Therapeutic Applications of Solid Dispersions for Drugs and New Molecules: In Vitro and In Vivo Activities

 , and
, and
Abstract
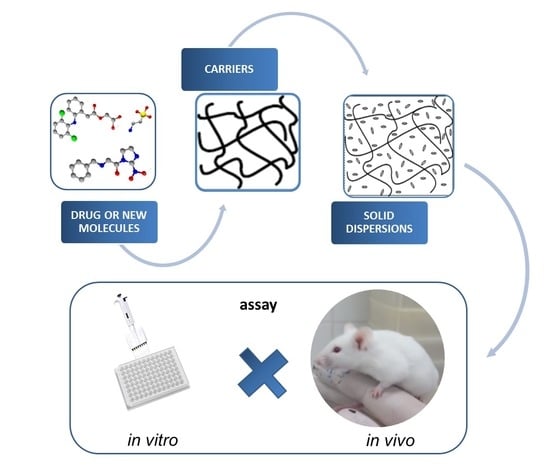
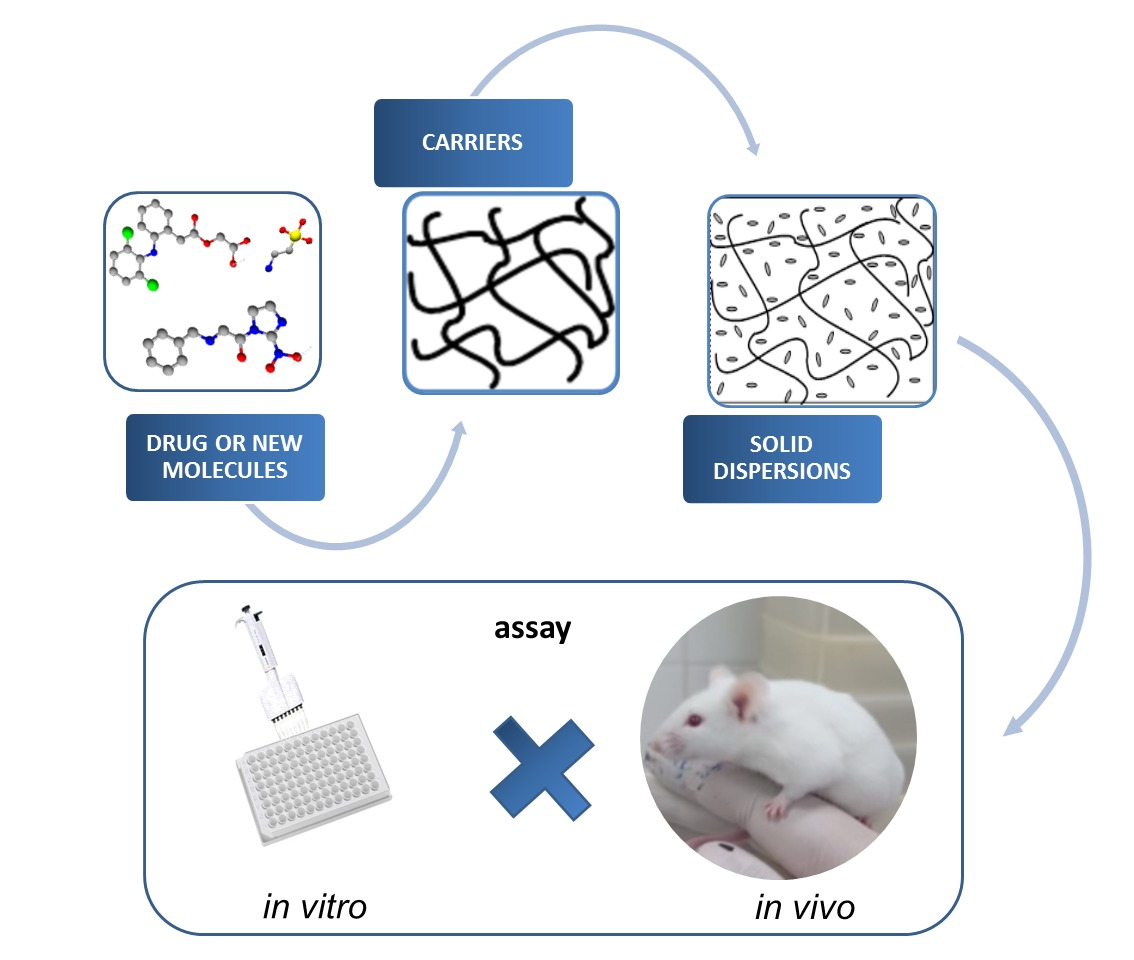
:
1. Introduction
2. Methods
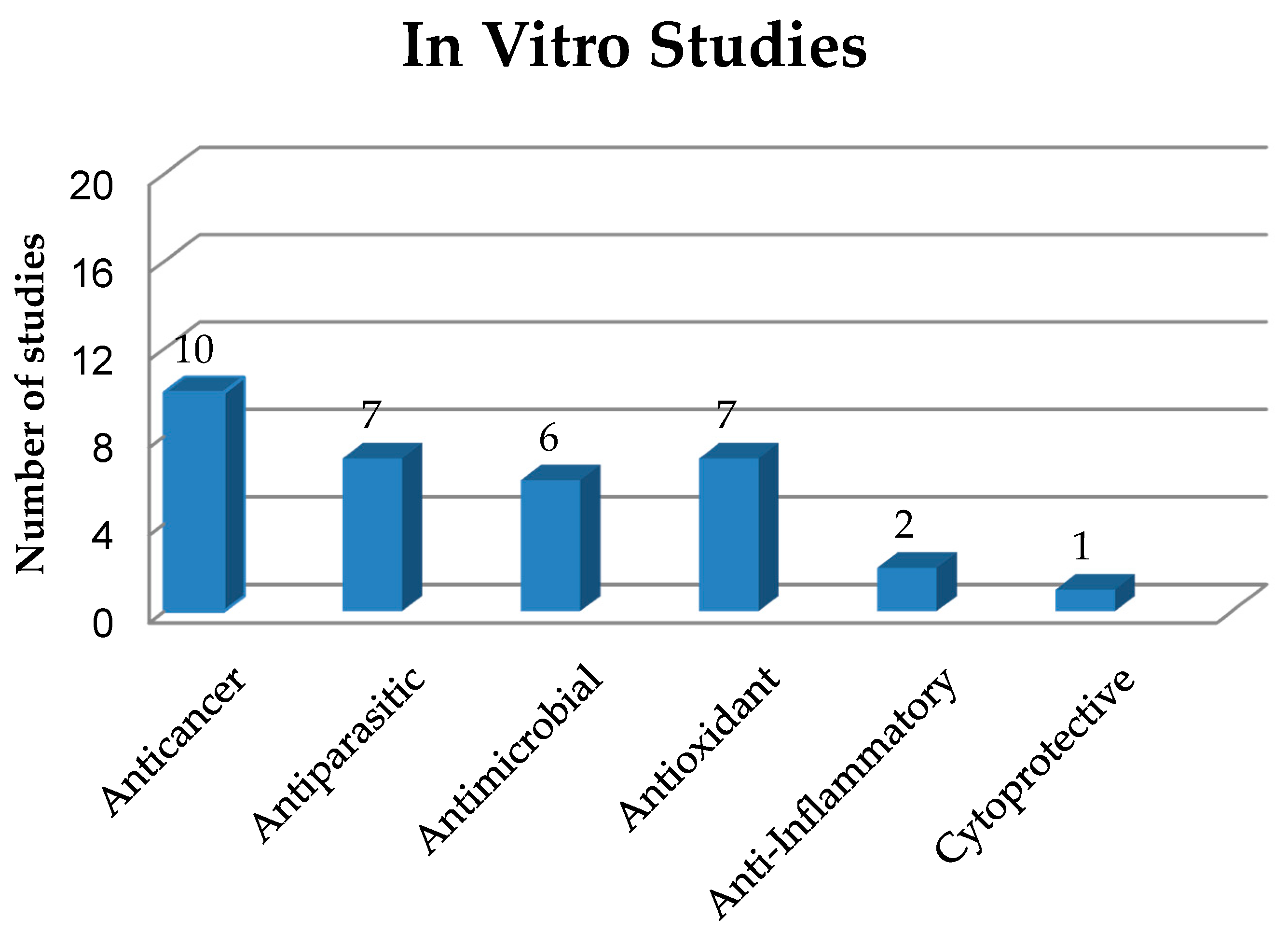
3. In Vitro Study of Solid Dispersions in Polymeric Matrices
3.1. Anticancer Activities of Solid Dispersions
3.2. Antiparasitic Activity of Solid Dispersions
3.2.1. Antichagasic Activity of Solid Dispersions
3.2.2. Antischistosomal Activity of Solid Dispersions
3.2.3. Antimalarial Activity of Solid Dispersions
3.3. Antimicrobial Activity of Solid Dispersions
3.4. Antioxidant Activity of Solid Dispersions
3.5. Anti-Inflammatory Activity of Solid Dispersions
3.6. Cytoprotective Activity on Liver Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Carrier Type | Substance | Cell Type | Activity | Improved Characteristics | Reference |
|---|---|---|---|---|---|
| OHPP | Niclosamide | PC-3, HeLa, A549 | Anticancer | SDs showed higher cytotoxicity to target cells (lower IC50) than the niclosamide solution. | [11] |
| OHPP | Paclitaxel | PC-3, HeLa, A549 | Anticancer | SDs showed significantly higher cytotoxicity to target cells (lower IC50) than the paclitaxel solution. | [13] |
| PVP/VA TPGS | Paclitaxel | BT-474, MCF-7, SK-BR-3 | Anticancer | SDs showed higher cytotoxicity against cancer cells compared to the pure drug. | [14] |
| Brij®L4 | Chrysin | HT29 | Anticancer | The higher solubility of chrysin in SDs compared to water solution increased cytotoxicity. | [15] |
| Poloxamer 407 | Curcumin (CM) | NCIH460, HeLa, HepG2, MCF-7 and PLP2 | Anticancer | SDs showed cytotoxicity against all tumor cell lines tested, but no toxic effects on non-tumor cells. | [16] |
| AChE, BChE, GST, MAO A-B | Enzyme inhibitory /Antioxidant | SD was able to inhibit the activities of AChE, BChE and GST in aqueous medium. | |||
| LPS-stimulated murine macrophages (RAW 264.7) | Anti-inflammatory | IC50 (inhibitory concentration of 50% NO production by macrophages) > 400 μg/mL. | |||
| PVP K30 | Zn(II)-curcumin complex | HepG2, SK-HEP1 | Anticancer | SD of Zn(II)-curcumin complex had a potent anticancer effect. | [17] |
| HPMC, PVP K30, PEG 6000 | Telaprevir | HepG2 | Anticancer | The antitumor activity was dose dependent and even with the addition of the polymer the drug maintained its efficacy. | [18] |
| Soluplus® | Angelica gigas Nakai | HeLa, HEK 293 | Anticancer | SD at the concentration of 200 μg/mL showed a significant decrease (to only 17.37%) in cell viability. There was no toxicity to normal cells. | [20] |
| Eudragit S-100 | Berberine hydrochloride (HB) | SW480, HCT116, Caco-2 | Anticancer | The release of HB from SDs was effective and cell viability was reduced in a dose and time dependent manner. | [21] |
| PVP K30 | IIIM-290 | Ehrlich ascites carcinoma cells | Cytotoxic | Despite the reduced amount of IIIM-290 in SD, the IC50 value of SD was lower than that of IIIM-290 alone. | [24] |
| Poloxamer 407 | Benznidazole | Epimastigotes of Trypanosoma cruzi | Antichagasic | SDs enhanced drug solubility, release kinetics and parasitic activity | [25] |
| Low-substituted HPC | Benznidazole | Epimastigotes and intracellular amastigotes of T. cruzi (CL-B5) | Antichagasic | SDs had higher antiparasitic activity against amastigotes than epimastigotes. | [28] |
| Gelucire 50/13 | Ursolic acid | Trypomastigotes of T. cruzi Y | Antichagasic | Increased antiparasitic activity. | [29] |
| PVP K30,PVP/VA, Kollidon-CL-M, sodium starch glycolate | Praziquantel | Adult Schistosomes of Schistosoma mansoni | Antischistosomal | Increased solubility, better bioavailability and stronger antiparasitic activity. | [30] |
| PVP K30 | Praziquantel | Newly transformed schistosomula of S. mansoni and adults | Antischistosomal | Increased solubility, reduced dosage especially for children and increased antiparasitic activity. | [31] |
| Soluplus, PEG 400, Lutrol F127 and Lutrol F68 | Artemether | Schizonts of Plasmodium falciparum 3D7 | Antimalarial | Increased dissolution rate, amorphous form, increased solubility and, mainly, increased antimalarial activity. | [34] |
| Soluplus, Kollidon VA64, Plasdone S630 | Lumefantrine | ITG cells | Antimalarial | Increased antiparasitic activity. | [35] |
