
Abstract
The disease caused by SARS-CoV-2—CoVID-19—is a global pandemic that has brought severe changes worldwide. Approximately 80% of the infected patients are largely asymptomatic or have mild symptoms such as fever or cough, while rest of the patients display varying degrees of severity of symptoms, with an average mortality rate of 3–4%. Severe symptoms such as pneumonia and acute respiratory distress syndrome may be caused by tissue damage, which is mostly due to aggravated and unresolved innate and adaptive immune response, often resulting from a cytokine storm. Here, we discuss how an intricate interplay among infected cells and cells of innate and adaptive immune system can lead to such diverse clinicopathological outcomes. Particularly, we discuss how the emergent nonlinear dynamics of interaction among the components of adaptive and immune system components and virally infected cells can drive different disease severity. Such minimalistic yet rigorous mathematical modeling approaches are helpful in explaining how various co-morbidity risk factors, such as age and obesity, can aggravate the severity of CoVID-19 in patients. Furthermore, such approaches can elucidate how a fine-tuned balance of infected cell killing and resolution of inflammation can lead to infection clearance, while disruptions can drive different severe phenotypes. These results can help further in a rational selection of drug combinations that can effectively balance viral clearance and minimize tissue damage.
Similar content being viewed by others


1 Introduction
The world has been severely impacted by the ongoing CoVID-19 pandemic on multiple perspectives: economic, social, and public health. Various candidates for vaccines are in different stages of development, and as we await to learn about the efficacy and longevity of these targeted outcomes, it becomes imperative to understand the underlying biology of the disease that can inform the rational design of treatment options for the people who have already contracted the virus. In this perspective, we aim to summarize in one part the biology of the SARS-CoV-2 infection, which relates to responses of adaptive and innate arms of our immune system. In the second part, we discuss how mathematical modeling of the interactions between immune cells and virally infected cells can help inform rational design of treatment options.
2 What is SARS-CoV-2?
Human coronaviruses, first identified in the 1960s1, commonly infect humans. Most coronavirus infections are respiratory in nature and primarily affect the upper respiratory tract and the lungs2. There are 7 identified coronaviruses that have been documented to infect humans—229E, NL63, OC43, HKU1, SARS-CoV, MERS-CoV and SARS-CoV-2. Most human coronaviruses are zoonotic in nature, i.e., these coronaviruses initially infected animals and later gained the ability to infect humans3. The SARS-CoV pandemic that plagued the world in 2002–2003 infected around 8000 people and claimed more than 770 lives across 27 countries4. The current pandemic SARS-CoV-2 has a significant amount of similarity to the previous SARS virus both in terms of the genome sequence and disease progression5. Comorbidities such as diabetes, hypertension, cancer, etc. have had an important role to play in defining the risks and fatality rates for a more severe manifestation of the SARS-CoV and the MERS-CoV virus6, 7. Even in the current pandemic, it is quite clear that comorbidities such as diabetes mellitus, cancer, lung disease, hypertension, etc. can increase the risk of the disease progressing to a relatively severe stage8,9. Specifically, a high blood glucose level has been shown to favor SARS-CoV-2 infection and increase the cytokine responses in monocytes leading to more severe clinical manifestations of the disease10. Similar to the previous SARS pandemic, the severity of the infection increases with age9, with older people displaying more severe manifestations of the disease.
SARS-CoV-2 infection of the human body can give rise to a myriad of clinical phenotypes ranging from completely asymptomatic to extremely severe manifestations of the disease. A large proportion of the population diagnosed with CoVID-19 (about 80%) suffers from a very mild symptoms or are completely asymptomatic in nature. Such patients do not require any significant medical interventions and usually recover on their own. Symptoms if any, manifest primarily as cough, fever or a loss of smell and taste in such patients. However, in a subset of patients, symptoms such as shortness of breath or pneumonia develop indicating a severe manifestation of the disease and, hence, require subsequent hospitalization. In a small percent of cases, severe pneumonia further develops into acute respiratory distress syndrome (ARDS), septic shock and multiple organ failure. Such patients often need ventilator support at Intensive Care Units (ICUs)11.
