
Abstract
Active accumulation of the data on new amyloids continuing nowadays dissolves boundaries of the term “amyloid”. Currently, it is most often used to designate aggregates with cross-β structure. At the same time, amyloids also exhibit a number of other unusual properties, such as: detergent and protease resistance, interaction with specific dyes, and ability to induce transition of some proteins from a soluble form to an aggregated one. The same features have been also demonstrated for the aggregates lacking cross-β structure, which are commonly called “amyloid-like” and combined into one group, although they are very diverse. We have collected and systematized information on the properties of more than two hundred known amyloids and amyloid-like proteins with emphasis on conflicting examples. In particular, a number of proteins in membraneless organelles form aggregates with cross-β structure that are morphologically indistinguishable from the other amyloids, but they can be dissolved in the presence of detergents, which is not typical for amyloids. Such paradoxes signify the need to clarify the existing definition of the term amyloid. On the other hand, the demonstrated structural diversity of the amyloid-like aggregates shows the necessity of their classification.
Similar content being viewed by others


INTRODUCTION. BRIEF OVERVIEW OF AMYLOID RESEARCH METHODS DEVELOPMENT
The term “amyloid” was introduced to scientific literature by the German botanist Matthias Schleiden. One of his ideas was the use the iodine-starch test to study chemical and anatomical composition of a plant cell. Originally described in 1814 by Jean-Jacques Colin and Henri-François Gaultier de Claubry, this test is based on detection of a blue stain produced in the course of reaction of starch with iodine in the presence of sulfuric acid. When applying this technique to analyze plant preparations, Schleiden was the first to use the term “amyloid” (from the Latin word “amylum,” starch) in the meaning of “starch-like” to refer to “a normal amylaceous component in plants” [1].
In medical literature, the term “amyloid” was first used by German pathologist Rudolf Virchow. He used this term in 1854 to describe pathological deposits in the nervous system that showed color reactions with iodine and sulfuric acid. For this reason, Virchow believed these structures to be identical to starch [1]. Later Virchow used the test with iodine and sulfuric acid on other tissues with amyloid deposits [2]. In later amyloid studies, histological dyes such as Congo red (1922) and thioflavin (1959) were used instead of iodine [3].
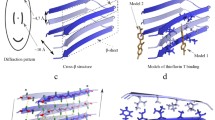
Structural studies of amyloids started in the 1930s using X-ray diffraction. In 1935, William Thomas Astbury and Sylvia Dickinson described a typical X-ray diffraction pattern, later referred to as “cross-β” structure. In 1959, using an electron microscope, Alan Cohen and Evan Calkins observed amyloid deposits with a fibrillar structure (~7.5-14 nm in width and ~100-1600 nm in length) in rabbit and human tissues [3]. Later it was demonstrated that β-strands in the aggregates are oriented perpendicular to the fibril axis and form intermolecular β-sheets. The gap between β-strands in a single β-sheet is 4.7-4.8 Å, and protofibrils have at least two β-sheets, with a distance of about 10 Å between them. Experiments on electron or X-ray diffraction show that this regular structure forms a typical pattern with two meridional and two equatorial reflections [4].
Other unusual properties of amyloid aggregates are described later. For example, some of them are less sensitive to the action of proteases than the same protein in the native form [5], as well as they are very stable and do not dissolve in the presence of detergents. Methods based on various modifications of electrophoresis were proposed to study yeast amyloids. Nowadays, these methods are successfully applied for demonstrating resistance of amyloids of diverse origins to detergents or proteases [6].
Most amyloids are capable of inducing aggregation of the soluble protein molecules they are made of. Dynamics of this process is commonly analyzed in vitro by using purified proteins and amyloid-specific dyes [7]. This characteristic is essential (but not sufficient) for the existence of infectious amyloids or prions, with PrP being the first reported example [8]. The discovery of yeast prions, which can be detected in the cell by their growth on selective media [9], allowed developing versatile systems to search for proteins with similar properties in vivo [10, 11] and identify new amyloids [12]. The latter task can also be accomplished by the C-DAG (curli-dependent amyloid generator) technique; it is used to evaluate formation of the amyloid aggregates by the protein investigated due to the fibrils appearing on the surface of bacterial cells [13].
There are some examples of aggregates demonstrating some of the properties described above: fibrillar morphology, interaction with thioflavin T or its analogs, ability to induce aggregation, resistance to detergents and proteases. Such aggregates will be further referred to as amyloid-like, while the proteins, for which at least in vitro Congo red staining or presence of cross-β structure is shown – as amyloids [4, 14-17].
AMYLOID DIVERSITY
To illustrate the variety of proteins that can form amyloid or amyloid-like aggregates, we have collected the data on the properties of over two hundred proteins involved. We have sought to include as many controversial examples as possible to demonstrate diversity of the phenomenon and how well it has been studied. For complete information on the proteins being considered, the reader is referred to Table S in the Supplement. It provides the data on fragments, isoforms, and substitutions in the proteins under analysis, but in our data analysis information on the individual proteins was considered without taking into account production levels in cells and other differences in model systems. This list can probably be extended, but we consider this selection representative enough to assess whether the amyloid properties of different proteins are fully described from the methodological point of view. We have considered amyloid properties to include the ability of a protein or its fragment to form aggregates in vitro or in vivo, the presence of a fibrillar morphology of these aggregates, the ability to induce aggregation of similar proteins, the increased resistance of aggregates to detergents and proteases, their interaction with amyloid-specific dyes (thioflavin and Congo red), as well as the presence of a cross-β structure. These properties have been described for many amyloids to varying degrees [17-19]. Since it is believed that under correctly chosen conditions, it is possible to obtain an amyloid form of absolutely any protein [20], we excluded from our consideration the proteins aggregates of which have been studied exclusively in vitro, meaning that none of the listed amyloid properties was observed in a living organism. Also, we did not analyze the proteins detected by various screenings techniques but without targeted examination.
Numerous examples of the amyloid-like aggregates are available. However, their investigations are extremely heterogeneous in terms of methodological approaches. To illustrate this, we have collected information on eight properties typical for protein aggregates considered in this section (Fig. 1):
– presence of a cross-β structure (a characteristic pattern of electron or X-rays diffraction with two reflections, or determination of the aggregate structure using cryo-electron microscopy or nuclear magnetic resonance methods);
– staining with Congo red (including birefringence in polarized light);
– staining with thioflavin T or analogs;
– fibrillar morphology of aggregates;
– ability to induce aggregation of the similar protein;
– protease resistance;
– detergent resistance;
– aggregate formation (as shown by any method, except those used to demonstrate previous properties).

Methodological approaches and their combinations in studying amyloid and amyloid-like aggregates (a) or just amyloids (b). Only the experiments that successfully demonstrated these properties were taken into account. Cross-β, demonstration of the cross-β structure of aggregates (based on electron diffraction or X-ray diffraction with two reflections, or an aggregate structure deciphered by cryo-electron microscopy or nuclear magnetic resonance methods); CR+, Congo red staining and birefringence in polarized light; ThT+– staining with thioflavin T or analogs; Fibr., fibrillar morphology of aggregates, Seed., ability to induce aggregation; Protr, protease resistance; Detr, detergent resistance; Aggr., aggregate formation (as shown by any method, except those used to demonstrate previous properties). The same abbreviations are used in Figs. 2-4. Literature references are presented in Table S in the Supplement. (Colored versions of Figs. 1-4 are available in online version of the article and can be accessed at: https://www.springer.com/journal/10541)
Most proteins included in our analysis were successfully shown to have two to five of the listed properties (Fig. 1a). It is worth noting that staining with thioflavin T or its analogs is the most popular among the tests (65% of all proteins). At the same time, the Congo red staining called the “golden standard” for demonstrating amyloid properties in some works is used much less frequently (45% of proteins). Staining with Congo red is considered to be a stronger proof of amyloid properties and is also used to demonstrate cross-β structure. However, this technique could yield false-positive results. In particular, some monomer proteins with globular or β-solenoid structure (one of the variants of amyloid aggregates structure [4]), as well as partially unstructured ones, bind Congo red in vitro [3, 21]. Moreover, the dye can interact with collagen fibers, extracellular fibrils, and cytoskeletal proteins and even demonstrate birefringence when used for staining tissue samples [21, 22]. Interaction of the aggregates with thioflavin T is not to be considered as a universal proof of the cross-β structure, as, for example, the PSMα3 aggregates with cross-α structure bind this dye [23].
Biophysical approaches have shown the cross-β structure for less than one-third of the proteins under analysis (27%). Nevertheless, combination of the two sets (staining with Congo red and a cross-β structure) includes 53% of the analyzed proteins (120 of 226 examples). In terms of the most common definition, they can be called amyloids. In this subset, the most popular test series was combination of the amyloid-specific dyes and demonstration of fibrils (Fig. 1b). Curiously, the second place is taken by the examples that used only staining with Congo red. Whether such test is sufficient in itself remains unclear. It is also worth noting that most of the experiments were performed on different model systems, and there is usually no evidence that native protein forms amyloid aggregates in living organisms under natural conditions and at the basal level of production. This problem exists even for the well-studied proteins such as amyloid yeast prions.
For clarity of description, we divided the whole variety of aggregates into three groups: (i) pathological, (ii) functional, and (iii) amyloids with undefined biological roles, with each group involving several subgroups. Despite similar classification being frequently used, it is very arbitrary, as indicated by the number of examples we reviewed.
PATHOLOGICAL AMYLOIDS
In clinical practice, the term “amyloid” is often used to refer to a homogeneous extracellular deposit, which is stained with Congo red and demonstrates a yellow-green birefringence in polarized light with a characteristic fibrillar ultrastructure [15]. At the same time, many authors do not distinguish between extracellular and intracellular aggregates that exhibit amyloid properties. Furthermore, even the inclusion bodies, which are stained with Congo red, tend to be called amyloids. Intranuclear aggregates in Huntington’s disease and Lewy bodies in Parkinson’s disease are examples of such inclusions [24].
Currently, about 50 different proteins and peptides are known to form amyloids and amyloid-like aggregates associated with human diseases, including Alzheimer’s, Huntington’s, and Parkinson’s diseases, type 2 diabetes, and a number of amyloidoses resulting from the systemic or local deposition of amyloid fibrils in extracellular spaces of tissues and organs [15, 25]. Many of these diseases are fatal and incurable, with the risk of developing them increasing with age [26].
Amyloidogenic proteins form aggregates that accumulate as extracellular plaques and intracellular inclusions [4]. A large amount of amyloid material can destroy the tissue structure and mechanically impact functions of the affected organs [27]. Nevertheless, a hypothesis exists that oligomers and non-fibrillar amyloid deposits are toxic in amyloid diseases [28]. Among the examples are tetramers of transthyretin (TTR) [29], amyloid β (Aβ) [30], immunoglobulin light chain (Ig-LC) [31], and PrP [32], which are toxic both to cell cultures and to living organisms. Conformational changes in the prefibrillar aggregates result in exposure of the surface groups that usually are inside the folded proteins or are scattered in the natively unstructured proteins. This is regarded as a potential factor of amyloid toxicity [28], and in vitro studies confirm this hypothesis [33]. The intracellular amyloid inclusions could affect cell physiology, for example, by inhibiting protein and RNA transport and disrupting proteasome functions [4].
As a rule, there are differences in pathogenesis between localized and systemic amyloidoses: in the first case, the amyloidogenic protein is synthesized close to the place of deposition, while in the second case the protein is synthesized in one or several organs and then transported in a soluble form by blood plasma to the place where amyloid fibrils are formed [28]. A large group of neurodegenerative diseases associated with formation of amyloid and amyloid-like aggregates can be identified in particular. Shown in Fig. 2 is the list of proteins considered in this subsection, as well as information on the properties of their aggregates.

Properties of pathological amyloids and amyloid-like aggregates. The designations are the same as in to Fig. 1. References are given in Table S in the Supplement.
SYSTEMIC AMYLOIDOSES
Immunoglobulin light chain amyloidosis (AL) is the most common form of systemic amyloidosis, accounting for about 70% of all cases [34]. This disease occurs typically in people with monoclonal gammopathy, a disorder characterized by proliferation of the plasma cell clones. The increased Ig-LC production results in amyloid formation and organ damage [35]. Less is known about the rarer amyloidosis of light and heavy chain (Ig-HC) and heavy chain amyloidosis (AHL- and AH-amyloidosis, respectively) [36], which are clinically similar to the AL-amyloidosis. It should be noted that the above-mentioned diseases have also localized forms.
Transthyretin amyloidosis is the most common hereditary amyloidosis caused by mutations destabilizing the transthyretin tetramer, responsible for the transport of thyroxine hormone and vitamin A [37]. Senile systemic amyloidosis (SSA), an acquired disorder caused by the wild-type TTR deposits, is also classified as transthyretin amyloidosis affecting mainly men over 60 [37]. Another common systemic amyloidosis is the reactive or AA-amyloidosis, a disease associated with stable high concentrations of serum amyloid A (SAA) in plasma or specific tissues in inflammatory processes leading to extracellular protein deposits [38]. Patients with kidney failure or those on dialysis have amyloid deposits due to elevated levels of β2-microglobulin (β2M) in blood [39]. Dialysis-related amyloidosis is caused by full-length β2M deposits, mostly of the wild type. There is also an inherited form of the disease that develops due to mutations in the β2M coding gene, resulting in the synthesis of a protein with an increased tendency to aggregation [39].
