Circulating Metabolites as Potential Biomarkers for Neurological Disorders—Metabolites in Neurological Disorders


 ,
,  ,
, 
Abstract
:1. Introduction
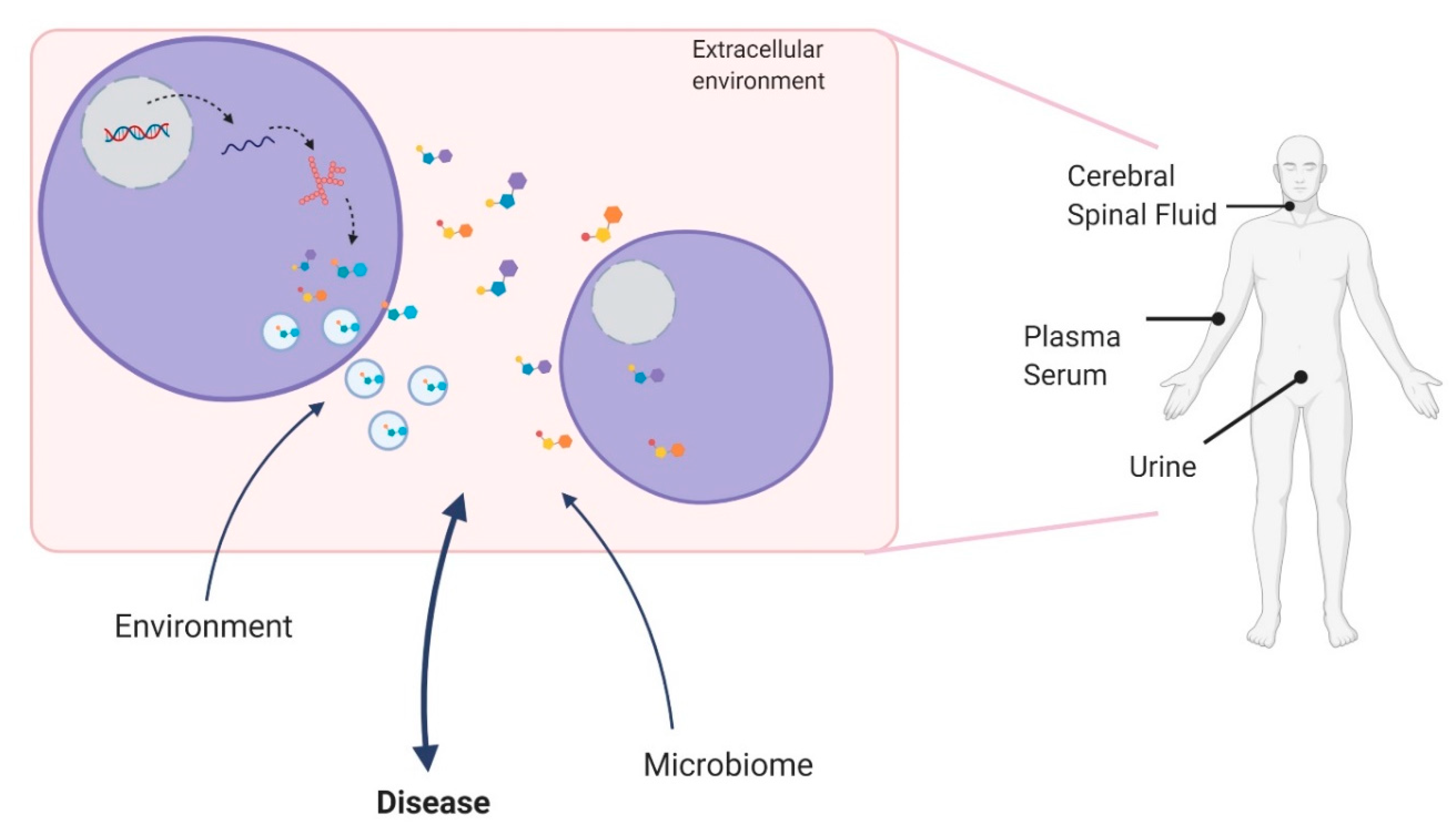
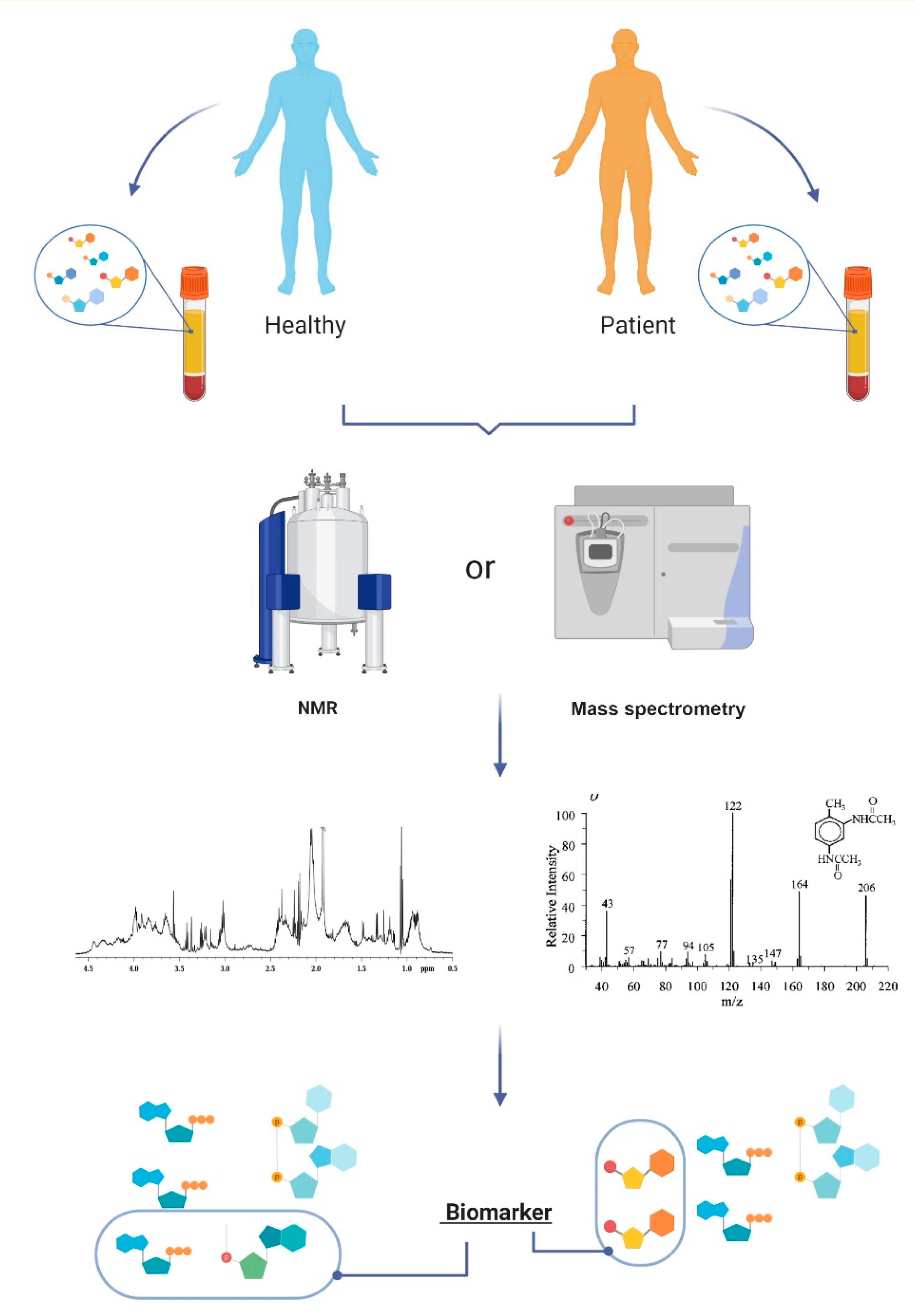
1.1. Metabolites and Metabolomics
1.2. Metabololites as Clinical Biomarkers
2. Metabolites in Specific Neurological Diseases
2.1. Alzheimer’s Disease
2.2. Amyotrophic Lateral Sclerosis
2.3. Epilepsy
2.4. Multiple Sclerosis
2.5. Parkinson’s Disease
2.6. Stroke

{kind=link}
{kind=link}
| Condition | Biological Fluid | Type of Analysis | Metabolite | Biomarker for | Related Mechanisms | Ref. |
|---|---|---|---|---|---|---|
| AD | CSF | LC-MS/ GC-MS | Aminoadipic acid | AD prediction | Not clear for this condition | [69] |
| CSF | LC-MS/ GC-MS | Tyrosine | AD prediction | Neurotransmitter synthesis | [69] | |
| CSF | LC-MS/ GC-MS | Sphingomyelin | AD prediction | Membrane Constitution | [69] | |
| CSF | LC-MS/ GC-MS | Lysophosphatidic acid C18:2 | AD prediction | Oxidative stress | [69] | |
| Plasma | FIA/ MS/MS | Acylcarnitine | AD and MCI prediction | Cascade of neurodegeneration | [68] | |
| Plasma | FIA/ MS/MS | Phosphatidylcholine | AD and MCI prediction | Cascade of neurodegeneration | [68] | |
| Plasma | FIA/ MS/MS | Sphingomyelin | AD and MCI prediction | Not clear for this condition | [68] | |
| Plasma | FIA/ MS/MS | Lysophospholipids | Differentiate AD from MCI | Not clear for this condition | [68] | |
| Plasma | FIA/ MS/MS | Dodecanedioyl carnitine | Differentiate AD from MCI from healthy subjects | Not clear for this condition | [68] | |
| Plasma | FIA/ MS/MS | Dodecanoylcarnitine | Differentiate AD from MCI from healthy subjects | Not clear for this condition | [68] | |
| Plasma | FIA/ MS/MS | PCaaC26:0 | Differentiate AD from MCI from healthy subjects | Not clear for this condition | [68] | |
| Urine | LC | Arginine | aMCI prediction | Protein homeostasis, taurine metabolism, glutathione metabolism | [63] | |
| Plasma | UPLC-MS/MS | Lysophosphatidyl ethanolamine | MCI-AD prediction | Membrane Constitution | [66] | |
| Plasma | UPLC-MS/MS | Choline | MCI-AD prediction | Neurotransmitter synthesis | [66] | |
| Plasma | UPLC-MS/MS | Soraphen A | MCI-AD prediction | It can interfere in the fatty acid elongation | [66] | |
| ALS | Serum/plasma; CSF; Plasma | NMR-based/MS-target; NMR-based; FIA/LC-MS/MS/NMR-based | Glutamate | ALS prediction; Differentiation from other neurological disorders; Drug responsiveness | Glutamate excitotoxicity | [17,71,72,78,81,82] |
| Serum; CSF; plasma | NMR-based; CG-MS; FIA/ LC-MS/MS | Glutamine | ALS prediction; Familial ALS prediction (SOD1 mutation); Drug responsiveness | Imbalance in glutamate–glutamine cycle | [72,74,78] | |
| Serum | NMR-based | Formate | ALS prediction | Increased levels may cause cell death | [78] | |
| CSF | NMR-based | Acetate | ALS prediction | Energy metabolism dysfunction | [5] | |
| CSF | NMR-based | Acetone | ALS prediction | Energy metabolism dysfunction | [5] | |
| CSF | NMR-based | Pyruvate | ALS prediction | Energy metabolism dysfunction | [5] | |
| CSF | NMR-based | Ascorbate | ALS prediction; Differentiation from other neurological disorders | Oxidative stress | [5,71] | |
| CSF | CG-MS | Creatinine | Familial ALS prediction - SOD1 mutation | Energy metabolism dysfunction | [74] | |
| Plasma | MS-target | Homocysteine | ALS prediction | Not clear for this condition | [17,81] | |
| Plasma | FIA/LC-MS/MS | Creatinine | Drug responsiveness | Not clear for this condition | [72] | |
| Plasma | FIA/LC-MS/MS/NMR-based | Glycine | Drug responsiveness | Changes in its levels can affect the activity of the NMDA receptor | [72,82] | |
| Plasma | LC-MS/MS | Acylcarnitines | Protective function | Not clear for this condition | [83] | |
| Plasma | LC-MS/MS | Diacylglicerols | Protective function | Not clear for this condition | [83] | |
| Plasma | LC-MS/MS | Triacylglicerols | Protective function | Not clear for this condition | [83] | |
| Plasma | LC-MS/MS | Phosphatidylcholine | Protective function | Not clear for this condition | [83] | |
| Epilepsy | Plasma | LC-MS | N8-acetylspermidine | Snyder–Robinson syndrome | Alterations in its levels may cause an imbalance of excitatory and inhibitory mechanisms | [90] |
| Serum; Brain tissue; Serum | CG-MS; HR-MAS¹H MRS; NMR-based | Lactate | Different types of seizures; Epileptic activity; Drug responsiveness | Energy metabolism dysfunction | [91,92,93] | |
| Serum; Brain tissue | CG-MS; HR-MAS¹H MRS | Glutamate | Different types of seizures; Epileptic activity | Glutamate excitotoxicity and hyperexcitability | [91,92] | |
| Brain tissue | HR-MAS¹H MRS | Choline | Epileptic activity | Alterations in its levels may suggest heightened cell membrane turnover in high-spiking tissue | [92] | |
| Brain tissue | HR-MAS¹H MRS | Glycerophosphorylcholine | Epileptic activity | Alterations in its levels may suggest heightened cell membrane turnover in high-spiking tissue | [92] | |
| Brain tissue | HR-MAS¹H MRS | Glutamine | Epileptic activity | Not clear for this condition | [92] | |
| Serum | NMR-based | Glucose | Drug responsiveness | Energy metabolism dysfunction | [93] | |
| Plasma | LC-HRMS | Neurosteroids | Effect of medicines in fetal development | Neurodevelop- mental functions | [93] | |
| Plasma | LC-HRMS | Progesterone | Effect of medicines in fetal development | Reduced levels may be related to a risk factor for miscarriage | [93] | |
| Plasma | LC-HRMS | 3β-androstanediol | Effect of medicines in fetal development | Not clear for this condition | [93] | |
| Plasma | LC-HRMS | 5-methyltetrahydrofolate | Effect of medicines in fetal development | AED-induced effect on folate uptake or metabolism | [93] | |
| Plasma | LC-HRMS | Tetrahydrofolate | Effect of medicines in fetal development | AED-induced effect on folate uptake or metabolism | [93] | |
| MuS | CSF; Serum | NMR-based | Acetate | MuS prediction; Differentiate Neuromyelitis optica from MuS and healthy subjects | The decrease may lead to myelination dysfunction; Neurotransmitter synthesis and suggested as a marker of astrocyte metabolism | [101,104,107] |
| CSF | NMR-based | N-Methyl metabolites | Demyelination process | Impairment in the choline-glycine cycle and myelin synthesis | [101] | |
| CSF | NMR-based | Sarcosine (N-methyl-glycine) | Demyelination process | Impairment in the choline-glycine cycle and myelin synthesis | [101] | |
| CSF | NMR-based | Formate | Demyelination process | Impairment in the choline-glycine cycle and myelin synthesis | [101,102] | |
| CSF | NMR-based | Lactate | MuS prediction | The increase was related to CSF mononuclear cells in MS patients and demyelinating areas | [102] | |
| CSF | NMR-based | N-acetyl aspartate (NAA) | Differentiate chronic lesions from healthy subjects | The decrease may be related to chronic demyelinating plaques | [102,103] | |
| CSF | NMR-based | Choline | Differentiate acute from chronic plaques and normal-appearing white matter | Increase related to active demyelinating plaques | [102] | |
| CSF | NMR-based | Citrate | MuS prediction | The decrease can be related to the disruption of the TCA cycle through the pyruvate pathway and the formation of myelin | [102,103,104] | |
| CSF | NMR-based | Threonate | MuS prediction | Not clear for this condition | [103] | |
| CSF | NMR-based | Myo-inositol | MuS prediction | Not clear for this condition | [103] | |
| CSF | NMR-based | Mannose | MuS prediction | Not clear for this condition | [103] | |
| CSF | NMR-based | Phenylalanine | MuS prediction | Not clear for this condition | [103] | |
| CSF | NMR-based | 3-hydroxybutyrate | MuS prediction | Not clear for this condition | [103] | |
