
Abstract
Ecologically relevant genes generally show patchy distributions among related bacterial genomes. This is commonly attributed to lateral gene transfer, whereas the opposite mechanism—gene loss—has rarely been explored. Pseudogenization is a major mechanism underlying gene loss, and pseudogenes are best characterized by comparing closely related genomes because of their short life spans. To explore the role of pseudogenization in microbial ecological diversification, we apply rigorous methods to characterize pseudogenes in the 279 newly sequenced Ruegeria isolates of the globally abundant Roseobacter group collected from two typical coastal habitats in Hong Kong, the coral Platygyra acuta and the macroalga Sargassum hemiphyllum. Pseudogenes contribute to ~16% of the accessory genomes of these strains. Ancestral state reconstruction reveals that many pseudogenization events are correlated with ancestral niche shifts. Specifically, genes related to resource scavenging and energy acquisition were often pseudogenized when roseobacters inhabiting carbon-limited and energy-poor coral skeleton switched to other resource-richer niches. For roseobacters inhabiting the macroalgal niches, genes for nitrogen regulation and carbohydrate utilization were important but became dispensable upon shift to coral skeleton where nitrate is abundant but carbohydrates are less available. Whereas low-energy-demanding secondary transporters are more favorable in coral skeleton, ATP-driven primary transporters are preferentially kept in the energy-replete macroalgal niches. Moreover, a large proportion of these families mediate organismal interactions, suggesting their rapid losses by pseudogenization as a potential response to host and niche shift. These findings illustrate an important role of pseudogenization in shaping genome content and driving ecological diversification of marine roseobacters.
Similar content being viewed by others


Introduction
Gene gains and losses play a major role in shaping genome content and ecological diversification of bacteria [1]. In bacteria, gene acquisition through lateral gene transfer (LGT) has been well studied [2, 3]. Compared with gene gains, the mechanisms and roles of gene losses in microbial evolution and ecological diversification are less clear. One of the most common ways of gene loss is pseudogenization [4]. Pseudogenes, first discovered in Xenopus laevis [5], have long been considered as a paradigm of neutral evolution [6]. They represent genes lost as a result of disabling mutations, fixed in the population by genetic drift because the effected gene becomes dispensable in the organism’s current niche and thus not subject to strong functional constraints [7]. Recently, accumulating evidence implies pseudogenization as an important contributor to genetic variation and evolution of microbial genomes [4, 8]. Despite being predominantly neutral, there are cases in which gene losses via pseudogenization increase fitness and are thus adaptive [9, 10]. A few characterized pseudogenization events are associated with increases in virulence [11], such as the marT gene in Salmonella Typhi, which likely contributes to the surV-dependent survival to oxidative stress imposed by abundant H2O2 inside human macrophages [12]. Further, pseudogenes may serve as raw materials from which novel genes and noncoding RNAs arise [13]. From the evolutionary perspective, pseudogenes provide a rapid way for ecological adaptation because (i) a single mutation could lead to the inactivation of a gene, and (ii) pseudogenes have the potential to be re-activated by back mutation or recombination, particularly when the functionality of the gene is selectively favored in a new environment [14]. Besides, pseudogenes represent unequivocal cases of ongoing gene loss, which is often inaccurately predicted by canonical methods [15, 16] based on the presence/absence pattern of a functional gene across a phylogenetic tree.
Despite their prevalence in eukaryotic genomes, pseudogenes are relatively infrequent in prokaryotes [17, 18]. Pseudogenes are known to be absent or depleted in streamlined free-living marine bacterioplankton lineages, such as Prochlorococcus, SAR11 and SAR86 among others [19, 20]. However, they are rarely explored in metabolically versatile lineages with larger and more fluid genomes. Members of the Roseobacter group comprise up to 20% of bacterial communities in coastal marine habitats [21]. In addition to their well-established free-living and patch-associated lifestyles in the pelagic environments [22], roseobacters often represent a significant fraction in the microbiomes associated with various marine algae and invertebrates [21, 23]. For instance, on average 19% of the epiphytic bacterial cells of a green Ulvacean alga fall within the Roseobacter group [24]. In coral species Acropora tenuis on the Great Barrier Reef, Roseobacter-affiliated sequences make up to 63% of the clone library [25].
Roseobacters and other marine bacteria are found in three unique niches in corals: the surface mucus layer, coral tissue, and skeleton [26, 27]. Coral nutrition is largely supported by Symbiodinium in coral tissue through translocation of photosynthates, and up to 50% of the net fixed carbon is released as mucus by corals [28]. Coral skeleton is characterized as a separate and relatively stable environment well-protected from external surroundings. Endolithic algae, shown as green bands beneath the coral tissue layer, have been reported in a wide array of coral species [29], including those in the genus Platygyra [30] sampled in the present study. While the photosynthetic rate is considerably lower in coral skeleton than that in coral tissue due to the extremely low intensity of light that reaches skeleton, the coral skeleton is subject to diurnal fluctuations in oxygen and pH levels as coral tissue [29, 31]. Members of the Roseobacter group are also commonly found in the macroalgal ecosystems, capable of degrading a variety of algal osmolytes [32]. For mutualistic relationship between macroalgae and bacteria, the algae provide nutrients and oxygen for epiphytic bacteria, which in turn mineralize organic substrates and provide CO2, essential vitamins, minerals, and growth factors to the host [33, 34]. Macroalgae release a large portion of their fixed carbon in the form of carbohydrates to the surrounding environment [35]. Therefore, the ambient seawater and sediments are typically rich in organic carbon [36, 37].
In line with their habitat diversity, roseobacters exhibit tremendous genomic diversity and metabolic versatility [38]. Strikingly, even between closely related members sharing nearly identical 16S rRNA gene sequences, the genome contents can vary greatly, as shown in 42 globally distributed strains of the roseobacter species Epibacterium mobile (formerly known as Ruegeria mobilis) [39]. Such intraspecific variation could be associated with habitat diversity, as illustrated by two Sulfitobacter sp. strains (NAS-14.1 and EE-36) and two Phaeobacter gallaeciensis strains [40, 41]. In fact, a patchy distribution of ecologically relevant genes is often observed among roseobacters [23, 42]. This may, to some extent, reflect their adaptation to the diverse ecological niches. However, the evolutionary mechanisms underlying the tremendous amount of diversity, especially how the genome content diverged among closely related lineages, are poorly understood.