A large group of systemic amyloidoses comprises disorders associated with apolipoprotein deposits. For example, apolipoprotein A-I (ApoA-I)-associated amyloidosis can occur both in a non-hereditary form with deposits of the wild type proteins in atherosclerotic plaques and an inherited form with deposits of mutant protein [40]. In rare cases, systemic hereditary amyloidoses can be caused by mutations of gelsolin, fibrinogen α-chain (FGA), cystatin C (CST3), or lysozyme [15, 25, 28]. Preeclampsia is characterized by aggregation and presence of toxic deposits of improperly folded Aβ, Ig-LC, TTR, α-1-antitrypsin, albumin, and ceruloplasmin in the placenta and physiological fluids. However, it is unknown which protein plays a key role in the development of this disease [41]. Familial British dementia is an autosomal dominant disease characterized by amyloid ABri deposits [42], with the main component being a peptide encoded by the BRI2 gene (also known as ITM2B). A BRI2 gene mutation leads to a stop codon being substituted by arginine resulting in protein elongation [43]. Amyloid peptide ABri includes 34 C-terminal amino acids of the mutant protein precursor and induces apoptotic cell death, while the wild type protein is not toxic to cells [44]. Interestingly enough, another mutation in this gene (duplications of ten nucleotides just before the stop codon) leads to the formation of amyloid peptide ADan [45]. Accumulation and deposition of the ADan are no longer related to the systemic amyloidosis but to the localized one: familial Danish dementia [46].
LOCALIZED AMYLOIDOSES
In this section we consider examples of localized amyloidoses that do not affect the central nervous system (CNS). The most common case among them is aortic amyloidosis associated with aggregation of medin, a product of lactadherin glycoprotein cleavage [47]. Localized amyloid and amyloid-like aggregates are also detected in tumor tissues. Medullary thyroid carcinoma is caused by transformation of the parafollicular C-cells and increase of the level of calcitonin thyroid hormone, thus resulting in amyloid deposits of this protein [48]. Nonfunctional amyloid-like oligomers of p53 [49], spherical prolactin deposits [50], and odontogenic ameloblast-associated protein (ODAM) deposits are also identified in tumors [15]. The islet amyloid polypeptide (IAPP), or amylin, is another hormone that aggregates causing pathology. This polypeptide got its name due to its tendency to form insoluble amyloid fibrils, a characteristic feature of the islets of Langerhans detected in the most people with type 2 diabetes. [51]. An example of age-related disease is isolated atrial amyloidosis caused by the presence of amyloid fibrils, the main component of which is an atrial natriuretic factor (ANF) [52]. Aggregation of semenogelin-1 (SEMG1) in epithelial cells of the seminal vesicles occurs during the senile localized amyloidosis [53]. In middle-aged and older men, amyloid cells are detected in the prostate, and they contain heterodimers of proteins S100A8 and S100A9 (S100A8/A9) [54]. The skin diseases are caused by amyloid deposits of corneodesmosin (CDSN) [55], galectin-7 [56], and keratins (CK5, CK14) [57]. Mutations in the TGFBI gene encoding kerato-epithelin (TGFBI), an extracellular matrix protein, are associated with various forms of corneal dystrophy [58]. Lactoferrin, an iron-binding glycoprotein, is another protein aggregation of which leads to the same pathology [59]. Insulin can form amyloid fibrils in a spot of medication injection in patients with diabetes, causing injection-localized amyloidosis, with insulin fibrils forming a solid subcutaneous mass at the injection site [60]. A similar effect can be produced by injections of enfuvirtide, a synthetic peptide that blocks the fusion of HIV-1 with the host cell [61].
NEURODEGENERATIVE DISEASES
We have put amyloidoses affecting the CNS in a separate group because they were studied more thoroughly. The peptide Aβ, a fragment of the amyloid precursor protein (APP), was first isolated as a major component of amyloid deposits in patients with Alzheimer’s disease. Although the function of APP remains to be understood in detail, its processing is well-studied, and the β- and γ-secretases are known to be responsible for it. The cleavage by γ-secretase is inaccurate, resulting in formation of the peptides with 36-43 amino acids in length, including amyloidogenic peptides Aβ40, Aβ42, and Aβ43 [51, 62]. The largest fraction (~80-90%) is represented by the variant Aβ40, with the second being the variant Aβ42 (about 5-10% of all variants). Aβ42 is the most hydrophobic and more prone to aggregation [63]. At the same time, Aβ peptides exhibit different toxicity, with variant Aβ43 being the most cytotoxic, but Aβ40 showing less toxic effect [51]. It is remarkable that Aβ40 and Aβ42 are capable of forming polymorphic fibrils, the structure of which could determine the rate of Alzheimer’s disease progression [64]. Associated with this disease are also amyloids, formed by Tau, a neuron-specific protein [65]. In the brain of Alzheimer’s patients, Tau is separated from microtubules, to which it is normally bound, loses the ability to stabilize them, and forms neurofibrillary tangles [66]. The α-synuclein protein forms intracellular amyloid aggregates found in the Lewy bodies in Parkinson’s disease and in Lewy body dementia, in glial cytoplasmic inclusions in patients with multiple system atrophy, and in axonal spheroids in neuroaxonal dystrophies [51, 67]. The α-synuclein inclusions are also detected in patients with Alzheimer’s disease [68].
Prion diseases are characterized with the ability of amyloid to induce its own assembly, to be transmitted from cell to cell or even from body to body, causing the disease to spread. In humans, they are associated with accumulation of the prion isoform (PrPSc) of the cellular prion protein PrPC in the tissue. These diseases include Creutzfeldt-Jacob disease, kuru, fatal familial insomnia, and Gerstmann–Sträussler–Scheinker syndrome (a hereditary variant of the Creutzfeldt–Jacob disease) [4]. Hereditary prion diseases are caused by mutations in the prion protein gene (PRNP). In particular, more than 60 mutations are known to be associated with a variety of clinical syndromes [69]. In animals, prion infections cause scrapie in sheep, bovine spongiform encephalopathy in cattle, and chronic wasting disease in deer and elk [4].
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disease. There are more than 180 known mutations in the gene encoding cytoplasmic superoxide dismutase (SOD1) [70] accounting for about 20% of the hereditary ALS cases [51]. Toxicity of the SOD1 mutant forms in ALS is attributed to incorrect folding and tendency to aggregation. It should be noted that in some ALS cases, the presence of cytoplasmic aggregates of TDP-43 and FUS proteins is detected [51].
FUNCTIONAL AMYLOIDS
This section provides information on aggregates that exhibit amyloid properties and formation of which is closely related to a particular function. Unfortunately, we could not find solid proof of the aggregate functionality for all the examples, as recently proposed [16]. Nevertheless, we do consider here the proteins for which the hypothesis about the biological significance of their aggregation dominates in the literature. Figure 3 provides information on the properties of functional amyloids and amyloid-like aggregates.

Properties of functional amyloids and amyloid-like aggregates. Designations are the same as in Fig. 1. References are given in Table S in the Supplement.
AMYLOIDS IN CELL WALL COMPOSITION AND CELL ADHESION PROCESSES
A number of proteins that are either part of or associated with the cell wall form amyloid and amyloid-like aggregates. These proteins contribute to the maintenance of the structure and integrity of the cell wall or other surface structures and participate in the adhesion of cells to each other or to the substrate.
Numerous amyloids and amyloid-like aggregates are formed by proteins that are part of the cell wall of yeast Saccharomyces cerevisiae. Glucan transferase Bgl2 was one of the first of such proteins to be recognized. This protein was shown to form amyloid fibrils in vitro [16]. Further, fibrils were found to form also by the cell adhesion proteins, Flo1 and Muc1. Some amyloid properties of these proteins were demonstrated in vivo. Mass spectrometry-based proteomic screening (PSIA-LC-MALDI) aimed at searching for potentially amyloidogenic proteins demonstrated that other proteins also form the detergent-resistant aggregates in the cell wall: Gas1, Gas3, Gas5, Toh1, and Ygp1. However, only three of them were studied in detail: Gas1, Toh1, and Ygp1. The functions of Toh1 and Ygp1 proteins have not been identified, while GAS1 was found to encode beta-1,3-glucanosyltransferase. The Toh1 protein is bound to the cell membrane by the GPI-anchor and presumably contributes to stabilization of the cell wall structure due to fibril formation [16]. Finally, amyloid-like aggregates of the cell wall proteins can participate in the adhesion of cells to each other.
Another example of the functional amyloids localized in the cell wall is associated with the cells of Streptomyces coelicolor bacteria forming an aerial hyphae. Eight proteins, which were the first to be identified in these structures (ChpA-H), were commonly referred to as chaplins [71]. ChpD-H proteins have been proved in vivo to have a number of amyloid properties; however, these results were obtained from mixtures of proteins from the detergent-resistant fractions. Formation of the S. coelicolor aerial hyphae also involves RdlA and RdlB proteins, forming an additional protein layer on top of chaplins [71]. Despite the fact that these two proteins have high sequence similarity (>90%), only RdlB can form fibrils in vitro [71].
Amyloid aggregates in aerial hyphae also make their surfaces hydrophobic. A similar function is performed by some proteins of fungi, which are commonly referred to as hydrophobins. The most known amyloid examples are EAS proteins (in Neurospora crassa) and SC3 (in Schizophyllum commune) [16, 19]. Similar proteins (RodA, MPG1, NC2, DewA, Rep1) from different types of fungi have the following properties: they have been shown to participate in the formation of fibrillar structures on the cell surface, to be highly resistant to detergents, and to interact with thioflavin T [72-75]. Among eukaryotes, similar examples have also been found in Candida albicans (Als1, Als5, Eap1) [76, 77].
To date, a number of bacterial proteins are known, aggregation of which is associated with biofilm formation and substrate adhesion. The most popular example is the protein CsgA in Escherichia coli, as well as its numerous orthologs [78], that are required for the fibrils called curli to be formed on the cell surface and an extracellular matrix to be built in bacterial biofilms [19, 79]. A similar function has been suggested for other proteins. In particular, TasA proteins in Bacillus subtilis, Bap in Staphylococcus aureus, and FapC proteins in Pseudomonas fluorescens were found to form fibrils on the bacterial surface, which were shown to have amyloid properties [19, 79, 80]. MTP protein also forms fibrillar aggregates on the surface of Mycobacterium tuberculosis [81]. Inhibition of the fibrillation of proteins P1, WapA, and SMU_63c in the bacterium Streptococcus mutans prevents biofilm formation [82]. AgfA and AgfB proteins form the detergent-resistant aggregates in the Salmonella enteritidis cells [83, 84]. Finally, the protein Sbp of Staphylococcus epidermidis is also assumed to be amyloid, since it forms fibrils, which are stained with amyloid-specific dyes [85].
In general, organization of the cell wall or interactions of cells with each other or with the external environment is an expected niche of functional amyloids. Stability and rigidity of the amyloid fibrils provide additional strength to cell coats and intercellular contacts.
AMYLOIDS IN NATURAL FIBERS
It was shown in 1968 by using X-ray diffraction for studying Chrysopa lacewing silk that the distance between the β-strands along the fibril axis was ~4.7 Å [4], which was characteristic for the cross-β structure. However, it is worth noting that the fibroin exuded by the mulberry silkworm Bombyx mori, forms in vitro amyloid-like fibrils with an unusual structure in which β-strands are parallel to the axis of the fibrils [86]. Another example of protein fiber is spider silks or spidroins, natural polymers characterized by exceptional strength and elasticity. They were observed to transition from the conformation based on α-helices and disordered regions to the structure enriched with β-sheets, which was similar to the formation of amyloid fibrils. Spidroin fibrils were observed in Nephila edulis using electron microscopy [16].
Minispidroin eADF-4(C16), based on the repetitive ADF-4 Araneus diadematus regions, forms fibrils with cross-β structure [16] and is capable of triggering formation of the amyloid-like fibrils in vitro [87]. It should be mentioned that REF (Hevb1) [88], one of the latex proteins from the rubber tree (Hevea brasiliensis) forms amyloid aggregates under physiological conditions in vitro [89].
AMYLOIDS IN SIGNALING PROCESSES
Changes in protein conformation provide the basis for intracellular signal transduction. Amyloid formation is also a conformational change, so it is hardly surprising that proteins whose aggregation represents an essential part of the signaling cascades have been found. A pair of human proteins RIPK1 and RIPK3 is a well-studied example, with their joint aggregation (co-aggregation) required to trigger necroptosis [90, 91]. These proteins have almost all amyloid properties shown in vitro, and there is also evidence of their aggregation in vivo. The regions required for both the aggregation and the signaling cascade contain RHIM sequences (Rip homotypic interaction motif) [90].
Similar motifs were found in various proteins from numerous organisms [92], including the well-studied amyloid HET-s from Podospora anserina. The aggregates of this protein have infectious properties, resulting in the formation of the [Het-s] prion. This cytoplasmic factor is required to trigger a heterokaryon incompatibility reaction. During the fusion of hyphae of different organisms, with the one carrying this prion and the other not carrying, the programmed cell death can occur. This process is initiated by the interaction of amyloid aggregates HET-s with the monomer protein HET-S (products of different alleles of the same gene), which appeared in one heterokaryon following the cell fusion [93]. Subsequent aggregation of HET-S leads to the change of its conformation facilitating its incorporation into the cell membrane disrupting its integrity [94].
The subsequent search for RHIM among other P. anserina proteins has revealed a new participant in this signaling cascade, NWD2, which can trigger HET-s aggregation and hence lead to the cell death [95]. HELLP, SBP, and PNT1 proteins of Chaetomium globosum fungus also contain RHIM and, possibly, are functional analogs of HET-s, HET-S, NWD2 system [96].
Cryptic RHIMs (cRHIM) were found in the Drosophila melanogaster PGRP-LC, PGRP-LE, and Imd proteins, which were involved in activation of the antimicrobial response. A model was proposed suggesting that aggregation of these proteins is the key event in signal transduction [97]. Since RHIM were found among different proteins related to signaling processes [98], one can expect discovery of new functional amyloids.