| CSF | NMR-based | 2-hydroxyisovalerate | MuS prediction | Not clear for this condition | [103] | |
| CSF | NMR-based | 2-hydroxybutyrate | MuS prediction | The increase may be related to raised lipid oxidation and oxidative stress | [104] | |
| CSF; Serum | NMR-based; GC-MS | Pyroglutamate | MuS prediction | The increase may be related to impairment in antioxidant pathways and leads to central nervous system dysfunction | [104,109] | |
| CSF | NMR-based | Acetone | MuS prediction | The increase may be related to impairment in energetic metabolism | [104] | |
| CSF; Serum | NMR-based | Glucose | MuS prediction | The decrease can be related to disturbed energy generation and progress of MS | [104,106] | |
| CSF | HRMS | Kynurenate | Differentiate SPMuS from RRMuS patients | Tryptophan metabolism | [105] | |
| CSF | HRMS | 5-hydroxytryptophan | Differentiate SPMuS from RRMuS patients | Tryptophan metabolism | [105] | |
| CSF | HRMS | 5-hydroxyindoleacetate | Differentiate SPMuS from RRMuS patients | Tryptophan metabolism | [105] | |
| CSF | HRMS | N-acetylserotonin | Differentiate SPMuS from RRMuS patients | Tryptophan metabolism | [105] | |
| CSF | HRMS | Uridine | Differentiate SPMuS from RRMuS patients | Pyrimidine metabolism; Significantly associated with disability, disease activity, and brain atrophy | [105] | |
| CSF | HRMS | Deoxyuridine | Differentiate SPMuS from RRMuS patients | Pyrimidine metabolism; Significantly associated with disability, disease activity, and brain atrophy | [105] | |
| CSF | HRMS | Thymine | Differentiate SPMuS from RRMuS patients | Pyrimidine metabolism; Significantly associated with disability, disease activity, and brain atrophy | [105] | |
| CSF | HRMS | Glutamine | Differentiate SPMuS from RRMuS patients | Pyrimidine metabolism; Significantly associated with disability, disease activity, and brain atrophy | [105] | |
| Serum | NMR-based | Selenium | MuS prediction | The decrease may be related to oxidative stress | [106] | |
| Serum | NMR-based | Valine | MuS prediction | The decrease may be related to myelination dysfunction of the neurons | [106] | |
| Serum | NMR-based | Scyllo-inositol | Differentiate MuS from Neuromyelitis optica and healthy subjects | May be related to diffuse glial proliferation, demyelination, and neuronal damages | [107] | |
| Serum | UHPLC-MS | Sphingomyelin | MuS prognosis | One of the main lipid class in myelin; influence the immune response | [108] | |
| Serum | UHPLC-MS | Lysophosphatidyl ethanolamine | MuS prognosis | Modulates the immune response | [108] | |
| Serum | UHPLC-MS | Hydrocortisone | MuS severity | Not clear for this condition | [108] | |
| Serum | UHPLC-MS | Tryptophan | MuS severity | Not clear for this condition | [108] | |
| Serum | UHPLC-MS | Glutamate | MuS severity | Related to excitatory neurotransmitter and oligodendrocyte death in the white matter | [108] | |
| Serum | UHPLC-MS | Eicosapentaenoic acid | MuS severity | Related to the activation of the immune system | [108] | |
| Serum | UHPLC-MS | 13S-hydroxyoctadecadienoic acid | MuS severity | Not clear for this condition | [108] | |
| Serum | UHPLC-MS | Lysophosphatidyl cholines | MuS severity | Present in the cell membrane; role in proliferative growth and apoptosis | [108] | |
| Serum | UHPLC-MS | Lysophosphatidyl ethanolamines | MuS severity | Not clear for this condition | [108] | |
| Serum | GC-MS | Laurate | Differentiate MuS from healthy subjects | Saturated fatty acid, may be related to immune response | [109] | |
| Serum | GC-MS | N-methylmaleimide | Differentiate MuS from healthy subjects | May be related to mitochondrial function and energy metabolism | [109] | |
| Serum | GC-MS | Acylcarnitine C14:1 | Differentiate MuS from healthy subjects | Related to mitochondrial function and energy metabolism | [109] | |
| Serum | GC-MS | Phosphatidylcholine | Differentiate MuS from healthy subjects | Present in cell membrane and myelin | [109] | |
| PD | CSF; Urine; Brain of goldfish homogenate | GC-MS/LC-MS; NMR-based | BCAA | Differentiate PD from healthy subjects; Idiopathic PD prediction; PD Goldfish model | Protein synthesis, energy production, and synthesis of the neurotransmitter glutamate | [6,143,145] |
| Serum | UPLC-MS/MS | Caffeine | Differentiate PD from healthy subjects | Regulate the release of neurotransmitters (glutamate and dopamine) | [133] | |
| Serum | UPLC-MS/MS | Tryptophan | Differentiate PD from healthy subjects | The decrease may be associated with psychiatric problems in advanced PD | [133] | |
| Serum | UPLC-MS/MS | Ergothioneine | Differentiate PD from healthy subjects | A decrease may suggest elevated oxidative stress | [133] | |
| Serum | UPLC-MS/MS | Bilirubin/Biliverdin ratio | Differentiate PD from healthy subjects | A decrease may suggest elevated oxidative stress | [133] | |
| Serum; Plasma | Enzymatic Methods | Uric acid | PD prediction | Antioxidant. An increase may suggest a potential protective effect | [136,137,138] | |
| Serum | MS-based | FA metabolism (acyl carnitine pathway) | PD prognosis and MCI development | Medium-long chain FA derived from beta-oxidation. Related to mitochondrial dysfunction and neuronal loss | [142] | |
| Urine | HPLC-HRMS | Steroidogenesis metabolism | PD progression | May be related to oxidative stress, inflammation, and neuron injury | [143] | |
| Urine | HPLC-HRMS | Fatty acid beta-oxidation | PD progression | May be related to mitochondrial dysfunction, oxidative stress, and impaired energy metabolism | [143] | |
| Urine | HPLC-HRMS | Histidine metabolism | PD progression | Suppressive neurotransmitter effects, and hormone secretion | [143] | |
| Urine | HPLC-HRMS | Phenylalanine metabolism | PD progression | Not clear for this condition | [143] | |
| Urine | HPLC-HRMS; GC-MS /LC-MS | Tryptophan metabolism | PD progression; Idiopathic PD prediction | Related to mitochondrial disturbances and impairment of brain energy metabolism | [143,144] | |
| Urine | HPLC-HRMS; GC-MS/ LC-MS | Glycine derivation | PD progression; Idiopathic PD prediction | Stimulate the release of dopamine and acetylcholine | [143,144] | |
| Urine | HPLC-HRMS | Nucleotide metabolism | PD progression | Not clear for this condition | [143] | |
| Urine | HPLC-HRMS | Tyrosine metabolism | PD progression | Not clear for this condition | [143] | |
| Urine | GC-MS/ LC-MS | Steroid hormone biosynthesis | Idiopathic PD prediction | Related to oxidative stress, and dopamine cell degeneration in PD | [144] | |
| Urine | GC-MS/ LC-MS | Phenylalanine metabolism | Idiopathic PD prediction | Precursor for dopamine | [144] | |
| Brain of goldfish homogenate | NMR-based | Myo-inositol | PD Goldfish model | Glial marker. An increase may suggest disruptive cell functions in the brain | [145] | |
| Brain of goldfish homogenate | NMR-based | N-acetylaspartate | PD Goldfish model | The decrease may suggest neuronal dysfunction or cell loss | [145] | |
| Brain of goldfish homogenate | NMR-based | Betaine | PD Goldfish model | Reduced may suggest a reduced antioxidant capacity | [145] | |
| Brain of goldfish homogenate | NMR-based | Phosphatidylcholines | PD Goldfish model | Component of cellular membranes. Decrease related to membrane damage | [145] | |
| Brain of goldfish homogenate | NMR-based | Creatine and phosphocreatine | PD Goldfish model | The decrease can be related to severe oxidative damage and energy impairment | [145] | |
| Brain of goldfish homogenate | NMR-based | Cholesterol | PD Goldfish model | The decrease may be related to elevated oxidative stress; impaired brain mitochondria | [145] | |