3 How does CoVID-19 Infect the Human Body?
The CoVID-19 virus, similar to the 2003 SARS-CoV, uses the cell surface receptor ACE2 to gain access to the cells12. Interestingly, there have been reports that the cell surface receptor TMPRSS2 is also required in combination with the ACE2 receptor to gain access to the cells13. For viral entry into the cell, the viral spike protein first needs to be primed by the host cell proteases, in this case TMPRSS2, after which the viral spike protein can interact with the ACE2 protein and can be internalized by the cell. Hence, it is not surprising that both the ACE2 and the TMPRSS2 proteins have been identified as potential candidates that can be targeted for anti-viral interventions12,13. It should be noted though that ACE2 and TMPRSS2 provide possible entry routes to cells of different organs; however, a network of host factors is required to facilitate viral entry, viral RNA synthesis and virus release. Thus, an understanding of cell-type-specific host machinery that mediates viral entry as well as enables replication and budding from the host cell is important to decode SARS-CoV-2 pathogenesis.
ACE2 expression is essential in various cell types across multiple organs in the body as, under homeostatic conditions, it is involved in maintaining the blood pressure levels in humans via the renin–angiotensin system14. Furthermore, ACE2 expression plays a critical role in providing protection to the lung from severe acute lung injury15. However, the presence of ACE2 on the cellular surfaces also makes it easier for the virus to infect the cells by providing a possible route of entry. Due to these seemingly contradictory roles of the ACE2 protein during the course of the infection and to maintain body homeostasis, the use of hypertensive drugs, that generally act on ACE pathways, either to treat CoVID-19 or its continued use in hypertensive patients with CoVID-19 is still debated16,17.
The SARS-CoV-2 virus can affect multiple organs like the heart, kidney, gut, etc.—as these organs harbor cells which contain the ACE2 receptor in significant amounts18. The ACE2 levels have been detected in various cell types in different organs both at the transcriptomic as well as proteomic levels using various high-throughput methods. Furthermore, the mRNA levels have been quantified at both a bulk and a single-cell level. ACE2 protein expression was mainly observed in enterocytes, renal tubules, gallbladder, cardiomyocytes, male reproductive cells, placental trophoblasts, ductal cells, eye, and vasculature with a limited expression in the respiratory system19. Using single-cell RNA sequencing, ACE2 has been detected in lungs20, bronchus21 and nasal mucosa21. In the lung, the SARS-CoV-2 primarily targets the alveolar type 2 cells22,23 a phenomenon similar to the pathogenesis of the previous SARS-CoV24. Hence, it is not surprising to find that the extent of lung damage is a robust marker of disease severity and is often associated with acute respiratory distress syndrome (ARDS)25.
4 How does Our Immune System Respond to SARS-CoV-2 Infection?
The human body has robust mechanisms for clearing infections. Through a series of well-coordinated steps various immune cells are recruited to the site of the infection to clear out the virus and the virally infected cells. The first step of the response to a SARS-CoV-2 infection is the infected epithelial cells secreting inflammatory cytokines 27. These molecules result in the recruitment of circulating innate immune cells as well as tissue-resident dendritic cells.
Innate immune cells, such as neutrophils and monocytes, clear virally infected apoptotic cells via phagocytosis. These cells also secrete a variety of proteases and produce large amounts of reactive oxygen species that help in neutralizing viruses. Additionally, they also help with the recruitment of additional immune cells through the secretion of cytokines and chemokines. These responses occur immediately following recruitment, and generally continue as long as virally infected cells are present27. As viral titers at the primary site of infection go down, these cells are also likely to reduce in numbers, thus indicating a self-inhibitory mechanism to resolve the buildup of cytokines and chemokines.