When a cell recognizes a viral RNA, it initiates a signal transduction cascade that triggers the antiviral response. MAVS protein is one of the newly discovered participants in this signaling, shown to perform its function in an aggregated form. MAVS molecule contains the C-terminal transmembrane domain anchoring it in the mitochondrial membrane, the N-terminal caspase recruiting prionogenic domain (CARD) exposed inside the cytosol, and an intermediate site that recruits signalling molecules from the underlying signaling pathway. The CARD domain interacts with the same region of RIG-1 and MDA5 receptor proteins that recognize the alien RNA by triggering an antiviral response cascade. In this case, MAVS forms complexes, which initiate aggregation of other MAVS molecules [99].
While demonstrating a number of properties characteristic of amyloids, MAVS aggregates have a unique structure, so they do not interact with Congo red and thioflavin T dyes. When turning into an aggregated form, MAVS protein does not undergo conformational transitions of α-helices into β-sheets. Based on the cryo-electron microscopy data, two models of aggregates of this protein were proposed. In both cases, MAVS filaments consist of separate subunits of the protein, stacked on top of each other forming a central pore [99].
TOXINS AND ANTIMICROBIAL PEPTIDES
Antimicrobial peptides (AMPs) are protective peptides that are part of innate immunity found in many organisms. Most AMPs are cationic and amphiphilic α-helical protein molecules. Their primary mechanism is binding to a negatively charged bacterial membrane and disrupting its integrity [100]. Some AMPs were shown to have amyloid properties that are supposed to be important for their functioning. In most cases, these properties have been analyzed only in vitro, so many of these peptides are not shown in Fig. 3 (see Table S in the Supplement).
The aggregation tendency has been shown for cecropins, but not enough data have been collected so far to support amyloid formation by them [101]. It is believed that the cecropin P1 forms a local “carpet-like” structure on the membrane of a pathogen, leading to membrane deformation and then to destruction when the concentration of AMP exceeds the critical one [102].
LL-37 is the first antimicrobial cationic peptide discovered in humans [103]. During the bacterial invasion, it is released by proteases from its predecessor hCAP-18. LL-37 forms fibrous aggregates in vitro, which are stained with Congo red and characterized by apple-green birefringence in cross-polarized light [103]. The fibrils formed by a short oligopeptide from the LL-37 sequence are not stained with thioflavin T and have a structure unusual for amyloid: they consist of densely packed amphipathic α-helices [104]. Aggregation of the LL-37 protein was shown to be directly related to its cytotoxicity [103].
Dermaseptins Drs S9 and aDrs PD-3-7 were isolated from the frog skin. They form β-sheet-rich fibrils that have shown a number of amyloid properties [105-107]; however, similar to many AMPs, these properties have not been confirmed in vivo. Magainins were found in the skin of Xenopus laevis frog and exhibited antimicrobial [108, 109] and antitumor effects [110, 111] and also demonstrated amyloid properties. An antimicrobial mechanism with the formation of toroidal pores was proposed for them for the first time. Magainins bind to the membrane and, after reaching the threshold concentration, lead to its deformation and destabilization. After that, the peptides are incorporated into the membrane, forming a toroidal pore [112]. Human AMP protegrin-1 (PG-1) forms fibrils in vitro, which bind thioflavin T. The protegrins are in an insoluble state in neutrophil and macrophage pellets. If a pathogen enters the cell, the granules merge with the vacuole, releasing peptides forming the channels in the membrane of the pathogen, leading to its death [113]. The known protegrins with amyloid properties also include PG-4, isolated from pig leukocytes. PG-4 was shown to form fibrillar aggregates in vitro, which were stained with Congo red and thioflavin T. Both monomers and aggregates show antimicrobial activity against B. subtilis [114]. Microcin E492 is a known pore-forming bacterial toxin produced by Klebsiella pneumoniae RYC492. It was shown to be capable of forming amyloid-like β-sheet-rich fibrils in vivo and in vitro [115]. Another example of a bacterial toxin is the S. aureus protein PSMα3, which forms fibrils with a cross-α structure that can bind thioflavin T. There is some evidence of the toxicity of fibrils to human cells [23].
Paneth cells of the human small intestine produce α-defensin 6 (HD6), which provides protection against invasion of pathogenic microorganisms in the intestine [116]. When binding to the surface proteins of bacteria, the HD6 undergoes self-assembly to form fibrils and nanonets that surround and entangle pathogens, thus providing an obstacle to the physical contact of bacteria with epithelial cells that is necessary for attachment or invasion [116].
Amyloid-like AMPs have also been found in plants. Cn-AMP2, obtained from the liquid endosperm of Cocos nucifera, forms visible aggregates in the aqueous buffer solution that bind Congo red, thioflavin T, and also have a fibrillar morphology. However, at present, it has not been proven directly that Cn-AMP2 exhibits an antibacterial effect only in the aggregated form [117].
It is important to note that proteins associated with human amyloidoses can behave like toxins. For example, Aβ exhibits antimicrobial properties that target pathogenic bacteria and fungi in vitro and in model systems: in Caenorhabditis elegans and mice [118, 119]. Antimicrobial properties have been demonstrated for several other amyloidogenic proteins. For example, in addition to enzymatic properties, lysozyme manifests itself as AMP. Its antimicrobial activity is associated with permeabilization of the cell membrane of the pathogen, presumably through pore formation [120].
AMYLOIDS IN MEMORY PROCESSES
The presence of amyloids in the CNS has been for decades associated exclusively with pathologies. But everything changed with the discovery of the functional amyloids of CPEB protein in the mollusk Aplysia californica and its homologs: Orb2 in D. melanogaster and CPEB3 in mice. CPEB proteins are RNA-binding, and they bind RNA more efficiently when oligomerized [121]. In the brain of studied animals, oligomerization of CPEB proteins occurs in response to neuronal stimulation [122, 123]. When these proteins are unable to form aggregates, it negatively affects maintenance of the long-term memory, at least in A. californica and D. melanogaster [121, 122]. Taken together, these facts allow concluding that CPEB proteins are indeed functional in an aggregated state. These proteins are also referred to by the researchers as functional prions [123], probably without sufficient justification, since the in vivo infectivity of CPEB and CPEB3 was demonstrated only in the yeast model [123, 124]. So far, no direct evidence had been presented that this group of proteins formed amyloid particles in living organisms without artificial superproduction. It was not until recently that amyloid nature has been strictly proven for Orb2 fibrils isolated from Drosophila heads. Also, for the first time, it was possible to reconstruct the structure of amyloid fibrils ex vivo in high resolution using cryo-electron microscopy [125]. The Orb2 fibrils demonstrate a three-fold symmetry similar to the known structure of Aβ40 [125]. Thus, it is evident that the amyloid structure itself could not be the cause of the development of CNS pathologies. In order to understand the real reasons for the development of neurodegenerative amyloidoses, a thorough study of the factors affecting the amyloidogenesis of the particular protein is necessary.
CPEB/Orb2 is not the only example of amyloids associated with memory processes. Recently, FXR1 protein involved in the development of long-term memory and emotions has been shown to be present in the rat brain in an amyloid form. Moreover, its monomeric form was almost undetectable. Meanwhile, the detected aggregates demonstrated amyloid properties [126]. FXR1 is an RNA-binding protein, which has been shown to be able to bind mRNA in amyloid form, protecting it from RNAse digestion [126]. It likely affects translation of the specific mRNAs in this way and, consequently, influences differential expression in neurons. The fact that FXR1 is detected in the brain of rats exclusively as part of the detergent-resistant aggregates, together with the data that FXR1 is important for regulation of memory and emotions, allow one to classify FXR1 as a functional amyloid. The amyloidogenic fragment of FXR1 is highly conservative in mammals [126], making it possible to extrapolate the obtained results to the orthologs of this protein in other organisms.
Amyloid involvement in the memory processes is likely to be not limited to animals and their nervous systems. Alternative amyloid conformation can bear specific information that can somehow be “deciphered” at the cellular level, so the use of such aggregates for long-term storage of information can be a universal mechanism that could be repeatedly and independently realized in evolution. One of the examples of a similar mechanism of cellular memory is the existence of so-called “mnemons”. These are Whi3 protein aggregates in yeast that are formed in response to unsuccessful fusion of cells of opposite mating types and remain in the yeast cells for life as a memory of the past event [127]. In contrast to prions, “mnemons” are not transferred to daughter cells when divided. Nevertheless, it is quite possible that the known prions of the lower eukaryotes (see below) may also be the units of inheritance of such “memory,” which, however, transfer information, not limited to the life of one cell, but for many cell generations. Taking into account that some prions nowadays are known to be able to arise in response to certain stressful effects, it is possible that prions for unicellular organisms represent such “ancestral memory” of the difficulties experienced.
AMYLOIDS RELATED TO REPRODUCTION
Functional amyloid fibrils have been found in structures associated with sexual reproduction, such as the sperm acrosome. These fibrils are believed to contribute to the controlled release of proteins during the acrosomal reaction, a key event in oocyte fertilization in which the acrosome content is poured out. The amyloids formed by the proteins of the subgroup CRES (Cystatin-related epididymal spermatogenic) – CRES, CRES2, CRES3, and cystatin E2 (mCSTE2) are present in the mouse epididymis, while in humans the same organ is found to have cystatin C (CST3) aggregates. The role of amyloids formed by CRES proteins is not quite clear, but there are assumptions about their functional significance. They are known to have antimicrobial activity, are necessary for the acrosomal reaction, normal functioning of lysosomes in the epididymis, and also play a role in sperm maturation. Semen fluid also contains amyloids and amyloid-like fibrils formed by fragments of the peptide from prostatic acid phosphatase (PAP) and semenogelin proteins (SEMG1 and SEMG2), respectively. These fibrils, originally identified as viral infection enhancers, bind to and immobilize damaged spermatozoa, ensuring their destruction by immune cells. Amyloids formed by the protein SEMG1 have also been found in some pathologies.
Oocytes of most vertebrates contain Balbiani bodies. This compartment matrix in X. laevis consists of a fibrillar network formed by the protein Xvelo. These fibrils are stained with thioflavin T and are resistant to detergent treatment [128]. Amyloid properties have also been demonstrated for proteins involved in the formation of the oocyte shell in insects, fish, and mammals. Data from X-ray scattering experiments show that amyloid structures are present in the chorion of the butterfly Antheraea polyphemus [129]. It should be noted that a significant amount of amyloid properties was demonstrated for the individual peptides of these proteins corresponding to the section of tandem repeats (cA-peptides) [130, 131]. It is interesting that the assembly of amyloid fibrils from such peptides occurs through the intermediate liquid crystal phase [132]. Bonefish have proteins of the ZPB group in the chorion, individual peptides of which also tend to form amyloid aggregates [133]. Amyloid properties were also shown for the proteins in the zona pellucida of mammalian oocytes. Thus, the proteins in this membrane in mice in vivo exhibit characteristic morphology of the aggregates, as well as the pattern of X-ray diffraction and binding to amyloid-specific dyes typical for the cross-β structure [134]. The same properties were shown for the aggregates of peptide analogs of human ZP1-ZP4 protein fragments in vitro [135]. Formation of the amyloid fibrils in the membranes of oocytes and embryos in various animals is believed to help prevent external influences such as high temperatures, mechanical pressure and others.
Some amyloid-like proteins act as cell cycle regulators and affect sexual reproduction of yeast S. cerevisiae. The protein Rim4 forms detergent-resistant aggregates during the G1 meiosis phase, leading to the Clb3 cyclin translation repression and subsequent induction of the division. Disassembly of the Rim4 aggregates is triggered by phosphorylation of this protein [136].
HORMONES AND AMYLOIDS
Discovery of the first functional mammalian amyloids – melanosome fibrils, formed by the protein PMel17 – is associated with hormones. PMel17 was found to be involved in melanin synthesis in the amyloid form [137]. PMel17 is subjected to proteolytic cleavage in melanocytes, in the first stage of which a fragment of Mα is formed [138]. Amyloid properties are well demonstrated for the full-length Mα, with its conversion to amyloid form occurring at least four orders of magnitude faster than that of Aβ or α-synuclein [137, 139]. It could be explained by the fact that, in contrast to pathological amyloids, PMel17 was subjected to evolutionary selection towards more effective amyloid formation. However, in the cells, Mα is subject to further proteolysis and is not the main component of melanosomal aggregates.
There were controversial data concerning the PMel17 domains directly associated with formation of amyloid fibrils in the cell [138-140]. Different PMel17 proteolysis products are supposed to form amyloids at different stages of the melanosome formation. Thus, the fibrils present in the cell comprise complex co-aggregates from different PMel17 fragments [141]. The monomeric precursor of melanin, indole-5,6-quinone (DHQ), is similar in structure to thioflavin T, and it is assumed that Pmel17 fibrils can serve as seeds in DHQ polymerization. Amyloid ability to function as such “catalyst” is evidenced by the fact that not only Mα, but also fibrils of α-synuclein or Aβ increase the rate of melanin synthesis in vitro [137]. Formation of the complex aggregates from PMel17 amyloid fibrils seems to provide maximum efficiency for melanin synthesis.
Not only were amyloids involved in the synthesis of hormones, but it was also shown that peptide hormones themselves could form amyloids that are supposed to be necessary for their storage in the secretory granules. Among 42 peptide hormones studied in vitro, 10 demonstrated spontaneous formation of the amyloid fibrils, and 31 hormones formed fibrils in the presence of glycosaminoglycan heparin [142], with prolactin forming them in the presence of chondroitin sulfate A, a glycosaminoglycan found earlier in the prolactin-containing granules.