Many previous studies have investigated the genetic diversity in members of the Roseobacter group and its relationship to ecological niches [23, 38, 39, 43, 44]. However, these studies were based on canonical methods of gene gain/loss inferences which ignored pseudogenes, or focused on distantly related species where traces of most pseudogenes have been eliminated from the genome. Here, we inferred the evolutionary history of pseudogenization events among closely related strains and linked them to the ancestral switches between niches of the coral and macroalgal ecosystems. Closely related genomes were analyzed because, according to previous studies, most bacterial pseudogenes are strain-specific, and of the few that are shared across strains, many may have been generated independently since pseudogenes are retained in bacterial genomes for only a short time on an evolutionary scale [45,46,47]. In terms of the ecosystem type, we chose a coral ecosystem represented by Platygyra acuta and a macroalgal ecosystem represented by Sargassum hemiphyllum. This is because these two species are ecologically significant and globally widespread throughout coastal environments including Hong Kong [48,49,50,51], serving as important nursery habitats for various marine organisms [52]. To minimize the effect of confounding biological and stochastic factors that may also explain the evolutionary changes of functional genes, we collected most samples at the same season and from locations nearby in Hong Kong. We isolated bacteria and sequenced 279 genomes of closely related strains affiliated with the genus Ruegeria of the Roseobacter group. Ruegeria is widely distributed among various habitats provided by marine eukaryotes [53], and is among the few bacterial genera associated with the greatest number of coral species according to a recent comprehensive review [53]. We tested the hypothesis that genes dispensable in new niches were likely lost via pseudogenization by showing the correlations between inferred pseudogenization events and corresponding niche shifts. We elaborated on specific ecologically relevant genes contributing to microbial diversification in these niches from the novel perspective of the gene loss processes.
Materials and methods
Bacterial isolation and pseudogene identification
We collected most microbial samples from the brown alga Sargassum hemiphyllum ecosystem and the coral Platygyra acuta ecosystem at the same season during 2016–2018 and from locations nearby in Hong Kong waters (Fig. 1b; Supplementary Table S1). Focusing on different niches in the macroalgal ecosystem (algal tissue, ambient seawater, and sediment) and the coral ecosystem (mucus, tissue, skeleton, and ambient seawater), we isolated and performed genome sequencing of 279 closely related strains affiliated with the roseobacter genus Ruegeria.

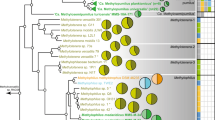
a The maximum likelihood phylogenomic tree based on core single-copy gene families identified by OrthoFinder was constructed using IQ-TREE. Black circles on the nodes denote those with ultrafast bootstrap values <95%. Clustering of isolates based on phylogenetic depth is shown in the outer layers. Different groups of genomes clustered using each phylogenetic depth cutoff are indicated by the differences in the height of the layer. The ecological niche and isolation method of each strain are marked on the tree. The two major marine habitats are represented by green (macroalga) and purple (coral), respectively. Niches within each habitat are marked with different shapes. Outgroups composed of public roseobacter genomes are collapsed into triangles and labeled with the numbers one and two. b Four different sites for the macroalgal and coral sample collection in Hong Kong. c Kernel density estimates of whole-genome ANI and pairwise 16S rRNA gene identity. The values above the plots indicate the ANI or 16S rRNA gene identity of the peak.
Next, we used the program suite Psi-Phi [45] for pseudogene identification. This program uses a conservative criterion considering a pseudogene only when it lost >20% of its original length, and has been widely employed in other studies [46, 47, 54]. Applying a comparative method, this approach enhances pseudogene recognition among closely related strains both in annotated regions by identifying incorrectly annotated open reading frames (ORFs) and in intergenic regions by detecting new pseudogenes [45]. We further applied the following three steps to filter out misidentified pseudogenes. First, Psi-Phi only works on complete chromosomes. However, all of our newly sequenced genomes contain contigs (Supplementary Dataset S1). We therefore modified the original scripts to make them capable of searching for pseudogenes based on merged contigs followed by removing identified pseudogenes whose locations spanned contigs (Supplementary Fig. S1A). Second, we removed those identified by a query of less than 100 amino acids, since the vast majority of such short annotated ORFs are less likely to be genuine genes [47, 55], which accounted for 19.7% of the original output. Last, as noted by Lerat and Ochman [45], if the query gene is annotated as being longer than its actual length, some “real” genes could be misidentified by this query ORF as pseudogenes truncated by a premature stop codon. The logic is that, if an ORF identified a lot of (here we used 10 as an arbitrary cutoff) pseudogenes as a single query, then it was highly suspicious that the query was annotated to be longer than its actual length (see Supplementary Fig. S1B for an example). Applying this criterion further removed 18.8% of the pseudogenes originally identified by Psi-Phi.
For pseudogene identification, annotated proteins of each genome were queried against the complete nucleotide sequence of every other genome. For the vast majority of identified pseudogenes (>93%), all of the query ORFs of each pseudogene were from the same gene family, and these pseudogenes were therefore referred to as “strict-consistent” pseudogenes. Approximately 6% of the pseudogenes had less than half of their query ORFs from different families, and these were named as “relaxed-consistent” pseudogenes. Both strict-consistent and relaxed-consistent pseudogenes were retained for subsequent analyses (Fig. 2). Further technical details were provided in Supplementary Text S1.

Flowchart of pseudogene analysis. OG1, OG2, and OG3 refer to three different homologous gene family IDs assigned by OrthoFinder. Pseudogenes with all query ORFs from the same gene family are “strict-consistent” and those with >1/2 of their query ORFs clustered into the same gene family are “relaxed-consistent”. “Inconsistent” pseudogenes do not have >1/2 of query ORFs clustered into one family. Only strict- and relaxed-consistent pseudogenes were assigned to corresponding gene families. “Multi-state” gene families contain both ORFs and pseudogenes from the same genome, while in “single-state” gene families, either ORFs or pseudogenes are present in each of the involved genome. Only single-state gene families were selected for ancestral reconstructions of genetic and ecological traits.
Phylogenomic analysis and ancestral state reconstruction
Amino acid sequences from core single-copy gene families identified by OrthoFinder v2 [56] were concatenated for the maximum likelihood (ML) phylogenomic tree inference using IQ-TREE v1.6.2 [57]. To choose proper candidates for ancestral state reconstruction (ASR), we explored all homologous gene families with assigned pseudogenes. Gene families containing both ORFs and pseudogenes from the same genome were designated as “multi-state” gene families (Fig. 2). In contrast, gene families in which each of the involved genomes corresponds to only one functional state, containing either ORFs or pseudogenes, were called “single-state” gene families (Fig. 2). In the case of the multi-state gene families, since pseudogenes likely evolved from a functional ancestor originating from vertical descent, or gene duplication, or LGT, it is difficult to identify (i) how the pseudogenes were formed, and (ii) whether the remaining ORF copy has changed its function. Thus, only single-state gene families were used for ASR (Fig. 2).