| Chitosan | Abietic acid | Staphylococcus epidermidis | Antimicrobial | SD exhibited better MIC values against S. epidermidis than chitosan and abietic acid alone. | [36] |
| DPPH radical scavenging | Antioxidant | SD had higher antioxidant power (IC50 of 0.61 mg/mL) than abietic acid alone (IC50 of 11 mg/mL). | |||
| PVP K30 and HPMCAS | Griseofulvin | Dermatophytes of Trichophyton rubrum NCPF 935 | Antimicrobial | SDs significantly reduced biofilm formation when compared to the control. | [37] |
| Pluronic F127 | Gatifloxacin | Staphylococcus aureus | Antimicrobial | The gatifloxacin/Pluronic F127 system exhibited antimicrobial efficacy when compared to commercialized eye drops. | [39] |
| PVP K30 | Curcumin | Salmonella enteritidis | Antimicrobial | SD had a strong antimicrobial effect on S. enteritidis, while CM alone did not show antimicrobial activity in vitro. | [40] |
| HPMC | Curcumin | Escherichia coli | Antimicrobial | SD used to prepare phototoxic supersaturated solutions showed significant bactericidal activity against E. coli. | [41] |
| Polaxamer 407 | Curcumin | E. coli, Pseudomonas aeruginosa and S. aureus | Antimicrobial | The association between SD and silver nanoparticles increased CM antimicrobial and antioxidant activities. | [42] |
| DPPH radical scavenging | Antioxidant | ||||
| PVP K25 | Quercetin | DPPH radical scavenging | Antioxidant | Increased quercetin antioxidant activity in SD (0.61 ± 0.03 ≤ IC50 ≤ 1.00 ± 0.02 μg/mL). | [44] |
| Mannitol | Coenzyme Q10 | Intracellular ROS level | Antioxidant | The SD with the smallest particle size showed the greatest absorption of UVB radiation as well as the highest antioxidant activity in vitro. | [45] |
| PVP K30 | Usnic acid | DPPH radical scavenging | Antioxidant | Increased usnic acid solubility and antioxidant activity. | [46] |
| PEG 4000 | Luteolin | DPPH | Antioxidant | Polymers increased luteolin solubility and antioxidant activity. | [47] |
| PVP K30, PEG 6000 and HPMC | α,β-Amyrin | LPS-stimulated macrophages J774 | Anti-inflammatory | SDs enhanced the anti-inflammatory activity of α,β-amyrin. | [7] |
| HPMC | Curcumin | HepG2 | Cytoprotective | SDs showed better cytoprotective activity than pure CM and inhibited cell death induced by t-BHP. | [48] |
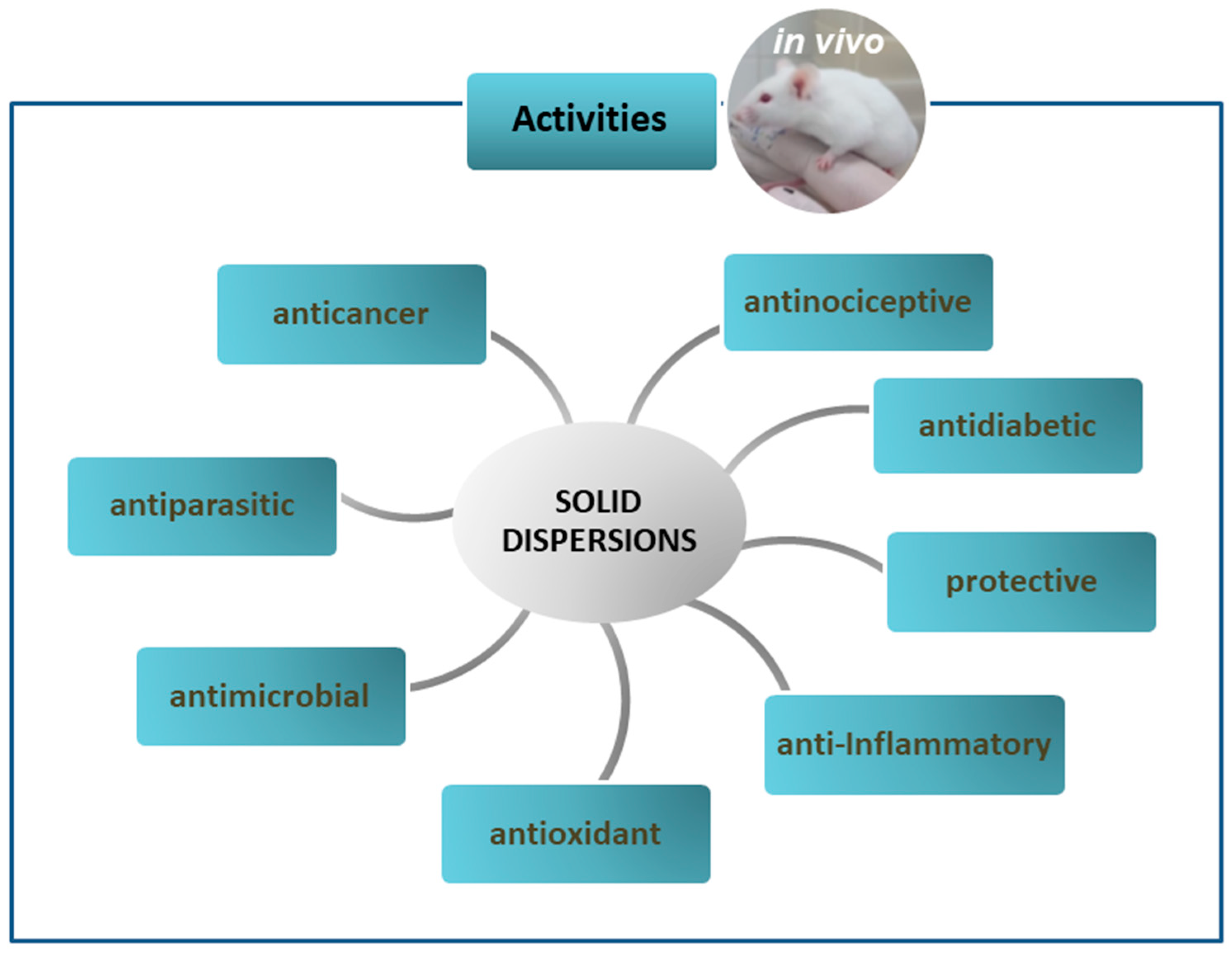
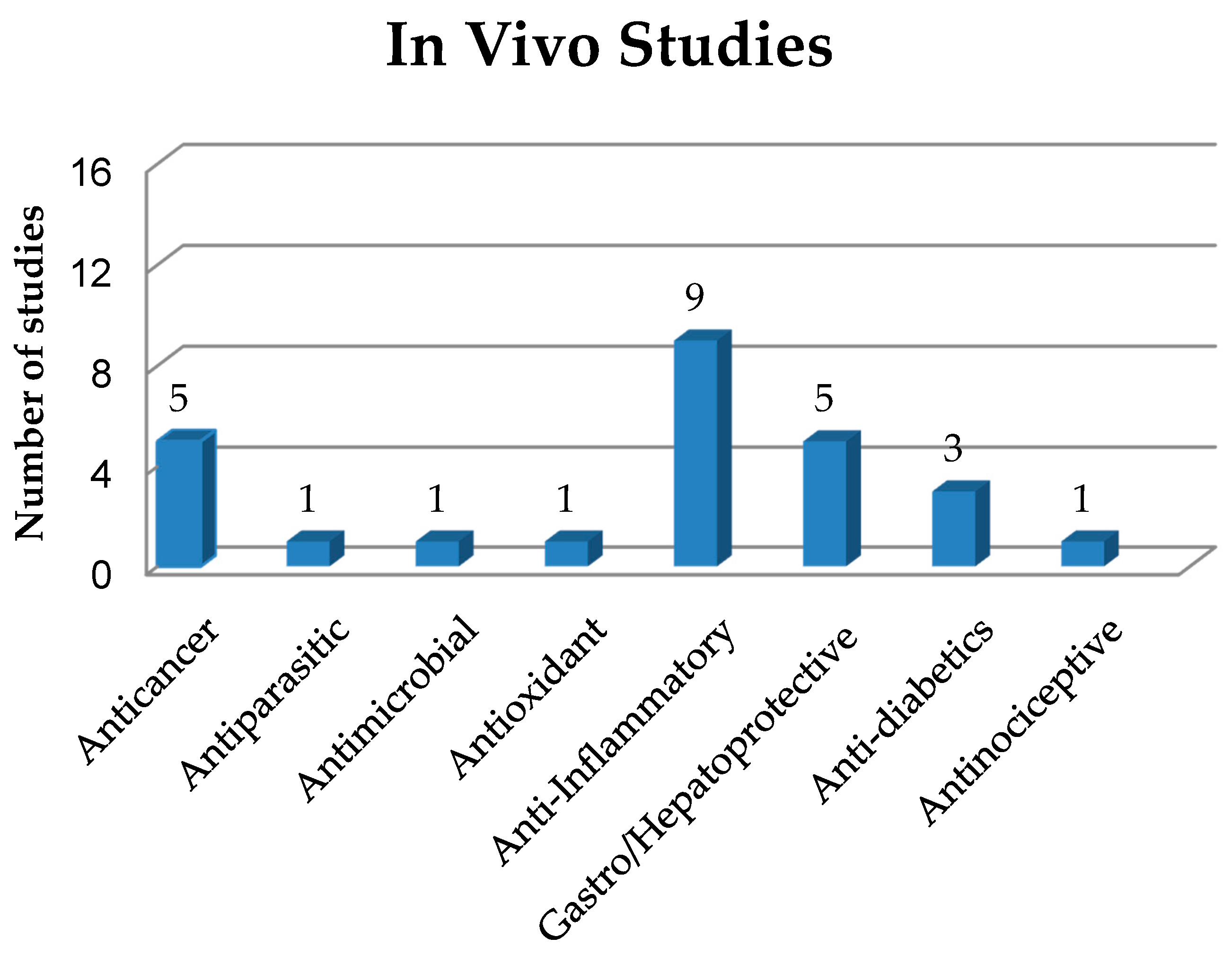
4. In Vivo Studies on Solid Dispersions in Polymeric Matrices
4.1. Anticancer Activity of Solid Dispersions
4.2. Antiparasitic Activity of Solid Dispersions
4.3. Antimicrobial Activity of Solid Dispersions
4.4. Antioxidant Activity of Solid Dispersions
4.5. Anti-Inflammatory Activity of Solid Dispersions
4.6. Gastro and Hepatoprotective Activity of Solid Dispersions
4.7. Antidiabetic Activity of Solid Dispersions
4.8. Antinociceptive Activity of Solid Dispersions
| Carrier Type | Substance | Animal | Dose | Activity | Improved Characteristics | References |
|---|---|---|---|---|---|---|
| PVP K30 | IIIM-290 | Swiss male mice (18–23 g) | 25, 50 and 75 mg/kg | Anticancer | SD was able to reduce the IIIM-290 dose by at least 1.5-fold thanks to its efficiency in Ehrlich solid tumor model. | [24] |
| Soluplus® | 9-Nitrocamptothecin | Male Sprague-Dawley rats (20 ± 2 g) | 4 mg/kg | Anticancer | SD showed higher tumor growth inhibitory rate than the pure compound due to improved oral bioavailability | [49] |
| (+)-Xylitol | (−)-Oleocanthal | Athymic nude mice | 10 mg/kg | Anticancer | Treatment with SD showed prevention, lower growth rate and less recurrence of tumor. | [50] |
| PVP K30 | Zinc(II)-curcumin complex | B-NDG, BALB/c mice | 100 mg/kg | Anticancer | SD reduced tumor size and weight in animals. | [17] |
| PVP K30 | Selaginella doederleinii Hieron | BALB/c mice | 200 mg/kg | Anticancer | SD reduced tumor size as well as the level of tumor angiogenesis. | [53] |
| Low substituted HPC | Benznidazole | Female NMRI mice (25 ± 2 g) | 25 mg/kg/day | Antichagasic | The best SD showed a 96.65% trypanocidal activity, expressed as percentage reduction in the area under the parasitic curve. | [28] |
| PVP K30 | Curcumin (CM) | Male Cobb-Vantress broiler chickens | 1 g/kg of feed | Antimicrobial | The synergistic effect of 0.05% CM/PVP SD with 0.05% boric acid reduced colonization of Salmonella enteritidis in crop and ceca-cecal tonsils. | [40] |