Oxytocin was shown to have a globular structure, which could bind thioflavin T. Adrenocorticotropic hormone (ACTH), was not able to form amyloids by itself and did it only in the presence of β-endorphin aggregates (both are the products of the same prohormone and are localized together in the secretory granules of the pituitary gland cells). These data were obtained using ACTH and human β-endorphin, but the amyloid properties of these proteins isolated from the mouse and rat secretory granules were confirmed in vivo [142].
A valuable property found in amyloids of some hormones is the ability to release monomers under conditions consistent with the release of peptides from the secretory granules. Moreover, amyloids of only seven hormones demonstrated toxicity to cells [142]. Both of these observations suggest that amyloids of some hormones could indeed have a storage function in the cell. Toxic effect of the glucagon amyloids was twice as strong as of Aβ40 [142], confirming the data that glucagon amyloids could be pathological [143]. It is interesting that another hormone, prolactin, was found in amyloid form in patients with adenoma [144]. However, taking into account the data that amyloids of prolactin could be functional, it is not clear whether the negative effect of amyloids of prolactin is associated with pathology. We have also considered the protein semenogelin in the previous subsection, whose aggregates were found both in amyloidoses and in normal spermatozoa. Thus, it is possible that the amyloids of some other proteins found in various pathologies may also be functional amyloids.
AMYLOIDS WITH AN UNDEFINED BIOLOGICAL ROLE
This section presents examples of protein aggregates whose biological role is subject to debate in the literature. In particular, there is still no consensus on the function of yeast prions, as there are arguments for and against their possible functionality [145, 146]. Proteins in the membraneless organelles can also form amyloid aggregates, but it is not always clear whether this feature is necessary for the functioning of these proteins. Many resident proteins of the membraneless organelles are also associated with pathogenesis of such diseases as ALS and several types of dementia. Finally, aggregation is shown for various enzymes and porins, functional significance of which is only presumed. Information on the properties of the aggregates of these proteins is presented in Fig. 4.

Properties of amyloids and amyloid-like aggregates with the undefined biological role. Designations are the same as in Fig. 1. References are given in Table S in the Supplement.
AMYLOID AND AMYLOID-LIKE PRIONS
A large number of amyloids with infectious properties, called prions, have been found in S. cerevisiae yeast. The phenotypic manifestation of most of them is associated with formation of the amyloid aggregates of a particular protein. In most cases, it leads to the loss or weakening of the function of the relevant protein. Proteins Sup35, Ure2, Swi1, Cyc8, Mot3 can be considered as examples [93]. Therefore, strains that propagate a particular prion have a phenotype similar to the strain with mutation in the gene encoding the corresponding protein. However, this phenotype demonstrates cytoplasmic inheritance because amyloid particles are localized in the cytoplasm [93]. Factors [URE3], [PSI+], and [PIN+]([RNQ+]), which are associated with the appearance of amyloid aggregates of Ure2, Sup35 and Rnq1, respectively, were the first to be described as yeast prions [93]. These proteins have become examples of amyloids, for which almost all properties have been demonstrated over the long history of their research. Later, more than ten additional yeast prions were identified (table).
The presence of properties specific to S. cerevisiae prions and convenient genetic tests allowing their detection made it possible to perform screenings in yeast to search for proteins with prion and amyloid properties. One screening [11] revealed such properties for dozens of yeast proteins, or, more precisely, for their potential prion domains detected in silico. In most cases, it remains unclear whether the detected proteins are capable of forming real prions in yeast. This is primarily due to the fact that the detected properties of a protein fragment cannot always be extrapolated to a full-length protein.
The [ISP+] factor, which is a prion form of the Sfp1 protein according to the genetic data [161], deserves special attention. However, its phenotype (Isp+) is caused by the changes in the chromosome II copy number resulting from the Sfp1 overproduction [162]. At the same time, Sfp1 can form detergent-resistant aggregates, which seem to have nothing to do with Isp+ [163].
The story of Isp+ makes it clear that conclusions about the amyloid nature of a particular prion, based on genetic analysis but not supported by substantial biochemical or structural data, cannot be considered sufficiently proved. The hereditary factors that are currently known to demonstrate prion-like behavior but not related to amyloids, such as [GAR+] [164], [SMAUG+] [165], and others [166], also confirm this point of view. In addition, proteins that tend to form liquid droplets (see below) are often able to form amyloids as well, such as Sup35 [167]. Such examples suggest that combining the in vivo genetic data with the evidence of amyloid formation in vitro is also insufficient to claim that a certain protein is capable of assuming amyloid conformation in living organisms. To justify this assumption, additional evidence of amyloid properties in vivo is needed, which is currently lacking in the case of most “classic” yeast amyloid prions.
MEMBRANELESS ORGANELLES AND PROTEIN AGGREGATES
Membraneless organelles are dynamic structures in the nucleus or cytoplasm of a cell formed by a large accumulation of interacting biological molecules that perform a specific set of functions. Many types of such compartments with various cellular functions have been described [168]. Nuclear membraneless organelles include the nucleolus, Cajal bodies, nuclear speckles or histone locus bodies. These organelles play an important role in the processes of transcription, maturation of various RNA species, and biogenesis of ribosomes [169]. In turn, cytoplasmic membraneless organelles are often associated with the processing, storage, and degradation of proteins and mRNA. These include such structures as P-bodies and stress granules [168], as well as recently discovered A-bodies [170].
Many membraneless organelles have liquid-like properties and are formed due to the liquid–liquid phase separation, which occurs during the interaction of protein molecules and leads to the formation of liquid droplet structures. A large number of proteins of membraneless organelles interact with RNA and contain the RNA recognition motifs in their structure. Intrinsically disordered regions play an important role in the formation of membraneless organelles, as they predetermine the ability of these proteins to enter into polyvalent intermolecular interactions leading to phase transitions [171-176]. Such segments tend to have a low complexity of amino acid composition and therefore are called LC-domains. The segments responsible for phase separation also often have an amino acid composition that is characteristic of prion domains, thereby being referred to as prion-like domains [168].
Normally, membraneless organelles at the initial stages of their formation are considered to contain no aggregates with classic amyloid structure. During maturation, the labile amyloid-like aggregates could first appear in such organelles followed by more stable amyloid fibrils [177-179]. At the same time, formation of the amyloid fibrils from some components of the membraneless organelles is associated with the development of such fatal human neurodegenerative diseases as ALS and frontotemporal dementia. Whether the formation of amyloid fibrils is a necessity or a side property of the proteins in question remains unclear. Moreover, the structure of aggregates formed by many proteins residing in the membraneless organelles has a number of important differences from the typical structure of amyloid fibrils (in particular, aggregates of a number of such proteins are sensitive to high temperatures and detergents [180, 181]).
The RNA-binding protein FUS, involved in the formation of P-bodies, is capable of aggregating and actively interacts with aggregates of other proteins [182], and its mutant forms are associated with ALS. The FUS aggregates are formed by its LC-domain. This fragment as a whole, as well as its separate segments named RAC (Reversible Amyloid Core), is capable of forming unbranched amyloid fibrils in vitro that bind amyloid-specific dyes and have characteristic structure [183, 184]. However, the structure of these fibrils has a number of notable differences from the typical cross-β-model: for example, FUS-LC aggregates are characterized by uniformity of the structure and absence of the regular interactions between the fragments outside the amyloid core [183]. The RAC1-fragment of FUS forms a structure based on ordered loops; the fibrils formed by RAC2 have a wet interface of β-sheets [184]. These properties are supposed to contribute to the reversible formation of amyloid aggregates (FUS-LC aggregates are shown not to be resistant to SDS, but resistant to other detergents [180, 185]).
The hnRNPA1 and hnRNPA2B1 proteins are nuclear ribonucleoproteins that play a significant role in the processing of heteronuclear RNA and regulation of gene expression [186]. Their amyloid properties were discovered when studying the effect of mutations in genes encoding these proteins in the familial forms of ALS. Both wild type proteins and their mutant forms are capable of forming fibrils in vitro, with ALS-associated mutations in prion-like domains hnRNPA1 and hnRNPA2B1 accelerating aggregation of these proteins [187]. Specific segments of hnRNPA1 (LC domain and RAC1/2) reversibly form amyloid fibrils when exposed to low temperatures in vitro. Their formation, similarly to FUS, is controlled by RAC elements that are capable of forming amyloid fibrils independently. However, their structure is slightly different from the typical cross-β structure. There is a residue of aspartic acid in the middle of the amyloid core, which is responsible for structure destabilization. Its mutations in RAC elements prevent protein solubilization and dissolution of the fibrils [181].
Aggregates of the TDP-43 protein were found in the brain inclusions in patients with ALS and frontotemporal lobar degeneration (FTLD). Despite the assumption of their prion-like properties, TDP-43 protein is not able to form long amyloid fibrils, producing only small oligomers. They can induce aggregation not only of normal TDP-43 but also of other amyloidogenic proteins such as Aβ [188]. Normally, TDP-43 is an RNA-binding protein regulating mRNA splicing; under stress, TDP-43 is a part of stress granules. Interestingly, TDP-43 oligomerization can also occur through different pathways involving interaction of monomers through their N-terminal segments, preventing interaction of LC-domains and pathological aggregation. Furthermore, such oligomerization is important for the biological function of TDP-43 [188]. Thus, TDP-43 aggregation can occur in two ways, but it is not clear whether each of them is functionally important and how they are related to pathological processes in ALS or FTLD.
Another important example of reversible aggregation is represented by A-bodies, formed in the cells under stress [170]. In contrast to the previously considered membraneless organelles, A-bodies seem to play a role in storage of amyloid aggregates, which are formed in cells during the stress response (such as acidification of cytoplasm and heat shock). These protein aggregates are stained with Congo red in vivo, resistant to protease treatment and contain fibrillar aggregates that interact with amyloid-specific antibodies. Proteomic analysis demonstrated that A-bodies contain a large number of different proteins, in particular, VHL and POLD1 proteins. Specific segments, called amyloid-converting motifs, have been identified as part of these proteins, which contribute to the recruitment of proteins to the A-body through interaction with ribosomal intergenic non-coding RNA. In the in vitro system, these segments form typical amyloid fibrils with characteristic structure. At the same time, full-length VHL and POLD1 proteins were not shown to be able to form complete amyloid fibrils, although structural features of their protein aggregates in A-bodies remain unexplored [170].
Recently, the discussed earlier Tau protein has been shown to be able to undergo phase separation and reside in the membraneless organelles in neurons. It is yet unclear whether the Tau recruitment to membraneless organelles is functionally important, but it has been shown that the phase separation can contribute to the formation of Tau aggregates in vivo and in vitro [189].
AGGREGATION-PRONE PORINS AND ENZYMES
Different protein porins can form amyloid and amyloid-like aggregates. The majority of known examples are prokaryotic proteins (RopA, RopB, OmpA, OmpC) [190-193] and one eukaryotic protein (MSP2) [19]. It is difficult to determine whether the process of porin aggregation is pathological or functional. However, there is a possibility that it could be widespread. Bioinformatic analysis suggested that the porin fragments forming the inner part of the pore were capable of forming amyloid structures [194].
In addition to porins, a number of enzymes are known that can form aggregates. Three proteins included in our analysis – CarD, HelD, and RepA proteins – physically interact with nucleic acids one way or another [195-200]. In addition, aggregation ability was shown for YghJ protease [201] and transglutaminase TGZ [202].
“AMYLOID” TERM CRISIS
The generally recognized term “amyloid” is considered to define the following: amyloids are fibrillar protein aggregates that have a cross-β structure [4, 14, 17, 51], it is also specified in this definition that the presence of apple-green birefringence after staining with Congo red is the proof of cross-β structure [15, 16]. However, a more precise definition also includes the requirement to demonstrate these properties in vivo for native proteins at natural production levels. In our review, we did not follow this clarification. When collecting and analyzing the data on the properties of different proteins, we have not found any fundamental contradictions to the definition of the term given above, although the authors do not always adhere to it, as noted earlier [17].
However, it is worth paying attention to the examples of protein aggregates with cross-β structure, which are thermolabile and not resistant to detergents. Such structure is shown in vitro for FUS peptides, its LC domain, and also LC fragment hnRNPA2. The authors refer to such aggregates as amyloid-like, but not amyloid [180]. Similar results were obtained for the full-length protein hnRNPA1 [181]. Thus, the question “Should we call these aggregates amyloids because they have a cross-β structure?” seems to us relevant.
Should the answer be positive we may have to admit that amyloid aggregates do not have to be stable. Such an approach is expected to increase the number of amyloids. In the case of FUS, it has been shown that a short fragment of the LC-domain called LARKS is responsible for forming the aggregates. Similar motifs of proteins NUP98 and hnRNPA1 also form thermolabile fibrils with cross-β structure. A large number of similar sequences among the proteins in various complexes formed by the phase separation principle have been identified with the help of bioinformatics [203]. However, this is only an assumption: the presence of similar sequences cannot guarantee the same properties for the full-length proteins. In addition, keeping the original definition based only on the structure of aggregates (without specifying the data on their stability) emphasizes limitation of the existing methods of searching for new amyloid candidates, which are based only on their resistance to detergents [204, 205] (these approaches cannot identify unstable aggregates with cross-β structure). Regardless of the chosen approach, there remains a problem to describe the vast number of amyloid-like aggregates, which are very diverse in terms of structure. Given that the structure criterion is the key for separating amyloids into a specific group, amyloid-like units should also be classified similarly. In our review, we have considered the examples of MAVS, PSMα3, SOD1, and LL-37 aggregates, which have some amyloid properties but also have various structural variants, such as β-corkscrew or cross-α [4]. Now they fall into a single group of amyloid-like units, although if the structure-based criterion is fundamental, such units must be classified separately.
