
Abstract
The ability to harness innate immunity is a promising solution for improving cancer immunotherapy. Interferon (IFN) induces expression of IFN-stimulated genes (ISGs) by activating the JAK-STAT signaling pathway to promote innate immunity and inhibit malignant tumor growth, but the functions and mechanisms of most ISGs in cancer regulation are unknown. As an innate immune effector, ISG12a promotes the innate immune response to viral infection. In this study, ISG12a was found to be expressed at low levels in gastrointestinal cancer, represented by hepatocellular cancer (HCC) and gastric cancer (GC), and it identified as a tumor suppressor that affects clinical prognosis. ISG12a silencing accelerated the malignant transformation and epithelial–mesenchymal transition of cancer cells. Mechanistically, ISG12a promoted β-catenin proteasomal degradation by inhibiting the degradation of ubiquitinated Axin, thereby suppressing the canonical Wnt/β-catenin signaling pathway. Notably, β-catenin was identified as a transcription factor for PD-L1. Inhibition of Wnt/β-catenin signaling by ISG12a suppressed expression of the immune checkpoint PD-L1, rendering cancer cells sensitive to NK cell-mediated killing. This study reveals a mechanism underlying the anticancer effects of IFN. Some ISGs, as represented by ISG12a, may be useful in cancer therapy and prevention. The identified interrelations among innate immunity, Wnt/β-catenin signaling, and cancer immunity may provide new insight into strategies that will improve the efficiency of immunotherapy.
Similar content being viewed by others


Introduction
Immuno-oncology, which mainly focuses on enhancing T-cell responses, is a great step forward in human cancer therapy. However, due to the limited efficacy of adaptive immunity for cancer therapy, researchers have increasingly focused on the crucial role of innate immunity.1,2 For example, activation of innate immunity may be necessary for T cell activation and accumulation.3 Interferons (IFNs) play important roles in defending against viral infections and cancer as well as in regulating immunity.4,5 Compared with nucleotide analogs, IFNs have more advantages in treating high-risk patients with HBV-related hepatocellular cancer (HCC).6 IFNs function by binding to specific receptors, activating JAK-STAT signaling and inducing expression of IFN-stimulated genes (ISGs),5 but the functions and mechanisms of most ISGs in preventing cancer are unknown. Moreover, the efficiency of chemotherapy, radiotherapy, and immunotherapy for cancer depends on IFN signaling.7,8 IFN signaling diminishes with age and is associated with the efficacy of immune checkpoint blockade in cancer therapy.9 As shown in our previous studies, ISG12a enhances the innate immune response to viral infection,10,11 functioning as an innate immune effector. In addition, ISG12a promotes cancer cell apoptosis in the context of TRAIL treatment.12 Published studies indicate a potential role for ISG12a in treating cancer and promoting cancer immunity.
Aberrations in the canonical Wnt/β-catenin signaling pathway usually cause carcinogenesis and cancer progression.13 Activated β-catenin (also known as CTNNB1) is translocated from the cytoplasm to the nucleus of cancer cells, where it subsequently interacts with the transcription factors TCF/LEF to induce transcription of oncogenes such as Cyclin D1 and c-Myc.14 Otherwise, β-catenin is degraded by the destruction complex composed of Axin, APC, CKIα, and GSK3β via the proteasome. Based on accumulating evidence, aberrant Wnt/β-catenin signaling may subvert cancer immunosurveillance,15 though the underlying mechanisms are unclear. The identification of the effect of the canonical Wnt/β-catenin signaling pathway on immune evasion may lead to progress in cancer immunotherapy.
In the present study, we investigated the regulatory role of ISG12a in cancer and examined the underlying mechanism. Suppression of ISG12a expression accelerates the malignant transformation of cancer cells and leads to a poor prognosis in gastrointestinal cancer, as represented by HCC and gastric cancer (GC). ISG12a promotes cancer immunity by suppressing the canonical Wnt/β-catenin signaling pathway. The results of this study highlight the significance and mechanisms of IFNs and IFN signaling in cancer therapy, which may assist with the development of new strategies designed to improve the efficacy of cancer immunotherapy.
Materials and methods
Cell culture
Human hepatic cell lines HLCZ01 (RRID: CVCL_1J92) and LH86 (RRID: CVCL_8889), and primary human hepatocytes (PHHs) were isolated and stored in our laboratory.16,17 The human hepatic cell line Huh7 (RRID: CVCL_0336) was obtained from the JCRB Cell Bank (Tokyo, Japan). The normal human gastric mucosal cell line GES-1 (RRID: CVCL_EQ22) was preserved in our laboratory. Human gastric cells BGC-823 (RRID: CVCL_1279) and HGC-27 (RRID: CVCL_3360) were obtained from the Stem Cell Bank, Chinese Academy of Sciences (Shanghai, China). The human hepatic cell line HepG2 (RRID: CVCL_0027) and the human embryonic kidney fibroblast line HEK293T (RRID: CVCL_0063) were purchased from ATCC (Manassas, VA, USA). NK cells were isolated from the peripheral blood of healthy individuals. HLCZ01 and LH86 cells were cultured in DMEM/F12 (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% (v/v) fetal bovine serum (FBS) (Thermo Fisher Scientific), penicillin–streptomycin (Thermo Fisher Scientific) and insulin–transferrin–selenium (Thermo Fisher Scientific). Other types of cells were cultivated in DMEM (Thermo Fisher Scientific) supplemented with 10% (v/v) FBS, MEM Non-Essential Amino Acids Solution (Thermo Fisher Scientific) and penicillin–streptomycin. Cells were used for experiments within ten passages after thawing from liquid nitrogen storage. All cells were authenticated by STR profiling at the time of receipt and periodically thereafter. Cells were passaged after culture for an average of three to four days and tested routinely for mycoplasma contamination before use. Cell transfection with plasmids was conducted using ViaFect™ Transfection Reagent (Promega, Madison, WI, USA) or Lipofectamine® 2000 (Thermo Fisher Scientific) in Opti-MEMTM medium (Thermo Fisher Scientific). For stable transfection or infection, monoclonal cells were screened using the antibiotic G418 (Thermo Fisher Scientific). To determine cell proliferation, 1 × 104 cells were seeded in each well of a 24-week cell culture plate, and the number of cells at different timepoints was determined using a blood counting chamber.
Plasmid construction
cDNA used for plasmid construction was synthesized using total cellular RNA isolated from HLCZ01 or Huh7 cells. Expression plasmids for ISG12a and SKP2 were constructed and reported in our previous study.10 The synthesized CTNNB1 sequence was cloned into the p3×Flag-CMV-14 vector (Sigma-Aldrich, St. Louis, MO, USA). The p3×Flag-Axin and Axin domain plasmids were constructed from the pEF-Flag-Axin plasmid (NM_181050.3, encoding Axin isoform b).18 Plasmids constructed using the p3×Flag vector express the Flag tag, and plasmids constructed using the His/pcDNA3.1a vector express the V5 tag. The PD-L1 promoter was amplified using HLCZ01 genomic DNA and inserted into the pGL3 Basic Vector (Promega). Sequences used for plasmid construction were obtained with the high-fidelity PCR kit KOD-Plus-Neo (TOYOBO, Tokyo, Japan). The primers used for plasmid construction are listed in Supplementary Table S1. The target sequences used for shRNA gene-silencing plasmids were as follows: ISG12a (CTGGACTCTCCGGATTGAC), CTNNB1 (#1, GCTTATGGCAACCAAGAAA; #2, GCAGCTGGAATTCTTTCTA), and negative control (ACTACCGTTGTTATAGGTG). The sequences were assembled with the loop sequence (TTCAAGAGA) and inserted into the pRNAT-U6.1/neo vector (GenScript, Piscataway, NJ, USA) according to the manufacturer’s instructions. The plasmids were amplified in Escherichia coli DH5α competent cells, sequenced at Sangon Biotech (Shanghai, China), and isolated using QIAprep Spin Miniprep Kit (Qiagen, Dusseldorf, Germany).
qRT-PCR
Tissue specimens were obtained immediately after surgery and maintained in liquid nitrogen. Total RNA was extracted from tissue samples and cells using TRIzol reagent (Thermo Fisher Scientific). Reverse transcription PCR of total RNA was performed using PrimeScript™ RT reagent Kit with gDNA Eraser (Takara, Kusatsu, Japan) to avoid genomic DNA contamination. qRT-PCR was conducted using SYBR® Premix Ex Taq™ II (Tli RNaseH Plus) (Takara) or FastStart Universal SYBR Green Master (ROX) (Roche, Basel, Switzerland) with an ABI7500 thermocycler (Thermo Fisher Scientific). GAPDH was used as the internal control. The primers used for qRT-PCR are listed in Supplementary Table S2.
Immunoblots
Cells or tissue samples were lysed with RIPA buffer (Thermo Fisher Scientific) supplemented with a proteinase inhibitor cocktail (Thermo Fisher Scientific). After incubation on ice for 30 min, the lysates were centrifuged at 16 100 g for 15 min at 4 °C, and the protein-containing supernatant was collected for immunoblotting. Nuclear and cytoplasmic proteins were isolated using NE-PER Nuclear and Cytoplasmic Extraction Reagents (Thermo Fisher Scientific). After determining the concentration using the BCA method, proteins were electrophoresed along with the EasySee® Western Marker or EasySee® II Western Marker (TRANSGEN BIOTECH, Beijing, China) on SDS-PAGE gels and transferred to PVDF membranes (Merck Millipore, Darmstadt, Germany). The PVDF membranes were then blocked with 5% skim milk, sequentially incubated with primary antibodies at 4 °C overnight and secondary antibodies for 1 h at room temperature, and detected using SuperSignal® West Pico Chemiluminescent Substrate (Thermo Fisher Scientific). GAPDH served as the internal control for total and cytoplasmic proteins, and Lamin B1 served as the internal control for nuclear proteins. The investigators were blinded to the identity of the tissue specimens during the experiments.
Wound-healing assay
Similar numbers of stably transfected cells in the logarithmic phase were seeded in a six-well plate at 50–60% confluence. After cultivation in a cell incubator with 5% CO2 at 37 °C for 24 h, the cells were incubated with serum-free medium for another 24 h. A wound was generated in the adherent cell monolayer using a standard 10-µl pipette tip and a ruler. Then, the cells were continually cultivated in serum-free medium for 24 to 48 h. Images of the wound-healing process at different timepoints were captured using an IX71 inverted light microscope (Olympus, Tokyo, Japan).