| PVP K30 | Taurine-zinc complex | Female Sprague-Dawley rats (240–260 g) | 100 and 200 mg/kg/day | Antioxidant | SDs protected rat gastric mucosa from ethanol-induced injury and increased SOD activity and glutathione level. | [58] |
| Gastroprotective | ||||||
| Kollidon (VA64) | Triacetylated andrographolide (TA) | Male Kunming mice | 50, 100 and 200 mg/kg/day | Anti-inflammatory | TA-SD prepared with VA64 significantly improved the drug activity against ulcerative colitis. | [59] |
| PVP K30, Poloxamer 188 | Curcumin | Female CD-1 mice | 100 mg/kg oral doses | Anti-inflammatory | CM-SD prepared with PVP decreased matrix metallo-peptidase 9 expression and levels of IL-1β and IL-6 cytokines. | [60] |
| Gelucire®50/13-Aerosil® | Curcumin | Rat | 10 to 100 mg/kg | Anti-inflammatory | A CM-SD dose of 100 mg/kg was more effective than 5 mg/kg indomethacin in reducing edema. | [61] |
| HPMC, lecithin and isomalt | Curcumin | Male Sprague-Dawley rats | 5 mg/kg | Anti-inflammatory | A CM-SD dose of 5 mg/kg had greater anti-inflammatory activity than 50 mg/kg curcumin alone. | [62] |
| Crospovidone | Aceclofenac | Male Sprague-Dawley rats | 1 g/cm2 (topical) | Anti-inflammatory | The enhanced drug permeation increased the intensity of the anti-inflammatory response. | [65] |
| PEG 8000 | Ibuprofen | Wistar rats | 20 mg/kg | Anti-inflammatory | All SDs showed better anti-inflammatory activity than the pure drug, allowing up to 90% edema inhibition after 6 h. | [66] |
| Urea and mannitol | Flurbiprofen | Rat | 11.69 mg/kg | Anti-inflammatory | SD showed better inhibition of rat paw edema up to 16 h. | [67] |
| Paracetamol | Meloxicam | Rat | - | Anti-inflammatory | SDs reduced by more than 50% the volume of carrageenan-induced tail edema compared to the physical mixture. | [68] |
| PVP K30 | Chelerythrine (CHE) | Mice | 10 mg/kg | Anti-inflammatory | SD enhanced CHE anti-inflammatory effect by reducing the levels of TNF-α, IL-6 and NO in mice serum. | [69] |
| HPMC | Curcumin | Male BABL/c mice | 200 and 400 mg/kg | Hepatoprotective | The best SD increased the hepatoprotective efficacy of CM. | [48] |
| HPC | Nobiletin | Male Sprague-Dawley rats(220 g) | 2 mg of drug/kg | Hepatoprotective | SD was more effective than the crystalline drug in rats with acute liver injury. | [70] |
| PVP K30 | Silymarin | Adult male albino rats (150–200 g) | 25 mg/kg | Hepatoprotective | The best SD improved biomarker rates and had a significantly better hepatoprotective effect than the commercial extract. | [71] |
| PVP K30 | Silymarin | Male Sprague-Dawley rats (190–210 g) | 50 mg/kg | Hepatoprotective | SD improved drug solubility and hepatoprotective activity, reducing the AST levels. | [72] |
| Poloxamer 188 | Repaglinide | Wistar rats (150–250 g) | (1 mg of drug) | Antihyperglycemic | SD obtained by the microwave method improved the drug anti-hyperglycemic activity. | [74] |
| Soluplus1 and PEG 4000 | Glimepiride | Albino Wistar rats (200–250 g) | 0.0285 mg of drug/kg | Anti-diabetic | SD reduced the glucose level in rats more than the pure drug and a commercial product. | [5] |
| PVP K17 | Pioglitazone | Male Swiss albino mice (25–30 g) | 30 mg/kg SD | Antihyperglycemic | SD reduced the mean glucose level in mice more than the pure drug and a commercial product. | [76] |
| HPMC | Hecogenin acetate | Male Swiss mice (28–35 g) | 40 mg/kg | Antinociceptive | Both the drug alone and its SD with HPMC-reduced mechanical and thermal hyperalgesia induced by crushing of the sciatic nerve in mice. | [77] |
5. Critical Analysis
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Kambayashi, A.; Kiyota, T.; Fujiwara, M.; Dressman, J.B. PBPK modeling coupled with biorelevant dissolution to forecast the oral performance of amorphous solid dispersion formulations. Eur. J. Pharm. Sci. 2019, 135, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Jermain, S.V.; Brough, C.; Williams, R.O. Amorphous solid dispersions and nanocrystal technologies for poorly water-soluble drug delivery—An update. Int. J. Pharm. 2018, 535, 379–392. [Google Scholar] [CrossRef] [PubMed]
- Loftsson, T.; Brewster, M.E. Pharmaceutical applications of cyclodextrins: Basic science and product development. J. Pharm. Pharmacol. 2010, 62, 1607–1621. [Google Scholar] [CrossRef] [PubMed]