| Brain of goldfish homogenate | NMR-based | Polyunsaturated fatty acid | PD Goldfish model | The decrease may be associated with elevated oxidative stress | [145] | |
| CSF | UHPLC/ GC-MS | Benzoate | PD progression | Derived from the catabolism of phenylalanine | [139] | |
| Plasma | UHPLC/ GC-MS | Theobromine | PD progression | Phenylalanine metabolism | [139] | |
| Plasma | UHPLC/GC-MS | Theophylline | PD progression | Metabolites of the purine compound caffeine | [139] | |
| Plasma | UHPLC/GC-MS | Paraxanthine | PD progression | Metabolites of the purine compound caffeine | [139] | |
| Plasma | UHPLC/GC-MS | 1-methylxanthine | PD progression | Metabolites of the purine compound caffeine | [139] | |
| Plasma | UHPLC/GC-MS | 5-dodecanoate | PD progression | Fatty acid metabolism | [139] | |
| Plasma | UHPLC/GC-MS | 3-hydroxydecanoate | PD progression | Fatty acid metabolism | [139] | |
| Plasma | UHPLC/GC-MS | Docosadienoate | PD progression | Fatty acid metabolism | [139] | |
| Plasma | UHPLC/GC-MS | Docosatrienoate | PD progression | Fatty acid metabolism | [139] | |
| Stroke | Serum | GC-MS/ LC-MS | Isoleucine | Differentiate AIS from healthy subjects | Signaling molecule to regulate the growth, repair, and maintenance of the brain functions | [9] |
| Serum | GC-MS/ LC-MS | Serine | Differentiate AIS from healthy subjects | Signaling molecule to regulate the growth, repair, and maintenance of the brain functions | [9] | |
| Serum | GC-MS/ LC-MS | Phosphatidylcholine | Differentiate AIS from healthy subjects | Component of cellular membrane | [9] | |
| Serum | GC-MS/LC-MS | Betaine | Differentiate AIS from healthy subjects | Part of the choline pathway; part of the antioxidant process | [9] | |
| Serum | GC-MS/LC-MS | Lysophosphatidylethanolamine | Differentiate AIS from healthy subjects | Component of cellular membrane | [9] | |
| Serum | GC-MS/LC-MS | Carnitine | Differentiate AIS from healthy subjects | Help the catabolism of lipids and energy conversion | [9] | |
| Serum; Plasma/Urine | GC-MS; NMR-based | Lactate | AIS prediction Small vessel disease prediction | An increase may indicate anaerobic glycolysis, hypoxia, and ischemia | [10,170] | |
| Serum | GC-MS | Tyrosine | AIS prediction | A low level can lead to oxidative stress and inflammation | [10] | |
| Serum; CSF | GC-MS | Tryptophan | AIS prediction; Long-term outcome of subarachnoid hemorrhage | A low level can reduce serotonin | [10,175] | |
| Plasma | HPLC | Dimethylarginine | Early-onset stroke | Inhibitor of nitric oxide synthase, part of the pathogenesis of atherosclerosis | [161] | |
| Plasma | NMR-based | Choline | Carotid artery stenosis pathogenesis | Its reduction increases the homocysteine methylation pathway | [162] | |
| Plasma | NMR-based | Homocysteine | Carotid artery stenosis pathogenesis | The increase could be associated with oxidative stress in vascular cells and platelet adhesion | [162] | |
| Plasma | LC-MS | Lysophosphatidylcholine | Stroke recurrence; Large artery atherosclerosis | It may be a potential trigger of the brain inflammation processes | [163,171] | |
| Serum | LC-MS/MS | Acetyl-L-lysine | Thrombotic ischemic prediction | The decrease may suggest elevated lysine catabolism and excitotoxic activity | [165] | |
| Serum | LC-MS/MS | Cadaverine | Thrombotic ischemic prediction | The decrease may suggest elevated lysine catabolism and excitotoxic activity | [165] | |
| Serum | LC-MS/MS | 2-oxoglutarate | Thrombotic ischemic prediction | The decrease may suggest elevated lysine catabolism and excitotoxic activity | [165] | |
| Serum | LC-MS/MS | Nicotinamide | Thrombotic ischemic prediction | The decrease may suggest elevated lysine catabolism and excitotoxic activity | [165] | |
| Serum | LC-MS/MS | Valine | Thrombotic ischemic prediction | A decrease may suggest an excitotoxic activity | [165] | |
| Plasma; CSF | LC-MS; GC-MS | BCAA | Stroke outcome and severity; Long-term outcome of subarachnoid hemorrhage | Decreased may influence the bioenergetic homeostasis and impair the citric acid cycle pathways | [168,175] | |
| Plasma/ Urine | NMR-based | Pyruvate | Small vessel disease prediction | The increase may be related to anaerobic glycolysis | [170] | |
| Plasma/ Urine | NMR-based | Glycolate | Small vessel disease prediction | The increase may be related to folic acid deficiency and hyperhomocysteinemia | [170] | |
| Plasma/ Urine | NMR-based | Formate | Small vessel disease prediction | The increase may be related to folic acid deficiency and hyperhomocysteinemia | [170] | |
| Plasma/ Urine | NMR-based | Glutamine | Small vessel disease prediction | The decrease may be related to elevating of glial fibrillary acidic protein and brain damage | [170] | |
| Plasma/ Urine | NMR-based | Methanol | Small vessel disease prediction | The decrease may be related to hyperhomocysteinemia | [170] | |
| Plasma | HPLC | Taurine | Stroke prognosis and recovery | Osmoregulator and neuromodulator. The increase may be related to brain tissue damage | [172] | |
| Blood | Mobile Photometric - Enzyme-kinetic Analyzer | Lactate:Pyruvate ratio | Hemorrhagic stroke prognosis | Reduced pyruvate may be related to impairment in energetic and repair functions | [174] | |
| CSF | GC-MS | 2-hydroxyglutarate | Long-term outcome of subarachnoid hemorrhage | The increase was related to adverse outcome and death, while the decrease was related to low disability outcomes | [175] | |
| CSF | GC-MS | Glycine | Long-term outcome of subarachnoid hemorrhage | Not clear for this condition | [175] | |
| CSF | GC-MS | Proline | Long-term outcome of subarachnoid hemorrhage | Not clear for this condition | [175] |
2.7. Overview of Relevant Metabolites in Neurological Disorders
3. Perspectives and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lanznaster, D.; de Assis, D.R.; Corcia, P.; Pradat, P.-F.; Blasco, H. Metabolomics Biomarkers: A Strategy Toward Therapeutics Improvement in ALS. Front. Neurol. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Strimbu, K.; Tavel, J.A. What are biomarkers? Curr. Opin. HIV AIDS 2010, 5, 463–466. [Google Scholar] [CrossRef]
- Vu, L.T.; Bowser, R. Fluid-based biomarkers for amyotrophic lateral sclerosis. Neurotherapeutics 2017, 14, 119–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thysell, E.; Surowiec, I.; Hörnberg, E.; Crnalic, S.; Widmark, A.; Johansson, A.I.; Stattin, P.; Bergh, A.; Moritz, T.; Antti, H.; et al. Metabolomic characterization of human prostate cancer bone metastases reveals increased levels of cholesterol. PLoS ONE 2010, 5, e14175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rozen, S.; Cudkowicz, M.E.; Bogdanov, M.; Matson, W.R.; Kristal, B.S.; Beecher, C.; Harrison, S.; Vouros, P.; Flarakos, J.; Vigneau-Callahan, K.; et al. Metabolomic analysis and signatures in motor neuron disease. Metabolomics 2005, 1, 101–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wuolikainen, A.; Jonsson, P.; Ahnlund, M.; Antti, H.; Marklund, S.L.; Moritz, T.; Forsgren, L.; Andersen, P.M.; Trupp, M. Multi-platform mass spectrometry analysis of the CSF and plasma metabolomes of rigorously matched amyotrophic lateral sclerosis, Parkinson’s disease and control subjects. Mol. Biosyst. 2016, 12, 1287–1298. [Google Scholar] [CrossRef] [PubMed]
- Goodacre, R.; Vaidyanathan, S.; Dunn, W.B.; Harrigan, G.G.; Kell, D.B. Metabolomics by numbers: Acquiring and understanding global metabolite data. Trends Biotechnol. 2004, 22, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, M.S.; Carvalho, M.; Bastos, M.L.; Guedes de Pinho, P. Metabolomics Analysis for Biomarker Discovery: Advances and Challenges. Available online: http://www.eurekaselect.com/105961/article (accessed on 1 June 2020).
