Development and Validation of an LC-MS/MS Assay to Quantitate 2′,4′,6′-Trihydroxyacetophenone in Rat and Dog Plasma and its Application to a Pharmacokinetic Study

 , , and
, , and
Abstract
:1. Introduction
2. Results and Discussion
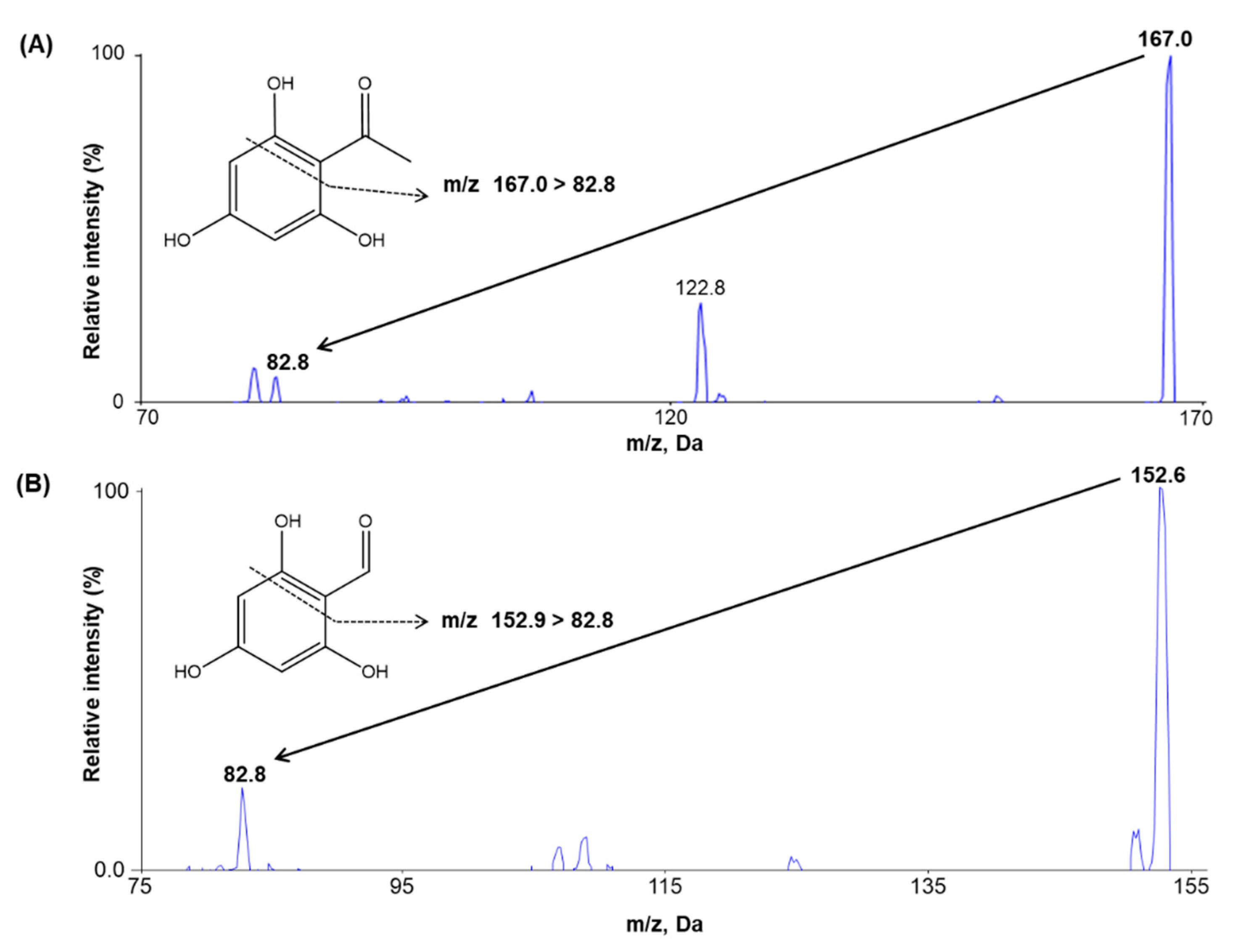
2.1. Method Development
2.2. Method Validation
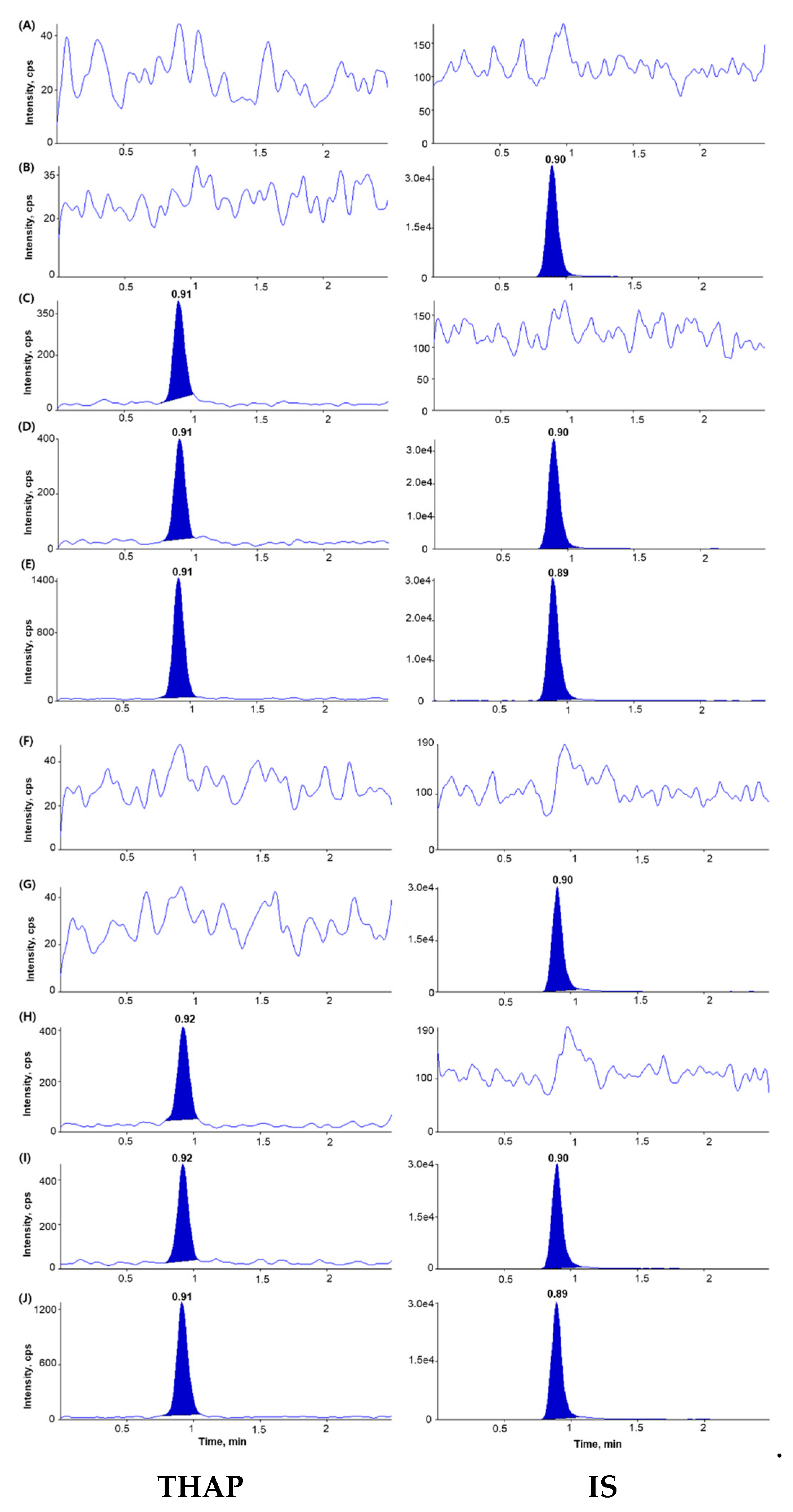
2.2.1. Selectivity and Sensitivity
2.2.2. Linearity and Carry-over
2.2.3. Precision and Accuracy
2.2.4. Extraction Recovery and Matrix Effects
2.2.5. Stability
2.2.6. Validation of Dilution
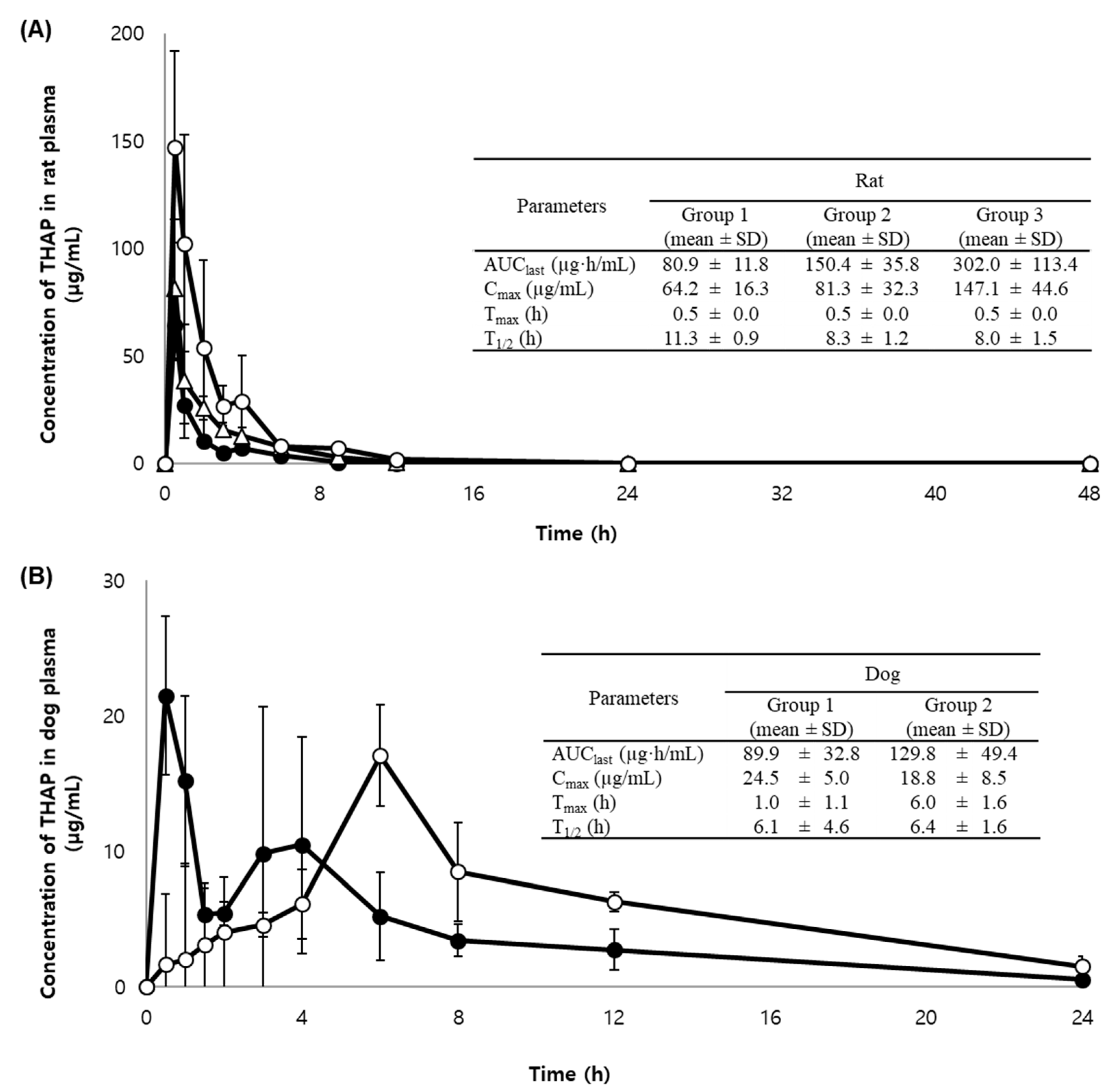
2.3. Application to a Pharmacokinetic Study in Rats and Dogs
2.4. Incurred Sample Reanalysis (ISR)
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Animals
3.3. Instruments and Analytical Conditions
3.4. Preparation of Calibration Standards and QC Samples
3.5. Sample Preparation
3.6. Method Validation in Rat and Dog Plasma
3.6.1. Selectivity and LLOQ
3.6.2. Linearity and carry-over
3.6.3. Precision and Accuracy
3.6.4. Extraction Recovery and Matrix Effects
3.6.5. Stability
3.6.6. Dilution Integrity in Rat Plasma
3.7. Application to a Pharmacokinetic Study
3.8. ISR