Transwell assay
Transwell chambers with 8.0-μm PET membrane pores (Corning Incorporated, Corning, NY, USA) were inserted into 24-well plates and used to evaluate the migration of cancer cells. After starvation with serum-free DMEM for 24 h, cancer cells were dispersed at a density of 1 × 106 per ml in serum-free DMEM. Then, 650 μl DMEM supplemented with 10% (v/v) FBS was added to the lower chamber of the Transwell insert, and 100 μl of serum-free DMEM containing cancer cells was added to the upper chamber. After cultivation in a cell incubator with 5% CO2 at 37 °C for 24 h, the cells were fixed with methyl alcohol and stained with 0.2% (m/v) crystal violet (Sigma-Aldrich); the cells remaining in the upper chamber were gently removed with a medical cotton swab. Images of migrated cells were captured under an IX71 inverted light microscope. The migrated cells were counted for further analysis.
Colony formation in agarose
Cells were dispersed into 2× DMEM (DMEM supplemented with 20% (v/v) FBS and 2× penicillin–streptomycin). A total of 250 µl of 1.2% sterilized low melting point agarose (Sigma-Aldrich) and 250 µl of 2× DMEM were mixed and added to each well of a 12-well cell culture plate. After the gel solidified, 250 µl of 0.8% agarose solution and 250 µl of 2× DMEM containing 3000 cells were mixed and added to the top of the agarose gel. After the gel solidified again, 500 µl of DMEM supplemented with 10% (v/v) FBS and penicillin–streptomycin was added on top of the upper agarose layer. The culture plates were then transferred to a cell incubator with 5% CO2 at 37 °C and cultivated for 3 weeks. The culture medium above the agarose gel was refreshed every 3 days. 3 weeks later, the number of colonies that formed in the soft agarose was recorded for analysis. Images of the colonies were captured under an IX71 inverted optical microscope.
Adhesion assay
A 96-well cell culture plate was coated with 40 μg/ml Collagen I (Thermo Fisher Scientific) for 12 h, rinsed with PBS and dried at room temperature. Cells were subjected to a four-step procedure: (a) starved with serum-free DMEM for 8 h, (b) detached from culture plates using 0.02% EDTA, (c) washed with DMEM to remove the EDTA, and (d) resuspended in DMEM supplemented with 0.1% (m/v) BSA at a density of 2 × 105 cells/ml. Then, 100 μl of cell suspension was added to the coated well and cultivated at 37 °C in an atmosphere containing 5% CO2 for 20 min. The nonadherent cells were gently washed away using serum-free DMEM. The adherent cells were cultured in DMEM supplemented with 10% (v/v) FBS at 37 °C in the presence of 5% CO2 for 4 h and then analyzed by the MTT assay using CellTiter 96® AQueous One Solution Cell Proliferation Assay (Promega).
Cultivation of cancer stem cell spheres
Cancer cells were digested with trypsin and dispersed in DMEM/F12 supplemented with EGF (20 ng/ml) (Thermo Fisher Scientific), FGF (20 ng/ml) (Thermo Fisher Scientific) and B27 (2%) (Thermo Fisher Scientific). A sample of 1 × 104 cells in 2 ml of medium was added to a 35-mm low-attachment cell culture plate (Corning Incorporated) and cultivated at 37 °C in the presence of 5% CO2 for 3 weeks. The culture medium was refreshed every 3 days by centrifuging the cells. Images of the cancer stem spheres were captured under an IX71 inverted fluorescence microscope.
Animal experiments
Six-week-old BALB/c nude mice (SLAC, Changsha, China) were fed under standard conditions in a pathogen-free environment at the animal care facility of Hunan University. The mice were housed in groups of up to three to five per cage on a 12-h light/dark cycle.
To assess the effect of ISG12a on tumor growth formed by Huh7 cells, mice were arbitrarily assigned to two groups (n = 6 for shCtrl, n = 6 shISG12a). Next, a sample of 1 × 107 cells in 200 μl of sterile PBS was subcutaneously injected. The size of the tumors formed was recorded every two days. Twenty-three days later, the mice were sacrificed, and the tumor tissues were harvested. The weight and size of the tumors were recorded for further analysis. The tumor volume was calculated using the following formula: tumor volume (mm3) = 0.5 × length × width2.
To assess the effect of ISG12a on the metastasis of Huh7 cells in vivo, six-week-old male athymic BALB/c nude mice were arbitrarily assigned into two groups (n = 6 for shCtrl, n = 5 for shISG12a). Next, a sample of 1.5 × 106 cells in 150 μl of sterile PBS was injected via the tail vein. Fluorescence in the lung regions of live mice was detected using the IVIS Lumina XR live animal imager (PerkinElmer, Waltham, MA, USA). Forty-five days later, the mice were sacrificed, and the lungs were collected for analysis. The tumor nodules that formed in the lungs were examined using H&E staining. No blinding was performed with regard to the animal experiments.
H&E staining
Tissue samples were routinely embedded in paraffin, followed by slicing and dewaxing. The slices were stained with hematoxylin (BOSTER, Wuhan, China) for 5 min, rinsed with PBS, and counterstained with eosin (BOSTER) for 1 min. Images were obtained using the Pannoramic MIDI II tissue biopsy scan system (3DHISTECH Ltd., Budapest, Hungary).
Immunohistochemistry (IHC)
Tissue slices used for IHC were identified by two pathologists at Hunan Cancer Hospital. The tissues were routinely embedded in paraffin, followed by slicing and dewaxing. After antigen retrieval by boiling in a sodium citrate solution (0.01 M and pH = 6.0), the slices were immersed in 2% H2O2 for 30 min, blocked with normal goat serum for 1 h, incubated with the primary antibody at 4 °C overnight and the second antibody for 1 h at room temperature, and stained with DAB (BOSTER) for 5 min. Hematoxylin was used to counterstain the nucleus in PBS for 15 min. The stained slices were sealed with neutral resin and scanned to obtain images using the Pannoramic MIDI II tissue biopsy scanning system. The investigators were blinded during the experiments.
To determine the correlation between ISG12a expression and immune infiltration, ISG12a, CD3, and CD8 in HCC and GC cancer tissues were examined by IHC with continuous tissue sections. For ISG12a expression, both membranous and cytoplasmic staining was evaluated based on the extent of staining (percentage of positive cells on a scale of 0–3: 0, negative or <1%; 1, 1–10%; 2, 10–50%; and 3, >50%) and the intensity of IHC staining (0, no staining; 1, weak staining; 2, moderate staining; or 3, strong staining). The evaluation of immune infiltration represented by CD3- and CD8-positive T cells was conducted according to a previous report.19 The number of cells with a positive reaction to antibodies was counted using ImageJ software (National Institutes of Health, MD, USA). The mean numbers of positive cells stained in each area were recorded for analysis. The total number of positive cells per high-power field (×400) was calculated (the mean number of positive cells in the five areas was added and divided by 5) for each antibody (CD3 and CD8).
Immunofluorescence staining
To examine β-catenin and PD-L1 expression in cancer cells, an average sample of 1.5 × 105 cells was seeded into a 35-mm confocal dish. After cultivation for 48 h, the cells were rinsed with PBS, fixed with 4% paraformaldehyde at 4 °C for 30 min, permeabilized with 0.2% Tween-20 for 10 min, blocked with normal goat serum (Thermo Fisher Scientific) for 1 h, and sequentially incubated with primary and secondary antibodies at room temperature for 1 h. Nuclei were counterstained with DAPI (Vector Laboratories, Burlingame, CA, USA) for 5 min. Images were captured using a TI-E + A1 SI confocal microscope (Nikon, Tokyo, Japan).
To assess F-actin expression in cancer cells, Alexa Fluor® 555 Phalloidin (8953S, CST) was commercially obtained and used according to the manufacturer’s instructions. In brief, the staining procedures of F-actin are consistent with the staining of β-catenin and PD-L1 mentioned above. Cells were incubated with 0.33 μM phalloidin at room temperature for 15 min and then rinsed with PBS three times for 5 min each before staining the cell nucleus with DAPI. Images were captured using a TI-E + A1 SI confocal microscope (Nikon, Tokyo, Japan).
RNA sequencing
RNA sequencing of three independent biological replicates was conducted at BGI (Shenzhen, Guangzhou, China). Investigators were blinded during the experiments. The procedures are briefly described below. The RNA concentration, RIN/RQN, 28S/18S value and fragment length distribution were determined using a Standard Sensitivity RNA Analysis kit (15 nt) (DNF-471, Bio-Thing, Sunnyvale, CA, USA) with a Fragment Analyzer. RNA sequencing was conducted using the BGISEQ platform. SOAPnuke V1.5.2 was applied for filtering reads and obtaining clean reads; reads with adapters, more than 5% of unknown bases and of low quality were removed. The clean reads were mapped to the human reference genome GRCh38.p12 using HISAT2 V2.0.4. The gene expression level was calculated using Bowtie2 V2.2.5 and RSEM V1.2.12. DEGs were analyzed using hierarchical clustering analysis, Gene Ontology analysis and pathway analysis. Images were obtained using the Dr. Tom system. RNA sequencing data that support the findings of this study were deposited in NCBI SRA under accession number PRJNA554221.
Bioinformatics analysis
Bioinformatics analyses of protein-protein interactions were conducted in a threefold procedure.20 First, we searched RCSB Protein Data Bank (https://www.rcsb.org/). As only partial structures of Axin and SKP2 has been crystallized as of August 8, 2020, the full-length protein structures were predicted using the I-TASSER online server (https://zhanglab.ccmb.med.umich.edu/I-TASSER/). This also led to information on protein functional domains. Second, in silico models of Axin-SKP2 and SKP2-ISG12a interactions were estimated using Cluspro V2.0 (http://cluspro.bu.edu/), and the possible models that matched with literature findings from protein interaction databases such as BioGrid (http://thebiogrid.org/) and IntAct (http://www.ebi.ac.uk/intact) were considered. Third, a structural visualization and movies were produced using PyMOL V1.7 (http://www.pymol.org/).
Co-IP
Cells were lysed with Pierce™ IP Lysis Buffer (Thermo Fisher Scientific) supplemented with a proteinase inhibitor cocktail and incubated on ice for 30 min. For analysis of ubiquitination, cells were cultivated with 25 μM MG132 (APExBIO, Houston, TX, USA) for 6 h before protein collection. The protein-containing supernatant was obtained by centrifugation at 16 100 g for 15 min at 4 °C. After determining input protein expression using immunoblots, lysates were diluted to 2 μg/μl with PBS, immunoprecipitated with primary antibodies at 4 °C overnight, and incubated with Protein G Agarose (Merck Millipore) at 4 °C for 2 h. The precipitated beads were washed with PBS five times and boiled at 100 °C in 2 × Laemmli Sample Buffer (Bio-Rad, Hercules, CA, USA) for 5 min. The supernatant was collected and examined by immunoblotting. Specifically, our previous study demonstrated that ISG12a targets the viral NS5A protein for degradation through a proteasome-dependent pathway in HCV-infected cells, revealing protein interactions among ISG12a, SKP2, and the viral protein NS5A.10 The interactions of SKP2-ISG12a and SKP2-Axin demonstrated in the current study are a continuous work.