ASR was performed for functional state and ecological trait separately using the “MPR” (most-parsimonious reconstruction) algorithm implemented in the Phangorn R package [58]. Based on the inferred functional states for ancestral nodes, pseudogenization, where an ancestral ORF became a pseudogene, or a back event, which refers to the functional restoration from an ancestral pseudogene to an ORF, was identified for each single-state gene family. By mapping the inferred functional transitions and niche shifts along the phylogeny for each single-state gene family, we identified cases where these two were matched on the same branch, i.e., an ORF became a pseudogene (or vice versa) during a niche shift. Gene families with at least one match were summarized (see details in Supplementary Text S1).
Results and discussion
Phylogenomic analysis of the 279 newly isolated Ruegeria strains
Our phylogenomic tree of the 279 new strains together with the 166 publicly available roseobacter genomes (Fig. 1a; Supplementary Dataset S2) showed that the new strains were in general related to several described species of the genus Ruegeria, which together formed a monophyletic group phylogenetically distinct from the model roseobacter R. pomeroyi DSS-3 (Fig. 1a; Supplementary Dataset S1). The mean pairwise average 16S rRNA gene identity and the mean whole-genome average nucleotide identity (ANI) were around 98% and 82%, respectively (Fig. 1c), strengthening the argument that these new isolates were members of multiple different species within the Ruegeria. Even closely related strains did not necessarily share the same ecological niche (Fig. 1a), indicating their rapid shift in habitat. Overall, the 279 newly sequenced isolates greatly expanded the genetic and ecological diversity of Ruegeria than previously appreciated (Supplementary Dataset S1).
Overview of the identified pseudogenes
In an attempt to explore the correlation of pseudogene formation with niche shifts, we first performed genome-wide pseudogene identification among all of the 279 isolates (Fig. 2). We tested whether increasing the number of analyzed genomes could identify more pseudogenes by grouping genomes using different cutoffs of the phylogenetic depth (which considers phylogenetic distance and topology to cluster all isolates into subclades; see Supplementary Fig. S2 for a schematic illustration). As shown in Fig. 3a, the number of genomes within each cluster increased as the phylogenetic depth gradually increased, and accordingly, the number of both the identified pseudogenes and gene families per genome became larger (Fig. 3a). Importantly, the trend suggests that the identified pseudogenes represent a conservative set of the pseudogene repertoire within surveyed genomes, which awaits a more complete sampling of Ruegeria species in future. Hence, in the following analyses, we pooled all 279 genomes together for pseudogene identification. This showed that the number of pseudogenes per genome ranged from 70 to 365 (median: 177). Approximately 16% of the families of the accessory genomes of the newly sequenced isolates contained pseudogenes. No significant differences in the number of pseudogenes per genome across niches were detected (p > 0.05 for both ANOVA and Kruskal–Wallis test; Fig. 3b).

a The number of pseudogenes per genome (left) and the number of pseudogene-containing gene families (right) plotted against the phylogenetic depth (shown in Fig. 1a). Genomes are first divided into different groups according to the phylogenetic depth cutoff. Pseudogenes are estimated using genomes within the same group. b The number of pseudogenes per genome in different ecological niches. Pseudogenes are estimated using the whole data set (i.e., 279 genomes). C-mu coral mucus, C-sk coral skeleton, C-sw coral ambient seawater, C-tis coral tissue, M-tis macroalgal tissue, M-sw macroalgal ambient seawater, M-sd macroalgal ambient sediment. c The pan-genome of the 279 sequenced Ruegeria isolates for the chromosome and the plasmid, respectively. Genes are sorted according to the genome of the model roseobacter Ruegeria pomeroyi DSS-3 (the innermost circle). The chromosome and the plasmid pan-genomes are not plotted in proportion to their number of nucleotides. d Proportions of different COG functional categories for the total pseudogenes and ORFs. The asterisk denotes significant difference (p < 0.05; Fisher’s exact test).
Approximately 99% of the total pseudogenes (strict- and relaxed-consistent; see “Materials and methods”) were assigned to corresponding homologous gene families according to their query ORFs. A proportion of 62% of the gene families with assigned pseudogenes occurred as either ORFs or pseudogenes in a given genome. These gene families were thereafter referred to as “single-state” families, which make 9.6% of the accessory genomes. The remaining families, where both ORFs and pseudogenes were found within a single genome, were termed as “multi-state” gene families. Due to the uncertainties in multi-state gene families, only single-state gene families were used in ancestral state reconstruction (ASR; see “Materials and methods”). Occasionally, pseudogenes can be converted to ORFs by back mutation, nonsense suppression or site-specific recombination [14]. Such events, referred to as “back events”, may reflect that a pseudogene in an ancestral niche became “activated” in a new habitat. Overall, pseudogenization events exceeded back events by a factor of four. The “transposable elements” appeared to be the only functional category overrepresented in pseudogenes compared to ORFs across the 279 genomes (Fisher’s exact test, p < 0.001, Bonferroni correction; Fig. 3d), agreeing with the result of a previous study [59]. A further look at the distribution of pseudogenes on the pan-genomes of the 279 isolates (Fig. 3c) revealed that mobile genetic elements (MGEs), which consist of genomic islands, insertion sequences, prophages and plasmid genes, carried a significantly higher proportion of pseudogenes than the remaining regions of the genome (12.4% vs. 3.0%; p < 0.001, χ2 test). This could be because MGEs are more likely to be dispensable for the cell, but might also suggest selection against “junk DNA” whose activity could be harmful under certain conditions (although some genes carried by genomic islands might provide adaptive functions).
There were 343 gene families whose members had undergone pseudogenization or back events during ecological niche shifts (Fig. 4, Supplementary Dataset S3). For such families, pseudogenization was in line with niche shift, thus we hypothesized that the functional transitions of these genes might be important in the ancestral environment and became dispensable in the new niche (and vice versa for the back events), potentially contributing to ecological adaptation. In 43 of these families, pseudogenization or back events occurred during the niche change in the same direction for at least twice (see those marked with an asterisk in Table 1 as examples and an illustrated example in Fig. 4a), indicating convergent evolution in response to niche shift in different roseobacter strains (we also provided a summary of families experiencing repeated pseudogenization irrespective of niche shift in Supplementary Fig. S3 and Supplementary Dataset S4). A vast majority of these families were transposons, hypothetical proteins, or those with unknown or ecologically irrelevant functions, and after excluding them there remained 41 families that were of particular interest. As follows, we elaborated on potential implications of pseudogenization (or back events) in these families for ecological adaptation of Ruegeria (Table 1, Supplementary Table S2; see also Supplementary Text S2).