- Liu, P.; Li, R.; Antonov, A.A.; Wang, L.; Li, W.; Hua, Y.; Guo, H.; Wang, L.; Liu, P.; Chen, L.; et al. Discovery of metabolite biomarkers for acute ischemic stroke progression. J. Proteome Res. 2017, 16, 773–779. [Google Scholar] [CrossRef]
- Wang, D.; Kong, J.; Wu, J.; Wang, X.; Lai, M. GC–MS-based metabolomics identifies an amino acid signature of acute ischemic stroke. Neurosci. Lett. 2017, 642, 7–13. [Google Scholar] [CrossRef]
- Pathan, M.; Fonseka, P.; Chitti, S.V.; Kang, T.; Sanwlani, R.; Van Deun, J.; Hendrix, A.; Mathivanan, S. Vesiclepedia 2019: A compendium of RNA, proteins, lipids and metabolites in extracellular vesicles. Nucleic Acids Res. 2019, 47, D516–D519. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.H.; Byun, J.; Pennathur, S. Analytical approaches to metabolomics and applications to systems biology. Semin. Nephrol. 2010, 30, 500–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beger, R.D.; Dunn, W.; Schmidt, M.A.; Gross, S.S.; Kirwan, J.A.; Cascante, M.; Brennan, L.; Wishart, D.S.; Oresic, M.; Hankemeier, T.; et al. Metabolomics enables precision medicine: “A White Paper, Community Perspective”. Metabolomics 2016, 12, 149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellis, D.I.; Dunn, W.B.; Griffin, J.L.; Allwood, J.W.; Goodacre, R. Metabolic fingerprinting as a diagnostic tool. Pharmacogenomics 2007, 8, 1243–1266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Germeys, C.; Vandoorne, T.; Bercier, V.; van den Bosch, L. Existing and emerging metabolomic tools for als research. Genes 2019, 10, 1011. [Google Scholar] [CrossRef] [Green Version]
- Alberich, R.; Castro, J.A.; Llabrés, M.; Palmer-Rodríguez, P. Metabolomics analysis: Finding out metabolic building blocks. PLoS ONE 2017, 12, e0177031. [Google Scholar] [CrossRef]
- Wittmann, C. Metabolic flux analysis using mass spectrometry. Adv. Biochem. Eng. Biotechnol. 2002, 74, 39–64. [Google Scholar] [CrossRef]
- Sauer, U. High-throughput phenomics: Experimental methods for mapping fluxomes. Curr. Opin. Biotechnol. 2004, 15, 58–63. [Google Scholar] [CrossRef]
- Griffiths, W.J.; Koal, T.; Wang, Y.; Kohl, M.; Enot, D.P.; Deigner, H.-P. Targeted metabolomics for biomarker discovery. Angew. Chem. Int. Ed. Engl. 2010, 49, 5426–5445. [Google Scholar] [CrossRef]
- Grandori, R.; Santambrogio, C.; Brocca, S.; Invernizzi, G.; Lotti, M. Electrospray-ionization mass spectrometry as a tool for fast screening of protein structural properties. Biotechnol. J. 2009, 4, 73–87. [Google Scholar] [CrossRef]
- Klassen, A.; Faccio, A.T.; Canuto, G.A.B.; da Cruz, P.L.R.; Ribeiro, H.C.; Tavares, M.F.M.; Sussulini, A. Metabolomics: Definitions and significance in systems biology. In Metabolomics: From Fundamentals to Clinical Applications; Advances in Experimental Medicine and Biology; Springer: Cham, Switzerland, 2017; Volume 965, pp. 3–17. [Google Scholar] [CrossRef]
- Psychogios, N.; Hau, D.D.; Peng, J.; Guo, A.C.; Mandal, R.; Bouatra, S.; Sinelnikov, I.; Krishnamurthy, R.; Eisner, R.; Gautam, B.; et al. The human serum metabolome. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [Green Version]
- Mastrangelo, A.; Barbas, C. Chronic diseases and lifestyle biomarkers identification by metabolomics. Adv. Exp. Med. Biol. 2017, 965, 235–263. [Google Scholar] [CrossRef] [PubMed]
- Barnes, S.; Benton, H.P.; Casazza, K.; Cooper, S.; Cui, X.; Du, X.; Engler, J.; Kabarowski, J.H.; Li, S.; Pathmasiri, W.; et al. Training in metabolomics research. II. Processing and statistical analysis of metabolomics data, metabolite identification, pathway analysis, applications of metabolomics and its future. J. Mass Spectrom. JMS 2016, 51, 535–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bales, J.R.; Bell, J.D.; Nicholson, J.K.; Sadler, P.J. 1H NMR studies of urine during fasting: Excretion of ketone bodies and acetylcarnitine. Magn. Reson. Med. 1986, 3, 849–856. [Google Scholar] [CrossRef] [PubMed]
- Novoa-Carballal, R.; Fernandez-Megia, E.; Jimenez, C.; Riguera, R. NMR methods for unravelling the spectra of complex mixtures. Nat. Prod. Rep. 2011, 28, 78–98. [Google Scholar] [CrossRef] [PubMed]
- Gebregiworgis, T.; Powers, R. Application of NMR metabolomics to search for human disease biomarkers. Comb. Chem. High Throughput Screen 2012, 15, 595–610. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, O.; Spraul, M. LC-NMR-MS in drug discovery. Drug Discov. Today 2003, 8, 624–631. [Google Scholar] [CrossRef]
- Lin, Y.; Schiavo, S.; Orjala, J.; Vouros, P.; Kautz, R. Microscale LC-MS-NMR platform applied to the identification of active cyanobacterial metabolites. Anal. Chem. 2008, 80, 8045–8054. [Google Scholar] [CrossRef] [Green Version]
- Pinnick, K.E.; Gunn, P.J.; Hodson, L. Measuring human lipid metabolism using deuterium labeling: In vivo and in vitro protocols. Methods Mol. Biol. Clifton NJ 2019, 1862, 83–96. [Google Scholar] [CrossRef]
- Buescher, J.M.; Antoniewicz, M.R.; Boros, L.G.; Burgess, S.C.; Brunengraber, H.; Clish, C.B.; DeBerardinis, R.J.; Feron, O.; Frezza, C.; Ghesquiere, B.; et al. A roadmap for interpreting 13C metabolite labeling patterns from cells. Curr. Opin. Biotechnol. 2015, 34, 189–201. [Google Scholar] [CrossRef]
- El-Maghrabey, M.; Kishikawa, N.; Kuroda, N. Novel isotope-coded derivatization method for aldehydes using 14n/15n-ammonium acetate and 9,10-phenanthrenequinone. Anal. Chem. 2018, 90, 13867–13875. [Google Scholar] [CrossRef]
- Broekaert, D.; Fendt, S.-M. Measuring in vivo tissue metabolism using 13c glucose infusions in mice. Methods Mol. Biol. Clifton NJ 2019, 1862, 67–82. [Google Scholar] [CrossRef]
- Schmid, B.; Hruscha, A.; Hogl, S.; Banzhaf-Strathmann, J.; Strecker, K.; van der Zee, J.; Teucke, M.; Eimer, S.; Hegermann, J.; Kittelmann, M.; et al. Loss of ALS-associated TDP-43 in zebrafish causes muscle degeneration, vascular dysfunction, and reduced motor neuron axon outgrowth. Proc. Natl. Acad. Sci. USA 2013, 110, 4986–4991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swaminathan, A.; Bouffard, M.; Liao, M.; Ryan, S.; Callister, J.B.; Pickering-Brown, S.M.; Armstrong, G.A.B.; Drapeau, P. Expression of C9orf72-related dipeptides impairs motor function in a vertebrate model. Hum. Mol. Genet. 2018, 27, 1754–1762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Giorgio, F.; Maduro, C.; Fisher, E.M.C.; Acevedo-Arozena, A. Transgenic and physiological mouse models give insights into different aspects of amyotrophic lateral sclerosis. Dis. Model. Mech. 2019, 12. [Google Scholar] [CrossRef] [Green Version]
- Torres, A.J.; Hill, A.S.; Love, J.C. Nanowell-based immunoassays for measuring single-cell secretion: Characterization of transport and surface binding. Anal. Chem. 2014, 86, 11562–11569. [Google Scholar] [CrossRef] [Green Version]
- Ungai-Salánki, R.; Gerecsei, T.; Fürjes, P.; Orgovan, N.; Sándor, N.; Holczer, E.; Horvath, R.; Szabó, B. Automated single cell isolation from suspension with computer vision. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [Green Version]
- Di Carlo, D.; Wu, L.Y.; Lee, L.P. Dynamic single cell culture array. Lab. Chip 2006, 6, 1445–1449. [Google Scholar] [CrossRef]
- Emara, S.; Amer, S.; Ali, A.; Abouleila, Y.; Oga, A.; Masujima, T. Single-cell metabolomics. Adv. Exp. Med. Biol. 2017, 965, 323–343. [Google Scholar] [CrossRef]
- Fletcher, J.S.; Kotze, H.L.; Armitage, E.G.; Lockyer, N.P.; Vickerman, J.C. Evaluating the challenges associated with time-of-fight secondary ion mass spectrometry for metabolomics using pure and mixed metabolites. Metabolomics 2013, 9, 535–544. [Google Scholar] [CrossRef]
- Lorenzo Tejedor, M.; Mizuno, H.; Tsuyama, N.; Harada, T.; Masujima, T. Direct single-cell molecular analysis of plant tissues by video mass spectrometry. Anal. Sci. Int. J. Jpn. Soc. Anal. Chem. 2009, 25, 1053–1055. [Google Scholar] [CrossRef] [Green Version]
- Laiko, V.V.; Baldwin, M.A.; Burlingame, A.L. Atmospheric pressure matrix-assisted laser desorption/ionization mass spectrometry. Anal. Chem. 2000, 72, 652–657. [Google Scholar] [CrossRef] [PubMed]
- Kaganman, I. SIMS for membranes. Nat. Methods 2006, 3, 962–963. [Google Scholar] [CrossRef] [PubMed]
- Chong, J.; Wishart, D.S.; Xia, J. Using metaboanalyst 4.0 for comprehensive and integrative metabolomics data analysis. Curr. Protoc. Bioinforma. 2019, 68, e86. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xiao, J.; Suzek, T.O.; Zhang, J.; Wang, J.; Bryant, S.H. PubChem: A public information system for analyzing bioactivities of small molecules. Nucleic Acids Res. 2009, 37, W623–W633. [Google Scholar] [CrossRef]