ChIP
SimpleChIP® Enzymatic Chromatin IP Kit (Agarose Beads) (CST, Danvers, MA, USA) based on enzymatic digestion of genomic DNA was used to conduct ChIP assays. Specific primers used for qRT-PCR and agarose electrophoresis analysis to determine enrichment of the PD-L1 promoter are listed in Supplementary Table S2; the RPL30 primers were provided in the kit.
Dual-luciferase reporter assay
To evaluate the influence of ISG12a and β-catenin on the transcriptional activity of the PD-L1 promoter, 250 ng of the promoter plasmid pGL3-CD274 (Firefly luciferase), 10 ng of the pRL-MCV plasmid (Renilla luciferase) (Promega) and the gene expression plasmids ISG12a and CTNNB1 were cotransfected with 3 μl of Lipofectamine® 2000 into cells in each well of a 12-well cell culture plate for 24 h. To evaluate the effect of ISG12a on TOP-Flash activity, 250 ng of the TOP-Flash plasmid (Firefly luciferase) and 10 ng of the pRL-MCV plasmid (Renilla luciferase) were cotransfected with 3 μl Lipofectamine® 2000 in cells in each well of a 12-well cell culture plate for 24 h. pRL-CMV was used as an internal control plasmid. Dual-Luciferase® Reporter 1000 Assay System (Promega) was used to determine the transcriptional activity of the promoter. The fold change in relative luciferase expression was calculated based on the ratio of the Firefly and Renilla luciferase activities.
Isolation of PBMCs and NK cells
PBMCs were freshly isolated from the peripheral blood of healthy individuals using Ficoll-PaqueTM PLUS (GE Health, Uppsala, Sweden), as described in our previous study.21 NK cells were purified from PBMCs by magnetic cell sorting with NK Cell Isolation Kit (Miltenyi Biotec, Bergisch Gladbach, Germany) based on the negative sorting method. The isolated NK cells were cultivated in RPMI 1640 supplemented with 10% FBS and penicillin–streptomycin. The purity of the isolated NK cells was no less than 95%, as examined by flow cytometry (Supplementary Fig. S11a).
Flow cytometry
For the analysis of surface PD-L1 expression, cancer cells in culture plates were digested with 0.02% EDTA. For analyzing PD-1, CD3, and CD56 expression, NK cells in a cell culture plate were collected by centrifuging at 400 g for 5 min. The collected cells were gently washed with PBS for 5 min, filtered through 200-mesh cell strainers, and sequentially incubated with PE-Cy™7 mouse anti-human PD-L1 in 100 μl of PBS at room temperature for 30 min. The cells were washed and resuspended in PBS for 5 min and examined using a BD FACSAria II flow cytometer (BD Bioscience, San Jose, CA, USA); the data were analyzed using FlowJo V10.0.7 software (FlowJo, LLC, San Jose, CA, USA). To determine the influence of IFN induction on PD-L1 expression, cancer cells were incubated with 100 U/ml IFNα (Thermo Fisher Scientific) or 20 ng/ml IFNγ (Thermo Fisher Scientific) for 24 h.
For the analysis of NK cell-mediated killing, 1.5 × 105 cancer cells were seeded into each well of a 12-well cell culture plate. The receptor–ligand interaction was blocked by cultivating the cancer cells in culture medium with 5 μg/ml anti-PD-L1 antibody or NK cells with 10 μg/ml anti-PD-1 antibody for 24 h. Normal goat IgG was used as an isotype control. At 20 h after the cells were seeded, 7.5 × 105 NK cells costimulated with 100 ng/ml IL-2 (R&D Systems) and/or 100 ng/ml IL-18 (R&D System) for 24 h were mixed and cocultivated with the cancer cells for 4 h. Then, the supernatant was collected and mixed with adherent cells that were detached using EDTA-free trypsin. The collected cells were washed with cold PBS and resuspended in 1× binding buffer (BD Biosciences). APC Annexin V (Biolegend, San Diego, CA, USA) and a propidium iodide (PI) staining solution (BD Biosciences) were added to the cells and incubated at 4 °C for 15 min in the dark. Finally, the cells in 200 µl of 1× binding buffer were examined using a BD FACSAria II flow cytometer. The collected data were analyzed using FlowJo V10.0.7 software. Gates for the green fluorescence protein (GFP), NK cells, and cancer cells were considered in the analysis. Information on the antibodies used for flow cytometry analysis is provided below.
Antibodies
Antibodies against the following proteins were obtained from commercial sources and used for immunoblots: ISG12a (SAB1408588, Sigma-Aldrich), GAPDH (6C5, MAB374, Merck Millipore), Flag (M2, F3165, Sigma-Aldrich), V5 (R960-25, Thermo Fisher Scientific), HA (6A1, AT0024, CMCTAG, Dover, DE, USA), Ubiquitin (3933S, CST, Danvers, MA, USA), E-cadherin (24E10, 3195S, CST), β-Catenin (D10A8, 8480S, CST), ZEB1 (D80D3, 3396S, CST), Claudin-1 (D5H1D, 13255S, CST), Slug (C19G7, 9585S, CST), Lamin B1 (D9V6H, 12586S, CST), GSK3β (27C10, 9315S, CST), Axin (C76H11, 2087S, CST), Cyclin D1 (92G2, 2978S, CST), SKP2 (A-2, sc-74477, SANTA CRUZ) and PD-L1 (17952-1-AP, Proteintech, Rosemont, IL, USA). Normal mouse IgG (sc-45123, SANTA CRUZ, Dallas, TX, USA), normal rabbit IgG (2729S, CST), goat anti-mouse IgG (HRP-linked) (AP124P, Merck Millipore), goat anti-rabbit IgG (HRP-linked) (AP132P, Merck Millipore) were also used.
The following antibodies were obtained from commercial sources and used for immunofluorescence staining: anti-β-catenin (D10A8, 8480S, CST), anti-PD-L1 (17952-1-AP, Proteintech), and donkey anti-rabbit IgG (H + L) highly cross-adsorbed secondary antibody conjugated with Alexa Fluor 594 (A-21207, Thermo Fisher Scientific).
The following antibodies were obtained from commercial sources and used for IHC: anti-ISG12a (HPA028875, Sigma-Aldrich), anti-CD3 (17617-1-AP, Proteintech), anti-CD8 (1G2B10, 66868-1-Ig, Proteintech), goat anti-mouse IgG (HRP-linked) (AP124P, Merck Millipore), and goat anti-rabbit IgG (HRP-linked) (AP132P, Merck Millipore).
The following antibodies were obtained from commercial sources and used for flow cytometry: PE-Cy™7 mouse anti-human PD-L1 (MIH1, 558017, BD Bioscience, San Jose, CA, USA), FITC mouse anti-human CD3 (UCHT1, 555332, BD Biosciences), PE-Cy™7 mouse anti-human CD56 (B159, 557747, BD Biosciences), and FITC mouse anti-human CD279 (also known as PD-1) (MIH4, 557860, BD Biosciences).
The following antibodies were obtained from commercial sources and used to block the receptor-ligand interaction: human PD-L1 antibody (AF156, R&D Systems, Minneapolis, MN, USA), human PD-1 antibody (AF1086, R&D Systems), and normal goat IgG control (AB-108-C, R&D Systems).
The following antibodies were obtained from commercial sources and used for ChIP assays: anti-β-Catenin (D10A8, 8480S, CST), anti-Histone H3 (D2B12, D2B12) (4620, CST), and normal rabbit IgG (2729, CST).
Patients
The human tissue specimens analyzed in the present study were obtained from 2013 to 2019. The HCC and GC samples used for qRT-PCR and immunoblots were obtained from Hunan Cancer Hospital, and the clinical data of the patients are presented in Supplementary Tables S3 and S4. HCC and GC samples used for IHC staining were obtained from Hunan Cancer Hospital. Peripheral blood from healthy individuals and the HCC samples used for flow cytometry analyses were obtained from the First Hospital of Jilin University.
Ethics approval
The protocols used in the animal experiments were approved by the Animal Care and Experiment Committee of Hunan University. All applicable international, national, and/or institutional guidelines for the care and use of animals were followed.
Human tissue specimens were obtained with informed consent and approved by the institutional review boards of Hunan Cancer Hospital and the First Hospital of Jilin University. The protocols used for the collection of human tissues and data analysis were performed in accordance with the Declaration of Helsinki.
Clinical data for human tissue specimens
Ninety-two HCC and 137 GC samples were collected to examine levels of the ISG12a and Axin proteins by immunoblotting (Supplementary Figs. S1–S3 and Supplementary Tables S3 and S4). The PVDF membranes used for immunoblotting were adjusted to a uniform background during the analysis to reduce error. The density values of the target protein bands were normalized to that of GAPDH, the loading control.18 For analysis of the expression profiles of target proteins, specimens in which the proteins (ISG12a, Axin, or GAPDH) were not detected and the deviation value was acquired according to Gaussian distribution and PauTa Criterion (μ ± 3σ) were removed; data are presented as log2 values. For analysis of the correlation between ISG12a level and prognosis or clinicopathological features, specimens missing associated clinical data were removed, and high and low expression groups were determined according to the median value.
Eighty-three tissue samples from patients with HCC were collected to analyze the ISG12a mRNA level by qRT-PCR (Supplementary Table 3). The relative level of ISG12a mRNA was calculated using the 2(−ΔΔCt) method. For analysis of the expression profile, specimens in which the mRNAs (ISG12a and GAPDH) were not detected and the deviation value acquired by Gaussian distribution and PauTa Criterion (μ ± 3σ) were removed; data are presented as log2 values. For analysis of the correlation between ISG12a mRNA level and prognosis, specimens with missing clinical data were removed, and high and low level groups were determined according to the median value.
Statistical analysis
Statistical analyses were performed using SPSS Statistics 22 (IBM, Armonk, NY, USA). GraphPad Prism 5.0 (GraphPad, San Diego, CA, USA) was employed to draw statistical graphs. Experiments were independently repeated two or three times, with similar results, and representative images are shown in figures. The correlation between two proteins examined by IHC or immunoblots was evaluated using Spearman or Pearson correlation analysis (two-tailed), respectively. Chi-square analysis (two-sided) was performed to determine the correlation between the ISG12a level and clinicopathological features. Kaplan–Meier analysis (log-rank test) was carried out to determine the correlation between the ISG12a level and overall survival. Data are presented as means ± SD. Two-sided Student’s t tests were performed to compare the significance of differences between two groups, and one-way ANOVA with the Tukey-Kramer or Games–Howell post hoc test was used to compare the significance of differences among multiple groups. Significance levels were set to *p < 0.05, **p < 0.01, ***p < 0.001 or nonsignificant (ns).