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Yoshikawa, M.; Shimada, H.; Nishida, N.; Li, Y.; Toguchida, I.; Yamahara, J.; Matsuda, H. Antidiabetic principles of natural medicines. II. Aldose reductase and alpha-glucosidase inhibitors from Brazilian natural medicine, the leaves of Myrcia multiflora DC. (Myrtaceae): Structures of myrciacitrins I and II and myrciaphenones A and B. Chem. Pharm. Bull. 1998, 46, 113–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piyachaturawat, P.; Tubtim, C.; Chuncharunee, A.; Komaratat, P.; Suksamrarn, A. Evaluation of the acute and subacute toxicity of a choleretic phloracetophenone in experimental animals. Toxicol. Lett. 2002, 129, 123–132. [Google Scholar] [CrossRef]
- Piyachaturawat, P.; Srivoraphan, P.; Chuncharunee, A.; Komaratat, P.; Suksamrarn, A. Cholesterol lowering effects of a choleretic phloracetophenone in hypercholesterolemic hamsters. Eur. J. Pharmacol. 2002, 439, 141–147. [Google Scholar] [CrossRef]
- Ferreira, E.A.; Gris, E.F.; Rebello, J.M.; Correia, J.F.; de Oliveira, L.F.; Filho, D.W.; Pedrosa, R.C. The 2′,4′,6′-trihydroxyacetophenone isolated from Myrcia multiflora has antiobesity and mixed hypolipidemic effects with the reduction of lipid intestinal absorption. Planta Med. 2011, 77, 1569–1574. [Google Scholar] [CrossRef] [PubMed]