- Worku, Z.A.; Aarts, J.; Singh, A.; Mooter, G.V.D. Drug–Polymer Miscibility across a Spray Dryer: A Case Study of Naproxen and Miconazole Solid Dispersions. Mol. Pharm. 2014, 11, 1094–1101. [Google Scholar] [CrossRef]
- Reginald-Opara, J.N.; Attama, A.; Ofokansi, K.; Umeyor, C.; Kenechukwu, F. Molecular interaction between glimepiride and Soluplus®-PEG 4000 hybrid based solid dispersions: Characterisation and anti-diabetic studies. Int. J. Pharm. 2015, 496, 741–750. [Google Scholar] [CrossRef]
- Danda, L.J.D.A.; Batista, L.D.M.; Melo, V.C.S.; Sobrinho, J.L.S.; Soares, M.F.D.L.R. Combining amorphous solid dispersions for improved kinetic solubility of posaconazole simultaneously released from soluble PVP/VA64 and an insoluble ammonio methacrylate copolymer. Eur. J. Pharm. Sci. 2019, 133, 79–85. [Google Scholar] [CrossRef]
- Júnior, W.F.D.S.; Pinheiro, J.G.D.O.; De Menezes, D.L.B.; Silva, N.E.D.S.E.; De Almeida, P.D.O.; Lima, E.S.; Veiga-Junior, V.F.; De Azevedo, E.P.; Lima, E.S. Development, Physicochemical Characterization and In Vitro Anti-Inflammatory Activity of Solid Dispersions of α,β Amyrin Isolated from Protium Oilresin. Molecules 2017, 22, 1512. [Google Scholar] [CrossRef] [Green Version]
- Vasconcelos, T.; Sarmento, B.; Costa, P.C. Solid dispersions as strategy to improve oral bioavailability of poor water soluble drugs. Drug Discov. Today 2007, 12, 1068–1075. [Google Scholar] [CrossRef]
- Leuner, C. Improving drug solubility for oral delivery using solid dispersions. Eur. J. Pharm. Biopharm. 2000, 50, 47–60. [Google Scholar] [CrossRef]
- Zhang, Z.; Chen, Y.; Deng, J.; Jia, X.-B.; Zhou, J.; Lv, H. Solid dispersion of berberine–phospholipid complex/TPGS 1000/SiO2: Preparation, characterization and in vivo studies. Int. J. Pharm. 2014, 465, 306–316. [Google Scholar] [CrossRef]
- Xie, Y.; Yao, Y. Octenylsuccinate hydroxypropyl phytoglycogen enhances the solubility and in-vitro antitumor efficacy of niclosamide. Int. J. Pharm. 2018, 535, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Steed, H.; Sawyer, M.B. Pharmacology, pharmacokinetics and pharmacogenomics of paclitaxel. Pharmacogenomics 2007, 8, 803–815. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Yao, Y. Incorporation with Dendrimer-Like Biopolymer Leads to Improved Soluble Amount and In Vitro Anticancer Efficacy of Paclitaxel. J. Pharm. Sci. 2019, 108, 1984–1990. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.-S.; Cho, N.H.; Kim, D.-H.; Park, J.-S. Comparison of paclitaxel solid dispersion and polymeric micelles for improved oral bioavailability and in vitro anti-cancer effects. Mater. Sci. Eng. C 2019, 100, 247–259. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Lee, Y.-S.; Song, J.G.; Han, H.-K. Improved In vivo Effect of Chrysin as an Absorption Enhancer Via the Preparation of Ternary Solid Dispersion with Brij®L4 and Aminoclay. Curr. Drug Deliv. 2018, 16, 86–92. [Google Scholar] [CrossRef] [PubMed]
- De Sá, I.S.; Peron, A.P.; Leimann, F.V.; Bressan, G.N.; Krum, B.N.; Fachinetto, R.; Pinela, J.; Calhelha, R.C.; Barreiro, M.F.; Ferreira, I.C.; et al. In vitro and in vivo evaluation of enzymatic and antioxidant activity, cytotoxicity and genotoxicity of curcumin-loaded solid dispersions. Food Chem. Toxicol. 2019, 125, 29–37. [Google Scholar] [CrossRef] [Green Version]
- Wu, R.; Mei, X.; Ye, Y.; Xue, T.; Wang, J.; Sun, W.; Lin, C.; Xue, R.; Zhang, J.; Xu, D. Zn(II)-curcumin solid dispersion impairs hepatocellular carcinoma growth and enhances chemotherapy by modulating gut microbiota-mediated zinc homeostasis. Pharmacol. Res. 2019, 150, 104454. [Google Scholar] [CrossRef]
- Xiong, X.; Zhang, M.; Hou, Q.; Tang, P.; Suo, Z.; Zhu, Y.; Li, H. Solid dispersions of telaprevir with improved solubility prepared by co-milling: Formulation, physicochemical characterization and cytotoxicity evaluation. Mater. Sci. Eng. C 2019, 105, 110012. [Google Scholar] [CrossRef]
- Lewinska, A.; Adamczyk, J.; Pajak, J.; Stoklosa, S.; Kubis, B.; Pastuszek, P.; Slota, E.; Wnuk, M. Curcumin-mediated decrease in the expression of nucleolar organizer regions in cervical cancer (HeLa) cells. Mutat. Res. Toxicol. Environ. Mutagen. 2014, 771, 43–52. [Google Scholar] [CrossRef]