Results
ISG12a is expressed at low levels in cancer cells and cancer tissues
Due to chromatin remodeling, external stimulation and mutation, and a lack of innate immune signals, expression of some ISGs is decreased in tumors.22,23,24 We examined levels of the ISG12a protein in HCC and GC using immunoblotting to elucidate the role of ISG12a in cancer. The ISG12a protein was detected at high levels in PHHs and GES-1 normal human gastric mucosal cells, but it was detected at low levels in HCC and GC cells (Fig. 1a, b). The low expression of ISG12a in cancer cells indicates its potential anticancer effect. We then extended our research from cancer cells to human tissue specimens obtained from patients with HCC or GC. The level of the ISG12a protein in most representative cancer tissues (CAs) was lower than that in paired para-cancerous tissues (para-CAs) (Fig. 1c). Statistically, compared with para-CAs, 71 (83.53%) of the 85 HCC samples expressed the ISG12a protein at a low level (Fig. 1d and Supplementary Fig. S1). Similarly, a low level of the ISG12a protein was detected in 90 of 117 GC tissues, reaching 76.92% (Fig. 1e and Supplementary Figs. S2 and S3). Moreover, 59 of 82 HCC CAs expressed ISG12a mRNA at a low level, representing 71.95% (Fig. 1f). Thus, ISG12a is expressed at low levels in tumor tissues. Suppression of ISG12a expression may be an important indicator of carcinogenesis and cancer development. In addition, the level of the ISG12a protein examined by IHC was decreased in samples from patients with higher tumor node metastasis (TNM) stages and was almost undetectable in stage IV CAs (Fig. 1g, h), indicating that decreased ISG12a expression may promote the malignant transformation of cancer. Therefore, we infer that ISG12a might exert an anticancer effect.

Low expression of ISG12a in cancer cells and tissues. a, b Immunoblots showing levels of ISG12a in PHH, HLCZ01, LH86, Huh7, and HepG2 cells (a) or GES-1, BGC-823, and HGC-27 cells (b). Experiments were replicated three times, with similar results. c Immunoblots showing levels of ISG12a and Axin in representative CAs and para-CAs from patients with HCC or GC. d, e Scatterplots showing the relative ISG12a protein level between CA and para-CA from patients with HCC or GC. Protein expression in CAs and para-CAs examined by immunoblots is presented in Supplementary Figs. S1–S3. f Scatterplot showing the relative level of ISG12a mRNA between CA and para-CA from patients with HCC, as determined by qRT-PCR. g, h Representative images of IHC staining for ISG12a protein in CAs at different TNM stages (I, II, III, and IV) from patients with HCC or GC. Scale bar, 100 μm. Data were analyzed using two-sided Student’s t tests, and the results are presented as means ± SD (d–f)
ISG12a suppresses cancer phenotypes
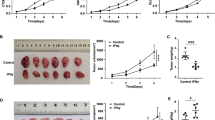
To verify our hypothesis, we silenced ISG12a in HCC cells represented by Huh7, HLCZ01, and LH86 and GC cells represented by HGC-27 (Fig. 2a). ISG12a knockdown promoted the proliferation of HCC and GC cells, including Huh7, HLCZ01, LH86, and HGC-27 cells (Fig. 2b and Supplementary Fig. S4a, b), and increased the number and size of Huh7 and HGC-27 cell colonies in soft agarose (Fig. 2c, d), suggesting that ISG12a inhibits cancer cell proliferation. Moreover, the weight and volume of tumors formed by Huh7 cells increased after ISG12a knockdown (Fig. 2e). Thus, ISG12a suppresses tumor growth in vivo. Based on the accelerated wound healing of ISG12a-silenced Huh7, LH86, and HGC-27 cells, ISG12a appears to restrict cancer cell migration (Supplementary Fig. S4c–e), as confirmed by the results of Transwell experiments using Huh7 and HGC-27 cells (Fig. 2f and Supplementary Fig. S4f). Moreover, the number of Huh7 and HGC-27 cells that adhered to the matrix was increased following ISG12a knockdown (Supplementary Fig. S4g). Thus, a decrease in ISG12a expression may allow cancer cells to adapt to the matrix microenvironment. Notably, the intensity of GFP staining in the lung regions of nude mice injected with Huh7 cells via the tail vein was increased in ISG12a-silenced groups (Fig. 2g), and more tumor nodules appeared in the mouse lungs following ISG12a knockdown (Fig. 2h). Therefore, silencing ISG12a may promote cancer metastasis in vivo. In addition, the number and size of cancer stem cell spheres formed by Huh7 and HGC-27 cells increased following ISG12a knockdown (Fig. 2i and Supplementary Fig. S4h), suggesting an inhibitory effect of ISG12a on cancer stemness. Interestingly, great differences in the morphology of cancer stem cell spheres formed by different cancer cells were noted. All the data support the antitumor effect of ISG12a.

Change in malignant phenotypes of cancer cells with ISG12a knockdown. a Immunoblots showing levels of ISG12a in Huh7, HLCZ01, LH86 and HGC-27 cells with ISG12a knockdown. b Proliferation of Huh7, HLCZ01, LH86 and HGC-27 cells with ISG12a knockdown. c, d Colony formation of Huh7 and HGC-27 cells in soft agarose with ISG12a knockdown. Scale bar, 200 μm. e Image of the tumors that formed in BALB/c nude mice at 23 days after subcutaneous injection of Huh7 cells with ISG12a knockdown (n = 6 per group). The minimum scale on the left of the ruler represents 1 mm. f Images and number of Huh7 cells with ISG12a knockdown that migrated through Transwells in 24 h. Scale bar, 100 μm. g Fluorescence in the lung regions of BALB/c nude mice (n = 6 for the shCtrl group, n = 5 for the shISG12a group) at 45 days after tail vein injection of Huh7 cells with ISG12a knockdown. h Representative images of H&E staining in the lung tissue sections from the animals shown in g. Scale bar, 200 μm. i The stem cell spheres formed by Huh7 cells with ISG12a knockdown in serum-free medium within 3 weeks. Cells transfected with gene silencing plasmids coding GFP. Scale bar, 100 μm. Experiments were independently replicated two (d) or three (a–c, e–i) times, with similar results. Two-tailed Student’s t tests were performed to analyze the data, and the data are presented as means ± SD of at least three biological replicates. *p < 0.05, **p < 0.01, and ***p < 0.001
ISG12a inhibits the canonical Wnt/β-catenin signaling pathway
To elucidate the regulatory mechanism of ISG12a in cancer, we conducted RNA sequencing using total cellular RNA extracted from Huh7 cells and identified 1002 upregulated and 806 downregulated differentially expressed genes (DEGs) (Supplementary Fig. S5a, b). KEGG pathway classification of DEGs suggested roles for ISG12a in signal transduction, viral infection, immunity, etc. (Supplementary Fig. S5c). Notably, ISG12a may affect signal transduction pathways, including Wnt/β-catenin signaling, extracellular matrix receptor interaction and focal adhesion (Fig. 3a), indicating the potential mechanism by which ISG12a regulates cancer. This finding may partially explain the accelerated malignant transformation of cancer cells lacking ISG12a.

ISG12a suppresses canonical Wnt/β-catenin signaling. a Enrichment analysis of 261 DEGs related to signal transduction pathways in Huh7 cells with ISG12a knockdown, as assayed by RNA sequencing. The log2(Fold Change) ≥ 1.00 and FDR ≤ 0.01 from three biological replicates. b Immunoblots showing levels of EMT-associated markers in Huh7 and HGC-27 cells with ISG12a knockdown. c Immunofluorescence staining for F-actin using phalloidin in Huh7 and HGC-27 cells with ISG12a knockdown. Nuclei were stained with DAPI. Scale bar, 10 μm. d Immunoblots showing levels of β-catenin and Axin in HLCZ01, LH86, Huh7, BGC-823, and HGC-27 cells. e Immunoblots showing levels of β-catenin and ISG12a in the cytoplasm and nucleus of Huh7 and HGC-27 cells with ISG12a knockdown. GAPDH and Lamin B1 served as loading controls for cytoplasmic and nuclear proteins, respectively. Cy Cytoplasm, Nu Nucleus. f Immunofluorescence staining for β-catenin in Huh7 and HGC-27 cells with ISG12a knockdown. Nuclei were stained with DAPI. Scale bar, 10 μm. g qRT-PCR analysis of ISG12a, c-Myc, and Cyclin D1 expression in Huh7 cells with ISG12a knockdown. h Fold change of luciferase expression of the TOP-Flash plasmid in Huh7 and HGC-27 cells with ISG12a knockdown. Experiments were independently repeated two (h) or three (b–g) times, with similar results. Two-tailed Student’s t tests were performed to analyze the data, and data are presented as means ± SD of three biological replicates. **p < 0.01 and ***p < 0.001
Furthermore, we examined changes in the expression of epithelial–mesenchymal transition (EMT)-associated markers. The protein levels of epithelial markers (E-cadherin and Claudin-1) were reduced in ISG12a-silenced Huh7 and HGC-27 cells, whereas the those of mesenchymal markers (β-Catenin, ZEB1 and Slug) were increased (Fig. 3b). Similar results were obtained for LH86 and HLCZ01 cells in which gene silencing was induced through transfection (Supplementary Fig. S5d). Cytoskeletal reorganization is an important characteristic of cancer EMT.25 We observed cytoskeletal changes by F-actin staining using phalloidin and found increased lamellipodia and stress fibers in ISG12a-silenced Huh7 and HGC-27 cells (Fig. 3c). Based on these data, ISG12a inhibits EMT in tumor cells. Increased β-catenin levels may predict activation of the canonical Wnt/β-catenin signaling pathway in cancer cells,26 indicating a potential regulatory mechanism of ISG12a in cancer. Based on the expression of the ISG12a protein and important components of the Wnt/β-catenin signaling pathway in cancer cells (Figs. 1a, b and 3d), we chose Huh7 and HGC-27 cells as the main cell lines for use in subsequent experiments.
Immunoblots revealed increased levels of β-catenin in the nuclei of ISG12a-silenced Huh7 and HGC-27 cells (Fig. 3e), which was confirmed by immunofluorescence staining (Fig. 3f). The mRNA levels of the oncogenes c-Myc and Cyclin D1 in Huh7 cells, which are downstream of the Wnt/β-catenin signaling pathway, were upregulated after ISG12a knockdown (Fig. 3g). The results of TOP-Flash luciferase reporter assays showed that Huh7 and HGC-27 cells with low expression of ISG12a responded with a more robust activation of TOP-Flash activity (Fig. 3h). However, the difference in expression of β-catenin protein between the cytoplasm and nuclei in Huh7 and HGC-27 cells might lead to the observed difference in TOP-Flash activity. Therefore, ISG12a silencing leads to abnormal activation of the canonical Wnt/β-catenin signaling pathway in cancer cells. Furthermore, ZEB1 expression was increased in ISG12a-silenced cancer cells (Fig. 3b and Supplementary Fig. S5d), which further increased activity of the Wnt/β-catenin signaling pathway.27 Thus, ISG12a exerts its antitumor effect by suppressing canonical Wnt/β-catenin signaling pathway activity.