Ancestral state reconstructions were performed for functional state (left) and ecological trait (right) with the “MPR” algorithm. Ancestral states are indicated by circles with different colors. The internal nodes with multiple colors mean that there are multiple possibilities of niche states at these nodes. The matched branches are highlighted in red. a–d Nitrite-sensitive transcriptional repressor (NsrR), glycogen phosphorylase (OG0005684), membrane-bound lytic murein transglycosylase E (MltE), and Type VI secretion system protein (ImpB).
Resource recovery and energy acquisition in coral skeleton
The porous calcium carbonate skeleton is characterized as a separate and relatively stable environment protected from external surroundings. It is a harsh environment since light intensity is exceedingly low, with only <1% photosynthetically active radiation penetration due to the absorbance by zooxanthellae-rich coral tissues [60, 61]. We therefore hypothesized that genes involved in resource and energy utilization may be pseudogenized during the niche shift from coral skeleton to energy-richer niches as a result of the relaxation of energy limitation. In agreement with this idea, we found that two gene families involved in cell wall degradation and recycling were pseudogenized in the shifts from the energy-limited coral skeleton to energy-richer niches (Table 1). Cell wall recycling is a common process for resource recovery in bacteria [62], which can recycle up to 50% of the peptidoglycan (PG) components of their cell wall per generation to rebuild cell wall or to use as energy sources [63, 64]. One of the gene families encodes the N-acetylmuramic acid 6-phosphate etherase MurQ, which is required for the utilization of anhydro-N-acetylmuramic acid derived from cell wall murein [65]. The other encodes the membrane-bound lytic murein transglycosylase E, which is essential for non-hydrolytic cleavage of the glycan strands of bacterial cell wall [66]. Consistent with the increase in energy availability during the niche shift, pseudogenization of these families suggests the importance of cell wall recycling in recovering resources and preserving critical energy resources in coral skeleton, in a way similar to the low-light adaptation through PG recycling in two cyanobacteria species [67]. In addition, the family encoding the carbon monoxide (CO) dehydrogenase small chain CoxS was pseudogenized during the niche shift from coral skeleton to macroalgal tissue (Table 1). As this enzyme participates in the oxidation of CO into CO2, which is an energy supplement to roseobacters [38], the increase in energy availability during the niche shift may also relax the functional constraint of the gene involved in this pathway.
Pseudogenization events related to nitrogen regulation and carbohydrate utilization
Pore water within the coral skeleton is highly enriched in nitrogen (N) sources like ammonium, nitrite and nitrate, the concentrations of which are about ten times higher than that in the ambient seawater [68, 69]. Consequently, increase in N availability during the niche change might relax the functional constraint of genes induced under N starvation. This idea was supported by the pseudogenization event in the gene family encoding the nitrogen regulation sensor histidine kinase NtrY during the shift from macroalgal ambient sediment to coral skeleton (Table 1). Genes of the Ntr family were reported to be significantly upregulated in response to N starvation [70]. Hence, we speculated that NtrY was important for the survival of roseobacters in the ancestral N-limited niche, but became dispensable when they shifted to coral skeleton where N is more abundant.
As macroalgae release a large portion of their fixed carbon mainly in the form of carbohydrates to the surrounding environment [35, 71], the ambient seawater and sediments are both rich in organic matter [36, 37]. Interestingly, we found two pseudogene-containing families related to the carbohydrate utilization in the macroalgal ecosystem. During the shift from macroalgal ambient sediment to coral skeleton, one gene encoding the ribose 5-phosphate isomerase A was pseudogenized. RipA converts ribose 5-phosphate to ribulose 5-phosphate, and plays a pivotal role in the pentose phosphate pathway [72]. Pseudogenized during the shift from macroalgal ambient sediment to coral mucus, the other gene encodes the permease protein of a sugar ABC transporter. Considering the rich carbohydrates in the S. hemiphyllum ecosystem, it is therefore conceivable that roseobacters inhabiting macroalgal ambient sediment harbored plenty of genes to utilize the abundant carbohydrates in the environment. The pseudogenization of the two gene families suggests that they are more necessary for carbohydrate utilization in macroalgal ecosystem than in coral skeleton or mucus where carbohydrates may be less available [73].
Transport systems suited to coral skeleton and macroalgal ecosystem
Among the families undergoing pseudogenization or back events during niche shift, we found several cases related to transport systems that may enable bacteria to obtain nutrients from environments. Specifically, two gene families both encoding tripartite ATP-independent periplasmic (TRAP) transporter proteins were pseudogenized during the niche change from coral skeleton to macroalgal ambient sediment (Table 1). TRAP transporters are widely used in marine bacteria inhabiting high-salt and nutrient-poor environments, which has a lower energetic cost than ABC transporters [74]. The family encoding the secondary Na+/H+ antiporter was also pseudogenized during the shift from coral skeleton to macroalgal ambient sediment (Table 1). In addition, a pseudogenization event occurred in the gene family encoding Na+/solute symporter during the shift from coral skeleton to macroalgal tissue (Table 1). In agreement with the energy limitation in coral skeleton, functional changes of these secondary transporters upon niche shifts imply that they may be more favorable in coral skeleton than in macroalgal niches, serving as an extra energy-saving strategy.
Considering the above mentioned gene family encoding the permease protein of sugar ABC transporter (Table 1), we speculated that ATP-driven primary transporters are presumably kept in energy-rich niches while low-energy demanding secondary transporters are favorable in energy-limited environments. This idea was further supported by two gene families pseudogenized during the shift from macroalgal ambient sediment to coral skeleton (Table 1), which encode the ABC-type peptide/nickel transport system permease protein and the V/A-type H+/Na+-transporting ATPase subunit F, respectively [75].
While we interpreted the different preferences of transport systems in the context of energy sources, we were aware that the performance of transport systems is also correlated with other important factors such as substrate availability and ion gradients. It should be noted that contradictory cases might exist here, including the loss of functions of ATPase components (OG0005367) and oligopeptide transport ATP-binding protein OppF (OG0008986) upon shifts from coral skeleton to macroalgal ambient sediment (Supplementary Dataset S3). In these cases, the functional changes plausibly were associated with factors other than energy, such as the uncharacterized substrate levels in different niches.