- Horai, H.; Arita, M.; Kanaya, S.; Nihei, Y.; Ikeda, T.; Suwa, K.; Ojima, Y.; Tanaka, K.; Tanaka, S.; Aoshima, K.; et al. MassBank: A public repository for sharing mass spectral data for life sciences. J. Mass Spectrom. JMS 2010, 45, 703–714. [Google Scholar] [CrossRef]
- Fahy, E.; Sud, M.; Cotter, D.; Subramaniam, S. LIPID MAPS online tools for lipid research. Nucleic Acids Res. 2007, 35, W606–W612. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Goto, S.; Hattori, M.; Aoki-Kinoshita, K.F.; Itoh, M.; Kawashima, S.; Katayama, T.; Araki, M.; Hirakawa, M. From genomics to chemical genomics: New developments in KEGG. Nucleic Acids Res. 2006, 34, D354–D357. [Google Scholar] [CrossRef]
- Kanehisa, M. The KEGG database. Novartis Found. Symp. 2002, 247, 91–101; discussion 101–103, 119–128, 244–252. [Google Scholar]
- Caspi, R.; Foerster, H.; Fulcher, C.A.; Kaipa, P.; Krummenacker, M.; Latendresse, M.; Paley, S.; Rhee, S.Y.; Shearer, A.G.; Tissier, C.; et al. The MetaCyc Database of metabolic pathways and enzymes and the biocyc collection of pathway/genome databases. Nucleic Acids Res. 2008, 36, D623–D631. [Google Scholar] [CrossRef]
- Frolkis, A.; Knox, C.; Lim, E.; Jewison, T.; Law, V.; Hau, D.D.; Liu, P.; Gautam, B.; Ly, S.; Guo, A.C.; et al. SMPDB: The small molecule pathway database. Nucleic Acids Res. 2010, 38, D480–D487. [Google Scholar] [CrossRef] [Green Version]
- Haug, K.; Salek, R.M.; Conesa, P.; Hastings, J.; de Matos, P.; Rijnbeek, M.; Mahendraker, T.; Williams, M.; Neumann, S.; Rocca-Serra, P.; et al. MetaboLights—An open-access general-purpose repository for metabolomics studies and associated meta-data. Nucleic Acids Res. 2013, 41, D781–D786. [Google Scholar] [CrossRef] [PubMed]
- Mamas, M.; Dunn, W.B.; Neyses, L.; Goodacre, R. The role of metabolites and metabolomics in clinically applicable biomarkers of disease. Arch. Toxicol. 2011, 85, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Bujak, R.; Struck-Lewicka, W.; Markuszewski, M.J.; Kaliszan, R. Metabolomics for laboratory diagnostics. J. Pharm. Biomed. Anal. 2015, 113, 108–120. [Google Scholar] [CrossRef] [PubMed]
- Attard, J.A.; Dunn, W.B.; Mergental, H.; Mirza, D.F.; Afford, S.C.; Perera, M.T.P.R. Systematic review: Clinical metabolomics to forecast outcomes in liver transplantation surgery. OMICS J. Integr. Biol. 2019, 23, 463–476. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Sun, H.; Wang, X. Recent advances in metabolomics in neurological disease, and future perspectives. Anal. Bioanal. Chem. 2013, 405, 8143–8150. [Google Scholar] [CrossRef]
- Oeckl, P.; Otto, M. A Review on ms-based blood biomarkers for alzheimer’s disease. Neurol. Ther. 2019, 8, 113–127. [Google Scholar] [CrossRef] [Green Version]
- Wishart, D.S. Metabolomics for investigating physiological and pathophysiological processes. Physiol. Rev. 2019, 99, 1819–1875. [Google Scholar] [CrossRef]
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef]
- Dementia. Available online: https://www.who.int/news-room/fact-sheets/detail/dementia (accessed on 28 April 2020).
- Tanzi, R.E. The genetics of Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2. [Google Scholar] [CrossRef]
- Zhang, Y.; Tang, Y.; Dammer, E.; Liu, J.; Zhao, Y.; Zhu, L.; Ren, R.; Chen, H.; Wang, G.; Cheng, Q. Dysregulated urinary arginine metabolism in older adults with amnestic mild cognitive impairment. Front. Aging Neurosci. 2019, 11. [Google Scholar] [CrossRef] [Green Version]
- Ibáñez, C.; Valdés, A.; García-Cañas, V.; Simó, C. Chapter 10–metabolomics in the study of alzheimer’s disease. In Comprehensive Analytical Chemistry; Applications of Advanced Omics Technologies: From Genes to Metabolites; García-Cañas, V., Cifuentes, A., Simó, C., Eds.; Elsevier: Amsterdam, The Netherlands, 2014; Volume 64, pp. 249–278. [Google Scholar]
- Atri, A. The Alzheimer’s disease clinical spectrum: Diagnosis and management. Med. Clin. North Am. 2019, 103, 263–293. [Google Scholar] [CrossRef] [PubMed]
- How Is Alzheimer’s Disease Diagnosed? Available online: https://www.nia.nih.gov/health/how-alzheimers-disease-diagnosed (accessed on 11 August 2020).
- González-Domínguez, R.; Sayago, A.; Fernández-Recamales, Á. Metabolomics in Alzheimer’s disease: The need of complementary analytical platforms for the identification of biomarkers to unravel the underlying pathology. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2017, 1071, 75–92. [Google Scholar] [CrossRef] [PubMed]
- Peña-Bautista, C.; Roca, M.; Hervás, D.; Cuevas, A.; López-Cuevas, R.; Vento, M.; Baquero, M.; García-Blanco, A.; Cháfer-Pericás, C. Plasma metabolomics in early Alzheimer’s disease patients diagnosed with amyloid biomarker. J. Proteom. 2019, 200, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, J.M.; Trushina, E. Application of metabolomics in Alzheimer’s disease. Front. Neurol. 2018, 8. [Google Scholar] [CrossRef] [Green Version]
- Costa, A.C.; Joaquim, H.P.G.; Forlenza, O.V.; Gattaz, W.F.; Talib, L.L. Three plasma metabolites in elderly patients differentiate mild cognitive impairment and Alzheimer’s disease: A pilot study. Eur. Arch. Psychiatry Clin. Neurosci. 2019. [Google Scholar] [CrossRef]
- De Leeuw, F.A.; Peeters, C.F.W.; Kester, M.I.; Harms, A.C.; Struys, E.A.; Hankemeier, T.; van Vlijmen, H.W.T.; van der Lee, S.J.; van Duijn, C.M.; Scheltens, P.; et al. Blood-based metabolic signatures in Alzheimer’s disease. Alzheimers Dement. Amst. Neth. 2017, 8, 196–207. [Google Scholar] [CrossRef]
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; van den Berg, L.H. Amyotrophic lateral sclerosis. Nat. Rev. Dis. Primer 2017, 3, 1–19. [Google Scholar] [CrossRef]
- Blasco, H.; Corcia, P.; Pradat, P.-F.; Bocca, C.; Gordon, P.H.; Veyrat-Durebex, C.; Mavel, S.; Nadal-Desbarats, L.; Moreau, C.; Devos, D.; et al. Metabolomics in cerebrospinal fluid of patients with amyotrophic lateral sclerosis: An untargeted approach via high-resolution mass spectrometry. J. Proteome Res. 2013, 12, 3746–3754. [Google Scholar] [CrossRef]
- Blasco, H.; Patin, F.; Descat, A.; Garçon, G.; Corcia, P.; Gelé, P.; Lenglet, T.; Bede, P.; Meininger, V.; Devos, D.; et al. A pharmaco-metabolomics approach in a clinical trial of ALS: Identification of predictive markers of progression. PLoS ONE 2018, 13, e0198116. [Google Scholar] [CrossRef]
- Turner, M.R.; Hardiman, O.; Benatar, M.; Brooks, B.R.; Chio, A.; de Carvalho, M.; Ince, P.G.; Lin, C.; Miller, R.G.; Mitsumoto, H.; et al. Controversies and priorities in amyotrophic lateral sclerosis. Lancet Neurol. 2013, 12, 310–322. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Sayana, P.; Zhang, X.; Le, W. Genetics of amyotrophic lateral sclerosis: An update. Mol. Neurodegener. 2013, 8, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobson, C.M.; Knowles, T.P.J.; Vendruscolo, M. The Amyloid phenomenon and its significance in biology and medicine. Cold Spring Harb. Perspect. Biol. 2020, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wuolikainen, A.; Moritz, T.; Marklund, S.L.; Antti, H.; Andersen, P.M. Disease-related changes in the cerebrospinal fluid metabolome in amyotrophic lateral sclerosis detected by gc/tofms. PLoS ONE 2011, 6, e17947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, P.J. Molecular and cellular pathways of neurodegeneration in motor neurone disease. J. Neurol. Neurosurg. Psychiatry 2005, 76, 1046–1057. [Google Scholar] [CrossRef] [Green Version]
- Andersen, P.M. Amyotrophic lateral sclerosis associated with mutations in the CuZn superoxide dismutase gene. Curr. Neurol. Neurosci. Rep. 2006, 6, 37–46. [Google Scholar] [CrossRef]
- Kumar, A.; Bala, L.; Kalita, J.; Misra, U.K.; Singh, R.L.; Khetrapal, C.L.; Babu, G.N. Metabolomic analysis of serum by (1) H NMR spectroscopy in amyotrophic lateral sclerosis. Clin. Chim. Acta Int. J. Clin. Chem. 2010, 411, 563–567. [Google Scholar] [CrossRef]
- Blasco, H.; Corcia, P.; Moreau, C.; Veau, S.; Fournier, C.; Vourc’h, P.; Emond, P.; Gordon, P.; Pradat, P.-F.; Praline, J.; et al. 1H-NMR-based metabolomic profiling of CSF in early amyotrophic lateral sclerosis. PLoS ONE 2010, 5, e13223. [Google Scholar] [CrossRef]
- Cieslarova, Z.; Lopes, F.S.; do Lago, C.L.; França, M.C.; Colnaghi Simionato, A.V. Capillary electrophoresis tandem mass spectrometry determination of glutamic acid and homocysteine’s metabolites: Potential biomarkers of amyotrophic lateral sclerosis. Talanta 2017, 170, 63–68. [Google Scholar] [CrossRef]
- Patin, F.; Corcia, P.; Vourc’h, P.; Nadal-Desbarats, L.; Baranek, T.; Goossens, J.-F.; Marouillat, S.; Dessein, A.-F.; Descat, A.; Madji Hounoum, B.; et al. Omics to explore amyotrophic lateral sclerosis evolution: The central role of arginine and proline metabolism. Mol. Neurobiol. 2017, 54, 5361–5374. [Google Scholar] [CrossRef]