ISG12a promotes the proteasomal degradation of β-catenin by inhibiting the proteasomal degradation of Axin
ISG12a knockdown did not alter the transcription of the CTNNB1 gene examined by qRT-PCR and RNA sequencing (Fig. 4a, b), translation of CTNNB1 mRNA (Fig. 4c), or lysosomal degradation of β-catenin (Fig. 4d) in Huh7 cells. Interestingly, the level of β-catenin was increased in the presence of MG132 (Fig. 4e and Supplementary Fig. S6a), and that of ubiquitinated β-catenin was increased in cells overexpressing ISG12a (Fig. 4f). MG132 is a peptide aldehyde that can effectively block the proteolytic activity of the proteasome. Thus, ISG12a promotes the proteasomal degradation of β-catenin. Notably, the increase in β-catenin and Cyclin D1 in ISG12a-silenced Huh7 cells was reversed by overexpression of ISG12a (Fig. 4g), suggesting that ISG12a specifically inhibits the canonical Wnt/β-catenin pathway. In addition, ISG12a did not alter the mRNA level of genes involved in the β-catenin destruction complex, as examined by qRT-PCR, including Axin, APC, CKIα, and GSK3β (Fig. 4a), which was also identified by RNA sequencing (Fig. 4b). However, the Axin protein level was decreased after ISG12a knockdown (Fig. 4g and Supplementary Fig. S6a), and this change was reversed by ISG12a overexpression (Fig. 4g). Thus, ISG12a may protect the integrity of the β-catenin destruction complex by preserving the Axin protein. In addition, MG132 increased the Axin protein level (Fig. 4g and Supplementary Fig. S6a). Overall, we infer that ISG12a might block the proteasomal degradation of Axin.

ISG12a induces the proteasomal degradation of β-catenin. a qRT-PCR analysis of expression of ISG12a, CTNNB1, Axin, CKIα, APC, and GSK3β mRNAs in Huh7 cells with ISG12a knockdown. b RNA sequencing showing the expression change of mRNAs associated with the canonical Wnt/β-catenin signaling pathway in Huh7 cells with ISG12a knockdown. The scale bar shows the expression change of genes by the log2(value+1) method. c–e Immunoblots showing β-catenin levels in Huh7 cells with ISG12a knockdown after treatment with 100 μg/ml CHX for different times (c) or 25 mM NH4Cl (d) or 25 μM MG132 (e) for 6 h. CHX cycloheximide. f Co-IP of ubiquitinated β-catenin (HA) in HEK293T cells overexpressing ISG12a. Cell lysates were immunoprecipitated with an anti-Flag antibody and examined for HA levels by immunoblotting. g Immunoblots showing levels of Flag, β-catenin, GSK3β, Axin, and Cyclin D1 in Huh7 cells after treatment with 25 μM MG132 for 6 h. ISG12a-silenced cancer cells were further transfected with p3×Flag-ISG12a or an empty vector for 48 h. Experiments were independently repeated two (c, d) or three (a, e–g) times, with similar results. Two-sided Student’s t tests were performed to analyze the results, and data are presented as means ± SD of three biological replicates. ***p < 0.001
In fact, the level of ubiquitinated Axin in HEK293T and Huh7 cells was reduced following overexpression of ISG12a (Fig. 5a and Supplementary Fig. S6b), thus confirming our hypothesis. As the scaffold of the β-catenin destruction complex, Axin is degraded by E3 ubiquitin ligases. As shown in our previous study, the E3 ubiquitin ligase SKP2 interacts with ISG12a, thereby inducing proteasomal degradation of the viral protein NS5A.10 Interestingly, the level of ubiquitinated Axin was enhanced in cells overexpressing SKP2 (Fig. 5b) but decreased in cells with SKP2 knockdown (Supplementary Fig. S6c), and the ubiquitination of Axin induced by SKP2 was attenuated by ISG12a (Fig. 5c). Therefore, ISG12a obstructs the ability of SKP2 to induce the proteasomal degradation of Axin. Furthermore, co-IP experiments revealed interaction between SKP2 and Axin (Fig. 5d). Thus, SKP2 is an E3 ubiquitin ligase for Axin. When ISG12a expression was maintained at a high level, the ISG12a and SKP2 interaction was enhanced but that between Axin and SKP2 was reduced (Fig. 5e). Based on these findings, ISG12a determines the intensity of the interaction with SKP2 and the stability of the Axin scaffold. In addition, the ISG12a protein level was not affected by SKP2 knockdown in Huh7 cells (Supplementary Fig. S6d), and that of the SKP2 protein was not altered by ISG12a knockdown (Supplementary Fig. S6e). Overexpression of SKP2 in ISG12a-silenced Huh7 cells has limited effects on β-catenin and Axin protein levels (Supplementary Fig. S6f), indicating the dominant role of ISG12a in regulating the canonical Wnt/β-catenin pathway.

ISG12a and SKP2 regulate the proteasomal degradation of Axin. a–c Co-IP of ubiquitinated exogenous Axin (HA) in HEK293T cells overexpressing ISG12a (a), SKP2 (b) or ISG12a and SKP2 (c). The expression level of HA relative to GAPDH among multiple groups was compared by grayscale analysis (c). Cell lysates were immunoprecipitated with an anti-Flag antibody and examined for HA levels using immunoblotting. d Co-IP showing SKP2 and Axin interaction in HEK293T cells. Cell lysates were immunoprecipitated with an anti-Flag antibody and examined for SKP2 levels using immunoblotting. e Co-IP showing competitive interactions among ISG12a, SKP2 and Axin in HEK293T cells. Cell lysates were immunoprecipitated with an anti-SKP2 antibody and examined for Flag levels using immunoblotting. ND: not detected. f Bioinformatics analysis predicting the key amino acids in domains responsible for protein interactions among SKP2, ISG12a and Axin. Green represents Axin, brown represents SKP2, and blue represents ISG12a. Experiments were independently repeated three (a–e) times, with similar results. One-way ANOVA with the Tukey-Kramer or the Games–Howell post hoc test was performed to analyze the results, and data are presented as means ± SD of three independent experiments. **p < 0.01 and ***p < 0.001
As shown in our previous study, the CTD domain (amino acids 402–424) of SKP2 and the DII-C domain of ISG12a are essential for the SKP2–ISG12a interaction.10 Co-IP experiments confirmed the interaction between SKP2 leucine repeat region (LRR) (amino acids 220–401) and Axin (Supplementary Fig. S7a, b). SKP2 LRR induced substantial proteasomal degradation of Axin (Supplementary Fig. S7c). Moreover, the Axin N-terminal domain (NTD) (amino acids 1–87) was responsible for its interaction with SKP2 (Supplementary Fig. S7d–f). In conclusion, the SKP2 LRR interacts with the Axin NTD, causing the proteasomal degradation of Axin. Structural analyses further verified that helix–helix interactions might be responsible for SKP2–Axin and SKP2–ISG12a interactions (Fig. 5f). The helices located in the LRR of SKP2 might interact with the helices in the NTD of Axin, and the helices in the SKP2 CTD might interact with the helix in the ISG12a DII-C. Based on these data, ISG12a promotes the proteasomal degradation of β-catenin by inhibiting the proteasomal degradation of Axin.
ISG12a suppresses the immune checkpoint PD-L1 in cancer cells
Considering the role of ISG12a in promoting the innate immune response to viral infection, we speculated that ISG12a might enhance cancer immunity. The results of RNA sequencing identified upregulation of PD-L1 mRNA after ISG12a knockdown (log2(shISG12a/shCtrl) = 4.5070, Q-value = 0.0017). Levels of PD-L1 (also known as CD274 and B7-H1) and TGFβ mRNA, as examined by qRT-PCR, were also increased in Huh7 and HGC-27 cells with ISG12a knockdown (Supplementary Fig. S8a, b), indicating the immunosuppressed state of cancer cells with low levels of ISG12a. Therefore, ISG12a may exert anticancer effects by inhibiting PD-L1 expression in cancer cells. Moreover, the increased level of glycosylated PD-L1 protein observed by immunoblotting might be enriched on the surface of ISG12a-silenced cancer cells (Fig. 6a), which was also observed under the fluorescence microscope (Fig. 6b and Supplementary Fig. S8c), and flow cytometry data confirmed the increased level of PD-L1 on the surface of ISG12a-silenced Huh7 and HGC-27 cells (Fig. 6c, d). However, the level of surface PD-L1 was reduced on Huh7 cells overexpressing ISG12a (Supplementary Fig. S8d). Nevertheless, IFNα and IFNγ were still able to induce expression of PD-L1 in Huh7 and HGC-27 cells with ISG12a knockdown (Fig. 6e, f and Supplementary Fig. S8e, f). Specifically, the expression tendency of PD-L1 with ISG12a was not counteracted by IFNα and IFNγ. These data suggest the strong and specific regulation of ISG12a on surface PD-L1 of cancer cells. Surface PD-L1 contributes to the immune evasion of cancer cells.28 ISG12a sustains cancer immunogenicity by repressing immune checkpoints represented by PD-L1, which may be helpful for cancer immunosurveillance.

ISG12a suppresses PD-L1 expression by controlling β-catenin. a Immunoblots showing PD-L1 and ISG12a levels in Huh7 and HGC-27 cells with ISG12a knockdown. b Immunofluorescence staining for PD-L1 in Huh7 cells with ISG12a knockdown. IgG was used as an isotype control. Nuclei were stained with DAPI. Scale bar, 10 μm. c-f Flow cytometry analysis of the level of PD-L1 on the surface of Huh7 (c, e) or HGC-27 (d, f) cells with ISG12a knockdown. Huh7 cells were stimulated with 100 U/ml IFNα for 24 h (e), and HGC-27 cells were stimulated with 20 ng/ml IFNγ for 24 h (f). Experiments were independently repeated two (d, f) or three (a–d) times, with similar results. Two-sided Student’s t tests were performed to analyze the results, and data are presented as means ± SD of three independent experiments. **p < 0.01
ISG12a inhibits the transcriptional activity of PD-L1 by controlling the transcription factor β-catenin
As canonical Wnt/β-catenin signaling controls the transcription of downstream oncogenes, we postulated that it might participate in regulating PD-L1 expression. Interestingly, we observed specific binding of β-catenin to the PD-L1 promoter in ISG12a-silenced Huh7 and HGC-27 cells (Fig. 7a, b). Indeed, ISG12a silencing increased the strength of the interaction between β-catenin and the PD-L1 promoter in cancer cells (Fig. 7c). According to the primers used in ChIP assays, we speculate that β-catenin binds to the PD-L1 promoter in the regions −837 bp to −636 bp and −493 bp to –393 bp. Therefore, β-catenin may be a transcription factor responsible for regulating expression of PD-L1. Moreover, the transcriptional activity of the PD-L1 promoter was increased following the introduction of CTNNB1 into HEK293T cells (Fig. 7d), though this increase in transcriptional activity induced by CTNNB1 was decreased by ISG12a in Huh7 cells (Fig. 7e), suggesting that β-catenin is a transcription factor inducing expression of PD-L1. Thus, the inhibitory effect of ISG12a on the Wnt/β-catenin signaling pathway suppresses expression of the immune checkpoint PD-L1. These findings provide a reasonable explanation for the increased PD-L1 in cells overexpressing CTNNB1 (Fig. 7f), and the increased PD-L1 level induced by ISG12a knockdown was reduced with β-catenin suppression (Fig. 7g and Supplementary Fig. S8g).