Stress response genes coping with environmental changes
Coral and macroalgal ecosystems are both exposed to changing physiochemical conditions such as oxygen, osmolarity, temperature and pH [76]. We identified several matched cases associated with stress responses, involving functional transitions between macroalgal and coral ecosystems. For example, three gene families were pseudogenized during the shift from coral skeleton to macroalgal ambient sediment (Table 1), including a thioredoxin (a key antioxidant in defense against oxidative stress), a Na+/H+ antiporter involved in pH homeostasis, and a potassium efflux system protein PhaA for pH adaptation. Given the diurnal fluctuation in oxygen and pH levels in coral skeleton, which drop sharply during the night [29, 31], these pseudogenization events may suggest the roles of these genes in oxidative stress response and pH regulation during habitat change. When the niche shifted from macroalgal ambient sediment to coral skeleton, the nitrite-sensitive transcriptional repressor NsrR and the general stress protein A were both pseudogenized (Table 1). NsrR likely contains an [Fe–S] cluster, and is known as a key regulator to cope with oxidative and nitrosative stresses [77, 78]. These two genes may help resist stress conditions in macroalgal ambient sediment.
In addition, the gene family encoding glutathione S-transferase, a protein responsible for protection against oxidative stress [79], was pseudogenized during the shift from macroalgal ambient sediment to coral mucus (Table 1). Members of another gene family encoding the same function were repeatedly pseudogenized during the shifts from coral skeleton to macroalgal tissue and macroalgal ambient seawater (Table 1). The frequent functional changes of this gene likely reflect the ability of roseobacters inhabiting different ecological niches to sense and rapidly respond to oxidative stresses through pseudogenization.
Genes mediating organismal interactions correlated with environmental changes
We found that pseudogenization and back events frequently occurred in gene families encoding the components of secretion systems and flagella, potentially mediating bacteria-host and bacteria-bacteria interactions (Table 1). For example, genes encoding the components of Type II, IV, and VI secretion systems were pseudogenized during niche shifts between coral and macroalgal ecosystems or between niches within each ecosystem (Table 1). Particularly, T4SS and T6SS were demonstrated to be important in the interactions with host and other bacteria [80]. Considering the diversity and specificity of bacterial secretion systems, secreting effector proteins or cytotoxins into certain environments may represent a competitive strategy that helps roseobacters exploit the host or compete with other bacteria inhabiting the same niche [80].
Further, pseudogenization or back events occurred in gene families associated with bacterial flagellar assembly and export (TipF, FlaA, FlgD, and FliI) during niche shifts between macroalgal ambient sediment and coral skeleton (Table 1). Besides enabling motility, bacterial flagella play additional roles in surface adhesion and biofilm formation, which may enhance the resistance to antimicrobial agents and enable the embedded cells to outcompete unrelated neighbors for both space and resources [81]. As a classic example are genes encoding components of the flagellar (mentioned above), whose mutant can lead to defective adhesion to coral [82] and the loss of the ability of biofilm formation and swarming motility (which may be relevant to roseobacters’ activities in the sediment) [83]. These events suggest the potential importance of dynamic regulation of flagellar assembly in host colonization and bacterial survival during niche shifts.
As each niche can be further divided into multiple physiochemical microenvironments, which differs from time to time, the nature of each niche is likely much more complex and heterogeneous than we can characterize. Accordingly, the resident microbial communities can be even more diverse and dynamic. Recently, a surprisingly greater microbiome diversity was revealed in coral skeleton than in coral mucus and tissue [84, 85], which may have been shaped by multiple physiochemical gradients across depth layers and limited dispersal of microbes in the skeletal matrix [86]. It was therefore speculated that roseobacters inhabiting coral skeleton may harbor some “niche factors” aiding competition with other members in the community or serving as cell-cell communication strategy [87]. One explanation for the pseudogenization events upon shift from coral skeleton to coral mucus could be that the microbiome diversity decreased as a result of niche shift, which leads to reduced extent of microbial interactions. Likewise, the functional transitions between coral and macroalgal ecosystems may reflect the different composition and diversity of microbial community in different niches. As microbial communities on algal surface differ remarkably from the free-living bacteria in the surrounding seawater [33, 88], it is possible that bacteria inhabiting distinct niches of macroalgal ecosystem are equipped with various weapons to defend against competitors and persist in the niche. However, the knowledge about specific niche factors in different environments at the current stage of research is still very limited. Therefore, future studies are necessary to address these important questions.
Caveats and future research needs
Similar to a recent study [89], we defined the ecological niche of roseobacters according to their isolation sites. Yet, this could lead to several caveats and future research needs. Importantly, the relationship between isolation sites and habitats is not simple, which could further be complicated by potential disruption in sampling (e.g., a reliable method to separate coral tissue from skeleton and mucus has not been available [90]). In the present study, although we presented interesting examples regarding the correlation between pseudogenization events and niche shifts, it is possible that some of these occurred simply by chance. Most of the pseudogenization or back events occurring during niche shifts were annotated as hypothetical proteins (Supplementary Dataset S3). For those with an assigned function, due to their small number (n = 41) of ecologically matched cases (Table 1, Supplementary Table S2), it is very difficult to perform rigorous statistical tests to examine whether specific functions are more likely to be associated with such events. As a matter of expediency, we elaborated on the relationship between their functions and the niche change, highlighting potential contributions of pseudogenization or back events to niche adaptation, which, however, could be speculative. In addition, except for a few cases (e.g., [91],), the knowledge of the adaptiveness of pseudogenes is still scarce. With the above caveats in mind, we suggest that function-testing experiments and population genetics analyses are needed before any conclusion of the adaptive role of pseudogenization are reached. This is beyond the scope of this study and awaits future work.
In addition, while most samples were collected in the same season and from locations nearby in Hong Kong, we cannot rule out the effect of sampling sites and dates on the evolutionary changes of functional genes. Since roseobacters isolated from samples collected before May 2017 and on Feb 25, 2018 (Ngo Mei Chau) together account for only ~14% of all isolates (Supplementary Table S1), we removed them from an updated analysis to minimize the effect of sampling sites and dates. A total of 306 gene families were identified, among which 262 families were consistent with the families identified with the dataset before removing the time-inconsistent isolates. Although the number of ecologically matched cases decreased from 41 to 31 in the updated analysis, 28 of them remained consistent with the previous ones (Supplementary Dataset S3). Among the rest three newly identified cases, two families encoding the multidrug ABC transporter ATP-binding protein and the flagellar biosynthesis protein FliR, respectively, were pseudogenized during the niche shift from macroalgal ambient sediment to coral skeleton. The remaining one encodes a catalase for mitigating oxidative stresses, and it was pseudogenized during the shift from coral skeleton to macroalgal ambient sediment.