Simultaneously, tissue-resident dendritic cells phagocytose apoptotic cells that were infected with the virus, and/or viral antigens that may have been shed, which they then present on their surface resulting in the activation of T cells28. The dendritic cell–T cell interaction occurs primarily in lymph nodes, where dendritic cells first migrate to following phagocytosis of antigens. Activation of specific helper T cell subsets results in B cell maturation and increased production of antibodies against viral antigens. Simultaneously, activation of other helper T cell subsets and cytotoxic T cells may result in killing of virally infected epithelial cells29. Together, these responses are termed as adaptive immune responses, and take about 4–7 days to develop following the initial phagocytosis by dendritic cells.
Alongside these inflammatory responses that assist in killing the virus and virally infected cells, the immune system also initiates responses that prevents excessive damage of self-tissue due to inflammation. Suppressive immune responses may include many cell types, but one of the most important are regulatory T cells, which have been reported to play an important role in the progression of the CoVID-19 in the body30. Their primary role is thought to be to control the hyper-activation of the immune system seen in the context of some CoVID-19 patients30.
5 How can Mathematical Models be Used to Understand the Crosstalk of Infected Cells and Immune System, and Its Connection to Diverse Patient Outcomes?
As discussed above, the interplay between infected cells and immune systems—adaptive and innate—happens at different length (at a cellular level, tissue level or over multiple organs) and time scales (refractory changes due to the presence of virus vs. more chronic effects of viral presence and/or unresolved inflammatory cytokine levels). To elucidate the emergent dynamics of any such multi-component and/or multi-scale phenomenon, mathematical models have been demonstrated to be a powerful tool. Mathematical models have been used extensively in the context of epidemiological studies of various infectious diseases like influenza31,32, HIV-AIDS33, hepatitis34, etc. Many mathematical models have been proposed elucidating the dynamics of spread of the SARS-CoV-2 across various communities35,36,37,–38, and they have been crucial in informing policy-making decisions in controlling the spread of various infectious diseases including the ongoing pandemic39,40,41,–42. These models describe how an infectious agent like a virus spreads in a community and what kind of intervention techniques can be designed to better combat the spread of the disease in a given population, but do not investigate the intra-host dynamics of SARS-CoV-2 infection that dictate the different patient outcomes in terms of disease severity.
A different class of mathematical models that focus on the intra-host dynamics of the spread of infectious agents have been studied extensively in the context of HIV-AIDS and influenza, among others43,44. These models, often using a set of coupled ordinary differential equations (ODEs), describe how the virus or the infective agent spreads within the host and the mechanisms within the host to resolve such infections. One of the key parameters tracked by such models is the viral load in the body of the patient. The viral load is one of the primary predictors of the disease progression in most infectious diseases. Mathematical models that track the viral kinetics in CoVID-19 patients have also been built recently45. However, very few models exist so far that incorporate the interactions of the virally infected cells with different types of immune responses in the body28,46. Such attempts can be powerful for understanding how the different clinical phenotypes appear as manifestations of underlying immune cell interactions with the infected cells.
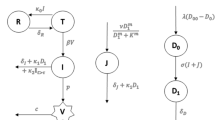
To elucidate the dynamics of CoVID-19 virus inside the human body and the response immune system as a result of this, we recently built a minimalistic model that can accurately capture various phenotypes generally seen in the context of CoVID-19 patients47. Such a model may help us understand the diversity of clinically observed phenotypes and provide insight into intervention strategies to reduce morbidity and mortality. In this model, we considered the primary interactions of the innate immune system, the adaptive immune system and the immuno-regulatory mechanisms in the body in response to the pool of infected cells in the body47. Here, no spatial aspects of lung architecture are considered, although lung is the primary site of infection of CoVID-19); thus our framework is generic enough to be adapted to any other site of infection such as the gut, kidneys, etc. We built this model in a modular manner increasing the complexity of the infected cells immune interactions in a step-wise manner. First, we assume that the infected cells can increase in number via a logistic model, i.e., the infected cells can release viral particles that can infect more susceptible cells and make them infected. This process occurs till there are no susceptible cells left to be infected. This process can be kept in check primarily by the action of the innate immune system or the adaptive immune system where these cell types can be activated at the site of infection and/or recruited by signals (cytokines or antigen based) sent by the site of infection. For simplicity, we have considered these two broad groups of cell types. In reality, the system is far more complicated with multiple immune and non-immune cell types involved in the immune reaction. The innate and the adaptive immune system can further activate/recruit more cells of their own type. We do not consider cross activation between the innate and the adaptive arms of the innate immune system again for simplicity. We also consider the fact that the innate immune cells have the capacity to suppress themselves in the absence of the virus and the adaptive immune cells can get exhausted if there is an excessive and prolonged presence of the viral antigen in the system. Finally, we consider the effect of regulatory T cells on this system. Regulatory T cells can be activated by the presence of the virus in the system and can actively suppress the number and/or activity of the innate and the adaptive immune cells (Fig. 1).