β-Catenin is a transcription factor targeting the PD-L1 gene. a, b qRT-PCR and agarose gel electrophoresis showing interaction of the PD-L1 promoter fragment with β-catenin in ISG12a-silenced Huh7 and HGC-27 cells, as determined using the ChIP assay. The immunoprecipitated DNA is presented as the signal relative to the total amount of input chromatin (a). Histone H3 ChIP for RPL30 served as the positive control, and IgG ChIP served as the negative control. The data are presented on the log2 scale. c Fold enrichment of the interaction fragment of the PD-L1 promoter with β-catenin in Huh7 and HGC-27 cells with ISG12a knockdown, as determined by qRT-PCR using the ChIP assay. d, e Transcriptional activity of the PD-L1 promoter (pGL3-CD274) was modulated by CTNNB1 in HEK293T cells (d) or ISG12a and CTNNB1 in Huh7 (e) cells, as examined using dual-luciferase reporter assays. f Immunoblots for PD-L1 and Flag in Huh7 cells, and the expression level of PD-L1 relative to GAPDH between the two groups was compared by grayscale analysis. Cells were transfected with p3×Flag-CTNNB1 or vector for 48 h. g Immunoblots showing levels of PD-L1 and β-catenin in HGC-27 cells. Cells stably transfected with plasmids for silencing ISG12a were further transfected with plasmids for 48 h to silence CTNNB1. Experiments were independently repeated two (a–e, g) or three (f) times, with similar results. A two-sided Student’s t test (f) or one-way ANOVA with the Tukey–Kramer or Games-Howell post hoc test (a, c–e) was performed to analyze the results, and data are presented as means ± SD of three biological replicates. *p < 0.05, **p < 0.01, and ***p < 0.001
Tumor-intrinsic ISG12a contributes to anticancer immunity
To determine the regulatory role of ISG12a in cancer immunity, we analyzed the correlation between ISG12a expression and immune infiltration in HCC and GC tissues by IHC staining for CD3 and CD8. Spearman’s correlation (ρ) showed no significant expression profile between ISG12a and CD3 or ISG12a and CD8 in HCC and GC, except for that between CD3 and CD8 (Supplementary Fig. S9). Thus, expression of ISG12a may have no correlation with T cell infiltration in tumor tissues.
NK cells are innate lymphoid cells expressing PD-1 that contribute to cancer immunotherapy.29,30 The cytolytic activities of NK cells promote CD8+ T cell cross-priming, and NK cell-derived cytokines and chemokines attract and activate DCs, macrophages, and neutrophils, improving the prognosis of patients with cancer.31,32 Specifically, IFN secreted by intratumor NK cells alters the tumor architecture and controls metastasis.33 The results of RNA sequencing showed that expression of NKG2D or DNAM-1 ligands was almost unchanged with ISG12a knockdown (Supplementary Fig. S10a); expression of DNAM-1 ligand CD155 (data not shown) and classical HLA-I molecules (Supplementary Fig. S10b) was also unchanged. Interestingly, the PD-1/PD-L1 axis is important for inhibiting NK cell responses in vivo.29 Considering the increased level of PD-L1 with ISG12a knockdown, we speculated that the PD-1/PD-L1 interaction might be an important mechanism for NK cell-mediated killing of cancer cells. To determine the relationship between ISG12a and NK cell-mediated immunity, isolated NK cells from the peripheral blood of healthy individuals were stimulated with IL-2, IL-18, or both to activate NK cells. The results of flow cytometry showed that although the level of PD-1 in untreated NK cells was low, expression of surface PD-1 was significantly increased and abundant on NK cells treated with IL-2, IL-18, or both (Fig. 8a and Supplementary Fig. S11a), with the combination resulting in the highest expression level. Thus, activated NK cells express high levels of PD-1. PD-1 expression is more abundant on NK cells with an activated and more responsive phenotype and does not mark NK cells with an exhausted phenotype.29 We next conducted NK-mediated killing of HGC-27 cells by setting the ratio of NK cells (effector cells) to cancer cells (target cells) (E:T) to 5:1. NK cells with high levels of PD-1 exhibited the best killing effect, and NK cells preferred to kill cancer cells with high levels of ISG12a (Supplementary Fig. S11b). Therefore, the ISG12a-mediated inhibition of PD-L1 expression might disrupt the PD-1/PD-L1 axis, thus promoting anticancer immunity via NK cells.

ISG12a promotes NK cell-mediated killing of cancer cells. a Flow cytometry analysis of the level of PD-1 on the surface of NK cells stimulated with IL-2, IL-18 or combined treatment for 24 or 48 h. b–d Flow cytometry analysis of apoptotic Huh7 (b, d) and HGC-27 (c) cells with ISG12a knockdown resulting from NK cell-mediated killing. After incubation with 5 μg/ml isotype control (normal goat IgG) or an anti-PD-L1 antibody (αPD-L1) for 24 h, Huh7 cells were cocultured with NK cells for 4 hours (d). The NK cells used in these experiments were isolated from the peripheral blood of different healthy individuals and stimulated with IL-2 and IL-18 for 24 h. e Flow cytometry analysis of apoptotic HGC-27 cells with ISG12a knockdown resulting from NK cell-mediated killing. NK cells stimulated with IL-2 and IL-18 were incubated with 10 μg/ml isotype control (normal goat IgG) or an anti-PD-1 antibody (αPD-1) for 24 h and then cocultured with HGC-27 cells for 4 hours. Experiments were independently repeated two (e) or three (a–d) times, with similar results. One-way ANOVA with the Tukey–Kramer or the Games-Howell post hoc test was used to analyze the results, and data are presented as means ± SD of three independent experiments. **p < 0.01, and ***p < 0.001
To determine a reasonable experimental model, we set a series of E:T ratios (5:1, 10:1, and 20:1) and tested the killing effect of NK cells toward Huh7 cells. The results showed that although high E:T ratios (10:1 and 20:1) induced better killing effects, the killing speed was too fast, and the killing efficacy was too high to conduct follow-up analysis. In addition, the lower E:T ratio of 2.5:1 did not well reflect the difference due to the change in ISG12a expression (data not shown). Therefore, we chose the E:T ratio of 5:1 for the follow-up study. NK cells exhibited significantly strong efficacy in killing Huh7 and HGC-27 cells expressing high levels of ISG12a (Fig. 8b, c). Therefore, ISG12a promotes anticancer immunity through NK cell-mediated killing, and NK cells possess great value in cancer immunotherapy. Notably, administration of an anti-PD-L1 antibody (αPD-L1) further enhanced the NK cell-mediated killing of Huh7 cells expressing ISG12a at low levels (Fig. 8d), whereas use of the antibody alone did not have an apparent cytotoxic effect (Supplementary Fig. S11d). Thus, PD-L1 blockade sensitizes cancer cells to NK cell-mediated death. The effect of PD-L1 blockade also reflects the specific regulatory effect of ISG12a on cancer immunity. In addition, the results of flow cytometry revealed that the anti-PD-1 antibody (αPD-1) promoted NK-mediated killing toward HGC-27 cells but could not reverse the influence of ISG12a knockdown in cancer cells (Fig. 8e). In conclusion, ISG12a promotes cancer immunity represented by NK cell killing by suppressing PD-L1 expression.
The protein level of PD-L1 on the surface of hepatic cells isolated from clinical tissue specimens increased with the occurrence and progression of HCC (Supplementary Fig. S11e). As ISG12a is expressed at low levels in tumors (Fig. 1), these findings indicate the strong possibility that cancer cells evade immunosurveillance in vivo. In general, a decreased level of ISG12a in cancer cells may be a clinical indicator of immune evasion.
Clinical importance of ISG12a in cancer
To further evaluate the clinical importance of ISG12a in cancer research, we examined the level of Axin protein in HCC and GC. Axin was generally expressed at lower levels in most representative CAs than in paired para-CAs (Fig. 1c). Compared with para-CAs, the proportion of CAs with low levels of Axin protein reached 71.76% (n = 61) in HCC (Fig. 9a and Supplementary Fig. S1) and 58.97% (n = 69) in GC (Fig. 9b and Supplementary Figs. S2 and S3), indicating the negative regulation of Axin expression in cancer. Pearson correlation (r) analyses revealed a similar expression profile between ISG12a and Axin protein in cancer tissue specimens (Fig. 9c, d), especially for HCC (r = 0.5185, p < 0.0001, n = 85), suggesting that ISG12a specifically regulates the canonical Wnt/β-catenin signaling pathway in vivo. These findings underscore the important role of ISG12a in inhibiting cancer growth. Notably, the level of ISG12a protein in CAs was significantly associated with the overall survival of patients with cancer. In fact, the 5-year overall survival rate of patients with HCC expressing ISG12a at a high level was ~60% in CAs, but the value decreased to ~30% in the low level group (p = 0.0387, Fig. 9e). Patients with GC presenting higher levels of the ISG12a protein in CAs exhibited a more than 20% increase in 5-year overall survival rates (p = 0.0328, Fig. 9f). A similar phenomenon was observed in patients with HCC who were analyzed based on the level of ISG12a mRNA (p = 0.0206, Fig. 9g). Therefore, a high level of ISG12a may improve the prognosis of patients with cancer. Moreover, the ISG12a protein level in CAs correlated negatively with TNM stage in HCC (I + II & III + IV, p = 0.0431, n = 69, Supplementary Table S5) and GC (I + II + III and IV, p = 0.0234, n = 113, Supplementary Table S6). Thus, a decrease in ISG12a expression may promote the malignant transformation of cancer cells in vivo, resulting in metastasis and a poor prognosis. These findings also verify our observations from IHC and animal experiments. ISG12a may be a valuable indicator of cancer metastasis and prognosis.