- Jiang, Y.; Piao, J.; Cho, H.-J.; Kang, W.-S.; Kim, H. Improvement in antiproliferative activity of Angelica gigas Nakai by solid dispersion formation via hot-melt extrusion and induction of cell cycle arrest and apoptosis in HeLa cells. Biosci. Biotechnol. Biochem. 2015, 79, 1635–1643. [Google Scholar] [CrossRef]
- Guo, S.; Wang, G.; Wu, T.; Bai, F.; Xu, J.; Zhang, X. Solid dispersion of berberine hydrochloride and Eudragit® S100: Formulation, physicochemical characterization and cytotoxicity evaluation. J. Drug Deliv. Sci. Technol. 2017, 40, 21–27. [Google Scholar] [CrossRef]
- Wang, L.; Li, H.; Wang, S.; Liu, R.; Wu, Z.; Wang, C.; Wang, Y.; Chen, M. Enhancing the Antitumor Activity of Berberine Hydrochloride by Solid Lipid Nanoparticle Encapsulation. AAPS PharmSciTech 2014, 15, 834–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patil, J.B.; Kim, J.; Jayaprakasha, G.K. Berberine induces apoptosis in breast cancer cells (MCF-7) through mitochondrial-dependent pathway. Eur. J. Pharmacol. 2010, 645, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Mintoo, M.J.; Mondhe, D.M.; Bharate, S.B.; Vishwakarma, R.A.; Bharate, S.S. Binary and ternary solid dispersions of an anticancer preclinical lead, IIIM-290: In vitro and in vivo studies. Int. J. Pharm. 2019, 570, 118683. [Google Scholar] [CrossRef]
- Simonazzi, A.; Davies, C.; Cid, A.G.; Gonzo, E.; Parada, L.; Bermúdez, J.M. Preparation and Characterization of Poloxamer 407 Solid Dispersions as an Alternative Strategy to Improve Benznidazole Bioperformance. J. Pharm. Sci. 2018, 107, 2829–2836. [Google Scholar] [CrossRef] [PubMed]
- Lima, Á.A.; Soares-Sobrinho, J.L.; Silva, J.L.; Corrêa-Júnior, R.A.; Lyra, M.A.; Santos, F.L.; Oliveira, B.G.; Hernandes, M.Z.; Rolim, L.A.; Rolim-Neto, P.J. The Use of Solid Dispersion Systems in Hydrophilic Carriers to Increase Benznidazole Solubility. J. Pharm. Sci. 2011, 100, 2443–2451. [Google Scholar] [CrossRef]
- Lima, Á.A.N.; Sobrinho, J.L.S.; De Lyra, M.A.M.; Dos Santos, F.L.A.; Figueirêdo, C.B.M.; Neto, P.J.R. Evaluation of in vitro dissolution of benznidazole and binary mixtures: Solid dispersions with hydroxypropylmethylcellulose and β-cyclodextrin inclusion complexes. Int. J. Pharm. Pharm. Sci. 2015, 7, 371–375. [Google Scholar]
- Fonseca-Berzal, C.; Palmeiro-Roldán, R.; Escario, J.A.; Torrado, S.; Arán, V.J.; Torrado-Santiago, S.; Gómez-Barrio, A. Novel solid dispersions of benznidazole: Preparation, dissolution profile and biological evaluation as alternative antichagasic drug delivery system. Exp. Parasitol. 2015, 149, 84–91. [Google Scholar] [CrossRef] [Green Version]
- Eloy, J.O.; Saraiva, J.; De Albuquerque, S.; Marchetti, J.M. Solid Dispersion of Ursolic Acid in Gelucire 50/13: A Strategy to Enhance Drug Release and Trypanocidal Activity. AAPS PharmSciTech 2012, 13, 1436–1445. [Google Scholar] [CrossRef] [Green Version]
- Perissutti, B.; Passerini, N.; Trastullo, R.; Keiser, J.; Zanolla, D.; Zingone, G.; Voinovich, D.; Albertini, B. An explorative analysis of process and formulation variables affecting comilling in a vibrational mill: The case of praziquantel. Int. J. Pharm. 2017, 533, 402–412. [Google Scholar] [CrossRef]
- Albertini, B.; Perissutti, B.; Bertoni, S.; Zanolla, D.; Franceschinis, E.; Voinovich, D.; Lombardo, F.; Keiser, J.; Passerini, N. Combining Mechanochemistry and Spray Congealing for New Praziquantel Pediatric Formulations in Schistosomiasis Treatment. Int. J. Mol. Sci. 2019, 20, 1233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, J.; Rathod, P.K. Indispensable malaria genes. Science 2018, 360, 490–491. [Google Scholar] [CrossRef] [PubMed]
- Abreu, T.P.; Silva, L.D.S.; Takiya, C.M.; Souza, M.C.; Henriques, M.D.G.M.O.; Pinheiro, A.A.S.; Caruso-Neves, C. Mice Rescued from Severe Malaria Are Protected against Renal Injury during a Second Kidney Insult. PLoS ONE 2014, 9, e93634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fule, R.A.; Meer, T.A.; Sav, A.R.; Amin, P. Artemether-Soluplus Hot-Melt Extrudate Solid Dispersion Systems for Solubility and Dissolution Rate Enhancement with Amorphous State Characteristics. J. Pharm. 2013, 2013, 1–15. [Google Scholar] [CrossRef]
- Fule, R.; Meer, T.; Sav, A.; Amin, P. Solubility and dissolution rate enhancement of lumefantrine using hot melt extrusion technology with physicochemical characterisation. J. Pharm. Investig. 2013, 43, 305–321. [Google Scholar] [CrossRef]