A simplified schematic of infected cell–immune cell interactions in a SARS-CoV-2-infected individual. Black arrows indicate activation/recruitment of a specific cell type, while the red links indicate exhaustion/repression/death of specific cell types
The interactions of the innate immune system with the virally infected cells give a variety of possible phenotypes. We found that depending on the relative strengths of the innate immune system in clearing the virally infected cells (dmv) and the self-suppression strength (resolution of inflammation) (dvm) of the innate immune system, the system can exist in qualitatively 3 distinct states (Fig. 2a): a parameter regime where the infected cells are cleared and the disease regresses (high dmv and moderate to low dvm), a region where the infected cells keep growing continuously (low dmv) and a region where the infection can persist with intermediate values of infected cells(high dmv and high dvm). We further quantified the levels of immunopathology (overactive immune system causing damage to the tissues) in the system as function of the number of immune cells (innate and adaptive) and the number of infected cells present at the site of infection weighted by the ability of each of these cell types to create an inflammatory environment. This metric is useful to gauge the levels of tissue damage that might occur due to the collective inflammatory environment created by both infected and immune cells, thus providing a metric for disease severity.

Heatmaps showing the infected number of cells present at the end of 30 days in individuals as a function of a the self-suppression strength (dvm) and the killing strength of the innate immune system (dmv) b the killing strength of the innate (dmv) and the adaptive (dcv) immune system. c Dynamic profiles of infected cell numbers and the immunopathology in the system in the presence and absence of the regulatory T cell arm of the immune system. Note that the blue broken line (infected cells w/o regulatory cells) is completely overlapping with the solid blue line (infected cells with regulatory cells) indicating that the introduction of the regulatory T cells does not have a significant impact on the viral clearance dynamics (Reference: Sahoo et. al. bioRxiv47)
Interestingly, the levels of immunopathology in the system are the lowest at intermediate values of the self-suppression strength of the innate immune system, indicating that over-activation or under-activation of this parameter can result in poorer outcome for the body. Furthermore, we show that depending on the proliferation rate of the virus in the system and the strength of the innate immune system in clearing out virally infected cells, the time taken for viral clearance can vary greatly (see Sahoo et al. for details47).
We also observe that in specific cases where the innate immune system was not able to clear the virally infected cells, the introduction of the adaptive system was able to clear such cells with a significant reduction in the immunopathology in the system. Additionally, we found that just as in the case of innate immune system, a very strong adaptive immune response was enough to clear the virus from the system within a short span of time. As expected, we observed that viral clearance took place when either the adaptive or the innate immune system or both were acting strongly to clear the virus (regions where either dmv or dcv or both are high) (Fig. 2b). Next, we introduced the regulatory T cells in our framework that can suppress adaptive and/or innate response, thus resolving relatively higher levels of unresolved immunopathology in a subset of cases, thereby underscoring the importance of such cells in regulation of the inflammation levels in the body. Thus, including the effect of regulatory T cells helps to reduce immunopathology as well as the number of infected cells (Fig. 2c).