- Charoenteeraboon, J.; Nithipatikom, K.; Campbell, W.B.; Piyachaturawat, P.; Wilairat, P.; Rongnoparut, P. Induction of human cholesterol 7alpha-hydroxylase in HepG2 cells by 2,4,6-trihydroxyacetophenone. Eur. J. Pharmacol. 2005, 515, 43–46. [Google Scholar] [CrossRef] [PubMed]
- Piyachaturawat, P.; Chai-ngam, N.; Chuncharunee, A.; Komaratat, P.; Suksamrarn, A. Choleretic activity of phloracetophenone in rats: Structure-function studies using acetophenone analogues. Eur. J. Pharmacol. 2000, 387, 221–227. [Google Scholar] [CrossRef]
- Tradtrantip, L.; Piyachaturawat, P.; Soroka, C.J.; Harry, K.; Mennone, A.; Mahagita, C.; Ballatori, N.; Boyer, J.L. Phloracetophenone-induced choleresis in rats is mediated through Mrp2. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 293, G66–G74. [Google Scholar] [CrossRef] [PubMed]
- Suksamrarn, A.; Ponglikitmongkol, M.; Wongkrajang, K.; Chindaduang, A.; Kittidanairak, S.; Jankam, A.; Yingyongnarongkul, B.E.; Kittipanumat, N.; Chokchaisiri, R.; Khetkam, P.; et al. Diarylheptanoids, new phytoestrogens from the rhizomes of Curcuma comosa: Isolation, chemical modification and estrogenic activity evaluation. Bioorganic Med. Chem. 2008, 16, 6891–6902. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.Y.; Park, S.Y.; Park, S.; Lee, Y.R.; Han, G.D.; Kim, J.A. Geranyl derivative of phloroacetophenone induces cancer cell-specific apoptosis through Bax-mediated mitochondrial