- Crucitti, V.C.; Migneco, L.M.; Piozzi, A.; Taresco, V.; Garnett, M.; Argent, R.H.; Francolini, I. Intermolecular interaction and solid state characterization of abietic acid/chitosan solid dispersions possessing antimicrobial and antioxidant properties. Eur. J. Pharm. Biopharm. 2018, 125, 114–123. [Google Scholar] [CrossRef] [Green Version]
- Al-Obaidi, H.; Kowalczyk, R.M.; Kalgudi, R.; Zariwala, M.G. Griseofulvin solvate solid dispersions with synergistic effect against fungal biofilms. Colloids Surfaces B Biointerfaces 2019, 184, 110540. [Google Scholar] [CrossRef]
- Gorle, A.P.; Gattani, S.G. Development and evaluation of ocular drug delivery system. Pharm. Dev. Technol. 2010, 15, 46–52. [Google Scholar] [CrossRef]
- Kanoujia, J.; Kushwaha, P.S.; Saraf, S.A. Evaluation of gatifloxacin pluronic micelles and development of its formulation for ocular delivery. Drug Deliv. Transl. Res. 2014, 4, 334–343. [Google Scholar] [CrossRef]
- Hernandez-Patlan, D.; Solis-Cruz, B.; Pontin, K.P.; Latorre, J.D.; Baxter, M.F.A.; Hernandez-Velasco, X.; Merino-Guzman, R.; Méndez-Albores, A.; Hargis, B.M.; Lopez-Arellano, R.; et al. Evaluation of a Solid Dispersion of Curcumin With Polyvinylpyrrolidone and Boric Acid Against Salmonella Enteritidis Infection and Intestinal Permeability in Broiler Chickens: A Pilot Study. Front. Microbiol. 2018, 9, 1289. [Google Scholar] [CrossRef]
- Hegge, A.B.; Vukićević, M.; Bruzell, E.; Kristensen, S.; Tønnesen, H. Solid dispersions for preparation of phototoxic supersaturated solutions for antimicrobial photodynamic therapy (aPDT). Eur. J. Pharm. Biopharm. 2013, 83, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Alves, T.; Chaud, M.; Grotto, D.; Jozala, A.F.; Pandit, R.; Rai, M.; Dos Santos, C.A. Association of Silver Nanoparticles and Curcumin Solid Dispersion: Antimicrobial and Antioxidant Properties. AAPS PharmSciTech 2017, 19, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Gunes, H.; Gulen, D.; Mutlu, R.; Gumus, A.; Tas, T.; Topkaya, A.E. Antibacterial effects of curcumin. Toxicol. Ind. Health 2013, 32, 246–250. [Google Scholar] [CrossRef] [PubMed]
- Costa, A.R.D.M.; Marquiafável, F.S.; Vaz, M.M.D.O.L.L.; Rocha, B.A.; Bueno, P.C.P.; Amaral, P.L.M.; Barud, H.D.S.; Berreta-Silva, A.A. Quercetin-PVP K25 solid dispersions. J. Therm. Anal. Calorim. 2010, 104, 273–278. [Google Scholar] [CrossRef]
- Ge, Y.; Zhao, X.; Wang, S.; Zu, Y.; Zhao, T.; Sang, M.; Sui, X.; Wang, K. 10 (CoQ10) Prepared by Heat Melt and High-pressure Homogenization Method. Curr. Nanosci. 2014, 10, 292–296. [Google Scholar] [CrossRef]
- Fitriani, L.; Rismawati, E.; Umar, S.; Zaini, E. Solid dispersion of usnic acid-PVP K30 and evaluation of antioxidant activity. Rasayan J. Chem. 2018, 11, 1643–1648. [Google Scholar] [CrossRef]
- Alshehri, S.; Imam, S.S.; Altamimi, M.A.; Hussain, A.; Shakeel, F.; Elzayat, E.M.; Mohsin, K.; Ibrahim, M.; Alanazi, F. Enhanced Dissolution of Luteolin by Solid Dispersion Prepared by Different Methods: Physicochemical Characterization and Antioxidant Activity. ACS Omega 2020, 5, 6461–6471. [Google Scholar] [CrossRef] [Green Version]
- Shin, M.-S.; Yu, J.S.; Lee, J.; Ji, Y.S.; Joung, H.J.; Han, Y.-M.; Yoo, H.H.; Kang, K.; Shin, Y. A Hydroxypropyl Methylcellulose-Based Solid Dispersion of Curcumin with Enhanced Bioavailability and its Hepatoprotective Activity. Biomolecules 2019, 9, 281. [Google Scholar] [CrossRef] [Green Version]
- Lian, X.; Dong, J.; Zhang, J.; Teng, Y.; Lin, Q.; Fu, Y.; Gong, T. Soluplus® based 9-nitrocamptothecin solid dispersion for peroral administration: Preparation, characterization, in vitro and in vivo evaluation. Int. J. Pharm. 2014, 477, 399–407. [Google Scholar] [CrossRef]
- Qusa, M.H.; Siddique, A.B.; Nazzal, S.; El Sayed, K. Novel olive oil phenolic (−)-oleocanthal (+)-xylitol-based solid dispersion formulations with potent oral anti-breast cancer activities. Int. J. Pharm. 2019, 569, 118596. [Google Scholar] [CrossRef]