It is also worth noting that some of the criteria that are used to prove amyloid properties are artificial and do not explicitly reflect properties that are relevant to biological systems. For example, staining with specific dyes does not make obvious biological sense as these substances are normally absent from the cell or organism. Nor do we know of cellular proteins or systems that would be able to specifically recognize the cross-β structure, but not other arrangements with amyloid-like properties. There are amyloid-specific antibodies that could be able to differentiate between different arrangements [206], but this example is difficult to consider as universal. At the same time, stable fibrillar aggregates are often mentioned as one of the examples of the formation of different protein complexes based on phase separation [207]. In this regard, the principal differences between these aggregates are their morphology (fibrils) and stability (weak exchange of molecules between the aggregate and the surrounding solution). From the biological point of view, these criteria are relevant, since they reflect biophysical state of specific molecules in the cell, possibility for them to move freely and, consequently, participation in specific processes.
Abbreviations
- β2M:
-
β2-microglobulin
- Aβ:
-
amyloid β
- ALS:
-
amyotrophic lateral sclerosis
- AMP:
-
antimicrobial peptide
- CRES:
-
cystatin-related epididymal spermatogenic (amyloid)
- Ig-LC:
-
immunoglobulin light chain
- LC:
-
low-complexity (domain)
- RAC:
-
reversible amyloid core
- PrP:
-
prion protein
- RHIM:
-
Rip homotypic interaction motif
- TTR:
-
transthyretin
References
Kyle, R. A. (2001) Amyloidosis: a convoluted story, Br. J. Haematol., 114, 529-538.
Tanskanen, M. (2013) “Amyloid” – historical aspects, in Amyloidosis (Feng, D. ed.) 8th Edn., InTech, Rijeka, Croatia, pp. 3-24.
Yakupova, E. I., Bobyleva, L. G., Vikhlyantsev, I. M., and Bobylev, A. G. (2019) Congo red and amyloids: history and relationship, Biosci. Rep., 39, BSR20181415.
Iadanza, M. G., Jackson, M. P., Hewitt, E. W., Ranson, N. A., and Radford, S. E. (2018) A new era for understanding amyloid structures and disease, Nat. Rev. Mol. Cell Biol., 19, 755-773.
Kushnirov, V. V., Dergalev, A. A., and Alexandrov, A. I. (2020) Proteinase K resistant cores of prions and amyloids, Prion, 14, 11-19.
Kushnirov, V. V., Alexandrov, I. M., Mitkevich, O. V., Shkundina, I. S., and Ter-Avanesyan, M. D. (2006) Purification and analysis of prion and amyloid aggregates, Methods, 39, 50-55.
Dear, A. J., Michaels, T. C. T., Meisl, G., Klenerman, D., Wu, S., Perrett, S., Linse, S., Dobson, C. M., and Knowles, T. P. J. (2020) Kinetic diversity of amyloid oligomers, Proc. Natl. Acad. Sci. USA, 117, 12087-12094.
Prusiner, S. B. (1987) Prions and neurodegenerative diseases, N. Engl. J. Med., 317, 1571-1581.
Wickner, R. (1994) [URE3] as an altered URE2 protein: evidence for a prion analog in Saccharomyces cerevisiae, Science, 264, 566-569.
Osherovich, L. Z., and Weissman, J. S. (2001) Multiple Gln/Asn-rich prion domains confer susceptibility to induction of the yeast [PSI+] prion, Cell, 106, 183-194.
Alberti, S., Halfmann, R., King, O., Kapila, A., and Lindquist, S. (2009) A systematic survey identifies prions and illuminates sequence features of prionogenic proteins, Cell, 137, 146-158.
Chandramowlishwaran, P., Sun, M., Casey, K. L., Romanyuk, A. V., Grizel, A. V., Sopova, J. V., Rubel, A. A., Nussbaum-Krammer, C., Vorberg, I. M., and Chernoff, Y. O. (2018) Mammalian amyloidogenic proteins promote prion nucleation in yeast, J. Biol. Chem., 293, 3436-3450.
Sivanathan, V., and Hochschild, A. (2013) A bacterial export system for generating extracellular amyloid aggregates, Nat. Protoc., 8, 1381-1390.
Nizhnikov, A. A., Antonets, K. S., and Inge-Vechtomov, S. G. (2015) Amyloids: from pathogenesis to function, Biochemistry (Moscow), 80, 1127-1144.
Benson, M. D., Buxbaum, J. N., Eisenberg, D. S., Merlini, G., Saraiva, M. J. M., Sekijima, Y., Sipe, J. D., and Westermark, P. (2018) Amyloid nomenclature 2018: recommendations by the International Society of Amyloidosis (ISA) nomenclature committee, Amyloid, 25, 215-219.
Sergeeva, A. V., and Galkin, A. P. (2020) Functional amyloids of eukaryotes: criteria, classification, and biological significance, Curr. Genet., doi: https://doi.org/10.1007/s00294-020-01079-7 .
Galkin, A. P., Velizhanina, M. E., Sopova, Y. V., Shenfeld, A. A., and Zadorsky, S. P. (2018) Prions and non-infectious amyloids of mammals – similarities and differences, Biochemistry (Moscow), 83, 1184-1195.
Baxa, U. (2008) Structural basis of infectious and non-infectious amyloids, Curr. Alzheimer Res., 5, 308-318.
Shewmaker, F., McGlinchey, R. P., and Wickner, R. B. (2011) Structural insights into functional and pathological amyloid, J. Biol. Chem., 286, 16533-16540.
Chiti, F., and Dobson, C. M. (2009) Amyloid formation by globular proteins under native conditions, Nat. Chem. Biol., 5, 15-22.
Khurana, R., Uversky, V. N., Nielsen, L., and Fink, A. L. (2001) Is Congo red an amyloid-specific dye? J. Biol. Chem., 276, 22715-22721.
Westermark, G. T., Johnson, K. H., and Westermark, P. (1999) Staining methods for identification of amyloid in tissue, Methods Enzymol., 309, 3-25.
Tayeb-Fligelman, E., Tabachnikov, O., Moshe, A., Goldshmidt-Tran, O., Sawaya, M. R., Coquelle, N., Colletier, J.-P., and Landau, M. (2017) The cytotoxic Staphylococcus aureus PSMα3 reveals a cross-α amyloid-like fibril, Science, 355, 831-833.
Brundin, P., Melki, R., and Kopito, R. (2010) Prion-like transmission of protein aggregates in neurodegenerative diseases, Nat. Rev. Mol. Cell Biol., 11, 301-307.
Chiti, F., and Dobson, C. M. (2017) Protein misfolding, amyloid formation, and human disease: a summary of progress over the last decade, Annu. Rev. Biochem., 86, 27-68.
Knowles, T. P. J., Vendruscolo, M., and Dobson, C. M. (2014) The amyloid state and its association with protein misfolding diseases, Nat. Rev. Mol. Cell Biol., 15, 384-396.
Pepys, M. B. (2006) Amyloidosis, Annu. Rev. Med., 57, 223-241.
Chiti, F., and Dobson, C. M. (2006) Protein misfolding, functional amyloid, and human disease, Annu. Rev. Biochem., 75, 333-366.
Andersson, K., Olofsson, A., Nielsen, E. H., Svehag, S.-E., and Lundgren, E. (2002) Only amyloidogenic intermediates of transthyretin induce apoptosis, Biochem. Biophys. Res. Commun., 294, 309-314.
Hartley, D. M., Walsh, D. M., Ye, C. P., Diehl, T., Vasquez, S., Vassilev, P. M., Teplow, D. B., and Selkoe, D. J. (1999) Protofibrillar intermediates of amyloid β-protein induce acute electrophysiological changes and progressive neurotoxicity in cortical neurons, J. Neurosci., 19, 8876-8884.
Shi, J., Guan, J., Jiang, B., Brenner, D. A., del Monte, F., et al. (2010) Amyloidogenic light chains induce cardiomyocyte contractile dysfunction and apoptosis via a non-canonical p38 MAPK pathway, Proc. Natl. Acad. Sci. USA, 107, 4188-4193.
Simoneau, S., Rezaei, H., Salès, N., Kaiser-Schulz, G., Lefebvre-Roque, M., Vidal, C., Fournier, J.-G., Comte, J., Wopfner, F., Grosclaude, J., Schätzl, H., and Lasmézas, C. I. (2007) In vitro and in vivo neurotoxicity of prion protein oligomers, PLoS Pathog., 3, e125.
Campioni, S., Mannini, B., Zampagni, M., Pensalfini, A., Parrini, C., Evangelisti, E., Relini, A., Stefani, M., Dobson, C. M., Cecchi, C., and Chiti, F. (2010) A causative link between the structure of aberrant protein oligomers and their toxicity, Nat. Chem. Biol., 6, 140-147.
Milani, P., Merlini, G., and Palladini, G. (2018) Light chain amyloidosis, Infect. Dis., 10, e2018022.
Merlini, G., Dispenzieri, A., Sanchorawala, V., Schönland, S. O., Palladini, G., Hawkins, P. N., and Gertz, M. A. (2018) Systemic immunoglobulin light chain amyloidosis, Nat. Rev. Dis. Prim., 4, 38.
Nasr, S. H., Said, S. M., Valeri, A. M., Sethi, S., Fidler, M. E., et al. (2013) The diagnosis and characteristics of renal heavy-chain and heavy/light-chain amyloidosis and their comparison with renal light-chain amyloidosis, Kidney Int., 83, 463-470.
Ando, Y., Coelho, T., Berk, J. L., Cruz, M. W., Ericzon, B.-G., et al. (2013) Guideline of transthyretin-related hereditary amyloidosis for clinicians, Orphanet J. Rare Dis., 8, 31.
Lu, J., Yu, Y., Zhu, I., Cheng, Y., and Sun, P. D. (2014) Structural mechanism of serum amyloid A-mediated inflammatory amyloidosis, Proc. Natl. Acad. Sci., 111, 5189-5194.
Morris, A. D., Smith, R. N., and Stone, J. R. (2019) The pathology and changing epidemiology of dialysis-related cardiac beta-2 microglobulin amyloidosis, Cardiovasc. Pathol., 42, 30-35.
Eriksson, M., Schönland, S., Yumlu, S., Hegenbart, U., von Hutten, H., Gioeva, Z., Lohse, P., Büttner, J., Schmidt, H., and Röcken, C. (2009) Hereditary apolipoprotein AI-associated amyloidosis in surgical pathology specimens, J. Mol. Diagnostics, 11, 257-262.
Gerasimova, E. M., Fedotov, S. A., Kachkin, D. V., Vashukova, E. S., Glotov, A. S., Chernoff, Y. O., and Rubel, A. A. (2019) Protein misfolding during pregnancy: new approaches to preeclampsia diagnostics, Int. J. Mol. Sci., 20, 6183.
Srinivasan, R., Jones, E. M., Liu, K., Ghiso, J., Marchant, R. E., and Zagorski, M. G. (2003) pH-dependent amyloid and protofibril formation by the ABri peptide of familial British dementia, J. Mol. Biol., 333, 1003-1023.
Ghiso, J. A., Holton, J., Miravalle, L., Calero, M., Lashley, T., Vidal, R., Houlden, H., Wood, N., Neubert, T. A., Rostagno, A., Plant, G., Révész, T., and Frangione, B. (2001) Systemic amyloid deposits in familial British dementia, J. Biol. Chem., 276, 43909-43914.
Srinivasan, R., Marchant, R. E., and Zagorski, M. G. (2004) ABri peptide associated with familial British dementia forms annular and ring-like protofibrillar structures, Amyloid, 11, 10-13.
Coomaraswamy, J., Kilger, E., Wölfing, H., Schäfer, C., Kaeser, S. A., et al. (2010) Modeling familial Danish dementia in mice supports the concept of the amyloid hypothesis of Alzheimer’s disease, Proc. Natl. Acad. Sci. USA, 107, 7969-7974.
Vidal, R., Revesz, T., Rostagno, A., Kim, E., Holton, J. L., Bek, T., Bojsen-Moller, M., Braendgaard, H., Plant, G., Ghiso, J., and Frangione, B. (2000) A decamer duplication in the 3′ region of the BRI gene originates an amyloid peptide that is associated with dementia in a Danish kindred, Proc. Natl. Acad. Sci. USA, 97, 4920-4925.
Davies, H. A., Wilkinson, M. C., Gibson, R. P., and Middleton, D. A. (2014) Expression and purification of the aortic amyloid polypeptide medin, Protein Expr. Purif., 98, 32-37.
Khurana, R., Agarwal, A., Bajpai, V. K., Verma, N., Sharma, A. K., Gupta, R. P., and Madhusudan, K. P. (2004) Unraveling the amyloid associated with human medullary thyroid carcinoma, Endocrinology, 145, 5465-5470.
Pedrote, M. M., Motta, M. F., Ferretti, G. D. S., Norberto, D. R., Spohr, T. C. L. S., Lima, F. R. S., Gratton, E., Silva, J. L., and de Oliveira, G. A. P. (2020) Oncogenic gain of function in glioblastoma is linked to mutant p53 amyloid oligomers, iScience, 23, 100820.
Levine, S. N., Ishaq, S., Nanda, A., Wilson, J. D., and Gonzalez-Toledo, E. (2013) Occurrence of extensive spherical amyloid deposits in a prolactin-secreting pituitary macroadenoma: a radiologic-pathologic correlation, Ann. Diagn. Pathol., 17, 361-366.
Chuang, E., Hori, A. M., Hesketh, C. D., and Shorter, J. (2018) Amyloid assembly and disassembly, J. Cell Sci., 131, jcs189928.
Millucci, L., Ghezzi, L., Bernardini, G., Braconi, D., Tanganelli, P., and Santucci, A. (2012) Prevalence of isolated atrial amyloidosis in young patients affected by congestive heart failure, Sci. World J., 2012, 1-8.