In addition to these dynamical features, the model we developed depicts how the various observed clinicopathologic features emerge from the dynamical interactions between the infected and the immune cells (see Sahoo et. al. for details47). Specifically, the model demonstrates, depending on the relative strengths of the innate and the adaptive immune system, the two “non-severe” phenotypes, presumably mapping onto the asymptomatic or the mild cases as observed clinically. Both these phenotypes have relatively lower levels of peak immunopathology and the virus is cleared from the system within a time-period of 15 days from the model observations. This is corroborated by the clinical observations where the viral load in asymptomatic and mild cases drops to undetectable levels within 15 days48. Similarly, the model demonstrates that in the cases where the strength of the innate immune system was of intermediate strength and the adaptive immune system was not strong enough to clear the virus, the virus clearance time was much higher than mild/ asymptomatic cases (> 20 days). This scenario may explain the majority of the cases that show an agedependent severity in clinical manifestations of the disease, because the adaptive immune system is reported to be generally weaker in older populations49. Interestingly, the cases where the strength of both the innate and adaptive immune system were weak, the disease severity was the maximum with high viral loads at the end of 30 days. Such patients are likely to die from the unresolved infection and consequent high levels of immunopathology. Such a phenotype is likely seen in immunocompromised individuals who are at the highest risk of the infection50.
The model also predicts that the over-activation or an untimely early activation of regulatory T cells could result in the failure to clear the virus from the system with a suppressed immune system facilitating other opportunistic pathogens to infect the lungs. Secondary and co-infections have been observed in a subset of patients51, for which one possible explanation can be a dysregulation of the regulatory T cell arm of the immune system; however, further evidence is needed before one can strengthen this hypothesis mechanistically. In addition to demonstrating how various clinical phenotypes could arise from these underlying cell–cell interactions, the model also highlights that treatment with anti-viral agents might not be sufficient to resolve the severe phenotypes seen in many patients. Hence, in principle, the model can be used to design intervention strategies and test the outcomes of anti-virals and possibly the use of anti-inflammatory drugs in combination to better treat the more severe cases of CoVID-19 infection.
6 Does the Existing Clinical Data Support the Model Predictions?
With the advent of high-throughput technological advances, there has been a burst of data focusing on the clinicopathological aspects of CoVID-19. Interestingly, immune profiling has revealed an overall increase in innate cell lineages with a concomitant reduction in T cell number with disease progression52. Multiple studies have now reported the existence of an exhausted T cell population in more severe patients suffering from the coronavirus. This is in direct agreement with the readout of our model47 in cases of more severe phenotypes of the disease53,54. Furthermore, in a cohort of 100 patients with hyperinflammatory syndrome, a monoclonal antibody that targets the IL-6 receptor, called Tocilizumab, has been shown to improve or stabilize the condition in about 77% of the cases, whereas the condition worsened in remaining 23%55. This observation supports the fact that one can use immune suppressants for treatment of the severe patients, but it would be important to make sure that the infection in largely cleared before the treatment. If the infection is not cleared, then these treatments can be counterproductive can as it may increase the likelihood of secondary or co-infections. Furthermore, it could also lead to viral persistence in the system56 and might affect the antibody response in the body in undesirable ways57.
7 Conclusions
Mathematical modeling of viral dynamics and various interactions of the immune system holds promise in developing a better mechanistic understanding of the SARS-CoV-2 infection and the host response. Improved comprehension of both these aspects of the infection is likely to improve current treatment strategies and help in designing newer ones. An important goal of such models would be to act as a platform to better design therapies that can minimize CoVID-19-related physiological disruptions and alleviate immunopathology too.