- Yang, S.; Shi, P.; Huang, X.; Zhao, M.; Li, S.; Wu, Y.; Lin, X.-H.; Yao, H. Pharmacokinetics, Tissue Distribution and Protein Binding Studies of Chrysocauloflavone I in Rats. Planta Med. 2015, 82, 217–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, B.; Wang, X.; Zou, Y.; Chen, W.; Wang, G.; Yao, W.; Shi, P.; Li, S.; Lin, S.; Lin, X.-H.; et al. Simultaneous quantification of five biflavonoids in rat plasma by LC-ESI–MS/MS and its application to a comparatively pharmacokinetic study of Selaginella doederleinii Hieron extract in rats. J. Pharm. Biomed. Anal. 2018, 149, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Wang, X.; Zhang, Y.; Huang, K.; Liu, H.; Xu, D.; Li, S.; Liu, Q.; Huang, J.; Yao, H.; et al. Improved solubility, dissolution rate and oral bioavailability of main biflavonoids from Selaginella doederleinii extract by amorphous solid dispersion. Drug Deliv. 2020, 27, 309–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.-H.; Wu, X.-J.; Niu, J.-X.; Yan, H.; Wang, X.-Z.; Yin, X.-D.; Pang, Y. Serum zinc status and Helicobacter pylori infection in gastric disease patients. Asian Pac. J. Cancer Prev. 2012, 13, 5043–5046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, W.; Zhao, Y.; Sun, X.; Song, Z.; McClain, C.J.; Zhou, Z. Dietary Zinc Deficiency Exaggerates Ethanol-Induced Liver Injury in Mice: Involvement of Intrahepatic and Extrahepatic Factors. PLoS ONE 2013, 8, e76522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, N.; Sasaki, T.; Sakata-Haga, H.; Ohta, K.-I.; Gao, M.; Kawanishi, S.; Fukui, Y. Protective effect of taurine against nitrosative stress in the stomach of rat with water immersion restraint stress. In Advances in Experimental Medicine and Biology; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 2009; Volume 643, pp. 273–283. [Google Scholar]
- Marcinkiewicz, J.; Kontny, E. Taurine and inflammatory diseases. Amino Acids 2012, 46, 7–20. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.; Mei, X.-T.; Zheng, Y.-P.; Xu, D. Gastroprotective effect of taurine zinc solid dispersions against absolute ethanol-induced gastric lesions is mediated by enhancement of antioxidant activity and endogenous PGE2 production and attenuation of NO production. Eur. J. Pharmacol. 2014, 740, 329–336. [Google Scholar] [CrossRef]
- Gou, J.; Fei, S.; Xue, B.; Zhang, J.; Zhang, Y.; Wang, X.; Zhang, Y.; Yin, T.; He, H.; Tang, X. Triacetylated andrographolide solid dispersions: Preparation, stability study and in vivo anti-inflammation in mice ulcerative colitis model. J. Drug Deliv. Sci. Technol. 2019, 51, 91–100. [Google Scholar] [CrossRef]
- He, Y.; Liu, H.; Bian, W.; Liu, Y.; Liu, X.; Ma, S.; Zheng, X.; Du, Z.; Zhang, K.; Ouyang, D. Molecular Interactions for the Curcumin-Polymer Complex with Enhanced Anti-Inflammatory Effects. Pharmaceutics 2019, 11, 442. [Google Scholar] [CrossRef] [Green Version]
- Teixeira, C.C.C.; Mendonça, L.M.; Bergamaschi, M.M.; Queiroz, R.H.C.; Souza, G.E.P.; Antunes, L.M.G.; Freitas, L. Microparticles Containing Curcumin Solid Dispersion: Stability, Bioavailability and Anti-Inflammatory Activity. AAPS PharmSciTech 2015, 17, 252–261. [Google Scholar] [CrossRef] [Green Version]
- Chuah, A.M.; Jacob, B.; Jie, Z.; Ramesh, S.; Mandal, S.; Puthan, J.K.; Deshpande, P.; Vaidyanathan, V.V.; Gelling, R.W.; Patel, G.; et al. Enhanced bioavailability and bioefficacy of an amorphous solid dispersion of curcumin. Food Chem. 2014, 156, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Khandai, M.; Sharma, A.; Khanam, N.; Patra, C.; Dinda, S.; Sen, K. Preparation, in vitro and in vivo evaluation of algino-pectinate bioadhesive microspheres: An investigation of the effects of polymers using multiple comparison analysis. Acta Pharm. 2010, 60, 255–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujii, M.; Okada, H.; Shibata, Y.; Teramachi, H.; Kondoh, M.; Watanabe, Y. Preparation, characterization and tableting of a solid dispersion of indomethacin with crospovidone. Int. J. Pharm. 2005, 293, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Jana, S.; Ali, S.A.; Nayak, A.K.; Sen, K.K.; Basu, S.K. Development of topical gel containing aceclofenac-crospovidone solid dispersion by “Quality by Design (QbD)” approach. Chem. Eng. Res. Des. 2014, 92, 2095–2105. [Google Scholar] [CrossRef]
- Kenechukwu, F.C.; Ofokansi, K.C.; Ezugwu, R.O.; Attama, A.A. Improved dissolution and anti-inflammatory activity of ibuprofen-polyethylene glycol 8000 solid dispersion systems. Int. J. Pharm. Investig. 2016, 6, 139–147. [Google Scholar] [CrossRef] [Green Version]