Linke, R. P., Joswig, R., Murphy, C. L., Wang, S., Zhou, H., Gross, U., Rocken, C., Westermark, P., Weiss, D. T., and Solomon, A. (2005) Senile seminal vesicle amyloid is derived from semenogelin I, J. Lab. Clin. Med., 145, 187-193.
Yanamandra, K., Alexeyev, O., Zamotin, V., Srivastava, V., Shchukarev, A., et al. (2009) Amyloid formation by the pro-inflammatory S100A8/A9 proteins in the ageing prostate, PLoS One, 4, e5562.
Caubet, C., Bousset, L., Clemmensen, O., Sourigues, Y., Bygum, A., et al. (2010) A new amyloidosis caused by fibrillar aggregates of mutated corneodesmosin, FASEB J., 24, 3416-3426.
Miura, Y., Harumiya, S., Ono, K., Fujimoto, E., Akiyama, M., Fujii, N., Kawano, H., Wachi, H., and Tajima, S. (2013) Galectin-7 and actin are components of amyloid deposit of localized cutaneous amyloidosis, Exp. Dermatol., 22, 36-40.
Inoue, K., Takahashi, M., Hamamoto, Y., Muto, M., and Ishihara, T. (2000) An immunohistochemical study of cytokeratins in skin-limited amyloidosis, Amyloid, 7, 259-265.
Ozawa, D., Kaji, Y., Yagi, H., Sakurai, K., Kawakami, T., Naiki, H., and Goto, Y. (2011) Destruction of amyloid fibrils of keratoepithelin peptides by laser irradiation coupled with amyloid-specific thioflavin T, J. Biol. Chem., 286, 10856-10863.
Ando, Y., Nakamura, M., Kai, H., Katsuragi, S., Terazaki, H., Nozawa, T., Okuda, T., Misumi, S., Matsunaga, N., Hata, K., Tajiri, T., Shoji, S., Yamashita, T., Haraoka, K., Obayashi, K., Matsumoto, K., Ando, M., and Uchino, M. (2002) A novel localized amyloidosis associated with lactoferrin in the cornea, Lab. Invest., 82, 757-766.
Gupta, Y., Singla, G., and Singla, R. (2015) Insulin-derived amyloidosis, Indian J. Endocrinol. Metab., 19, 174.
D’Souza, A., Theis, J. D., Vrana, J. A., and Dogan, A. (2014) Pharmaceutical amyloidosis associated with subcutaneous insulin and enfuvirtide administration, Amyloid, 21, 71-75.
Chen, G., Xu, T., Yan, Y., Zhou, Y., Jiang, Y., Melcher, K., and Xu, H. E. (2017) Amyloid beta: structure, biology and structure-based therapeutic development, Acta Pharmacol. Sin., 38, 1205-1235.
Murphy, M. P., and LeVine, H. (2010) Alzheimer’s disease and the amyloid-β peptide, J. Alzheimer’s Dis., 19, 311-323.
Qiang, W., Yau, W.-M., Lu, J.-X., Collinge, J., and Tycko, R. (2017) Structural variation in amyloid-β fibrils from Alzheimer’s disease clinical subtypes, Nature, 541, 217-221.
Giasson, B. I., Lee, V. M.-Y., and Trojanowski, J. Q. (2003) Interactions of amyloidogenic proteins, Neuromolecular Med., 4, 49-58.
Lim, S., Haque, M. M., Kim, D., Kim, D. J., and Kim, Y. K. (2014) Cell-based models to investigate Tau aggregation, Comput. Struct. Biotechnol. J., 12, 7-13.
Kim, W. S., Kågedal, K., and Halliday, G. M. (2014) Alpha-synuclein biology in Lewy body diseases, Alzheimer’s Res. Ther., 6, 73.
Goedert, M., Jakes, R., and Spillantini, M. G. (2017) The synucleinopathies: twenty years on, J. Parkinson’s Dis., 7, S51-S69.
Mead, S., and Reilly, M. M. (2015) A new prion disease: relationship with central and peripheral amyloidoses, Nat. Rev. Neurol., 11, 90-97.
Pansarasa, O., Bordoni, M., Diamanti, L., Sproviero, D., Gagliardi, S., and Cereda, C. (2018) SOD1 in amyotrophic lateral sclerosis: “ambivalent” behavior connected to the disease, Int. J. Mol. Sci., 19, 1345.
Dragoš, A., Kovács, Á. T., and Claessen, D. (2017) The role of functional amyloids in multicellular growth and development of gram-positive bacteria, Biomolecules, 7, 60.
Valsecchi, I., Dupres, V., Stephen-Victor, E., Guijarro, J. I., Gibbons, J., Beau, R., Bayry, J., Coppee, J.-Y., Lafont, F., Latgé, J.-P., and Beauvais, A. (2017) Role of hydrophobins in Aspergillus fumigatus, J. Fungi, 4, 2.
Pham, C. L. L., Rey, A., Lo, V., Soulès, M., Ren, Q., Meisl, G., Knowles, T. P. J., Kwan, A. H., and Sunde, M. (2016) Self-assembly of MPG1, a hydrophobin protein from the rice blast fungus that forms functional amyloid coatings, occurs by a surface-driven mechanism, Sci. Rep., 6, 25288.
Lo, V. C., Ren, Q., Pham, C. L. L., Morris, V. K., Kwan, A. H., and Sunde, M. (2014) Fungal hydrophobin proteins produce self-assembling protein films with diverse structure and chemical stability, Nanomaterials, 4, 827-843.
Morris, V. K., Ren, Q., Macindoe, I., Kwan, A. H., Byrne, N., and Sunde, M. (2011) Recruitment of class I hydrophobins to the air:water interface initiates a multi-step process of functional amyloid formation, J. Biol. Chem., 286, 15955-15963.
Rauceo, J. M., Gaur, N. K., Lee, K.-G., Edwards, J. E., Klotz, S. A., and Lipke, P. N. (2004) Global cell surface conformational shift mediated by a Candida albicans adhesin, Infect. Immun., 72, 4948-4955.
Otoo, H. N., Lee, K. G., Qiu, W., and Lipke, P. N. (2008) Candida albicans Als adhesins have conserved amyloid-forming sequences, Eukaryot. Cell, 7, 776-782.
Dueholm, M. S., Albertsen, M., Otzen, D., and Nielsen, P. H. (2012) Curli functional amyloid systems are phylogenetically widespread and display large diversity in operon and protein structure, PLoS One, 7, e51274.
Blanco, L. P., Evans, M. L., Smith, D. R., Badtke, M. P., and Chapman, M. R. (2012) Diversity, biogenesis and function of microbial amyloids, Trends Microbiol., 20, 66-73.
Taglialegna, A., Navarro, S., Ventura, S., Garnett, J. A., Matthews, S., Penades, J. R., Lasa, I., and Valle, J. (2016) Staphylococcal Bap proteins build amyloid scaffold biofilm matrices in response to environmental signals, PLoS Pathog., 12, e1005711.
Alteri, C. J., Xicohténcatl-Cortes, J., Hess, S., Caballero-Olín, G., Girón, J. A., and Friedman, R. L. (2007) Mycobacterium tuberculosis produces pili during human infection, Proc. Natl. Acad. Sci. USA, 104, 5145-5150.
Besingi, R. N., Wenderska, I. B., Senadheera, D. B., Cvitkovitch, D. G., Long, J. R., Wen, Z. T., and Brady, L. J. (2017) Functional amyloids in Streptococcus mutans, their use as targets of biofilm inhibition and initial characterization of SMU_63c, Microbiology, 163, 488-501.
Collinson, S. K., Emödy, L., Müller, K. H., Trust, T. J., and Kay, W. W. (1991) Purification and characterization of thin, aggregative fimbriae from Salmonella enteritidis, J. Bacteriol., 173, 4773-4781.
Doran, J. L., Collinson, S. K., Burian, J., Sarlós, G., Todd, E. C., Munro, C. K., Kay, C. M., Banser, P. A., Peterkin, P. I., and Kay, W. W. (1993) DNA-based diagnostic tests for Salmonella species targeting agfA, the structural gene for thin, aggregative fimbriae, J. Clin. Microbiol., 31, 2263-2273.
Wang, Y., Jiang, J., Gao, Y., Sun, Y., Dai, J., Wu, Y., Qu, D., Ma, G., and Fang, X. (2018) Staphylococcus epidermidis small basic protein (Sbp) forms amyloid fibrils, consistent with its function as a scaffolding protein in biofilms, J. Biol. Chem., 293, 14296-14311.
Ling, S., Li, C., Adamcik, J., Shao, Z., Chen, X., and Mezzenga, R. (2014) Modulating materials by orthogonally oriented β-strands: composites of amyloid and silk fibroin fibrils, Adv. Mater., 26, 4569-4574.
Humenik, M., Smith, A. M., Arndt, S., and Scheibel, T. (2015) Ion and seed dependent fibril assembly of a spidroin core domain, J. Struct. Biol., 191, 130-138.
Berthelot, K., Lecomte, S., Estevez, Y., Coulary-Salin, B., and Peruch, F. (2014) Homologous Hevea brasiliensis REF (Hevb1) and SRPP (Hevb3) present different auto-assembling, Biochim. Biophys. Acta, 1844, 473-485.
Berthelot, K., Lecomte, S., Estevez, Y., Coulary-Salin, B., Bentaleb, A., Cullin, C., Deffieux, A., and Peruch, F. (2012) Rubber elongation factor (REF), a major allergen component in Hevea brasiliensis latex has amyloid properties, PLoS One, 7, e48065.
Li, J., McQuade, T., Siemer, A. B., Napetschnig, J., Moriwaki, K., Hsiao, Y.-S., Damko, E., Moquin, D., Walz, T., McDermott, A., Chan, F. K.-M., and Wu, H. (2012) The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis, Cell, 150, 339-350.
Wu, X.-N., Yang, Z.-H., Wang, X.-K., Zhang, Y., Wan, H., Song, Y., Chen, X., Shao, J., and Han, J. (2014) Distinct roles of RIP1-RIP3 hetero- and RIP3-RIP3 homo-interaction in mediating necroptosis, Cell Death Differ., 21, 1709-1720.
Kajava, A. V., Klopffleisch, K., Chen, S., and Hofmann, K. (2014) Evolutionary link between metazoan RHIM motif and prion-forming domain of fungal heterokaryon incompatibility factor HET-s/HET-s, Sci. Rep., 4, 7436.
Liebman, S. W., and Chernoff, Y. O. (2012) Prions in yeast, Genetics, 191, 1041-1072.
Seuring, C., Greenwald, J., Wasmer, C., Wepf, R., Saupe, S. J., Meier, B. H., and Riek, R. (2012) The mechanism of toxicity in HET-S/HET-s prion incompatibility, PLoS Biol., 10, e1001451.
Loquet, A., and Saupe, S. J. (2017) Diversity of amyloid motifs in NLR signaling in fungi, Biomolecules, 7, 38.
Daskalov, A., Habenstein, B., Sabaté, R., Berbon, M., Martinez, D., Chaignepain, S., Coulary-Salin, B., Hofmann, K., Loquet, A., and Saupe, S. J. (2016) Identification of a novel cell death-inducing domain reveals that fungal amyloid-controlled programmed cell death is related to necroptosis, Proc. Natl. Acad. Sci. USA, 113, 2720-2725.
Kleino, A., Ramia, N. F., Bozkurt, G., Shen, Y., Nailwal, H., Huang, J., Napetschnig, J., Gangloff, M., Chan, F. K.-M., Wu, H., Li, J., and Silverman, N. (2017) Peptidoglycan-sensing receptors trigger the formation of functional amyloids of the adaptor protein Imd to initiate Drosophila NF-κB signaling, Immunity, 47, 635-647.e6.
Gonçalves, A. P., Heller, J., Daskalov, A., Videira, A., and Glass, N. L. (2017) Regulated forms of cell death in fungi, Front. Microbiol., 8, 1837.
Hafner-Bratkovič, I. (2017) Prions, prionoid complexes and amyloids: the bad, the good and something in between, Swiss Med. Wkly., 147, w14424.
Mojsoska, B., and Jenssen, H. (2015) Peptides and peptidomimetics for antimicrobial drug design, Pharmaceuticals (Basel), 8, 366-415.
Lyu, Y., Fitriyanti, M., and Narsimhan, G. (2019) Nucleation and growth of pores in 1,2-Dimyristoyl-sn-glycero-3-phosphocholine (DMPC)/cholesterol bilayer by antimicrobial peptides melittin, its mutants and cecropin P1, Colloids Surf. B Biointerfaces, 173, 121-127.
Gazit, E., Miller, I. R., Biggin, P. C., Sansom, M. S., and Shai, Y. (1996) Structure and orientation of the mammalian antibacterial peptide cecropin P1 within phospholipid membranes, J. Mol. Biol., 258, 860-870.
Sood, R., Domanov, Y., Pietiäinen, M., Kontinen, V. P., and Kinnunen, P. K. J. (2008) Binding of LL-37 to model biomembranes: insight into target vs host cell recognition, Biochim. Biophys. Acta, 1778, 983-996.
Engelberg Y., and Landau, M. (2020) The human LL-37(17-29) antimicrobial peptide reveals a functional supramolecular nanostructure, bioRxiv, doi: https://doi.org/10.1101/2020.02.04.933432 .
Auvynet, C., El Amri, C., Lacombe, C., Bruston, F., Bourdais, J., Nicolas, P., and Rosenstein, Y. (2008) Structural requirements for antimicrobial versus chemoattractant activities for dermaseptin S9, FEBS J., 275, 4134-4151.
Caillon, L., Killian, J. A., Lequin, O., and Khemtémourian, L. (2013) Biophysical investigation of the membrane-disrupting mechanism of the antimicrobial and amyloid-like peptide dermaseptin S9, PLoS One, 8, e75528.