References
Kahn JS, McIntosh K (2005) History and recent advances in coronavirus discovery. Pediatr Infect Dis J. https://doi.org/10.1097/01.inf.0000188166.17324.60
Van Der Hoek L (2007) Human coronaviruses: what do they cause? Antivir Ther 12:651–658
Ye ZW, Yuan S, Yuen KS et al (2020) Zoonotic origins of human coronaviruses. Int J Biol Sci 16:1686–1697
De Wit E, Van Doremalen N, Falzarano D, Munster VJ (2016) SARS and MERS: Recent insights into emerging coronaviruses. Nat Rev Microbiol 14:523–534
Petrosillo N, Viceconte G, Ergonul O et al (2020) COVID-19, SARS and MERS: are they closely related? Clin Microbiol Infect 5:729–734
Chan JWM, Ng CK, Chan YH et al (2003) Short term outcome and risk factors for adverse clinical outcomes in adults with severe acute respiratory syndrome (SARS). Thorax. https://doi.org/10.1136/thorax.58.8.686
Yang YM, Hsu CY, Lai CC et al (2017) Impact of comorbidity on fatality rate of patients with middle east respiratory syndrome. Sci Rep. https://doi.org/10.1038/s41598-017-10402-1
Sanyaolu A, Okorie C, Marinkovic A et al (2020) Comorbidity and its impact on patients with COVID-19. SN Compr Clin Med. https://doi.org/10.1007/s42399-020-00363-4
Li X, Xu S, Yu M et al (2020) Risk factors for severity and mortality in adult COVID-19 inpatients in Wuhan. J Allergy Clin Immunol. https://doi.org/10.1016/j.jaci.2020.04.006
Codo AC, Davanzo GG, de Monteiro LB et al (2020) Elevated glucose levels favor SARS-CoV-2 infection and monocyte response through a HIF-1α/glycolysis-dependent axis. Cell Metab. https://doi.org/10.1016/j.cmet.2020.07.007
Richardson S, Hirsch JS, Narasimhan M et al (2020) Presenting characteristics, comorbidities, and outcomes among 5700 patients hospitalized with COVID-19 in the New York City area. J Am Med Assoc. https://doi.org/10.1001/jama.2020.6775
Zhang H, Penninger JM, Li Y et al (2020) Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: molecular mechanisms and potential therapeutic target. Intensive Care Med. https://doi.org/10.1007/s00134-020-05985-9
Hoffmann M, Kleine-Weber H, Schroeder S et al (2020) SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. https://doi.org/10.1016/j.cell.2020.02.052
Tikellis C, Thomas MC (2012) Angiotensin-converting enzyme 2 (ACE2) is a key modulator of the renin angiotensin system in health and disease. Int J Pept 2012:256294
Imai Y, Kuba K, Rao S et al (2005) Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature. https://doi.org/10.1038/nature03712
Yan T, Xiao R, Lin G (2020) Angiotensin-converting enzyme 2 in severe acute respiratory syndrome coronavirus and SARS-CoV-2: a double-edged sword? FASEB J 34:6017–6026
Singh AK, Gupta R, Misra A (2020) Comorbidities in COVID-19: outcomes in hypertensive cohort and controversies with renin angiotensin system blockers. Diabetes Metab Syndr Clin Res Rev. https://doi.org/10.1016/j.dsx.2020.03.016
Zaim S, Chong JH, Sankaranarayanan V, Harky A (2020) COVID-19 and multiorgan response. Curr Probl Cardiol
Hikmet F, Mear L, Uhlen M, Lindskog C (2020) The protein expression profile of ACE2 in human tissues. bioRxiv. https://doi.org/10.1101/2020.03.31.016048
Han X, Zhou Z, Fei L et al (2020) Construction of a human cell landscape at single-cell level. Nature. https://doi.org/10.1038/s41586-020-2157-4
Vieira Braga FA, Kar G, Berg M et al (2019) A cellular census of human lungs identifies novel cell states in health and in asthma. Nat Med. https://doi.org/10.1038/s41591-019-0468-5