pathway in MCF-7 human breast cancer cells. Biol. Pharm. Bull. 2012, 35, 98–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, K.W.; Oh, K.S.; Kim, Y.C.; Jeong, S.B. Pharmaceutical Composition Comprising 2,4,6-Trihydroxyacetophenone as an Active Ingredient for Treating Breast Cancer or Hormone-Refractory Breast Cancer. KR Patent KR101819509B1, 17 January 2018. [Google Scholar]
- Strebhardt, K.; Ullrich, A. Targeting polo-like kinase 1 for cancer therapy. Nat. Rev. Cancer 2006, 6, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Gutteridge, R.E.; Ndiaye, M.A.; Liu, X.; Ahmad, N. Plk1 Inhibitors in Cancer Therapy: From Laboratory to Clinics. Mol. Cancer Ther. 2016, 15, 1427–1435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basabe, P.; de Roman, M.; Marcos, I.S.; Diez, D.; Blanco, A.; Bodero, O.; Mollinedo, F.; Sierra, B.G.; Urones, J.G. Prenylflavonoids and prenyl/alkyl-phloroacetophenones: Synthesis and antitumour biological evaluation. Eur. J. Med. Chem. 2010, 45, 4258–4269. [Google Scholar] [CrossRef] [PubMed]
- Food and Drug Administration. Guidance for Industry: M3(R2) Nonclinical Safety Studies for the Conduct of Human Clinical Trials and Marketing Authorization for Pharmaceuticals. Available online: https://www.fda.gov/media/71542/download (accessed on 4 August 2020).