Gössler-Schöfberger, R., Hesser, G., Muik, M., Wechselberger, C., and Jilek, A. (2009) An orphan dermaseptin from frog skin reversibly assembles to amyloid-like aggregates in a pH-dependent fashion, FEBS J., 276, 5849-5859.
Zasloff, M. (1987) Magainins, a class of antimicrobial peptides from Xenopus skin: isolation, characterization of two active forms, and partial cDNA sequence of a precursor, Proc. Natl. Acad. Sci. USA, 84, 5449-5453.
Zasloff, M., Martin, B., and Chen, H. C. (1988) Antimicrobial activity of synthetic magainin peptides and several analogues, Proc. Natl. Acad. Sci. USA, 85, 910-913.
Cruciani, R. A., Barker, J. L., Zasloff, M., Chen, H. C., and Colamonici, O. (1991) Antibiotic magainins exert cytolytic activity against transformed cell lines through channel formation, Proc. Natl. Acad. Sci. USA, 88, 3792-3796.
Baker, M. A., Maloy, W. L., Zasloff, M., and Jacob, L. S. (1993) Anticancer efficacy of Magainin2 and analogue peptides, Cancer Res., 53, 3052-3057.
Ludtke, S., He, K., and Huang, H. (1995) Membrane thinning caused by magainin 2, Biochemistry, 34, 16764-16769.
Jang, H., Arce, F. T., Mustata, M., Ramachandran, S., Capone, R., Nussinov, R., and Lal, R. (2011) Antimicrobial protegrin-1 forms amyloid-like fibrils with rapid kinetics suggesting a functional link, Biophys. J., 100, 1775-1783.
Gour, S., Kumar, V., Singh, A., Gadhave, K., Goyal, P., Pandey, J., Giri, R., and Yadav, J. K. (2019) Mammalian antimicrobial peptide protegrin-4 self assembles and forms amyloid-like aggregates: Assessment of its functional relevance, J. Pept. Sci., 25, e3151.
Bieler, S., Estrada, L., Lagos, R., Baeza, M., Castilla, J., and Soto, C. (2005) Amyloid formation modulates the biological activity of a bacterial protein, J. Biol. Chem., 280, 26880-26885.
Chu, H., Pazgier, M., Jung, G., Nuccio, S.-P., Castillo, P. A., de Jong, M. F., Winter, M. G., Winter, S. E., Wehkamp, J., Shen, B., Salzman, N. H., Underwood, M. A., Tsolis, R. M., Young, G. M., Lu, W., Lehrer, R. I., Bäumler, A. J., and Bevins, C. L. (2012) Human α-defensin 6 promotes mucosal innate immunity through self-assembled peptide nanonets, Science, 337, 477-481.
Gour, S., Kaushik, V., Kumar, V., Bhat, P., Yadav, S. C., and Yadav, J. K. (2016) Antimicrobial peptide (Cn-AMP2) from liquid endosperm of Cocos nucifera forms amyloid-like fibrillar structure, J. Pept. Sci., 22, 201-207.
Wang, G. (2008) Structures of human host defense cathelicidin LL-37 and its smallest antimicrobial peptide KR-12 in lipid micelles, J. Biol. Chem., 283, 32637-32643.
Fahrner, R. L., Dieckmann, T., Harwig, S. S., Lehrer, R. I., Eisenberg, D., and Feigon, J. (1996) Solution structure of protegrin-1, a broad-spectrum antimicrobial peptide from porcine leukocytes, Chem. Biol., 3, 543-550.
De Felice, F. G., Vieira, M. N. N., Meirelles, M. N. L., Morozova-Roche, L. A., Dobson, C. M., and Ferreira, S. T. (2004) Formation of amyloid aggregates from human lysozyme and its disease-associated variants using hydrostatic pressure, FASEB J., 18, 1099-1101.
Majumdar, A., Cesario, W. C., White-Grindley, E., Jiang, H., Ren, F., Khan, M. R., Li, L., Choi, E. M.-L., Kannan, K., Guo, F., Unruh, J., Slaughter, B., and Si, K. (2012) Critical role of amyloid-like oligomers of Drosophila Orb2 in the persistence of memory, Cell, 148, 515-529.
Si, K., Choi, Y.-B., White-Grindley, E., Majumdar, A., and Kandel, E. R. (2010) Aplysia CPEB can form prion-like multimers in sensory neurons that contribute to long-term facilitation, Cell, 140, 421-435.
Stephan, J. S., Fioriti, L., Lamba, N., Colnaghi, L., Karl, K., Derkatch, I. L., and Kandel, E. R. (2015) The CPEB3 protein is a functional prion that interacts with the actin cytoskeleton, Cell Rep., 11, 1772-1785.
Heinrich, S. U., and Lindquist, S. (2011) Protein-only mechanism induces self-perpetuating changes in the activity of neuronal Aplysia cytoplasmic polyadenylation element binding protein (CPEB), Proc. Natl. Acad. Sci. USA, 108, 2999-3004.
Hervas, R., Rau, M. J., Park, Y., Zhang, W., Murzin, A. G., Fitzpatrick, J. A. J., Scheres, S. H. W., and Si, K. (2020) Cryo-EM structure of a neuronal functional amyloid implicated in memory persistence in Drosophila, Science, 367, 1230-1234.
Sopova, J. V., Koshel, E. I., Belashova, T. A., Zadorsky, S. P., Sergeeva, A. V., Siniukova, V. A., Shenfeld, A. A., Velizhanina, M. E., Volkov, K. V., Nizhnikov, A. A., Kachkin, D. V., Gaginskaya, E. R., and Galkin, A. P. (2019) RNA-binding protein FXR1 is presented in rat brain in amyloid form, Sci. Rep., 9, 18983.
Caudron, F., and Barral, Y. (2013) A super-assembly of Whi3 encodes memory of deceptive encounters by single cells during yeast courtship, Cell, 155, 1244-1257.
Boke, E., Ruer, M., Wühr, M., Coughlin, M., Lemaitre, R., Gygi, S. P., Alberti, S., Drechsel, D., Hyman, A. A., and Mitchison, T. J. (2016) Amyloid-like self-assembly of a cellular compartment, Cell, 166, 637-650.
Iconomidou, V. A., Vriend, G., and Hamodrakas, S. J. (2000) Amyloids protect the silkmoth oocyte and embryo, FEBS Lett., 479, 141-145.
Iconomidou, V. A., Chryssikos, G. D., Gionis, V., Vriend, G., Hoenger, A., and Hamodrakas, S. J. (2001) Amyloid-like fibrils from an 18-residue peptide analogue of a part of the central domain of the B-family of silkmoth chorion proteins, FEBS Lett., 499, 268-273.
Iconomidou, V. A., Chryssikos, G. D., Gionis, V., Galanis, A. S., Cordopatis, P., Hoenger, A., and Hamodrakas, S. J. (2006) Amyloid fibril formation propensity is inherent into the hexapeptide tandemly repeating sequence of the central domain of silkmoth chorion proteins of the A-family, J. Struct. Biol., 156, 480-488.
Hamodrakas, S. J., Hoenger, A., and Iconomidou, V. A. (2004) Amyloid fibrillogenesis of silkmoth chorion protein peptide-analogues via a liquid-crystalline intermediate phase, J. Struct. Biol., 145, 226-235.
Louros, N. N., Petronikolou, N., Karamanos, T., Cordopatis, P., Iconomidou, V. A., and Hamodrakas, S. J. (2014) Structural studies of “aggregation-prone” peptide-analogues of teleostean egg chorion ZPB proteins, Biopolymers, 102, 427-436.
Egge, N., Muthusubramanian, A., and Cornwall, G. A. (2015) Amyloid properties of the mouse egg zona pellucida, PLoS One, 10, e0129907.
Louros, N. N., Chrysina, E. D., Baltatzis, G. E., Patsouris, E. S., Hamodrakas, S. J., and Iconomidou, V. A. (2016) A common “aggregation-prone” interface possibly participates in the self-assembly of human zona pellucida proteins, FEBS Lett., 590, 619-630.
Carpenter, K., Bell, R. B., Yunus, J., Amon, A., and Berchowitz, L. E. (2018) Phosphorylation-mediated clearance of amyloid-like assemblies in meiosis, Dev. Cell, 45, 392-405.e6.
Fowler, D. M., Koulov, A. V., Alory-Jost, C., Marks, M. S., Balch, W. E., and Kelly, J. W. (2006) Functional amyloid formation within mammalian tissue, PLoS Biol., 4, e6.
Hoashi, T., Muller, J., Vieira, W. D., Rouzaud, F., Kikuchi, K., Tamaki, K., and Hearing, V. J. (2006) The repeat domain of the melanosomal matrix protein PMEL17/GP100 is required for the formation of organellar fibers, J. Biol. Chem., 281, 21198-21208.
Watt, B., van Niel, G., Fowler, D. M., Hurbain, I., Luk, K. C., Stayrook, S. E., Lemmon, M. A., Raposo, G., Shorter, J., Kelly, J. W., and Marks, M. S. (2009) N-terminal domains elicit formation of functional Pmel17 amyloid fibrils, J. Biol. Chem., 284, 35543-35555.
McGlinchey, R. P., Shewmaker, F., McPhie, P., Monterroso, B., Thurber, K., and Wickner, R. B. (2009) The repeat domain of the melanosome fibril protein Pmel17 forms the amyloid core promoting melanin synthesis, Proc. Natl. Acad. Sci. USA, 106, 13731-13736.
Leonhardt, R. M., Vigneron, N., Hee, J. S., Graham, M., and Cresswell, P. (2013) Critical residues in the PMEL/Pmel17 N-terminus direct the hierarchical assembly of melanosomal fibrils, Mol. Biol. Cell, 24, 964-981.
Maji, S. K., Perrin, M. H., Sawaya, M. R., Jessberger, S., Vadodaria, K., Rissman, R. A., Singru, P. S., Nilsson, K. P. R., Simon, R., Schubert, D., Eisenberg, D., Rivier, J., Sawchenko, P., Vale, W., and Riek, R. (2009) Functional amyloids as natural storage of peptide hormones in pituitary secretory granules, Science, 325, 328-332.
Onoue, S., Ohshima, K., Debari, K., Koh, K., Shioda, S., Iwasa, S., Kashimoto, K., and Yajima, T. (2004) Mishandling of the therapeutic peptide glucagon generates cytotoxic amyloidogenic fibrils, Pharm. Res., 21, 1274-1283.
Gul, S., Bahadir, B., Dusak, A., Kalayci, M., Edebali, N., and Acikgoz, B. (2009) Spherical amyloid deposition in a prolactin-producing pituitary adenoma, Neuropathology, 29, 81-84.
Wickner, R. B., Edskes, H. K., Kryndushkin, D., McGlinchey, R., Bateman, D., and Kelly, A. (2011) Prion diseases of yeast: amyloid structure and biology, Semin. Cell Dev. Biol., 22, 469-475.
Newby, G. A., and Lindquist, S. (2013) Blessings in disguise: biological benefits of prion-like mechanisms, Trends Cell Biol., 23, 251-259.
Cox, B. S. (1965) ψ, a cytoplasmic suppressor of super-suppressor in yeast, Heredity, 20, 505-521, doi: https://doi.org/10.1038/hdy.1965.65 .
King, C.-Y., and Diaz-Avalos, R. (2004) Protein-only transmission of three yeast prion strains, Nature, 428, 319-323.
Sondheimer, N., and Lindquist, S. (2000) Rnq1: an epigenetic modifier of protein function in yeast, Mol. Cell, 5, 163-172.
Derkatch, I. L., Bradley, M. E., Hong, J. Y., and Liebman, S. W. (2001) Prions affect the appearance of other prions: the story of [PIN+], Cell, 106, 171-182.
Du, Z., Park, K.-W., Yu, H., Fan, Q., and Li, L. (2008) Newly identified prion linked to the chromatin-remodeling factor Swi1 in Saccharomyces cerevisiae, Nat. Genet., 40, 460-465.
Saifitdinova, A. F., Nizhnikov, A. A., Lada, A. G., Rubel, A. A., Magomedova, Z. M., Ignatova, V. V., Inge-Vechtomov, S. G., and Galkin, A. P. (2010) [NSI+]: a novel non-Mendelian nonsense suppressor determinant in Saccharomyces cerevisiae, Curr. Genet., 56, 467-478.
Nizhnikov, A. A., Ryzhova, T. A., Volkov, K. V., Zadorsky, S. P., Sopova, J. V., Inge-Vechtomov, S. G., and Galkin, A. P. (2016) Interaction of prions causes heritable traits in Saccharomyces cerevisiae, PLoS Genet., 12, e1006504.
Suzuki, G., Shimazu, N., and Tanaka, M. (2012) A yeast prion, Mod5, promotes acquired drug resistance and cell survival under environmental stress, Science, 336, 355-359.
Patel, B. K., Gavin-Smyth, J., and Liebman, S. W. (2009) The yeast global transcriptional co-repressor protein Cyc8 can propagate as a prion, Nat. Cell Biol., 11, 344-349.
Holmes, D. L., Lancaster, A. K., Lindquist, S., and Halfmann, R. (2013) Heritable remodeling of yeast multicellularity by an environmentally responsive prion, Cell, 153, 153-165.
Halfmann, R., Wright, J. R., Alberti, S., Lindquist, S., and Rexach, M. (2012) Prion formation by a yeast GLFG nucleoporin, Prion, 6, 391-399.
Chernova, T. A., Kiktev, D. A., Romanyuk, A. V., Shanks, J. R., Laur, O., Ali, M., Ghosh, A., Kim, D., Yang, Z., Mang, M., Chernoff, Y. O., and Wilkinson, K. D. (2017) Yeast short-lived actin-associated protein forms a metastable prion in response to thermal stress, Cell Rep., 18, 751-761.