Chu H, Chan JFW, Wang Y et al (2020) Comparative replication and immune activation profiles of SARS-CoV-2 and SARS-CoV in human lungs: an ex vivo study with implications for the pathogenesis of COVID-19. Clin Infect Dis. https://doi.org/10.1093/cid/ciaa410
Li H, Liu L, Zhang D et al (2020) SARS-CoV-2 and viral sepsis: observations and hypotheses. Lancet 395:1517–1520
Mossel EC, Wang J, Jeffers S et al (2008) SARS-CoV replicates in primary human alveolar type II cell cultures but not in type I-like cells. Virology. https://doi.org/10.1016/j.virol.2007.09.045
Liu Y, Yang Y, Zhang C et al (2020) Clinical and biochemical indexes from 2019-nCoV infected patients linked to viral loads and lung injury. Sci China Life Sci. https://doi.org/10.1007/s11427-020-1643-8
Blanco-Melo D, Nilsson-Payant BE, Liu WC et al (2020) Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell. https://doi.org/10.1016/j.cell.2020.04.026
McKechnie JL, Blish CA (2020) The innate immune system: fighting on the front lines or fanning the flames of COVID-19? Cell Host Microbe 27:863–869
Du SQ, Yuan W (2020) Mathematical modeling of interaction between innate and adaptive immune responses in COVID-19 and implications for viral pathogenesis. J Med Virol. https://doi.org/10.1002/jmv.25866
Chen Z, John Wherry E (2020) T cell responses in patients with COVID-19. Nat Rev Immunol 20:529–536
Stephen-Victor E, Das M, Karnam A et al (2020) Potential of regulatory T cell-based therapies in the management of severe COVID-19. Eur Respir J. https://doi.org/10.1183/13993003.02182-2020
Hill EM, Petrou S, De Lusignan S et al (2019) Seasonal influenza: Modelling approaches to capture immunity propagation. PLoS Comput Biol. https://doi.org/10.1371/journal.pcbi.1007096
Sambaturu N, Mukherjee S, López-García M et al (2018) Role of genetic heterogeneity in determining the epidemiological severity of H1N1 influenza. PLoS Comput Biol. https://doi.org/10.1371/journal.pcbi.1006069
Li Z, Teng Z, Miao H (2017) Modeling and control for HIV/AIDS transmission in china based on data from 2004 to 2016. Comput Math Methods Med. https://doi.org/10.1155/2017/8935314
Liang P, Zu J, Zhuang G (2018) A literature review of mathematical models of hepatitis B virus transmission applied to immunization strategies from 1994 to 2015. J Epidemiol 28:221–229
Ivorra B, Ferrández MR, Vela-Pérez M, Ramos AM (2020) Mathematical modeling of the spread of the coronavirus disease 2019 (COVID-19) taking into account the undetected infections. The case of China. Commun Nonlinear Sci Numer Simul. https://doi.org/10.1016/j.cnsns.2020.105303
Kucharski AJ, Russell TW, Diamond C et al (2020) Early dynamics of transmission and control of COVID-19: a mathematical modelling study. Lancet Infect Dis. https://doi.org/10.1016/S1473-3099(20)30144-4
Lin Q, Zhao S, Gao D et al (2020) A conceptual model for the coronavirus disease 2019 (COVID-19) outbreak in Wuhan, China with individual reaction and governmental action. Int J Infect Dis. https://doi.org/10.1016/j.ijid.2020.02.058
Wu JT, Leung K, Leung GM (2020) Nowcasting and forecasting the potential domestic and international spread of the 2019-nCoV outbreak originating in Wuhan, China: a modelling study. Lancet. https://doi.org/10.1016/S0140-6736(20)30260-9
Shim E, Meyers LA, Galvani AP (2011) Optimal H1N1 vaccination strategies based on self-interest versus group interest. BMC Public Health. https://doi.org/10.1186/1471-2458-11-S1-S4
Kamyad AV, Akbari R, Heydari AA, Heydari A (2014) Mathematical modeling of transmission dynamics and optimal control of vaccination and treatment for hepatitis B virus. Comput Math Methods Med. https://doi.org/10.1155/2014/475451