Concluding remarks
Through comprehensive genomic analyses of 279 newly sequenced Ruegeria isolates collected from coral and macroalgal ecosystems nearby, we showed that gene loss via pseudogenization is likely an important mechanism driving genome content differentiation of this ecologically diverse and metabolically versatile marine bacterial lineage, and further identified a potential correlation between changes in genome content mediated by pseudogenization and shifts in ecological niches harbored in these two typical coastal ecosystems. Genes whose pseudogenization events may be correlated with niche switches include those involved in resource recovery and energy conservation, N metabolism and carbohydrate utilization, transport systems, stress response and organismal interactions (summarized in Fig. 5). Since gene loss by pseudogenization often requires only a single point-nonsense mutation, this mechanism may enable roseobacters to rapidly respond to environmental changes and adapt to new habitats. Overall, our study suggests that gene loss mediated by gene pseudogenization is an important contributor to the genetic variation and ecological diversification of the Roseobacter group. This mechanism may similarly act in other generalist bacteria.

The images of the coral and macroalgae, respectively, are kindly provided by CHUI Pui Yi and ANG Put On Jr. with permission to use here.
Data availability
Code availability
Codes used to generate the results are available at https://doi.org/10.6084/m9.figshare.12545984.
References
Nowell RW, Green S, Laue BE, Sharp PM. The extent of genome flux and its role in the differentiation of bacterial lineages. Genome Biol Evol. 2014;6:1514–29.
Ochman H, Lawrence JG, Groisman EA. Lateral gene transfer and the nature of bacterial innovation. Nature. 2000;405:299–304.
Wiedenbeck J, Cohan FM. Origins of bacterial diversity through horizontal genetic transfer and adaptation to new ecological niches. FEMS Microbiol Rev. 2011;35:957–76.
Albalat R, Cañestro C. Evolution by gene loss. Nat Rev Genet. 2016;17:379–91.
Jacq C, Miller JR, Brownlee GG. A pseudogene structure in 5S DNA of Xenopus laevis. Cell. 1977;12:109–20.
Li W-H, Gojobori T, Nei M. Pseudogenes as a paradigm of neutral evolution. Nature. 1981;292:237–9.
Ohta T. The nearly neutral theory of molecular evolution. Annu Rev Ecol Syst. 1992;23:263–86.
Bolotin E, Hershberg R. Gene loss dominates as a source of genetic variation within clonal pathogenic bacterial species. Genome Biol Evol. 2015;7:2173–87.
Hottes AK, Freddolino PL, Khare A, Donnell ZN, Liu JC, Tavazoie S. Bacterial adaptation through loss of function. PLoS Genet. 2013;9:e1003617.
Sharma V, Hecker N, Roscito JG, Foerster L, Langer BE, Hiller M. A genomics approach reveals insights into the importance of gene losses for mammalian adaptations. Nat Commun. 2018;9:1–9.
Sokurenko EV, Hasty DL, Dykhuizen DE. Pathoadaptive mutations: gene loss and variation in bacterial pathogens. Trends Microbiol. 1999;7:191–5.
Ortega AP, Villagra NA, Urrutia IM, Valenzuela LM, Talamilla-Espinoza A, Hidalgo AA, et al. Lose to win: marT pseudogenization in Salmonella enterica serovar Typhi contributed to the surV-dependent survival to H2O2, and inside human macrophage-like cells. Infect Genet Evol. 2016;45:111–21.
Goodhead I, Darby AC. Taking the pseudo out of pseudogenes. Curr Opin Microbiol. 2015;23:102–9.
Johnson LJ. Pseudogene rescue: an adaptive mechanism of codon reassignment. J Evol Biol. 2010;23:1623–30.
Librado P, Vieira FG, Rozas J. BadiRate: estimating family turnover rates by likelihood-based methods. Bioinformatics. 2012;28:279–81.
David LA, Alm EJ. Rapid evolutionary innovation during an Archaean genetic expansion. Nature. 2011;469:93–96.
Avni E, Montoya D, Lopez D, Modlin R, Pellegrini M, Snir S. A phylogenomic study quantifies competing mechanisms for pseudogenization in prokaryotes—the Mycobacterium leprae case. PLoS One. 2017;13:e0204322.
Ochman H. The nature and dynamics of bacterial genomes. Science. 2006;311:1730–3.
Grote J, Thrash JC, Huggett MJ, Landry ZC, Carini P, Giovannoni SJ, et al. Streamlining and core genome conservation among highly divergent members of the SAR11 clade. mBio. 2012;3:e00252–12.
Giovannoni SJ, Cameron Thrash J, Temperton B. Implications of streamlining theory for microbial ecology. ISME J. 2014;8:1553–65.
Buchan A, González JM, Moran MA. Overview of the marine Roseobacter lineage. Appl Environ Microbiol. 2005;71:5665–77.
Luo H, Moran MA. How do divergent ecological strategies emerge among marine bacterioplankton lineages? Trends Microbiol. 2015;23:577–84.
Luo H, Moran MA. Evolutionary ecology of the marine Roseobacter clade. Microbiol Mol Biol Rev. 2014;78:573–87.
Tujula NA, Crocetti GR, Burke C, Thomas T, Holmström C, Kjelleberg S. Variability and abundance of the epiphytic bacterial community associated with a green marine Ulvacean alga. ISME J. 2010;4:301–11.
Littman RA, Willis BL, Pfeffer C, Bourne DG. Diversities of coral-associated bacteria differ with location, but not species, for three acroporid corals on the Great Barrier Reef. FEMS Microbiol Ecol. 2009;68:152–63.
Rosenberg E, Koren O, Reshef L, Efrony R, Zilber-Rosenberg I. The role of microorganisms in coral health, disease and evolution. Nat Rev Microbiol. 2007;5:355–62.
Sweet MJ, Croquer A, Bythell JC. Bacterial assemblages differ between compartments within the coral holobiont. Coral Reefs. 2011;30:39–52.
Crossland CJ, Barnes DJ, Borowitzka MA. Diurnal lipid and mucus production in the staghorn coral Acropora acuminata. Mar Biol. 1980;60:81–90.
Shashar N, Stambler N. Endolithic algae within corals—life in an extreme environment. J Exp Mar Biol Ecol. 1992;163:277–86.
Highsmith RC. Lime-boring algae in hermatypic coral skeletons. J Exp Mar Biol Ecol. 1981;55:267–81.
Kühl M, Holst G, Larkum AWD, Ralph PJ. Imaging of oxygen dynamics within the endolithic algal community of the massive coral Porites lobata. J Phycol. 2008;44:541–50.