The tumor-intrinsic ISG12a level is associated with the prognosis of patients with HCC or GC. a, b Scatterplots showing the relative level of the Axin protein between CA and para-CA from patients with HCC or GC. Protein expression in CAs and para-CAs examined by immunoblots is presented in Supplementary Figs. S1–S3. c, d Pearson’s correlation coefficients for the relative levels of ISG12a and Axin proteins in CAs and para-CAs from patients with HCC or GC. Protein expression in CAs and para-CAs examined by immunoblots is presented in Supplementary Figs. S1–S3. e–g Kaplan–Meier analysis of the overall survival of patients with HCC and GC presenting high and low expression of the ISG12a protein (e, f) or mRNA (g) in CAs, as analyzed using the log-rank test. Protein expression in CAs examined by immunoblots is presented in Supplementary Figs. S1–S3. Two-sided Student’s t tests were used to analyze the results, and data are presented as means ± SD (a, b)
Discussion
IFNs are promising drugs for cancer therapy because they sensitize tumor cells to radiotherapy and chemotherapy.34,35 Genotoxic anticancer agents and some DNA methyltransferase inhibitors induce IFN and ISG expression to inhibit cancer growth.36,37 Here, we present evidence that ISG12a, an innate immune effector, exerts an anticancer effect in gastrointestinal cancer represented by HCC and GC by inhibiting canonical Wnt/β-catenin signaling and thereby promoting cancer immunity. Decreased levels of ISG12a induced EMT in tumor cells in the current study (Fig. 3b, c and Supplementary Fig. S5d). Unfortunately, EMT generally contributes to cancer invasion and metastasis, the induction of the cancer stem cell (CSC) phenotype, and resistance to traditional anticancer drugs and immunotherapies.38,39,40 Some of these malignant transformations were observed in cells with ISG12a knockdown (Fig. 2 and Supplementary Fig. S4). Interestingly, a study reported that EMT induces an antitumor immune response mediated by the NKG2D receptor in colorectal cancer,41 which seems to be the opposite of our findings. Nonetheless, the results of RNA sequencing showed that ISG12a had almost no influence on the level of expression of NKG2D or DNAM-1 ligands (Supplementary Fig. S10a), that of DNAM-1 ligand CD155 (data not shown), or that of classical HLA molecules in Huh7 cells with ISG12a knockdown (Supplementary Fig. S10b). These differences may lead to the observed inconsistency between our research and the previous report. In addition, another study found that EMT leads to NK cell-mediated metastasis-specific immunosurveillance in lung cancer.42 However, according to Human Protein Atlas (https://www.proteinatlas.org/), high expression of ISG12a may predict poor prognosis in lung cancer, which may be consistent with the previous report. Our current study focused on the regulatory effect of ISG12a on gastrointestinal cancer represented by HCC and GC, and it is possible that ISG12a has different roles in other cancers. Our previous study identified the role of ISG12a in sensitizing cancer cells to TRAIL treatment,12 and the present study elaborated a novel mechanism of the anticancer effects of ISG12a. CSCs and the dedifferentiated state cause metabolic changes to impair prodrug activation and drug uptake,43 promoting the acquisition of radioresistance and chemoresistance.44 The Wnt/β-catenin signaling pathway contributes to the maintenance of the CSC population and EMT in tumors.45,46 ISG12a suppresses the Wnt/β-catenin signaling pathway by maintaining the stability of the Axin protein, indicating the inhibitory effect of ISG12a on CSCs and EMT in tumors. Moreover, increased levels of ZEB1 (Fig. 3b) may reinforce the metastasis, stemness, colonization capacity, and phenotypic/metabolic plasticity of cancer cells.47,48 The significant correlation between cancer metastasis or prognosis and ISG12a in animal experiments and clinical tissue specimens underscores the clinical significance of the findings of this study.
The Axin scaffold determines the efficacy of the destruction complex in degrading β-catenin. Preclinical agents, including XAV-939, JW 55, G007-LK, and IWR-1, protect Axin from proteasomal degradation, thereby inhibiting the Wnt/β-catenin signaling pathway.45 By maintaining the stability of the Axin protein, ISG12a restricted Wnt/β-catenin signaling in the current study (Figs. 4 and 5 and Supplementary Figs. S6 and S7). Considering that almost no intracellular Axin protein was found in HCLZ01 cells by immunoblotting (Fig. 3d), we speculate that other mechanisms by which ISG12 regulates the Wnt/β-catenin pathway exist. However, expression of the Axin protein in most HCC cells and the significant Pearson correlation in clinical HCC specimens with ISG12a expression strongly support that ISG12a inhibits this pathway by maintaining the stability of the Axin protein. Indeed, the activity of the Wnt/β-catenin signaling pathway is generally increased in tumors, modulating expression of crucial oncogenes, transcription factors, and valuable neoantigens.49 Interestingly, neoantigens are promising targets for improving anticancer immunity.50,51,52 Therefore, the inhibitory effect of ISG12a on the Wnt/β-catenin pathway suggests a pivotal role for ISG12a in cancer immunotherapy. Tumor-intrinsic active Wnt/β-catenin signaling results in T cell exclusion and resistance to anti-PD-L1/anti-CTLA-4 antibody therapy.53 In our study, β-catenin was identified as a transcription factor regulating expression of PD-L1 (Fig. 7). The inhibitory effect of ISG12a on the Wnt/β-catenin signaling pathway suppresses expression of the immune checkpoint PD-L1 and increases the sensitivity of cancer cells to NK cell-mediated death, promoting cancer immunity, especially in gastrointestinal cancer represented by HCC and GC.
The discovery that PD-L1 suppresses cancer immunity prompted the FDA approval of PD-1/PD-L1 inhibitors,54,55 though the heterogeneity of immune responses and cancer has limited the efficacy of inhibitors in most patients.56,57,58 Activation of IFN signaling enhances anticancer immunity.59 Innate immunity, as represented by cGAS and STING, elicits the production of IFNs, and thus, it is an important pathway for tumor-associated antigen presentation.60 c-GAS and STING are expected to increase the efficacy of immunotherapy and reverse resistance to immune checkpoint inhibitors.61,62,63,64 Some ISGs, as represented by ISG12a, are typical products and effectors of innate immunity with great potential for improving cancer immunotherapy. In our study, NK cells preferentially killed HCC and GC cells expressing high levels of ISG12a, an effect that was intensified in combination with an anti-PD-L1 antibody (Fig. 8b–d). Combining the promoting effect of PD-1/PD-L1 blockade on NK cell killing (Fig. 8d, e), we conclude that ISG12a promotes cancer immunity by regulating the PD-1/PD-L1 axis. Innate immunity, as represented by IFNs and ISGs, exhibits great potential for cancer immunotherapy.
In summary, ISG12a is a tumor suppressor that is strongly associated with the prognosis of patients with cancer. ISG12a promotes the degradation of β-catenin by inhibiting the proteasomal degradation of Axin, inhibiting the Wnt/β-catenin signaling pathway and thereby suppressing the immune checkpoint PD-L1 to promote cancer immunity (Fig. 10). ISG12a is a key molecule linking innate immunity, the Wnt/β-catenin signaling pathway and cancer immunity. NK cells plus an anti-PD-1/PD-L1 antibody and IFN therapies may be promising solutions for cancer therapy. Due to the low level of ISG12a detected in tumors and its response to IFNs, ISG12a may be a promising target for cancer diagnosis and immunotherapy.

Relationships between ISG12a and innate immunity, the Wnt/β-catenin signaling pathway and cancer immunity in cancer. ISG12a promotes the proteasomal degradation of β-catenin by inhibiting the proteasomal degradation of Axin in cancer cells. By suppressing the canonical Wnt/β-catenin signaling pathway, ISG12a decreases expression of the immune checkpoint PD-L1 to promote the anticancer immunity represented by NK cell-mediated killing. Innate immune effector ISG12a promotes cancer immunity through suppressing canonical Wnt/β-catenin signaling pathway
References
Demaria, O. et al. Harnessing innate immunity in cancer therapy. Nature 574, 45–56 (2019).
Liu, Z., Han, C. & Fu, Y. X. Targeting innate sensing in the tumor microenvironment to improve immunotherapy. Cell. Mol. Immunol. 17, 13–26 (2020).
Corrales, L., Matson, V., Flood, B., Spranger, S. & Gajewski, T. F. Innate immune signaling and regulation in cancer immunotherapy. Cell Res. 27, 96–108 (2017).
Wu, S. et al. HER2 recruits AKT1 to disrupt STING signalling and suppress antiviral defence and antitumour immunity. Nat. Cell Biol. 21, 1027–1040 (2019).
Borden, E. C. Interferons alpha and beta in cancer: therapeutic opportunities from new insights. Nat. Rev. Drug Discov. 18, 219–234 (2019).
Ren, P. et al. Interferon-based treatment is superior to nucleos(t)ide analog in reducing HBV-related hepatocellular carcinoma for chronic hepatitis B patients at high risk. Exp. Opin. Biol. Ther. 18, 1085–1094 (2018).
Wang, W. et al. Effector T cells abrogate stroma-mediated chemoresistance in ovarian cancer. Cell 165, 1092–1105 (2016).
Zou, W., Wolchok, J. D. & Chen, L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: mechanisms, response biomarkers, and combinations. Sci. Transl. Med. 8, 328rv324 (2016).
Sceneay, J. et al. Interferon signaling is diminished with age and is associated with immune checkpoint blockade efficacy in triple-negative. Breast Cancer Cancer Discov. 9, 1208–1227 (2019).
Xue, B. et al. ISG12a restricts hepatitis C virus infection through the ubiquitination-dependent degradation pathway. J. Virol. 90, 6832–6845 (2016).
Yang, D. et al. MiR-942 mediates hepatitis C virus-induced apoptosis via regulation of ISG12a. PLoS ONE 9, e94501 (2014).
Liu, N. et al. miR-942 decreases TRAIL-induced apoptosis through ISG12a downregulation and is regulated by AKT. Oncotarget 5, 4959–4971 (2014).
Aoki, M., Hecht, A., Kruse, U., Kemler, R. & Vogt, P. K. Nuclear endpoint of Wnt signaling: neoplastic transformation induced by transactivating lymphoid-enhancing factor 1. Proc. Natl Acad. Sci. USA 96, 139–144 (1999).
Clevers, H. & Nusse, R. Wnt/beta-catenin signaling and disease. Cell 149, 1192–1205 (2012).
Galluzzi, L., Spranger, S., Fuchs, E. & Lopez-Soto, A. WNT signaling in cancer immunosurveillance. Trends Cell Biol. 29, 44–65 (2019).
Yang, D. et al. Complete replication of hepatitis B virus and hepatitis C virus in a newly developed hepatoma cell line. Proc. Natl Acad. Sci. USA 111, E1264–E1273 (2014).
Zhu, H. et al. Hepatitis C virus triggers apoptosis of a newly developed hepatoma cell line through antiviral defense system. Gastroenterology 133, 1649–1659 (2007).
Lu, Y. et al. Twa1/Gid8 is a beta-catenin nuclear retention factor in Wnt signaling and colorectal tumorigenesis. Cell Res. 27, 1422–1440 (2017).
Gao, Q. et al. Intratumoral balance of regulatory and cytotoxic T cells is associated with prognosis of hepatocellular carcinoma after resection. J. Clin. Oncol. 25, 2586–2593 (2007).