- Matuszewski, B.K.; Constanzer, M.L.; Chavez-Eng, C.M. Matrix effect in quantitative LC/MS/MS analyses of biological fluids: A method for determination of finasteride in human plasma at picogram per milliliter concentrations. Anal. Chem. 1998, 70, 882–889. [Google Scholar] [CrossRef] [PubMed]
- Fu, I.; Woolf, E.J.; Matuszewski, B.K. Effect of the sample matrix on the determination of indinavir in human urine by HPLC with turbo ion spray tandem mass spectrometric detection. J. Pharm. Biomed. Anal. 1998, 18, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Mazzeti, A.L.; Oliveira, L.T.; Goncalves, K.R.; Schaun, G.C.; Mosqueira, V.C.F.; Bahia, M.T. Benznidazole self-emulsifying delivery system: A novel alternative dosage form for Chagas disease treatment. Eur. J. Pharm. Sci. J. Eur. Fed. Pharm. Sci. 2020, 145, 105234. [Google Scholar] [CrossRef] [PubMed]
- Sawatdee, S.; Atipairin, A.; Sae Yoon, A.; Srichana, T.; Changsan, N.; Suwandecha, T. Formulation Development of Albendazole-Loaded Self-Microemulsifying Chewable Tablets to Enhance Dissolution and Bioavailability. Pharmaceutics 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Food and Drug Administration. Bioanalytical Method Validation Guidance for Industry. Available online: https://www.fda.gov/media/70858/download (accessed on 4 August 2020).
- Ministry of Food and Drug Safety. Guideline on Bioanalytical Method Validation. Available online: https://www.mfds.go.kr/brd/m_210/down.do?brd_id=data0010&seq=13054&data_tp=A&file_seq=1 (accessed on 4 August 2020).
- De Boer, T.; Wieling, J. Incurred sample accuracy assessment: Design of experiments based on standard addition. Bioanalysis 2011, 3, 983–992. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |




{kind=link}
{kind=link}
{kind=link}
| Theoretical Concentration (µg/mL) | Predicted Concentration (µg/mL) (Mean ± SD) | Precision (CV %) a | Accuracy (%) b | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Intra-Day | Inter-Day | Intra-Day | Inter-Day | Intra-Day | Inter-Day | ||||||
| Rat | 0.1 | 0.10 | ± | 0.01 | 0.09 | ± | 0.01 | 10.18 | 9.86 | 103.80 | 98.80 |
| 0.3 | 0.28 | ± | 0.01 | 0.30 | ± | 0.02 | 1.84 | 5.50 | 94.33 | 99.40 | |
| 25 | 25.06 | ± | 0.22 | 25.76 | ± | 0.62 | 0.86 | 2.39 | 100.24 | 103.02 | |
| 100 | 99.02 | ± | 1.83 | 98.49 | ± | 1.32 | 1.85 | 1.34 | 99.02 | 98.49 | |
| Dog | 0.1 | 0.10 | ± | 0.01 | 0.11 | ± | 0.01 | 3.86 | 9.73 | 97.00 | 105.00 |
| 0.3 | 0.31 | ± | 0.01 | 0.31 | ± | 0.01 | 4.00 | 4.18 | 102.60 | 103.76 | |
| 25 | 24.88 | ± | 0.19 | 25.74 | ± | 0.92 | 0.77 | 3.56 | 99.52 | 102.97 | |
| 100 | 94.46 | ± | 0.81 | 94.47 | ± | 1.07 | 0.86 | 1.14 | 94.46 | 94.07 | |
| Nominal Concentration (µg/mL) | Extraction Recovery a (%) | Matrix Effect b (%) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Mean ± SD | CV | Mean ± SD | CV | ||||||
| Rat | |||||||||