- Malipeddi, V.R.; Dua, K.; Awasthi, R. Development and characterization of solid dispersion-microsphere controlled release system for poorly water-soluble drug. Drug Deliv. Transl. Res. 2016, 6, 540–550. [Google Scholar] [CrossRef]
- Al-Remawi, M.; Ali, A.M.A.; Khames, A.; Hamaidi, M. Meloxicam-Paracetamol Binary Solid Dispersion Systems with Enhanced Solubility and Dissolution Rate:Preparation, Characterization and In Vivo Evaluation. J. Pharm. Innov. 2017, 12, 206–215. [Google Scholar] [CrossRef]
- Li, W.; Qing, S.; Zhi, W.; Yao, H.; Fu, C.; Niu, X. The pharmacokinetics and anti-inflammatory effects of chelerythrine solid dispersions in vivo. J. Drug Deliv. Sci. Technol. 2017, 40, 51–58. [Google Scholar] [CrossRef]
- Onoue, S.; Nakamura, T.; Uchida, A.; Ogawa, K.; Yuminoki, K.; Hashimoto, N.; Hiza, A.; Tsukaguchi, Y.; Asakawa, T.; Kan, T.; et al. Physicochemical and biopharmaceutical characterization of amorphous solid dispersion of nobiletin, a citrus polymethoxylated flavone, with improved hepatoprotective effects. Eur. J. Pharm. Sci. 2013, 49, 453–460. [Google Scholar] [CrossRef]
- Balata, G.; Shamrool, H. Spherical agglomeration versus solid dispersion as different trials to optimize dissolution and bioactivity of silymarin. J. Drug Deliv. Sci. Technol. 2014, 24, 478–485. [Google Scholar] [CrossRef]
- Hwang, D.H.; Kim, Y.-I.; Cho, K.H.; Poudel, B.K.; Choi, J.Y.; Kim, D.-W.; Shin, Y.-J.; Bae, O.-N.; Yousaf, A.M.; Yong, C.S.; et al. A novel solid dispersion system for natural product-loaded medicine: Silymarin-loaded solid dispersion with enhanced oral bioavailability and hepatoprotective activity. J. Microencapsul. 2014, 31, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Heydari, I.; Radi, V.; Razmjou, S.; Amiri, A. Chronic complications of diabetes mellitus in newly diagnosed patients. Int. J. Diabetes Mellit. 2010, 2, 61–63. [Google Scholar] [CrossRef] [Green Version]
- Zawar, L.R.; Bari, S.B. Preparation, characterization and in vivo evaluation of antihyperglycemic activity of microwave generated repaglinide solid dispersion. Chem. Pharm. Bull. 2012, 60, 482–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Ma, L.; Li, X.; Cui, X.; Yang, W.; Shen, S.; Chen, M. A simple and effective method to improve bioavailability of glimepiride by utilizing hydrotropy technique. Eur. J. Pharm. Sci. 2015, 77, 154–160. [Google Scholar] [CrossRef]
- Pokharkar, V.; Kutwal, M.; Mandpe, L. Pioglitazone Solid Dispersion System Prepared by Spray Drying Method: In Vitro and In Vivo Evaluation. PDA J. Pharm. Sci. Technol. 2013, 67, 23–34. [Google Scholar] [CrossRef]
- Moreira, C.D.L.D.F.A.; Pinheiro, J.G.D.O.; Da Silva-Júnior, W.F.; Barbosa, E.G.; Lavra, Z.M.M.; Pereira, E.W.M.; Resende, M.M.; De Azevedo, E.P.; Quintans-Júnior, L.J.; Araújo, A.A.D.S.; et al. Amorphous solid dispersions of hecogenin acetate using different polymers for enhancement of solubility and improvement of anti-hyperalgesic effect in neuropathic pain model in mice. Biomed. Pharmacother. 2018, 97, 870–879. [Google Scholar] [CrossRef]
- Gama, K.B.; Quintans, J.S.; Antoniolli, A.R.; Quintans-Júnior, L.J.; Santana, W.A.; Branco, A.; Soares, M.B.P.; Villarreal, C.F. Evidence for the Involvement of Descending Pain-Inhibitory Mechanisms in the Antinociceptive Effect of Hecogenin Acetate. J. Nat. Prod. 2013, 76, 559–563. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oliveira, V.d.S.; de Almeida, A.S.; Albuquerque, I.d.S.; Duarte, F.Í.C.; Queiroz, B.C.S.H.; Converti, A.; Lima, Á.A.N.d. Therapeutic Applications of Solid Dispersions for Drugs and New Molecules: In Vitro and In Vivo Activities. Pharmaceutics 2020, 12, 933. https://doi.org/10.3390/pharmaceutics12100933
Oliveira VdS, de Almeida AS, Albuquerque IdS, Duarte FÍC, Queiroz BCSH, Converti A, Lima ÁANd. Therapeutic Applications of Solid Dispersions for Drugs and New Molecules: In Vitro and In Vivo Activities. Pharmaceutics. 2020; 12(10):933. https://doi.org/10.3390/pharmaceutics12100933
Chicago/Turabian StyleOliveira, Verônica da Silva, Amanda Silva de Almeida, Ingrid da Silva Albuquerque, Fernanda Ílary Costa Duarte, Bárbara Cristina Silva Holanda Queiroz, Attilio Converti, and Ádley Antonini Neves de Lima. 2020. "Therapeutic Applications of Solid Dispersions for Drugs and New Molecules: In Vitro and In Vivo Activities" Pharmaceutics 12, no. 10: 933. https://doi.org/10.3390/pharmaceutics12100933