- Bjornevik, K.; Zhang, Z.; O’Reilly, É.J.; Berry, J.D.; Clish, C.B.; Deik, A.; Jeanfavre, S.; Kato, I.; Kelly, R.S.; Kolonel, L.N.; et al. Prediagnostic plasma metabolomics and the risk of amyotrophic lateral sclerosis. Neurology 2019, 92, e2089–e2100. [Google Scholar] [CrossRef] [Green Version]
- Fisher, R.S.; Acevedo, C.; Arzimanoglou, A.; Bogacz, A.; Cross, J.H.; Elger, C.E.; Engel, J.; Forsgren, L.; French, J.A.; Glynn, M.; et al. ILAE official report: A practical clinical definition of epilepsy. Epilepsia 2014, 55, 475–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pati, S.; Alexopoulos, A.V. Pharmacoresistant epilepsy: From pathogenesis to current and emerging therapies. Cleve. Clin. J. Med. 2010, 77, 457–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sander, J.W.A.S. Some Aspects of Prognosis in the Epilepsies: A Review. Epilepsia 1993, 34, 1007–1016. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.-Y.; Cheng, X.-L.; Vaziri, N.D.; Liu, S.; Lin, R.-C. UPLC-based metabonomic applications for discovering biomarkers of diseases in clinical chemistry. Clin. Biochem. 2014, 47, 16–26. [Google Scholar] [CrossRef] [Green Version]
- Cavus, I.; Kasoff, W.S.; Cassaday, M.P.; Jacob, R.; Gueorguieva, R.; Sherwin, R.S.; Krystal, J.H.; Spencer, D.D.; Abi-Saab, W.M. Extracellular metabolites in the cortex and hippocampus of epileptic patients. Ann. Neurol. 2005, 57, 226–235. [Google Scholar] [CrossRef]
- Al Zweiri, M.; Sills, G.J.; Leach, J.P.; Brodie, M.J.; Robertson, C.; Watson, D.G.; Parkinson, J.A. Response to drug treatment in newly diagnosed epilepsy: A pilot study of (1)H NMR- and MS-based metabonomic analysis. Epilepsy Res. 2010, 88, 189–195. [Google Scholar] [CrossRef]
- Abela, L.; Simmons, L.; Steindl, K.; Schmitt, B.; Mastrangelo, M.; Joset, P.; Papuc, M.; Sticht, H.; Baumer, A.; Crowther, L.M.; et al. N(8)-acetylspermidine as a potential plasma biomarker for snyder-robinson syndrome identified by clinical metabolomics. J. Inherit. Metab. Dis. 2016, 39, 131–137. [Google Scholar] [CrossRef]
- Wang, D.; Wang, X.; Kong, J.; Wu, J.; Lai, M. GC–MS–Based metabolomics discovers a shared serum metabolic characteristic among three types of epileptic seizures. Epilepsy Res. 2016, 126, 83–89. [Google Scholar] [CrossRef]
- Wu, H.C.; Dachet, F.; Ghoddoussi, F.; Bagla, S.; Fuerst, D.; Stanley, J.A.; Galloway, M.P.; Loeb, J.A. Altered metabolomic–genomic signature: A potential noninvasive biomarker of epilepsy. Epilepsia 2017, 58, 1626–1636. [Google Scholar] [CrossRef] [Green Version]
- Murgia, F.; Muroni, A.; Puligheddu, M.; Polizzi, L.; Barberini, L.; Orofino, G.; Solla, P.; Poddighe, S.; Del Carratore, F.; Griffin, J.L.; et al. Metabolomics as a tool for the characterization of drug-resistant epilepsy. Front. Neurol. 2017, 8. [Google Scholar] [CrossRef]
- Walker, D.I.; Perry-Walker, K.; Finnell, R.H.; Pennell, K.D.; Tran, V.; May, R.C.; McElrath, T.F.; Meador, K.J.; Pennell, P.B.; Jones, D.P. Metabolome-wide association study of anti-epileptic drug treatment during pregnancy. Toxicol. Appl. Pharmacol. 2019, 363, 122–130. [Google Scholar] [CrossRef]
- Bhargava, P.; Anthony, D.C. Metabolomics in multiple sclerosis disease course and progression. Mult. Scler. J. 2020. [Google Scholar] [CrossRef] [PubMed]
- Comi, G. Induction vs. escalating therapy in multiple sclerosis: Practical implications. Neurol. Sci. Off. J. Ital. Neurol. Soc. Ital. Soc. Clin. Neurophysiol. 2008, 29 (Suppl. 2), S253–S255. [Google Scholar] [CrossRef]
- International Multiple Sclerosis Genetics Consortium (IMSGC); Beecham, A.H.; Patsopoulos, N.A.; Xifara, D.K.; Davis, M.F.; Kemppinen, A.; Cotsapas, C.; Shah, T.S.; Spencer, C.; Booth, D.; et al. Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat. Genet. 2013, 45, 1353–1360. [Google Scholar] [CrossRef] [PubMed]
- Isobe, N.; Madireddy, L.; Khankhanian, P.; Matsushita, T.; Caillier, S.J.; Moré, J.M.; Gourraud, P.-A.; McCauley, J.L.; Beecham, A.H.; Piccio, L.; et al. An immunoChip study of multiple sclerosis risk in African Americans. Brain 2015, 138, 1518–1530. [Google Scholar] [CrossRef] [PubMed]
- Lynch, J.; Peeling, J.; Auty, A.; Sutherland, G.R. Nuclear magnetic resonance study of cerebrospinal fluid from patients with multiple sclerosis. Can. J. Neurol. Sci. J. Can. Sci. Neurol. 1993, 20, 194–198. [Google Scholar]
- Simone, I.L.; Federico, F.; Trojano, M.; Tortorella, C.; Liguori, M.; Giannini, P.; Picciola, E.; Natile, G.; Livrea, P. High resolution proton MR spectroscopy of cerebrospinal fluid in MS patients. Comparison with biochemical changes in demyelinating plaques. J. Neurol. Sci. 1996, 144, 182–190. [Google Scholar] [CrossRef]
- Reinke, S.N.; Broadhurst, D.L.; Sykes, B.D.; Baker, G.B.; Catz, I.; Warren, K.G.; Power, C. Metabolomic profiling in multiple sclerosis: Insights into biomarkers and pathogenesis. Mult. Scler. Houndmills Basingstoke Engl. 2014, 20, 1396–1400. [Google Scholar] [CrossRef]
- Kim, H.-H.; Jeong, I.H.; Hyun, J.-S.; Kong, B.S.; Kim, H.J.; Park, S.J. Metabolomic profiling of CSF in multiple sclerosis and neuromyelitis optica spectrum disorder by nuclear magnetic resonance. PLoS ONE 2017, 12, e0181758. [Google Scholar] [CrossRef]
- Herman, S.; Åkerfeldt, T.; Spjuth, O.; Burman, J.; Kultima, K. Biochemical differences in cerebrospinal fluid between secondary progressive and relapsing–remitting multiple sclerosis. Cells 2019, 8. [Google Scholar] [CrossRef] [Green Version]
- Mehrpour, M.; Kyani, A.; Tafazzoli, M.; Fathi, F.; Joghataie, M.-T. A metabonomics investigation of multiple sclerosis by nuclear magnetic resonance. Magn. Reson. Chem. MRC 2013, 51, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Moussallieh, F.-M.; Elbayed, K.; Chanson, J.B.; Rudolf, G.; Piotto, M.; De Seze, J.; Namer, I.J. Serum analysis by 1H nuclear magnetic resonance spectroscopy: A new tool for distinguishing neuromyelitis optica from multiple sclerosis. Mult. Scler. Houndmills Basingstoke Engl. 2014, 20, 558–565. [Google Scholar] [CrossRef] [PubMed]
- Villoslada, P.; Alonso, C.; Agirrezabal, I.; Kotelnikova, E.; Zubizarreta, I.; Pulido-Valdeolivas, I.; Saiz, A.; Comabella, M.; Montalban, X.; Villar, L.; et al. Metabolomic signatures associated with disease severity in multiple sclerosis. Neurol. Neuroimmunol. Neuroinflamm. 2017, 4, e321. [Google Scholar] [CrossRef] [Green Version]
- Andersen, S.L.; Briggs, F.B.S.; Winnike, J.H.; Natanzon, Y.; Maichle, S.; Knagge, K.J.; Newby, L.K.; Gregory, S.G. Metabolome-based signature of disease pathology in MS. Mult. Scler. Relat. Disord. 2019, 31, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Cicalini, I.; Rossi, C.; Pieragostino, D.; Agnifili, L.; Mastropasqua, L.; di Ioia, M.; de Luca, G.; Onofrj, M.; Federici, L.; del Boccio, P. Integrated lipidomics and metabolomics analysis of tears in multiple sclerosis: An insight into diagnostic potential of lacrimal fluid. Int. J. Mol. Sci. 2019, 20, 1265. [Google Scholar] [CrossRef] [Green Version]
- Havelund, J.F.; Heegaard, N.H.H.; Færgeman, N.J.K.; Gramsbergen, J.B. Biomarker research in parkinson’s disease using metabolite profiling. Metabolites 2017, 7, 42. [Google Scholar] [CrossRef] [Green Version]
- Beitz, J.M. Parkinson’s disease: A review. Front. Biosci. Sch. Ed. 2014, 6, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Marek, K.; Innis, R.; van Dyck, C.; Fussell, B.; Early, M.; Eberly, S.; Oakes, D.; Seibyl, J. [123I]beta-CIT SPECT imaging assessment of the rate of Parkinson’s disease progression. Neurology 2001, 57, 2089–2094. [Google Scholar] [CrossRef]
- Morrish, P.K.; Sawle, G.V.; Brooks, D.J. Clinical and [18F] dopa PET findings in early Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 1995, 59, 597–600. [Google Scholar] [CrossRef] [Green Version]
- Fearnley, J.M.; Lees, A.J. Ageing and Parkinson’s disease: Substantia nigra regional selectivity. Brain J. Neurol. 1991, 114 Pt 5, 2283–2301. [Google Scholar] [CrossRef]
- Parnetti, L.; Castrioto, A.; Chiasserini, D.; Persichetti, E.; Tambasco, N.; El-Agnaf, O.; Calabresi, P. Cerebrospinal fluid biomarkers in Parkinson disease. Nat. Rev. Neurol. 2013, 9, 131–140. [Google Scholar] [CrossRef]
- Parnetti, L.; Chiasserini, D.; Persichetti, E.; Eusebi, P.; Varghese, S.; Qureshi, M.M.; Dardis, A.; Deganuto, M.; de Carlo, C.; Castrioto, A.; et al. Cerebrospinal fluid lysosomal enzymes and alpha-synuclein in Parkinson’s disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2014, 29, 1019–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Dijk, K.D.; Persichetti, E.; Chiasserini, D.; Eusebi, P.; Beccari, T.; Calabresi, P.; Berendse, H.W.; Parnetti, L.; van de Berg, W.D.J. Changes in endolysosomal enzyme activities in cerebrospinal fluid of patients with Parkinson’s disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2013, 28, 747–754. [Google Scholar] [CrossRef] [PubMed]
