the threatened Mallorcan midwife toad Alytes muletensis. Mol Ecol 14:3307–3315
Lahoz-Monfort JJ, Guillera-Arroita G, Tingley R (2016) Statistical approaches to account for false-positive errors in environmental DNA samples. Mol Ecol Res 16:673–685
Leuenberger J, Gander A, Schmidt BR, Perrin N (2014) Are invasive marsh frogs (Pelophylax ridibundus) replacing the native P. lessonae/P. esculentus hybridogenetic complex in Western Europe? Genetic evidence from a field study. Conserv Genet 15:869–878
Lowe WH, Allendorf FW (2010) What can genetics tell us about population connectivity? Mol Ecol 19:3038–3051
Magurran AE (2003) Measuring biological diversity. Blackwell, Malden
Manel S, Holderegger R (2013) Ten years of landscape genetics. Trends Ecol Evol 28:614–621
Marsh DM, Page RB, Hanlon TJ, Corritone R, Little EC, Seifert DE, Cabe PR (2008) Effects of roads on patterns of genetic differentiation in red-backed salamanders, Plethodon cinereus. Conserv Genet 9:603–613
Marschalek DA, Jesu JA, Berres ME (2013) Impact of non-lethal genetic sampling on the survival, longevity and behaviour of the Hermes copper (Lycaena hermes) butterfly. Insect Conserv Div 6:658–662
Mathieu C, Hermans SM, Lear G, Buckley TR, Lee KC, Buckley HL (2020) A systematic review of sources of variability and uncertainty in eDNA data for environmental monitoring. Front Ecol Evol 8:135
Meirmans PG (2015) Seven common mistakes in population genetics and how to avoid them. Mol Ecol 24:3223–3231
Microsynth Ecogenics (2018) Nachweis von Amphibien-DNA mittels Barcoding und Deep-Sequencing. Rechtobler Gmäändsblatt 2018(8):22
Oyler-McCance SJ, Fedy BC, Landguth EL (2013) Sample design effects in landscape genetics. Conserv Genet 14:275–285
Pesch M-L, Jacquat O, Zürcher D (2016) Forschungskonzept Umwelt für die Jahre 2017–2020. BAFU, Berne
Peterson BK, Weber JN, Kay EH, Fisher HS, Hoekstra HE (2012) Double digest RADseq: an inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLoS ONE 7:e37135
Piry S, Alapetite A, Cornuet J-M, Paetkau D, Baudouin L, Estoup A (2004) GENECLASS2: a software for genetic assignment and first-generation migrant detection. J Hered 95:536–539
Papadopoulou L, McEntaggart K, Etienne J (2018) Communicating scientific uncertainty in advice provision to decision-makers: review of approaches and recommendations for UK statutory nature conservation bodies. JNCC Rep 617:1–37
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Puckett EE (2017) Variability in total project and per sample genotyping costs under varying study designs including with microsatellites or SNPs to answer conservation genetic questions. Conserv Genet Res 9:289–304
Puckett EE, Eggert LS (2016) Comparison of SNP and microsatellite genotyping panels for spatial assignment of individuals to natal range: a case study using the American black bear (Ursus americanus). Biol Conserv 193:86–93
Purfield DC, Berry DP, McParland S, Bradley DG (2012) Runs of homozygosity and population history in cattle. MBC Genet 13:70
Robinson JA, Räikkönen J, Vucetich LM, Vucetich JA, Peterson RO, Lohmueller KE, Wayne RK (2019) Genomic signatures of extensive inbreeding in Isle Royale wolves, a population on the threshold of extinction. Sci Adv 5:eaau9757
Sandström A, Lundmark C, Andersson K, Johannessson K, Laikre L (2019) Understanding and bridging the conservation genetics gap in marine conservation. Conserv Biol 33:725–728
Schmidt BR, Furrer S, Kwet A, Lötters S, Rödder D, Sztatecsny M, Tobler U, Zumbach S (2009) Desinfektion als Maßnahme gegen die Verbreitung der Chytridiomykose bei Amphibien. Z Feldherpetol Suppl 15:229–241