Kalhoefer D, Thole S, Voget S, Lehmann R, Liesegang H, Wollher A, et al. Comparative genome analysis and genome-guided physiological analysis of Roseobacter litoralis. BMC Genomics. 2011;12:324.
Lachnit T, Fischer M, Künzel S, Baines JF, Harder T. Compounds associated with algal surfaces mediate epiphytic colonization of the marine macroalga Fucus vesiculosus. FEMS Microbiol Ecol. 2013;84:411–20.
Singh RP, Reddy CRK. Seaweed–microbial interactions: key functions of seaweed-associated bacteria. FEMS Microbiol Ecol. 2014;88:213–30.
Khailov KM, Burlakova ZP. Release of dissolved organic matter by marine seaweeds and distribution of their total organic production to inshore communities. Limnol Oceanogr. 1969;14:521–7.
Wai TC, Ng JSS, Leung KMY, Dudgeon D, Williams GA. The source and fate of organic matter and the significance of detrital pathways in a tropical coastal ecosystem. Limnol Oceanogr. 2008;53:1479–92.
Braeckman U, Pasotti F, Vázquez S, Zacher K, Hoffmann R, Elvert M, et al. Degradation of macroalgal detritus in shallow coastal Antarctic sediments. Limnol Oceanogr. 2019;64:1423–41.
Moran MA, Belas R, Schell MA, Gonzalez JM, Sun F, Sun S, et al. Ecological genomics of marine roseobacters. Appl Environ Microbiol. 2007;73:4559–69.
Sonnenschein EC, Nielsen KF, D’Alvise P, Porsby CH, Melchiorsen J, Heilmann J, et al. Global occurrence and heterogeneity of the Roseobacter clade species Ruegeria mobilis. ISME J. 2017;11:569–83.
Slightom RN, Buchan A. Surface colonization by marine roseobacters: integrating genotype and phenotype. Appl Environ Microbiol. 2009;75:6027–37.
Thole S, Kalhoefer D, Voget S, Berger M, Engelhardt T, Liesegang H, et al. Phaeobacter gallaeciensis genomes from globally opposite locations reveal high similarity of adaptation to surface life. ISME J. 2012;6:2229–44.
Newton RJ, Griffin LE, Bowles KM, Meile C, Gifford S, Givens CE, et al. Genome characteristics of a generalist marine bacterial lineage. ISME J. 2010;4:784–98.
Brinkhoff T, Giebel H-A, Simon M. Diversity, ecology, and genomics of the Roseobacter clade: a short overview. Arch Microbiol. 2008;189:531–9.
Luo H, Löytynoja A, Moran MA. Genome content of uncultivated marine Roseobacters in the surface ocean. Environ Microbiol. 2012;14:41–51.
Lerat E, Ochman H. Ψ-Φ: Exploring the outer limits of bacterial pseudogenes. Genome Res. 2004;14:2273–8.
Lerat E, Ochman H. Recognizing the pseudogenes in bacterial genomes. Nucleic Acids Res. 2005;33:3125–32.
Kuo C-H, Ochman H. The extinction dynamics of bacterial pseudogenes. PLoS Genet. 2010;6:e1001050.
Umezaki I. Ecological studies of Sargassum hemiphyllum C. AGARDH in Obama Bay, Japan Sea. Nippon Suisan Gakkaishi. 1984;50:1677–83.
Tam TW, Ang PO. Repeated physical disturbances and the stability of sub-tropical coral communities in Hong Kong. China Aquat Conserv Mar Freshw Ecosyst. 2008;18:1005–24.
Cheang CC, Chu KH, Ang PO. Phylogeography of the marine macroalga Sargassum hemiphyllum (Phaeophyceae, Heterokontophyta) in northwestern Pacific. Mol Ecol. 2010;19:2933–48.
Raghunathan C, Venkataraman K. Diversity and distribution of corals and their associated fauna of Rani Jhansi Marine National Park, Andaman and Nicobar Islands. In: Venkataraman K, Raghunathan C, Sivaperuman C, (eds). Ecology of Faunal Communities on the Andaman and Nicobar Islands. Berlin, Heidelberg: Springer; 2012. p. 177–208.
Ang PO. Phenology of Sargassum spp. in Tung Ping Chau Marine Park, Hong Kong SAR, China. J Appl Phycol. 2006;18:629–36.
Huggett MJ, Apprill A. Coral microbiome database: Integration of sequences reveals high diversity and relatedness of coral-associated microbes. Environ Microbiol Rep. 2019;11:372–85.
Passel MWJ, van, Marri PR, Ochman H. The emergence and fate of horizontally acquired genes in Escherichia coli. PLoS Comput Biol. 2008;4:e1000059.
Ochman H. Distinguishing the ORFs from the ELFs: short bacterial genes and the annotation of genomes. Trends Genet. 2002;18:335–7.
Emms DM, Kelly S. OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 2019;20:238.
Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2015;32:268–74.
Schliep KP. Phangorn: phylogenetic analysis in R. Bioinformatics. 2011;27:592–3.
Liu Y, Harrison PM, Kunin V, Gerstein M. Comprehensive analysis of pseudogenes in prokaryotes: widespread gene decay and failure of putative horizontally transferred genes. Genome Biol. 2004;5:R64.
Halldal P. Photosynthetic capacities and photosynthetic action spectra of endozoic algae of the massive coral Favia. Biol Bull. 1968;134:411–24.
Shibata K, Haxo FT. Light transmission and spectral distribution through epi- and endozoic algal layers in the brain coral, Favia. Biol Bull. 1969;136:461–8.
Park JT, Uehara T. How bacteria consume their own exoskeletons (turnover and recycling of cell wall peptidoglycan). Microbiol Mol Biol Rev. 2008;72:211–27.
Mauck J, Chan L, Glaser L. Turnover of the cell wall of gram-positive bacteria. J Biol Chem. 1971;246:1820–7.
Goodell E. Recycling of murein by Escherichia coli. J Bacteriol. 1985;163:305–10.
Uehara T, Suefuji K, Jaeger T, Mayer C, Park JT. MurQ etherase is required by Escherichia coli in order to metabolize Anhydro-N-Acetylmuramic acid obtained either from the environment or from its own cell wall. J Bacteriol. 2006;188:1660–2.
Dik DA, Marous DR, Fisher JF, Mobashery S. Lytic transglycosylases: concinnity in concision of the bacterial cell wall. Crit Rev Biochem Mol Biol. 2017;52:503–42.