- Hong, Z.; Shi, M.; Chung, K.A.; Quinn, J.F.; Peskind, E.R.; Galasko, D.; Jankovic, J.; Zabetian, C.P.; Leverenz, J.B.; Baird, G.; et al. DJ-1 and alpha-synuclein in human cerebrospinal fluid as biomarkers of Parkinson’s disease. Brain J. Neurol. 2010, 133, 713–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, M.; Furay, A.R.; Sossi, V.; Aasly, J.O.; Armaly, J.; Wang, Y.; Wszolek, Z.K.; Uitti, R.J.; Hasegawa, K.; Yokoyama, T.; et al. DJ-1 and αSYN in LRRK2 CSF do not correlate with striatal dopaminergic function. Neurobiol Aging 2012, 33, 836.e5–836.e7. [Google Scholar] [CrossRef] [Green Version]
- Lesage, S.; Brice, A. Parkinson’s disease: From monogenic forms to genetic susceptibility factors. Hum. Mol. Genet. 2009, 18, R48–R59. [Google Scholar] [CrossRef]
- Cassarino, D.S.; Fall, C.P.; Swerdlow, R.H.; Smith, T.S.; Halvorsen, E.M.; Miller, S.W.; Parks, J.P.; Parker, W.D.; Bennett, J.P. Elevated reactive oxygen species and antioxidant enzyme activities in animal and cellular models of Parkinson’s disease. Biochim. Biophys. Acta 1997, 1362, 77–86. [Google Scholar] [CrossRef] [Green Version]
- Schildknecht, S.; Gerding, H.R.; Karreman, C.; Drescher, M.; Lashuel, H.A.; Outeiro, T.F.; Di Monte, D.A.; Leist, M. Oxidative and nitrative alpha-synuclein modifications and proteostatic stress: Implications for disease mechanisms and interventions in synucleinopathies. J. Neurochem. 2013, 125, 491–511. [Google Scholar] [CrossRef] [Green Version]
- Beal, M.F. Metabolic disorders and neurotoxicology. Curr. Opin. Neurol. 1995, 8, 467–468. [Google Scholar] [CrossRef]
- Hertzman, C.; Wiens, M.; Bowering, D.; Snow, B.; Calne, D. Parkinson’s disease: A case-control study of occupational and environmental risk factors. Am. J. Ind. Med. 1990, 17, 349–355. [Google Scholar] [CrossRef]
- Benecke, R.; Strümper, P.; Weiss, H. Electron transfer complexes I and IV of platelets are abnormal in Parkinson’s disease but normal in Parkinson-plus syndromes. Brain J. Neurol. 1993, 116 Pt 6, 1451–1463. [Google Scholar] [CrossRef]
- Haas, R.H.; Nasirian, F.; Nakano, K.; Ward, D.; Pay, M.; Hill, R.; Shults, C.W. Low platelet mitochondrial complex I and complex II/III activity in early untreated Parkinson’s disease. Ann. Neurol. 1995, 37, 714–722. [Google Scholar] [CrossRef] [PubMed]
- Heeman, B.; Van den Haute, C.; Aelvoet, S.-A.; Valsecchi, F.; Rodenburg, R.J.; Reumers, V.; Debyser, Z.; Callewaert, G.; Koopman, W.J.H.; Willems, P.H.G.M.; et al. Depletion of PINK1 affects mitochondrial metabolism, calcium homeostasis and energy maintenance. J. Cell Sci. 2011, 124, 1115–1125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krige, D.; Carroll, M.T.; Cooper, J.M.; Marsden, C.D.; Schapira, A.H. Platelet mitochondrial function in Parkinson’s disease. The royal kings and queens parkinson disease research group. Ann. Neurol. 1992, 32, 782–788. [Google Scholar] [CrossRef] [PubMed]
- Shoffner, J.M.; Watts, R.L.; Juncos, J.L.; Torroni, A.; Wallace, D.C. Mitochondrial oxidative phosphorylation defects in Parkinson’s disease. Ann. Neurol. 1991, 30, 332–339. [Google Scholar] [CrossRef]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.K.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef] [Green Version]
- Truban, D.; Hou, X.; Caulfield, T.R.; Fiesel, F.C.; Springer, W. PINK1, parkin, and mitochondrial quality control: What can we learn about parkinson’s disease pathobiology? J. Park. Dis. 2017, 7, 13–29. [Google Scholar] [CrossRef] [Green Version]
- Hatano, T.; Saiki, S.; Okuzumi, A.; Mohney, R.P.; Hattori, N. Identification of novel biomarkers for Parkinson’s disease by metabolomic technologies. J. Neurol. Neurosurg. Psychiatry 2016, 87, 295–301. [Google Scholar] [CrossRef]
- Chen, J.F.; Xu, K.; Petzer, J.P.; Staal, R.; Xu, Y.H.; Beilstein, M.; Sonsalla, P.K.; Castagnoli, K.; Castagnoli, N.; Schwarzschild, M.A. Neuroprotection by caffeine and A(2A) adenosine receptor inactivation in a model of Parkinson’s disease. J. Neurosci. Off. J. Soc. Neurosci. 2001, 21, RC143. [Google Scholar] [CrossRef] [Green Version]
- Palacios, N.; Gao, X.; McCullough, M.L.; Schwarzschild, M.A.; Shah, R.; Gapstur, S.; Ascherio, A. Caffeine and risk of Parkinson’s disease in a large cohort of men and women. Mov. Disord. Off. J. Mov. Disord. Soc. 2012, 27, 1276–1282. [Google Scholar] [CrossRef] [Green Version]
- De Lau, L.M.L.; Koudstaal, P.J.; Hofman, A.; Breteler, M.M.B. Serum uric acid levels and the risk of Parkinson disease. Ann. Neurol. 2005, 58, 797–800. [Google Scholar] [CrossRef] [PubMed]
- Annanmaki, T.; Muuronen, A.; Murros, K. Low plasma uric acid level in Parkinson’s disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2007, 22, 1133–1137. [Google Scholar] [CrossRef] [PubMed]
- Weisskopf, M.G.; O’Reilly, E.; Chen, H.; Schwarzschild, M.A.; Ascherio, A. Plasma urate and risk of Parkinson’s disease. Am. J. Epidemiol. 2007, 166, 561–567. [Google Scholar] [CrossRef] [PubMed]
- Schipper, H.M.; Song, W. A heme oxygenase-1 transducer model of degenerative and developmental brain disorders. Int. J. Mol. Sci. 2015, 16, 5400–5419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LeWitt, P.A.; Li, J.; Lu, M.; Guo, L.; Auinger, P. Parkinson study group–datatop investigators metabolomic biomarkers as strong correlates of parkinson disease progression. Neurology 2017, 88, 862–869. [Google Scholar] [CrossRef] [Green Version]
- Farmer, K.; Smith, C.A.; Hayley, S.; Smith, J. Major alterations of phosphatidylcholine and lysophosphotidylcholine lipids in the substantia nigra using an early stage model of parkinson’s disease. Int. J. Mol. Sci. 2015, 16, 18865–18877. [Google Scholar] [CrossRef] [Green Version]
- Burté, F.; Houghton, D.; Lowes, H.; Pyle, A.; Nesbitt, S.; Yarnall, A.; Yu-Wai-Man, P.; Burn, D.J.; Santibanez-Koref, M.; Hudson, G. Metabolic profiling of Parkinson’s disease and mild cognitive impairment. Mov. Disord. Off. J. Mov. Disord. Soc. 2017, 32, 927–932. [Google Scholar] [CrossRef]
- Luan, H.; Liu, L.-F.; Meng, N.; Tang, Z.; Chua, K.-K.; Chen, L.-L.; Song, J.-X.; Mok, V.C.T.; Xie, L.-X.; Li, M.; et al. LC-MS-based urinary metabolite signatures in idiopathic Parkinson’s disease. J. Proteome Res. 2015, 14, 467–478. [Google Scholar] [CrossRef]
- Luan, H.; Liu, L.-F.; Tang, Z.; Zhang, M.; Chua, K.-K.; Song, J.-X.; Mok, V.C.T.; Li, M.; Cai, Z. Comprehensive urinary metabolomic profiling and identification of potential noninvasive marker for idiopathic Parkinson’s disease. Sci. Rep. 2015, 5, 13888. [Google Scholar] [CrossRef] [Green Version]
- Lu, Z.; Wang, J.; Li, M.; Liu, Q.; Wei, D.; Yang, M.; Kong, L. (1)H NMR-based metabolomics study on a goldfish model of Parkinson’s disease induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Chem. Biol. Interact. 2014, 223, 18–26. [Google Scholar] [CrossRef]
- Valerio, A.; D’Antona, G.; Nisoli, E. Branched-chain amino acids, mitochondrial biogenesis, and healthspan: An evolutionary perspective. Aging 2011, 3, 464–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.; Xie, G.; Jia, W.; Jia, W. Insulin resistance and the metabolism of branched-chain amino acids. Front. Med. 2013, 7, 53–59. [Google Scholar] [CrossRef]
- Bogdanov, M.; Matson, W.R.; Wang, L.; Matson, T.; Saunders-Pullman, R.; Bressman, S.S.; Flint Beal, M. Metabolomic profiling to develop blood biomarkers for Parkinson’s disease. Brain J. Neurol. 2008, 131, 389–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernstrom, J.D. Large neutral amino acids: Dietary effects on brain neurochemistry and function. Amino Acids 2013, 45, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Pogson, C.I.; Knowles, R.G.; Salter, M. The control of aromatic amino acid catabolism and its relationship to neurotransmitter amine synthesis. Crit. Rev. Neurobiol. 1989, 5, 29–64. [Google Scholar]
- Eisenhofer, G.; Brown, S.; Peitzsch, M.; Pelzel, D.; Lattke, P.; Glöckner, S.; Stell, A.; Prejbisz, A.; Fassnacht, M.; Beuschlein, F.; et al. Levodopa therapy in Parkinson’s disease: Influence on liquid chromatographic tandem mass spectrometric-based measurements of plasma and urinary normetanephrine, metanephrine and methoxytyramine. Ann. Clin. Biochem. 2014, 51, 38–46. [Google Scholar] [CrossRef] [Green Version]