Shafer ABA, Wolf JBW, Alves PC, Bergström L, Bruford MW, Brännström I, Colling G, Dalen L, De Meester L, Ekblom R, Fawcett KD, Fior S, Hajibabaei M, Hill JA, Hoezel AR, Höglund J, Jensen EL, Krause J, Kristensen TN, Krützen M, McKay JK, Norman AJ, Ogden R, Östering EM, Ouborg NJ, Piccolo J, Popovic D, Primmer CR, Reed FA, Roumet M, Slamona J, Schenekar T, Schwartz MK, Segelbacher G, Senn H, Thaulev J, Valtonen M, Veale A, Vergeer P, Vijay N, Vila C, Weissensteiner M, Wennerström L, Wheat CW, Zielinski P (2015) Genomics and the challenging translation into conservation practice. Trends Ecol Evol 30:78–87
Shafer ABA, Peart CR, Tusso S, Maayan I, Brelsford A, Wheat CW, Wolf JBW (2017) Bioinformatic processing of RADseq data dramatically impacts downstream population genetic inference. Methods Ecol Evol 8:907–917
Smart AS, Tingley R, Weeks AR, van Rooyen AR, McCarthy MA (2015) Environmental DNA sampling is more sensitive than a traditional survey technique for detecting an aquatic invader. Ecol Appl 25:1944–1952
Stapfer A, Csencsics D, Holderegger R, Schmidt B (2019) Werkzeugkasten Naturschutzgenetik: eDNA Amphibien und Verbund. Boîte à outils de génétiques de la conservation: eDNA amphibens et connectivité. N + L Inside 2019(3):23
Taberlet P, Bonin A, Zinger L, Coissac E (2018) Environmental DNA for biodiversity research and monitoring. Oxford University Press, Oxford
Taylor HR, Dussex N, van Heezik Y (2017) Bridging the conservation genetics gap by identifying barriers to implementation for conservation practitioners. Global Ecol Conserv 10:231–242
Thomsen PF, Kielgast J, Iversen L, Wiuf C, Rasmussen M, Gilbert MTP, Orlando L, Willerslev E (2012) Monitoring endangered freshwater biodiversity using environmental DNA. Mol Ecol 21:2565–2573
Turner MG, Gardner RH, O’Neill RV (2001) Landscape ecology in theory and practice. Springer, New York
Valentini A, Ta M, Taberlet P, Miaud D, Civade R, Herder J, Thomson PF, Bellemain E, Besnard A, Coissac E, Boyer F, Gaboriaud C, Jean P, Poulet N, Roset N, Copp GH, Geniez P, Pont D, Argillier C, Baudoin J-M, Peroux T, Crivelli AJ, Olivier A, Acqueberge M, Le Brun M, Moller PR, Willerslev E, Dejean T (2016) Next-generation monitoring of aquatic biodiversity using environmental DNA metabarcoding. Mol Ecol 25:929–942
Whitlock MC, McCauley DE (1999) Indirect measures of gene flow and migration: Fst ≠ 1/(4 N m + 1). Heredity 82:117–125
Wildi O (2013) Data analysis in vegetation ecology. Wiley-Blackwell, Chichester
Wilson GA, Rannala B (2003) Bayesian inference of recent migration rates using multilocus genotypes. Genetics 163:1177–1191
Zemanova MA (2019) Poor implementation of non-invasive sampling in wildlife genetics studies. Rethinking Ecol 4:119–132
Acknowledgements
Open Access funding provided by Lib4RI – Library for the Research Institutes within the ETH Domain: Eawag, Empa, PSI & WSL. We thank InnoSuisse (Grant 19204.1 PFLS-LS), the Swiss Federal Office for the Environment BAFU, the Swiss cantons of Argovia, Appenzell Ausserrhoden, Berne, Geneva, Lucerne, Ob- and Nidwalden, Schaffhausen, Thurgau, Uri, Zug and Zurich and the Austrian state Salzburg for financial support. We thank Michael Buchalsky and Irene Biebach for the provision of microsatellite data, and two reviewers for helpful comments on an earlier version of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Project partners ARNAL and Microsynth Ecogenics GmbH are private sector companies making profit by offering conservation genetic services, including the workflows presented in this article. Public sector partners WSL, ETH Zurich, University of Zurich, info fauna-karch and HSR declare no conflict of interests. The relationship among project partners is regulated in a contract with Innosuisse (Grant 19204.1 PFLS-LS).
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Holderegger, R., Schmidt, B.R., Grünig, C. et al. Ready-to-use workflows for the implementation of genetic tools in conservation management. Conservation Genet Resour 12, 691–700 (2020). https://doi.org/10.1007/s12686-020-01165-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12686-020-01165-5