Chernova, T. A., Romanyuk, A. V., Karpova, T. S., Shanks, J. R., Ali, M., Moffatt, N., Howie, R. L., O’Dell, A., McNally, J. G., Liebman, S. W., Chernoff, Y. O., and Wilkinson, K. D. (2011) Prion induction by the short-lived, stress-induced protein Lsb2 is regulated by ubiquitination and association with the actin cytoskeleton, Mol. Cell, 43, 242-252.
Antonets, K. S., Belousov, M. V., Belousova, M. E., and Nizhnikov, A. A. (2019) The Gln3 transcriptional regulator of Saccharomyces cerevisiae manifests prion-like properties upon overproduction, Biochemistry (Moscow), 84, 441-451.
Rogoza, T., Goginashvili, A., Rodionova, S., Ivanov, M., Viktorovskaya, O., Rubel, A., Volkov, K., and Mironova, L. (2010) Non-Mendelian determinant [ISP+] in yeast is a nuclear-residing prion form of the global transcriptional regulator Sfp1, Proc. Natl. Acad. Sci. USA, 107, 10573-10577.
Drozdova, P., Mironova, L., and Zhouravleva, G. (2016) Haploid yeast cells undergo a reversible phenotypic switch associated with chromosome II copy number, BMC Genet., 17, 152.
Matveenko, A. G., Drozdova, P. B., Belousov, M. V., Moskalenko, S. E., Bondarev, S. A., Barbitoff, Y. A., Nizhnikov, A. A., and Zhouravleva, G. A. (2016) SFP1 – mediated prion-dependent lethality is caused by increased Sup35 aggregation and alleviated by Sis1, Genes Cells, 21, 1290-1308.
Brown, J. C. S., and Lindquist, S. (2009) A heritable switch in carbon source utilization driven by an unusual yeast prion, Genes Dev., 23, 2320-2332.
Chakravarty, A. K., Smejkal, T., Itakura, A. K., Garcia, D. M., and Jarosz, D. F. (2020) A non-amyloid prion particle that activates a heritable gene expression program, Mol. Cell, 77, 251-265.e9.
Chakrabortee, S., Byers, J. S., Jones, S., Garcia, D. M., Bhullar, B., Chang, A., She, R., Lee, L., Fremin, B., Lindquist, S., and Jarosz, D. F. (2016) Intrinsically disordered proteins drive emergence and inheritance of biological traits, Cell, 167, 369-381.e12.
Franzmann, T. M., Jahnel, M., Pozniakovsky, A., Mahamid, J., Holehouse, A. S., Nüske, E., Richter, D., Baumeister, W., Grill, S. W., Pappu, R. V., Hyman, A. A., and Alberti, S. (2018) Phase separation of a yeast prion protein promotes cellular fitness, Science, 359, eaao5654.
Gomes, E., and Shorter, J. (2019) The molecular language of membraneless organelles, J. Biol. Chem., 294, 7115-7127.
Mitrea, D. M., and Kriwacki, R. W. (2016) Phase separation in biology; functional organization of a higher order, Cell Commun. Signal., 14, 1.
Audas, T. E., Audas, D. E., Jacob, M. D., Ho, J. J. D., Khacho, M., et al. (2016) Adaptation to stressors by systemic protein amyloidogenesis, Dev. Cell, 39, 155-168.
Uversky, V. N., Kuznetsova, I. M., Turoverov, K. K., and Zaslavsky, B. (2015) Intrinsically disordered proteins as crucial constituents of cellular aqueous two phase systems and coacervates, FEBS Lett., 589, 15-22.
Uversky, V. N. (2017) Protein intrinsic disorder-based liquid-liquid phase transitions in biological systems: complex coacervates and membrane-less organelles, Adv. Colloid Interface Sci., 239, 97-114.
Uversky, V. N. (2019) Supramolecular fuzziness of intracellular liquid droplets: liquid–liquid phase transitions, membrane-less organelles, and intrinsic disorder, Molecules, 24, 3265.
Turoverov, K. K., Kuznetsova, I. M., Fonin, A. V., Darling, A. L., Zaslavsky, B. Y., and Uversky, V. N. (2019) Stochasticity of biological soft matter: emerging concepts in intrinsically disordered proteins and biological phase separation, Trends Biochem. Sci., 44, 716-728.
Darling, A. L., Liu, Y., Oldfield, C. J., and Uversky, V. N. (2018) Intrinsically disordered proteome of human membrane-less organelles, Proteomics, 18, e1700193.
Uversky, V. N. (2017) Intrinsically disordered proteins in overcrowded milieu: Membrane-less organelles, phase separation, and intrinsic disorder, Curr. Opin. Struct. Biol., 44, 18-30.
Babinchak, W. M., and Surewicz, W. K. (2020) Liquid–liquid phase separation and its mechanistic role in pathological protein aggregation, J. Mol. Biol., 432, 1910-1925.
Darling, A. L., Zaslavsky, B. Y., and Uversky, V. N. (2019) Intrinsic disorder-based emergence in cellular biology: physiological and pathological liquid–liquid phase transitions in cells, Polymers (Basel), 11, 990.
De Oliveira, G. A. P., Cordeiro, Y., Silva, J. L., and Vieira, T. C. R. G. (2019) Liquid–liquid phase transitions and amyloid aggregation in proteins related to cancer and neurodegenerative diseases, Adv. Protein Chem. Struct. Biol., 118, 289-331.
Kato, M., Han, T. W., Xie, S., Shi, K., Du, X., et al. (2012) Cell-free formation of RNA granules: low complexity sequence domains form dynamic fibers within hydrogels, Cell, 149, 753-767.
Gui, X., Luo, F., Li, Y., Zhou, H., Qin, Z., et al. (2019) Structural basis for reversible amyloids of hnRNPA1 elucidates their role in stress granule assembly, Nat. Commun., 10, 2006.
Chen, C., Ding, X., Akram, N., Xue, S., and Luo, S.-Z. (2019) Fused in sarcoma: properties, self-assembly and correlation with neurodegenerative diseases, Molecules, 24, 1622.
Murray, D. T., Kato, M., Lin, Y., Thurber, K. R., Hung, I., McKnight, S. L., and Tycko, R. (2017) Structure of FUS protein fibrils and its relevance to self-assembly and phase separation of low-complexity domains, Cell, 171, 615-627.e16.
Luo, F., Gui, X., Zhou, H., Gu, J., Li, Y., Liu, X., Zhao, M., Li, D., Li, X., and Liu, C. (2018) Atomic structures of FUS LC domain segments reveal bases for reversible amyloid fibril formation, Nat. Struct. Mol. Biol., 25, 341-346.
Fushimi, K., Long, C., Jayaram, N., Chen, X., Li, L., and Wu, J. Y. (2011) Expression of human FUS/TLS in yeast leads to protein aggregation and cytotoxicity, recapitulating key features of FUS proteinopathy, Protein Cell, 2, 141-149.
Chaudhury, A., Chander, P., and Howe, P. H. (2010) Heterogeneous nuclear ribonucleoproteins (hnRNPs) in cellular processes: focus on hnRNP E1’s multifunctional regulatory roles, RNA, 16, 1449-1462.
Kim, H. J., Kim, N. C., Wang, Y.-D., Scarborough, E. A., Moore, J., et al. (2013) Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS, Nature, 495, 467-473.
Prasad, A., Bharathi, V., Sivalingam, V., Girdhar, A., and Patel, B. K. (2019) Molecular mechanisms of TDP-43 misfolding and pathology in amyotrophic lateral sclerosis, Front. Mol. Neurosci., 12, 25.
Wegmann, S., Eftekharzadeh, B., Tepper, K., Zoltowska, K. M., Bennett, R. E., et al. (2018) Tau protein liquid–liquid phase separation can initiate tau aggregation, EMBO J., 37, e98049.
Kosolapova, A. O., Belousov, M. V., Sulatskaya, A. I., Belousova, M. E., Sulatsky, M. I., et al. (2019) Two novel amyloid proteins, RopA and RopB, from the root nodule bacterium Rhizobium leguminosarum, Biomolecules, 9, 694.
Danoff, E. J., and Fleming, K. G. (2015) Aqueous, unfolded OmpA forms amyloid-like fibrils upon self-association, PLoS One, 10, e0132301.
Antonets, K. S., Volkov, K. V., Maltseva, A. L., Arshakian, L. M., Galkin, A. P., and Nizhnikov, A. A. (2016) Proteomic analysis of Escherichia coli protein fractions resistant to solubilization by ionic detergents, Biochemistry (Moscow), 81, 34-46.
Sahaya Rajan, J. J., Chinnappan Santiago, T., Singaravel, R., and Ignacimuthu, S. (2016) Outer membrane protein C (OmpC) of Escherichia coli induces neurodegeneration in mice by acting as an amyloid, Biotechnol. Lett., 38, 689-700.
Villain, E., Nikekhin, A. A., and Kajava, A. V. (2019) Porins and amyloids are coded by similar sequence motifs, Proteomics, 19, e1800075.
Kaur, G., Kaundal, S., Kapoor, S., Grimes, J. M., Huiskonen, J. T., and Thakur, K. G. (2018) Mycobacterium tuberculosis CarD, an essential global transcriptional regulator forms amyloid-like fibrils, Sci. Rep., 8, 10124.
Higurashi, T., Yagi, H., Mizobata, T., and Kawata, Y. (2005) Amyloid-like fibril formation of co-chaperonin GroES: nucleation and extension prefer different degrees of molecular compactness, J. Mol. Biol., 351, 1057-1069.
Giraldo, R. (2007) Defined DNA sequences promote the assembly of a bacterial protein into distinct amyloid nanostructures, Proc. Natl. Acad. Sci. USA, 104, 17388-17393.
Gasset-Rosa, F., Coquel, A.-S., Moreno-Del Álamo, M., Chen, P., Song, X., Serrano, A. M., Fernández-Tresguerres, M. E., Moreno-Díaz de la Espina, S., Lindner, A. B., and Giraldo, R. (2014) Direct assessment in bacteria of prionoid propagation and phenotype selection by Hsp70 chaperone, Mol. Microbiol., 91, 1070-1087.
Fernández-Tresguerres, M. E., Moreno-Díaz de la Espina, S., Gasset-Rosa, F., and Giraldo, R. (2010) A DNA-promoted amyloid proteinopathy in Escherichia coli, Mol. Microbiol., 77, 1456-1469.
Torreira, E., Moreno-Del Álamo, M., Fuentes-Perez, M. E., Fernández, C., Martín-Benito, J., Moreno-Herrero, F., Giraldo, R., and Llorca, O. (2015) Amyloidogenesis of bacterial prionoid RepA-WH1 recapitulates dimer to monomer transitions of RepA in DNA replication initiation, Structure, 23, 183-189.
Belousov, M. V., Bondarev, S. A., Kosolapova, A. O., Antonets, K. S., Sulatskaya, A. I., Sulatsky, M. I., Zhouravleva, G. A., Kuznetsova, I. M., Turoverov, K. K., and Nizhnikov, A. A. (2018) M60-like metalloprotease domain of the Escherichia coli YghJ protein forms amyloid fibrils, PLoS One, 13, e0191317.
Villar-Piqué, A., Sabaté, R., Lopera, O., Gibert, J., Torne, J. M., Santos, M., and Ventura, S. (2010) Amyloid-like protein inclusions in tobacco transgenic plants, PLoS One, 5, e13625.
Hughes, M. P., Sawaya, M. R., Boyer, D. R., Goldschmidt, L., Rodriguez, J. A., Cascio, D., Chong, L., Gonen, T., and Eisenberg, D. S. (2018) Atomic structures of low-complexity protein segments reveal kinked β sheets that assemble networks, Science, 359, 698-701.
Nizhnikov, A. A., Alexandrov, A. I., Ryzhova, T. A., Mitkevich, O. V., Dergalev, A. A., Ter-Avanesyan, M. D., and Galkin, A. P. (2014) Proteomic screening for amyloid proteins, PLoS One, 9, e116003.
Kryndushkin, D., Pripuzova, N., Burnett, B. G., and Shewmaker, F. (2013) Non-targeted identification of prions and amyloid-forming proteins from yeast and mammalian cells, J. Biol. Chem., 288, 27100-27111.
Kayed, R., Head, E., Sarsoza, F., Saing, T., Cotman, C. W., et al. (2007) Fibril specific, conformation dependent antibodies recognize a generic epitope common to amyloid fibrils and fibrillar oligomers that is absent in prefibrillar oligomers, Mol. Neurodegener., 2, 18.
Alberti, S. (2017) The wisdom of crowds: regulating cell function through condensed states of living matter, J. Cell Sci., 130, 2789-2796.
Acknowledgements
The authors express their gratitude to Prof. Dr. Sci. Alexey P. Galkin (St. Petersburg State University; St. Petersburg Branch, Vavilov Institute of General Genetics, Russian Academy of Sciences) for critical reading of the manuscript.
Funding
The study was funded by the Russian Foundation for Basic Research (project no. 19-14-50590).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare no conflict of interest in financial or any other sphere. This article does not contain any studies with human participants or animals performed by any of the authors.
Electronic supplementary material
Electronic supplementary material
Rights and permissions
Open access. This article is distributed under the terms of the Creative Commons Attribution 4.0 International License ( http://creativecommons.org/licenses/by/4.0/ ), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Matiiv, A.B., Trubitsina, N.P., Matveenko, A.G. et al. Amyloid and Amyloid-Like Aggregates: Diversity and the Term Crisis. Biochemistry Moscow 85, 1011–1034 (2020). https://doi.org/10.1134/S0006297920090035
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0006297920090035