Jiang H, Kong R, Xu X. The N-acetylmuramic acid 6-phosphate etherase gene promotes growth and cell differentiation of cyanobacteria under light-limiting conditions. J Bacteriol. 2010;192:2239–45.
Ferrer LM, Szmant AM. Nutrient regeneration by the endolithic community in coral skeletons. In: Proceedings of the 6th International Coral Reef Symposium 1988. pp 1–4.
Risk MJ, Muller HR. Porewater in coral heads: evidence for nutrient regeneration. Limnol Oceanogr. 1983;28:1004–8.
Yu LJ, Wu JR, Zheng ZZ, Lin CC, Zhan XB. Changes in gene transcription and protein expression involved in the response of Agrobacterium sp. ATCC 31749 to nitrogen availability during curdlan production. Appl Biochem Microbiol. 2011;47:487–93.
Wada S, Aoki M, Mikami A, Komatsu T, Tsuchiya Y, Sato T, et al. Bioavailability of macroalgal dissolved organic matter in seawater. Mar Ecol Prog Ser. 2008;370:33–44.
Essenberg MK, Cooper RA. Two ribose-5-phosphate isomerases from Escherichia coli K12: partial characterisation of the enzymes and consideration of their possible physiological roles. Eur J Biochem. 1975;55:323–32.
Nelson CE, Goldberg SJ, Wegley Kelly L, Haas AF, Smith JE, Rohwer F, et al. Coral and macroalgal exudates vary in neutral sugar composition and differentially enrich reef bacterioplankton lineages. ISME J. 2013;7:962–79.
Mulligan C, Fischer M, Thomas GH. Tripartite ATP-independent periplasmic (TRAP) transporters in bacteria and archaea. FEMS Microbiol Rev. 2011;35:68–86.
Beyenbach KW, Wieczorek H. The V-type H+ ATPase: molecular structure and function, physiological roles and regulation. J Exp Biol. 2006;209:577–89.
Guadayol Ò, Silbiger NJ, Donahue MJ, Thomas FIM. Patterns in temporal variability of temperature, oxygen and pH along an environmental gradient in a coral reef. PLoS One. 2014;9:e85213.
Bodenmiller DM, Spiro S. The yjeB(nsrR) gene of Escherichia coli encodes a nitric oxide-sensitive transcriptional regulator. J Bacteriol. 2006;188:874–81.
Gilberthorpe NJ, Lee ME, Stevanin TM, Read RC, Poole RK. NsrR: a key regulator circumventing Salmonella enterica serovar Typhimurium oxidative and nitrosative stress in vitro and in IFN-γ-stimulated J774.2 macrophages. Microbiology. 2007;153:1756–71.
da Fonseca RR, Johnson WE, O’Brien SJ, Vasconcelos V, Antunes A. Molecular evolution and the role of oxidative stress in the expansion and functional diversification of cytosolic glutathione transferases. BMC Evol Biol. 2010;10:281.
Green ER, Mecsas J. Bacterial secretion systems—an overview. Microbiol Spectr. 2016;4:1–32.
Ansari MI, Schiwon K, Malik A, Grohmann E. Biofilm formation by environmental bacteria. In: Malik A, Grohmann E (eds). Environmental protection strategies for sustainable development. 2012. Springer Netherlands, Dordrecht, pp 341–77.
Meron D, Efrony R, Johnson WR, Schaefer AL, Morris PJ, Rosenberg E, et al. Role of flagella in virulence of the coral pathogen Vibrio coralliilyticus. Appl Environ Microbiol. 2009;75:5704–7.
Attmannspacher U, Scharf BE, Harshey RM. FliL is essential for swarming: motor rotation in absence of FliL fractures the flagellar rod in swarmer cells of Salmonella enterica. Mol Microbiol. 2008;68:328–41.
Fernando SC, Wang J, Sparling K, Garcia GD, Francini-Filho RB, de Moura RL, et al. Microbiota of the major south atlantic reef building coral Mussismilia. Micro Ecol. 2015;69:267–80.
Pollock FJ, McMinds R, Smith S, Bourne DG, Willis BL, Medina M, et al. Coral-associated bacteria demonstrate phylosymbiosis and cophylogeny. Nat Commun. 2018;9:4921.
Marcelino VR, van Oppen MJ, Verbruggen H. Highly structured prokaryote communities exist within the skeleton of coral colonies. ISME J. 2018;12:300–3.
Hill C. Virulence or niche factors: what’s in a name? J Bacteriol. 2012;194:5725–7.
Egan S, Harder T, Burke C, Steinberg P, Kjelleberg S, Thomas T. The seaweed holobiont: understanding seaweed–bacteria interactions. FEMS Microbiol Rev. 2013;37:462–76.
Levy A, Salas Gonzalez I, Mittelviefhaus M, Clingenpeel S, Herrera Paredes S, Miao J, et al. Genomic features of bacterial adaptation to plants. Nat Genet. 2018;50:138–50.
Koren O, Rosenberg E. Bacteria associated with mucus and tissues of the coral Oculina patagonica in summer and winter. Appl Environ Microbiol. 2006;72:5254–9.
Wang X, Grus WE, Zhang J. Gene losses during human origins. PLoS Biol. 2006;4:e52.
Acknowledgements
We thank Professor ANG Put On Jr. for providing the coral samples and LEUNG Yu Hin for assisting in macroalgal sample collection. We also thank FENG Xiaoyuan and LIAO Tianhua for assistance in bioinformatics analysis. This work was supported by the Shenzhen Science and Technology Committee (JCYJ20180508161811899), the National Natural Science Foundation of China (41776129), the Hong Kong Research Grants Council General Research Fund (14163917), the Hong Kong Environment and Conservation Fund (15/2016), the Hong Kong Research Grants Council Area of Excellence Scheme (AoE/M-403/16), and the Direct Grant of CUHK (4053437, 4053257, and 3132809).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Chu, X., Li, S., Wang, S. et al. Gene loss through pseudogenization contributes to the ecological diversification of a generalist Roseobacter lineage. ISME J 15, 489–502 (2021). https://doi.org/10.1038/s41396-020-00790-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41396-020-00790-0
This article is cited by
-
Phylogenomic and comparative analyses of Coffeeae alliance (Rubiaceae): deep insights into phylogenetic relationships and plastome evolution
BMC Plant Biology (2022)
-
Mechanisms driving genome reduction of a novel Roseobacter lineage
The ISME Journal (2021)
-
Pseudogenization of the chloroplast threonine (trnT-GGU) gene in the sunflower family (Asteraceae)
Scientific Reports (2021)
-
Population differentiation of Rhodobacteraceae along with coral compartments
The ISME Journal (2021)