Qin, Y. et al. NLRX1 Mediates MAVS degradation to attenuate the hepatitis C virus-induced innate immune response through PCBP2. J. Virol. 91, e01264-17 (2017).
Li, H. et al. Regulatory NK cells mediated between immunosuppressive monocytes and dysfunctional T cells in chronic HBV infection. Gut 67, 2035–2044 (2018).
Yang, Y. et al. Hepatic IFIT3 predicts interferon-alpha therapeutic response in patients of hepatocellular carcinoma. Hepatology 66, 152–166 (2017).
Walter, K. R. et al. Interferon-stimulated genes are transcriptionally repressed by PR in breast cancer. Mol. Cancer Res. 15, 1331–1340 (2017).
Au-Yeung, N. & Horvath, C. M. Transcriptional and chromatin regulation in interferon and innate antiviral gene expression. Cytokine Growth Factor Rev. 44, 11–17 (2018).
Han, M. L. et al. Cathepsin L upregulation-induced EMT phenotype is associated with the acquisition of cisplatin or paclitaxel resistance in A549 cells. Acta Pharmacol. Sin. 37, 1606–1622 (2016).
Nusse, R. & Clevers, H. Wnt/beta-catenin signaling, disease, and emerging therapeutic modalities. Cell 169, 985–999 (2017).
Kroger, C. et al. Acquisition of a hybrid E/M state is essential for tumorigenicity of basal breast cancer cells. Proc. Natl Acad. Sci. USA 116, 7353–7362 (2019).
Juneja, V. R. et al. PD-L1 on tumor cells is sufficient for immune evasion in immunogenic tumors and inhibits CD8 T cell cytotoxicity. J. Exp. Med. 214, 895–904 (2017).
Hsu, J. et al. Contribution of NK cells to immunotherapy mediated by PD-1/PD-L1 blockade. J. Clin. Investig. 128, 4654–4668 (2018).
Zhang, Q. et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat. Immunol. 19, 723–732 (2018).
Habif, G., Crinier, A., Andre, P., Vivier, E. & Narni-Mancinelli, E. Targeting natural killer cells in solid tumors. Cell. Mol. Immunol. 16, 415–422 (2019).
Tan, S. et al. Tim-3 hampers tumor surveillance of liver-resident and conventional NK cells by disrupting PI3K signaling. Cancer Res. 80, 1130–1142 (2020).
Glasner, A. et al. NKp46 receptor-mediated interferon-gamma production by natural killer cells increases fibronectin 1 to alter tumor architecture and control metastasis. Immunity 48, 107–119. e104 (2018).
Sistigu, A. et al. Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat. Med. 20, 1301–1309 (2014).
Parker, B. S., Rautela, J. & Hertzog, P. J. Antitumour actions of interferons: implications for cancer therapy. Nat. Rev. Cancer 16, 131–144 (2016).
Deng, L. et al. STING-dependent cytosolic DNA sensing promotes radiation-induced type i interferon-dependent antitumor. Immun. Immunogenic Tumors Immun. 41, 843–852 (2014).
Minn, A. J. & Wherry, E. J. Combination cancer therapies with immune checkpoint blockade: convergence on interferon signaling. Cell 165, 272–275 (2016).
Chen, H. A. et al. Angiopoietin-like protein 1 antagonizes MET receptor activity to repress sorafenib resistance and cancer stemness in hepatocellular carcinoma. Hepatology 64, 1637–1651 (2016).
Shibue, T. & Weinberg, R. A. EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 14, 611–629 (2017).
Qin, X. et al. The tumor biochemical and biophysical microenvironments synergistically contribute to cancer cell malignancy. Cell. Mol. Immunol. https://doi.org/10.1038/s41423-019-0282-5 (2019).
Lopez-Soto, A. et al. Epithelial-mesenchymal transition induces an antitumor immune response mediated by NKG2D receptor. J. Immunol. 190, 4408–4419 (2013).
Chockley, P. J. et al. Epithelial-mesenchymal transition leads to NK cell-mediated metastasis-specific immunosurveillance in lung cancer. J. Clin. Invest. 128, 1384–1396 (2018).
Del Vecchio, C. A. et al. De-differentiation confers multidrug resistance via noncanonical PERK-Nrf2 signaling. PLoS Biol. 12, e1001945 (2014).
Gupta, P. B., Pastushenko, I., Skibinski, A., Blanpain, C. & Kuperwasser, C. Phenotypic plasticity: driver of cancer initiation, progression, and therapy resistance. Cell Stem Cell 24, 65–78 (2019).
Takebe, N. et al. Targeting notch, hedgehog, and Wnt pathways in cancer stem cells: clinical update. Nat. Rev. Clin. Oncol. 12, 445–464 (2015).
Agliano, A., Calvo, A. & Box, C. The challenge of targeting cancer stem cells to halt metastasis. Semin. Cancer Biol. 44, 25–42 (2017).
Krebs, A. M. et al. The EMT-activator Zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nat. Cell Biol. 19, 518–529 (2017).
Caramel, J., Ligier, M. & Puisieux, A. Pleiotropic roles for ZEB1 in cancer. Cancer Res. 78, 30–35 (2018).
Aizarani, N. et al. A human liver cell atlas reveals heterogeneity and epithelial progenitors. Nature 572, 199–204 (2019).
Yarchoan, M., Johnson, B. A. 3rd, Lutz, E. R., Laheru, D. A. & Jaffee, E. M. Targeting neoantigens to augment antitumour immunity. Nat. Rev. Cancer 17, 209–222 (2017).
Wang, R. F. & Wang, H. Y. Immune targets and neoantigens for cancer immunotherapy and precision medicine. Cell Res. 27, 11–37 (2017).
Burrack, A. L. et al. Combination PD-1 and PD-L1 blockade promotes durable neoantigen-specific T cell-mediated immunity in pancreatic ductal adenocarcinoma. Cell Rep. 28, 2140–2155. e2146 (2019).
Spranger, S., Bao, R. & Gajewski, T. F. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature 523, 231–235 (2015).
Ansell, S. M. et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N. Engl. J. Med. 372, 311–319 (2015).
Topalian, S. L., Drake, C. G. & Pardoll, D. M. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell 27, 450–461 (2015).
Mandal, R. et al. Genetic diversity of tumors with mismatch repair deficiency influences anti-PD-1 immunotherapy response. Science 364, 485–491 (2019).
Thommen, D. S. & Schumacher, T. N. T cell dysfunction in cancer. Cancer Cell 33, 547–562 (2018).
Wang, Z. et al. Cancer-derived sialylated IgG promotes tumor immune escape by binding to Siglecs on effector T cells. Cell. Mol. Immunol. https://doi.org/10.1038/s41423-019-0327-9 (2019).
Sheng, W. et al. LSD1 ablation stimulates anti-tumor immunity and enables checkpoint blockade. Cell 174, 549–563. e519 (2018).
Woo, S. R. et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 41, 830–842 (2014).
Fu, J. et al. STING agonist formulated cancer vaccines can cure established tumors resistant to PD-1 blockade. Sci. Transl. Med. 7, 283ra252 (2015).
Wang, H. et al. cGAS is essential for the antitumor effect of immune checkpoint blockade. Proc. Natl Acad. Sci. USA 114, 1637–1642 (2017).
Zhu, Y. et al. STING: a master regulator in the cancer-immunity cycle. Mol. Cancer 18, 152 (2019).
Schadt, L. et al. Cancer-cell-intrinsic cGAS expression mediates tumor immunogenicity. Cell Rep. 29, 1236–1248. e1237 (2019).
Acknowledgements
We thank Dr. Zhengfan Jiang (Peking University) for providing the pHA-Ub plasmid, Dr. Tianhua Zhou (Zhejiang University) for providing the pEF-Flag-Axin plasmid, Dr. Chen Liu (Yale University) for providing the LH86 and Huh7 cells, and Dr. Weiping Zou (University of Michigan) for discussion consultation. This study was supported by grants from the National Natural Science Foundation of China (81730064 and 81571985 to H.Z.), National Science and Technology Major Project (2017ZX10202201 and 2009ZX10004-312 to H.Z.), National Natural Science Foundation of China (81601383 to S.T., 31571368 and 31871324 to G.L., 81902069 to B.X.), China Postdoctoral Science Foundation (2019M652760 to B.X.), and Hunan Natural Science Foundation (2018JJ3090 to H.Z., 2018JJ3091 to S.T., 2018JJ3713 and 2018RS3006 to G.L.).
Author information
Authors and Affiliations
Contributions
R.D. and H.Z. conceived the project and designed the experiments. R.D., Z.X., Y.G., Y.X., and S.C. performed the cell experiments. R.D., B.X., Z.X., Y.G., Y.X., R.T., S.C., Q.L., Jingjing Wang, X.H., H.L., M.G., Xintao Wang, J.M., and S.T. conducted the plasmid construction and molecular experiments. C.Z., Jinfeng Wang, and J.H. provided the human tissue samples and clinical information. R.D., B.X., and Xiaohong Wang performed the immunofluorescence assays. R.D., Xiaohong Wang, Jinfeng Wang, J.H., and H.Z. performed the IHC and H&E staining. R.D., Z.X., Y.G., Xiaohong Wang, Y.X., R.T., J.C., and S.T. performed the animal experiments. G.L. performed the bioinformatics analyses. R.D., Y.L., M.Y., Z.W., and Z.T. performed the NK cell isolation and flow cytometry. R.D., R.T., S.C., and Q.L. performed the dual-luciferase reporter and ChIP assays. R.D., S.T., and H.Z. conducted the RNA sequencing analysis. R.D., Y.L., B.X., S.T., Z.T., H.J., and H.Z. analyzed and interpreted the data. H.Z. supervised the overall execution of the experiments. The manuscript was written by R.D. with support from G.L., Z.T., H.J., and H.Z. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary information
Rights and permissions
About this article

Cite this article
Deng, R., Zuo, C., Li, Y. et al. The innate immune effector ISG12a promotes cancer immunity by suppressing the canonical Wnt/β-catenin signaling pathway. Cell Mol Immunol 17, 1163–1179 (2020). https://doi.org/10.1038/s41423-020-00549-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41423-020-00549-9
Keywords
This article is cited by
-
Carnosic acid nanocluster-based framework combined with PD-1 inhibitors impeded tumorigenesis and enhanced immunotherapy in hepatocellular carcinoma
Functional & Integrative Genomics (2024)
-
Extensive review on breast cancer its etiology, progression, prognostic markers, and treatment
Medical Oncology (2023)
-
Exosomal PIK3CB promotes PD-L1 expression and malignant transformation in esophageal squamous cell carcinoma
Medical Oncology (2023)
-
RasGRP1 promotes the acute inflammatory response and restricts inflammation-associated cancer cell growth
Nature Communications (2022)
-
Prognostic signature composed of transcription factors accurately predicts the prognosis of gastric cancer patients
Cancer Cell International (2021)