| THAP | 0.3 | 90.00 | ± | 5.08 | 5.64 | 109.29 | ± | 5.46 | 5.00 |
| 25 | 97.16 | ± | 1.69 | 1.74 | 102.34 | ± | 2.45 | 2.40 | |
| 80 | 97.47 | ± | 1.90 | 1.95 | 102.34 | ± | 1.82 | 1.78 | |
| IS | 2 | 82.39 | ± | 2.51 | 3.05 | 86.81 | ± | 2.86 | 3.29 |
| Dog | |||||||||
| THAP | 0.3 | 94.40 | ± | 2.54 | 2.69 | 113.59 | ± | 2.78 | 2.44 |
| 25 | 103.81 | ± | 2.43 | 2.34 | 103.64 | ± | 3.63 | 3.50 | |
| 80 | 102.51 | ± | 2.38 | 2.32 | 103.76 | ± | 2.57 | 2.48 | |
| IS | 2 | 70.75 | ± | 3.83 | 5.42 | 90.86 | ± | 3.62 | 3.98 |
| Nominal Concentration (µg/mL) | Plasma Stability (Mean %) | ||||||
|---|---|---|---|---|---|---|---|
| Species | Room Temperature (7 h) | 4 °C (7 h) | −70 °C (7 h) | Freeze-Thaw Stability (5 Cycles) | Autosampler (44 h, 10 °C) | Long-Term a (−70 °C) | |
| Rat | |||||||
| 0.3 | 98.22 | 99.22 | 98.11 | 101.00 | 105.00 | 111.67 | |
| 25 | 101.62 | 106.91 | 109.87 | 102.84 | 104.80 | 104.15 | |
| 80 | 100.47 | 100.98 | 101.61 | 99.62 | 100.58 | 99.39 | |
| Dog | |||||||
| 0.3 | 109.89 | 111.89 | 109.56 | 101.33 | 99.67 | 102.89 | |
| 25 | 106.66 | 105.57 | 105.88 | 105.31 | 106.75 | 103.95 | |
| 80 | 98.17 | 98.87 | 97.32 | 98.41 | 97.58 | 99.45 | |
| Selected Sample Numbers | Accepted Sample Numbers | Results (%) | |
|---|---|---|---|
| Rat | 17 | 16 | 94.12 |
| Dog | 17 | 17 | 100.00 |
| Composition | Function * | IR Tablet (mg) | SR Tablet (mg) |
|---|---|---|---|
| THAP | API | 500 | 500 |
| Hydroxypropyl cellulose (HPC) | Binder | 20 | 20 |
| Kollidon SR (Soluplus®) | SR material | - | 250 |
| Microcrystalline cellulose | Diluent | 200 | - |
| Sodium starch glycolate | Disintegrant | 23 | 23 |
| Colloidal silicon dioxide | Glidant | 15 | 15 |
| Sodium stearyl fumarate | Lubricant | 8 | 8 |
| Total Weight | 766 | 816 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yoo, H.J.; Hwang, S.-J.; Lee, J.-H.; Shim, W.-S.; Choi, Y.-W.; Cho, S.M.; Chung, E.K.; Park, J.-B.; Lee, K.-T. Development and Validation of an LC-MS/MS Assay to Quantitate 2′,4′,6′-Trihydroxyacetophenone in Rat and Dog Plasma and its Application to a Pharmacokinetic Study. Molecules 2020, 25, 4373. https://doi.org/10.3390/molecules25194373
Yoo HJ, Hwang S-J, Lee J-H, Shim W-S, Choi Y-W, Cho SM, Chung EK, Park J-B, Lee K-T. Development and Validation of an LC-MS/MS Assay to Quantitate 2′,4′,6′-Trihydroxyacetophenone in Rat and Dog Plasma and its Application to a Pharmacokinetic Study. Molecules. 2020; 25(19):4373. https://doi.org/10.3390/molecules25194373
Chicago/Turabian StyleYoo, Hee Jo, Se-Jung Hwang, Jeong-Hun Lee, Wang-Seob Shim, Yun-Woong Choi, Sang Min Cho, Eun Kyoung Chung, Jun-Bom Park, and Kyung-Tae Lee. 2020. "Development and Validation of an LC-MS/MS Assay to Quantitate 2′,4′,6′-Trihydroxyacetophenone in Rat and Dog Plasma and its Application to a Pharmacokinetic Study" Molecules 25, no. 19: 4373. https://doi.org/10.3390/molecules25194373