Tang B, Wang X, Li Q et al (2020) Estimation of the transmission risk of the 2019-nCoV and its implication for public health interventions. J Clin Med. https://doi.org/10.3390/jcm9020462
Leung K, Wu JT, Liu D, Leung GM (2020) First-wave COVID-19 transmissibility and severity in China outside Hubei after control measures, and second-wave scenario planning: a modelling impact assessment. Lancet. https://doi.org/10.1016/S0140-6736(20)30746-7
Perelson AS, Ribeiro RM (2013) Modeling the within-host dynamics of HIV infection. BMC Biol 11:96
Perelson AS (2002) Modelling viral and immune system dynamics. Nat Rev Immunol 2:28–36
Hernandez-Vargas EA, Velasco-Hernandez JX (2020) In-host modelling of COVID-19 kinetics in humans. medRxiv. https://doi.org/10.1101/2020.03.26.20044487
Baral S, Antia R, Dixit NM (2019) A dynamical motif comprising the interactions between antigens and CD8 T cells may underlie the outcomes of viral infections. Proc Natl Acad Sci USA. https://doi.org/10.1073/pnas.1902178116
Sahoo S, Hari K, Jhunjhunwala S, Jolly MK (2020) Mechanistic modeling of the SARS-CoV-2 and immune system interplay unravels design principles for diverse clinicopathological outcomes. bioRxiv. https://doi.org/10.1101/2020.05.16.097238
Kim SE, Jeong HS, Yu Y et al (2020) Viral kinetics of SARS-CoV-2 in asymptomatic carriers and presymptomatic patients. Int J Infect Dis. https://doi.org/10.1016/j.ijid.2020.04.083
Quinn KM, Fox A, Harland KL et al (2018) Age-related decline in primary CD8+ T cell responses is associated with the development of senescence in virtual memory CD8+ T cells. Cell Rep. https://doi.org/10.1016/j.celrep.2018.05.057
Fung M, Babik JM (2020) COVID-19 in immunocompromised hosts: what we know so far. Clin Infect Dis. https://doi.org/10.1093/cid/ciaa863
Langford BJ, So M, Raybardhan S et al (2020) Bacterial co-infection and secondary infection in patients with COVID-19: a living rapid review and meta-analysis. Clin Microbiol Infect. https://doi.org/10.1016/j.cmi.2020.07.016
Lucas C, Wong P, Klein J et al (2020) Longitudinal immunological analyses reveal inflammatory misfiring in severe COVID-19 patients. medRxiv. https://doi.org/10.1101/2020.06.23.20138289
Diao B, Wang C, Tan Y et al (2020) Reduction and functional exhaustion of T cells in patients with coronavirus disease 2019 (COVID-19). Front Immunol. https://doi.org/10.3389/fimmu.2020.00827
Zheng M, Gao Y, Wang G et al (2020) Functional exhaustion of antiviral lymphocytes in COVID-19 patients. Cell Mol Immunol 17:533–535
Toniati P, Piva S, Cattalini M et al (2020) Tocilizumab for the treatment of severe COVID-19 pneumonia with hyperinflammatory syndrome and acute respiratory failure: a single center study of 100 patients in Brescia. Italy. Autoimmun Rev 19:102568
Panichaya P, Thaweerat W, Uthaisan J (2020) Prolonged viral persistence in COVID-19 second trimester pregnant patient. Eur J Obstet Gynecol Reprod Biol 250:263
Huang C-TC-G, Dutta A et al (2020) Relative COVID-19 viral persistence and antibody kinetics. medRxiv. https://doi.org/10.1101/2020.07.01.20143917
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Sahoo, S., Jhunjhunwala, S. & Jolly, M.K. The Good, The Bad and The Ugly: A Mathematical Model Investigates the Differing Outcomes Among CoVID-19 Patients. J Indian Inst Sci 100, 673–681 (2020). https://doi.org/10.1007/s41745-020-00205-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s41745-020-00205-1