- Lei, S.; Zavala-Flores, L.; Garcia-Garcia, A.; Nandakumar, R.; Huang, Y.; Madayiputhiya, N.; Stanton, R.C.; Dodds, E.D.; Powers, R.; Franco, R. Alterations in energy/redox metabolism induced by mitochondrial and environmental toxins: A specific role for glucose-6-phosphate-dehydrogenase and the pentose phosphate pathway in paraquat toxicity. ACS Chem. Biol. 2014, 9, 2032–2048. [Google Scholar] [CrossRef]
- Shukla, A.K.; Ratnasekhar, C.; Pragya, P.; Chaouhan, H.S.; Patel, D.K.; Chowdhuri, D.K.; Mudiam, M.K.R. Metabolomic analysis provides insights on paraquat-induced parkinson-like symptoms in drosophila melanogaster. Mol. Neurobiol. 2016, 53, 254–269. [Google Scholar] [CrossRef]
- Ling, S.-C.; Polymenidou, M.; Cleveland, D.W. Converging mechanisms in ALS and FTD: Disrupted RNA and protein homeostasis. Neuron 2013, 79, 416–438. [Google Scholar] [CrossRef] [Green Version]
- Feigin, V.L.; Forouzanfar, M.H.; Krishnamurthi, R.; Mensah, G.A.; Connor, M.; Bennett, D.A.; Moran, A.E.; Sacco, R.L.; Anderson, L.; Truelsen, T.; et al. Global and regional burden of stroke during 1990–2010: Findings from the global burden of disease study 2010. Lancet 2014, 383, 245–255. [Google Scholar] [CrossRef]
- Caplan, L.R. Chapter 2–Basic Pathology, Anatomy, and Pathophysiology of Stroke. In Caplan’s Stroke, 4th ed.; Caplan, L.R., Ed.; W.B. Saunders: Philadelphia, PA, USA, 2009; pp. 22–63. ISBN 978-1-4160-4721-6. [Google Scholar]
- Markus, H.S. Stroke genetics: Prospects for personalized medicine. BMC Med. 2012, 10, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobkin, B.H.; Carmichael, S.T. The specific requirements of neural repair trials for stroke. Neurorehabil. Neural Repair 2016, 30, 470–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunz, A.; Dirnagl, U.; Mergenthaler, P. Acute pathophysiological processes after ischaemic and traumatic brain injury. Best Pract. Res. Clin. Anaesthesiol. 2010, 24, 495–509. [Google Scholar] [CrossRef] [PubMed]
- Boehme, A.K.; Esenwa, C.; Elkind, M.S.V. Stroke risk factors, genetics, and prevention. Circ. Res. 2017, 120, 472–495. [Google Scholar] [CrossRef] [PubMed]
- Mamatha, S.N.; Nagaraja, D.; Philip, M.; Christopher, R. Asymmetric dimethylarginine as a risk marker for early-onset ischemic stroke in indian population. Clin. Chim. Acta 2011, 412, 139–142. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.-H.; Cheng, M.-L.; Shiao, M.-S.; Lin, C.-N. Metabolomics study in severe extracranial carotid artery stenosis. BMC Neurol. 2019, 19. [Google Scholar] [CrossRef]
- Sidorov, E.; Sanghera, D.K.; Vanamala, J.K.P. Biomarker for ischemic stroke using metabolome: A clinician perspective. J. Stroke 2019, 21, 31–41. [Google Scholar] [CrossRef]
- Pilz, S.; Tomaschitz, A.; Meinitzer, A.; Drechsler, C.; Ritz, E.; Krane, V.; Wanner, C.; Bernhard, O.B.; März, W. Low serum homoarginine is a novel risk factor for fatal strokes in patients undergoing coronary angiography. Stroke 2011, 42, 1132–1134. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Khan, A.; Hong, S.; Jee, S.H.; Park, Y.H. A metabolomic study on high-risk stroke patients determines low levels of serum lysine metabolites: A retrospective cohort study. Mol. Biosyst. 2017, 13, 1109–1120. [Google Scholar] [CrossRef] [Green Version]
- Laborde, C.M.; Mourino–Alvarez, L.; Akerstrom, F.; Padial, L.R.; Vivanco, F.; Gil-Dones, F.; Barderas, M.G. Potential blood biomarkers for stroke. Expert Rev. Proteom. 2012, 9, 437–449. [Google Scholar] [CrossRef]
- Geng, L.-Y.; Qian, F.-Y.; Qian, J.-F.; Zhang, Z.-J. The combination of plasma glutamate and physical impairment after acute stroke as a potential indicator for the early-onset post-stroke depression. J. Psychosom. Res. 2017, 96, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Kimberly, W.T.; Wang, Y.; Pham, L.; Furie, K.L.; Gerszten, R.E. Metabolite profiling identifies a branched chain amino acid signature in acute cardioembolic stroke. Stroke J. Cereb. Circ. 2013, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, H.P.; Bendixen, B.H.; Kappelle, L.J.; Biller, J.; Love, B.B.; Gordon, D.L.; Marsh, E.E. Classification of subtype of acute ischemic stroke. Definitions for use in a multicenter clinical trial. TOAST. Trial of org 10172 in acute stroke treatment. Stroke 1993, 24, 35–41. [Google Scholar] [CrossRef] [Green Version]
- Jung, J.Y.; Lee, H.S.; Kang, D.G.; Kim, N.S.; Cha, M.H.; Bang, O.S.; Ryu, D.H.; Hwang, G.S. 1H-NMR-based metabolomics study of cerebral infarction. Stroke 2011, 42, 1282–1288. [Google Scholar] [CrossRef] [Green Version]
- Jové, M.; Mauri-Capdevila, G.; Suárez, I.; Cambray, S.; Sanahuja, J.; Quílez, A.; Farré, J.; Benabdelhak, I.; Pamplona, R.; Portero-Otín, M.; et al. Metabolomics predicts stroke recurrence after transient ischemic attack. Neurology 2015, 84, 36–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghandforoush-Sattari, M.; Mashayekhi, S.O.; Nemati, M.; Ayromlou, H. Changes in plasma concentration of taurine in stroke. Neurosci. Lett. 2011, 496, 172–175. [Google Scholar] [CrossRef]
- Burrell, C.; Avalon, N.E.; Siegel, J.; Pizzi, M.; Dutta, T.; Charlesworth, M.C.; Freeman, W.D. Precision medicine of aneurysmal subarachnoid hemorrhage–vasospasm and delayed cerebral ischemia. Expert Rev. Neurother. 2016, 16, 1251–1262. [Google Scholar] [CrossRef]
- Sarrafzadeh, A.; Haux, D.; Küchler, I.; Lanksch, W.R.; Unterberg, A.W. Poor-grade aneurysmal subarachnoid hemorrhage: Relationship of cerebral metabolism to outcome. J. Neurosurg. 2004, 100, 400–406. [Google Scholar] [CrossRef]
- Lu, A.Y.; Damisah, E.C.; Winkler, E.A.; Grant, R.A.; Eid, T.; Bulsara, K.R. Cerebrospinal fluid untargeted metabolomic profiling of aneurysmal subarachnoid hemorrhage: An exploratory study. Br. J. Neurosurg. 2018, 32, 637–641. [Google Scholar] [CrossRef]
- Trotti, D.; Aoki, M.; Pasinelli, P.; Berger, U.V.; Danbolt, N.C.; Brown, R.H.; Hediger, M.A. Amyotrophic lateral sclerosis-linked glutamate transporter mutant has impaired glutamate clearance capacity. J. Biol. Chem. 2001, 276, 576–582. [Google Scholar] [CrossRef] [Green Version]
- Bryant, A.S.; Li, B.; Beenhakker, M.P.; Huguenard, J.R. Maintenance of thalamic epileptiform activity depends on the astrocytic glutamate-glutamine cycle. J. Neurophysiol. 2009, 102, 2880–2888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arundine, M.; Tymianski, M. Molecular mechanisms of glutamate-dependent neurodegeneration in ischemia and traumatic brain injury. Cell. Mol. Life Sci. CMLS 2004, 61, 657–668. [Google Scholar] [CrossRef] [PubMed]
- Kooi, E.-J.; Prins, M.; Bajic, N.; Beliën, J.A.M.; Gerritsen, W.H.; van Horssen, J.; Aronica, E.; van Dam, A.-M.; Hoozemans, J.J.M.; Francis, P.T.; et al. Cholinergic imbalance in the multiple sclerosis hippocampus. Acta Neuropathol. (Berl.) 2011, 122, 313–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartus, R.T. On neurodegenerative diseases, models, and treatment strategies: Lessons learned and lessons forgotten a generation following the cholinergic hypothesis. Exp. Neurol. 2000, 163, 495–529. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Pedersen, O. Gut microbiota in human metabolic health and disease. Nat. Rev. Microbiol. 2020. [Google Scholar] [CrossRef]
- Gupta, A.; Osadchiy, V.; Mayer, E.A. Brain-gut-microbiome interactions in obesity and food addiction. Nat. Rev. Gastroenterol. Hepatol. 2020. [Google Scholar] [CrossRef]
- Hirschberg, S.; Gisevius, B.; Duscha, A.; Haghikia, A. Implications of diet and the gut microbiome in neuroinflammatory and neurodegenerative diseases. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef] [Green Version]
- Xu, G.; Li, J. Recent advances in mass spectrometry imaging for multiomics application in neurology. J. Comp. Neurol. 2019, 527, 2158–2169. [Google Scholar] [CrossRef]
- Sancesario, G.M.; Bernardini, S. Alzheimer’s disease in the omics era. Clin. Biochem. 2018, 59, 9–16. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Donatti, A.; Canto, A.M.; Godoi, A.B.; da Rosa, D.C.; Lopes-Cendes, I. Circulating Metabolites as Potential Biomarkers for Neurological Disorders—Metabolites in Neurological Disorders. Metabolites 2020, 10, 389. https://doi.org/10.3390/metabo10100389
Donatti A, Canto AM, Godoi AB, da Rosa DC, Lopes-Cendes I. Circulating Metabolites as Potential Biomarkers for Neurological Disorders—Metabolites in Neurological Disorders. Metabolites. 2020; 10(10):389. https://doi.org/10.3390/metabo10100389
Chicago/Turabian StyleDonatti, Amanda, Amanda M. Canto, Alexandre B. Godoi, Douglas C. da Rosa, and Iscia Lopes-Cendes. 2020. "Circulating Metabolites as Potential Biomarkers for Neurological Disorders—Metabolites in Neurological Disorders" Metabolites 10, no. 10: 389. https://doi.org/10.3390/metabo10100389